

Abstract
Simultaneous and accurate measurement of circulating vitamin D metabolites is critical to studies of the metabolic regulation of vitamin D and its impact on health and disease. To that end, we developed a specific LC-MS/MS method that permits the quantification of major circulating vitamin D3 metabolites in human plasma. Plasma samples were subjected to a protein precipitation, liquid-liquid extraction and Diels-Alder derivatization procedure prior to LC-MS/MS analysis. Importantly, in all human plasma samples tested, we identified a significant dihydroxyvitamin D3 peak that could potentially interfere with the determination of 1α,25-dihydroxyvitamin D3 [1α,25(OH)2D3] concentrations. This interfering metabolite has been identified as 4β,25-dihydroxyvitamin D3 [4β,25(OH)2D3] and was found at concentrations comparable to 1α,25(OH)2D3. Quantification of 1α,25(OH)2D3 in plasma required complete chromatographic separation of 1α,25(OH)2D3 from 4β,25(OH)2D3. An assay incorporating this feature was used to simultaneously determine the plasma concentrations of 25OHD3, 24R,25(OH)2D3, 1α,25(OH)2D3, and 4β,25(OH)2D3 in healthy individuals. The LC-MS/MS method developed and described here, could result in considerable improvement in the quantification of 1α,25(OH)2D3, as well as monitoring the newly identified circulating metabolite, 4β,25(OH)2D3.
Keywords: Vitamin D3; 25-hydroxyvitamin D3; 24R,25-dihydroxyvitamin D3; 1α,25-dihydroxyvitamin D3; 4β,25-dihydroxyvitamin D3; Plasma; LC-MS/MS
Vitamin D is critical for the regulation of calcium and phosphate homeostasis and has been implicated in a number of other important biological processes including immune function [1-3]. Insufficiency or deficiency of vitamin D is a risk factor for metabolic bone diseases such as rickets, osteoporosis and osteomalacia [2, 4]. Vitamin D exists naturally in two forms, namely vitamin D2 (ergocalciferol) and vitamin D3 (cholecalciferol). The major source of vitamin D in humans is the photoconversion of 7-dehydrocholesterol to pre-vitamin D3 in the epidermis, which isomerizes to vitamin D3 [5, 6]. Vitamin D3 undergoes 25-hydroxylation by 25-hydroxylase enzymes in the liver to produce the most abundant circulating form, 25-hydroxyvitamin D3 (25OHD3) [7]. 25OHD3 is typically used as a biomarker for vitamin D status, and a plasma level less than 20 ng/mL has been reported to be indicative of vitamin D deficiency [5]. Hydroxylation at the C-24 position is generally considered to be the main deactivating pathway for vitamin D [8]. One of the products, 24R,25-dihydroxyvitamin D3 [24R,25(OH)2D3], is the major circulating dihydroxyvitamin D3 metabolite (Figure 1). Alternatively, 25OHD3 can be converted into the most biologically active form, 1α,25-dihydroxyvitamin D3 [1α,25(OH)2D3], a reaction occurring predominantly in the kidney. The circulating plasma level of 1α,25(OH)2D3 is approximately 1/500 to 1/1000 lower than that of 25OHD3 [9, 10]. It has been proposed that the plasma concentration of 1α,25(OH)2D3 relative to that of 25OHD3 or 24R,25(OH)2D3 may give a better indication of vitamin D3 status [11]. However, due to its very low circulating concentrations (picomolar), methodological difficulties with the accurate measurement of 1α,25(OH)2D3 are considerable [12]. Moreover, the inter-laboratory variability in 1α,25(OH)2D3 measurement makes direct comparisons difficult. Thus, improvements in the accuracy and reliability of vitamin D metabolite measurement, especially 1α,25(OH)2D3 in plasma/serum, would be very valuable to translational research and clinical practice.
Figure 1.

Structures of 1α,25(OH)2D3, 24R,25(OH)2D3, 25OHD3 and 4β,25(OH)2D3.
Quantification of vitamin D metabolites in plasma/serum is a challenging task because the molecules are very lipophilic and bind tightly to vitamin D binding protein; moreover, some metabolites are found at extremely low levels [13]. A number of methodologies have been successfully developed for the quantification of vitamin D metabolites in plasma/serum [14]. These typically involve measurements by immunoassays [15-17], protein binding assays [18-21], HPLC-UV [22, 23] and LC-MS [24, 25]. Hospitals and clinical laboratories have relied on immunoassays because of their rapidity to execute. However, those methods require extensive sample preparation to remove cross-reactive interferences [15, 21, 22, 26]. HPLC-UV and LC-MS are suitable for 25OHD3 measurement; however, due to detection limits they can not quantitate metabolites found at very low concentrations. Accordingly, high performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS) methods have been developed to measure nonderivatized 25OHD3 [27-31] and 1α,25(OH)2D3 [32, 33], which provide much higher sensitivity and specificity [34]. Additionally, it was found that derivatization by Cookson-type triazolinediones and related reagents can enhance ionization efficiencies of vitamin D metabolites and thereby improve sensitivity [35-37]. Therefore, ultra-performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) with derivatization prior to MS analysis has been touted as more favorable analytical methodology [38-40]. These techniques allow for simultaneous quantification of concentrations for multiple endogenous vitamin D metabolites, including 1α,25(OH)2D3.
In the present study, we have also developed a sensitive LC-MS/MS method, but in the process identified a significant peak which interferes with quantifying 1α,25(OH)2D3 in human plasma extracts. Results being published elsewhere [41] show that the interfering compound is also a dihydroxyvitamin D3 metabolite with two vicinal hydroxyl groups on the A-ring, but hydroxylated at the 4β position, rather than the 1α position (Figure 1). Initially, the presence of this metabolite proved problematic since it co-eluted with 1α,25(OH)2D3 during chromatography. Upon further effort, we successfully developed a sensitive and specific LC-MS/MS method to simultaneously quantify 25OHD3, 24R,25(OH)2D3, 1α,25(OH)2D3 and this interfering metabolite [4β,25(OH)2D3]. The assay described here could result in considerable improvement in the quantification of 1α,25(OH)2D3.
Materials and Methods
Chemicals
Ethylenediaminetetraacetic acid (EDTA), human serum albumin (HSA), 25OHD3, 24R,25(OH)2D3, 4-phenyl-1,2,4-triazoline-3,5-dione (PTAD) and sodium periodate were purchased from Sigma-Aldrich. 1α,25(OH)2D3 was purchased from Calbiochem (San Diego, CA). Deuterated standards of vitamin D3 metabolites, d6-25OHD3 and d6-1α,25(OH)2D3 (containing six deuterium atoms at C-26 and C-27), were purchased from Medical Isotope Inc. (Pelham, NH). HPLC-grade solvents acetonitrile, methanol, ethyl acetate, formic acid were obtained from Fisher Chemicals. Deionized water was generated in-house for mobile phase preparation.
Standards and plasma samples
All standards were prepared as 50 μg/mL primary stock solutions in methanol and stored in amber vials. Quality control (QC) samples were made by serial dilutions from the stock solutions. Six different levels of calibration standards were prepared by serial 2X dilutions starting from the calibration standard mixture at the highest concentration [1α,25(OH)2D3, 24R,25(OH)2D3, 25OHD3; 0.8, 16, 80 ng/mL respectively]. A HAS solution (30 mg/mL) in phosphate-buffered saline was used as the blank matrix for generating calibration curves. A working internal standard solution was prepared containing 0.1 μg/mL d6-1α,25(OH)2D3 and 0.5 μg/mL d6-25OHD3. All standards were stored at -80 °C.
Outdated human plasma from the local Blood Bank was used for assay development and validation. We estimated the variability in plasma vitamin D metabolite concentrations in a group of 25 healthy, ethnically diverse adults. The University of Washington Institutional Review Board approved this study and all subjects gave written informed consent. We enrolled 12 females and 13 males between 20 and 40 years of age and their self-reported ethnicities were Blacks (n = 4), Asian (n = 5) and Caucasian (n = 16). Blood was collected in vacutainer tubes containing EDTA from subjects following an overnight fast and all plasma samples were stored at -80° C until analysis.
Sample preparation from plasma
Because vitamin D metabolites are light sensitive, the extraction procedure was conducted under low light to avoid unpredictable degradation. Different methods and conditions for sample preparation were evaluated and are further described in the “Results and Discussion” section. The optimized procedure is described below and was performed for all subsequent analyses.
After thawing at room temperature, human plasma (1 mL) was transferred to a 15 mL polypropylene tube (Sarstedt, Newton, NC), spiked with 10 μL of working internal standard solution, vortex-mixed, and allowed to equilibrate for 30 min at room temperature in the dark. Proteins were precipitated by adding 2 mL of acetonitrile, vortex-mixed, followed by centrifugation for 10 min at 2360 g (Jouan CR422, Rockville, MD). The supernatant was transferred to a glass tube, and the volume was reduced under a nitrogen stream. The remaining solution (~ 1 mL) was subjected to liquid-liquid extraction (LLE) by adding 5 mL ethyl acetate. After shaking vigorously for 10 min, samples were centrifuged for 20 min at 590 g (Jouan CR422), and the upper organic layer was transferred into a new glass tube. After complete evaporation of solvent under a nitrogen stream, the derivatization reagent, PTAD (150 μL, 1 mg/mL) in acetonitrile, was added to the residue, vortex-mixed, and left at room temperature for 1 hour to complete the reaction. The sample was then transferred to another glass tube, evaporated under a nitrogen stream and reconstituted in 40% acetonitrile (40 μL). The sample was transferred to an amber glass vial with a glass insert for analysis.
Chromatography and mass spectrometry
Chromatographic separation was performed using a Hypersil Gold (2.1 × 100 mm, 1.9 μm) column (Thermo Scientific) at 40 °C on an Agilent 1200 LC system using acetonitrile (B)/water (A, 0.1% formic acid) as a mobile phase. Starting gradient conditions were 40% B with 0.2 mL/min flow rate. The following gradient program was used: 0 to 3 min 40% B, 9 to 10.5 min 60% B, 11 to 14 min 90% B, 18 to 25 min 40% B. Under these chromatographic conditions, the interfering peak 4β,25(OH)2D3 was separated from 1α,25(OH)2D3. Samples were kept at 4 °C, and the injection volume was 5 μL. Mass spectrum (MS) analysis was carried out using a positive mode electrospray ionization method on an Agilent 6410 triple quadrupole tandem mass spectrometer. The ionization and fragmentation parameters were set as follows: capillary voltage, 5000 V; gas temperature, 300 °C; gas flow rate, 11 L/min; nebulizer: 35 psi; fragmentor: 140 V; collision energy: 14 V. Multiple Reaction Monitoring (MRM) channels of m/z 574 → 298, 574 → 314, 558 → 298, 580 → 314 and 564 → 298 were set to detect 24R,25(OH)2D3, 1α,25(OH)2D3, 25OHD3, d6-1α,25(OH)2D3 and d6-25OHD3, respectively. The interfering compound, 4β,25(OH)2D3, appeared in the same MRM channel as 1α,25(OH)2D3 but with a different retention time (RT).
Quantification and data analysis
Data acquisition was performed using the Agilent MassHunter Workstation software. Calibration curves were constructed by plotting the peak area ratio for each vitamin D3 metabolite and its internal standard, versus the corresponding concentration, and fitting a linear regression equation to the data. Calibration statistics are shown in Table 2. Deuterated 1α,25(OH)2D3 was used as an internal standard for the quantification of 1α,25(OH)2D3, 4β,25(OH)2D3 and 24R,25(OH)2D3. 4β,25(OH)2D3 was quantified using 1α,25(OH)2D3 for the standard curve because 4β,25(OH)2D3 is not commercially available. Data analysis was performed using MassHunter Qualitative Analysis software (Agilent) and Microsoft Excel 2003.
Table 2.
Sensitivity, quantification range and linearity.
Instrumental LOD and LOQ were estimated from a series of dilutions of standards in blank matrix. The linear range was determined by spiking six concentrations of standards into blank matrix. Experimental details are described in Materials and Methods.
| 1α,25(OH)2D3 | 24R,25(OH)2D3 | 25OHD3 | |
|---|---|---|---|
| LOD (pg) | 1.0 | 2.5 | 4.0 |
| LOQ (pg/mL) | 25 | 50 | 50 |
| R2 | 0.999 | 0.999 | 0.996 |
| Linear Range (pg/mL) | 25 – 800 | 500 – 16000 | 2500 – 80000 |
Stability and recovery
Previous studies have shown that vitamin D3 metabolites are stable unless exposed to intense light [40]. Consistent with those reports, we found metabolites were stable (residues ≥ 95%) at 4 °C (1 week) or -20 °C (2 months) and for 5 freeze-thaw cycles. The derivatized products of these metabolites were also stable in a cooled autosampler (4 °C) for 3 days. To determine recovery rates, we prepared samples in blank matrix (30 mg/mL HSA solution) at two concentrations [1α,25(OH)2D3: 50 and 200 pg/mL; 24R,25(OH)2D3: 1.0 and 4.0 ng/mL; 25OHD3: 2.5 and 10 ng/mL] and extracted according to the LLE procedure. In control experiments, solid phase extraction (SPE) was performed using Oasis HLB 1 cc (30 mg) cartridges. Cartridges were preconditioned by sequential washing with acetonitrile and water. After loading the samples, we washed the columns with 30% acetonitrile and eluted with 100% acetonitrile. The recovery of analytes was determined by comparison of LC-MS/MS signals against those generated for the corresponding standard solutions measured without the extraction process.
Linearity and detection limits
Linearity of each calibration curve was determined by spiking nine concentrations of standards in blank matrix [1α,25(OH)2D3: 0.0125 to 2 ng/mL; 24R,25(OH)2D3: 0.5 to 50 ng/mL; 25OHD3: 1.25 to 200 ng/mL]. Calibration curves were plotted as the peak area ratio of each vitamin D3 metabolite and its surrogate internal standard versus the corresponding concentration using linear regression. The limit of detection (LOD) was defined as a signal-to-noise ratio (S/N ratio) of 3:1, and limit of quantification (LOQ) as a S/N ratio of 10:1. Instrumental LOD were estimated as the lowest calculated injected amount on the column that produced a S/N ratio ≥ 3 after injecting a series of dilutions (5 to 160 pg/mL in 1 mL blank matrix). LOQ was determined by three replicate injections from serial dilutions of QC samples, and estimated as the lowest concentration which produced a mean S/N ratio ≥ 10.
Precision and accuracy
The intra-assay and inter-assay precision and accuracy were evaluated using QC samples at three nominal concentrations [1α,25(OH)2D3: 50, 100 and 200 pg/mL; 24R,25(OH)2D3: 0.5, 1.0 and 2.0 ng/mL; 25OHD3: 10, 20 and 40 ng/mL]. The QC samples were spiked into blank matrix for analysis and the endogenous concentrations of vitamin D3 metabolites in native plasma were measured in parallel. Intra-assay precision and accuracy were determined by measuring metabolite concentrations in the QCs from six replicates on the same day. Inter-assay precision and accuracy were evaluated based on the replicate measurements of the QCs made six times over a ten day period.
Addition of standards in plasma
Human plasma (1 mL) was spiked with 10 μL of a standard mixture of 1α,25(OH)2D3, 24R,25(OH)2D3 and 25OHD3 in methanol (initial concentrations given in parentheses): set 1 [2.5, 50 and 125 ng/mL, respectively], set 2 [5.0, 100 and 250 ng/mL, respectively] and set 3 [10, 200 and 500 ng/mL, respectively. After equilibrating for 30 min in the dark, samples were extracted and measured as described above.
Periodate cleavage of plasma extracts
Since the interfering peak contained a characteristic m/z 314 fragment, we used periodate cleavage to identify the regiochemistry of the two hydroxyl groups on the A-ring. Briefly, vitamin D3 metabolites were extracted from human plasma as described above. After PTAD derivatization, samples were evaporated and reconstituted in 300 μL methanol. Standard solutions of 1α,25(OH)2D3 (1 ng), 24R,25(OH)2D3 (1 ng) were used as controls in a parallel experiment. An aqueous solution of 5% sodium periodate (NaIO4, 30 μL) was added to the plasma extracts and water was added as a negative control in parallel plasma extracts. After incubation for 1 hour, 0.5 mL water was added to mixtures and the derivatized metabolites were extracted using 2 mL ethyl acetate. The extracts were then transferred to a glass tube, evaporated and reconstituted in 40% acetonitrile (40 μL) for LC-MS/MS analysis.
Results and Discussion
Optimization of extraction
Aronov et al. proposed a general strategy for the quantification of vitamin D metabolites from plasma/serum [38]. Following their approach, we subjected samples to a three-step protein precipitation, extraction (LLE/SPE) and Diels-Alder derivatization procedure, prior to LC-MS/MS analysis. We validated and modified each step when necessary, yielding an optimized procedure that is described in “Materials and Method”. Due to its similarity to previously published procedures [38, 39], we briefly highlight our optimized extraction conditions as a complement to the published work. 1) Protein precipitation. Because vitamin D3 metabolites are highly lipophilic, protein precipitation prior to extraction was necessary. We tested organic solvents such as acetonitrile, acetone and acetonitrile/methanol mixtures at various volume ratios. The addition of two volumes of acetonitrile was found to be efficient for protein precipitation. In addition, partial evaporation of acetonitrile prior to LLE increased the extraction efficiency. 2) Liquid-liquid extraction. Since both LLE and SPE have been used previously for the extraction step [38-40], we compared the efficiency of each by spiking known amounts of standards into blank matrices to evaluate the recovery rates. LLE resulted in the recovery of 84 ± 2% of 1α,25(OH)2D3, 85 ± 4% of 24R,25(OH)2D3 and 73 ± 2% of 25OHD3 which was comparable to previously reported results. However, we observed lower recoveries (50% to 70%) following SPE, especially for the dihydroxyvitamin D3 metabolites. In addition, SPE had slightly greater variability in a sample-to-sample comparison. Thus, we opted to use LLE rather than SPE for the isolation of metabolites, despite the longer processing time.
Optimization of LC-MS/MS conditions
The ionization and fragmentation parameters were optimized by the MassHunter software optimizer (Agilent), using injections of derivatized standards. Consistent with previous findings [38], the parent ions at m/z 592 [1α,25(OH)2D3, 24R,25(OH)2D3] and m/z 576 (25OHD3) lost one molecule of water at the C-25 position, generating the major ions at m/z 574 and 558, respectively. The collision-induced dissociation (CID) spectra of derivatized 1α,25(OH)2D3, 24R,25(OH)2D3 and 25OHD3 were acquired from the precursor ion [M-H2O+H]+. As shown in Figure 2, the predominant product ions (m/z 314 and 298) represent moieties that include the PTAD-attached A-ring. Since this product ion was detected at high sensitivity and selectivity, we used selected reaction monitoring (SRM) for each metabolite, as listed in Table 1. Moreover, the presence of the characteristic m/z 314 fragment was used as an indicator of hydroxylation on the A-ring.
Figure 2.
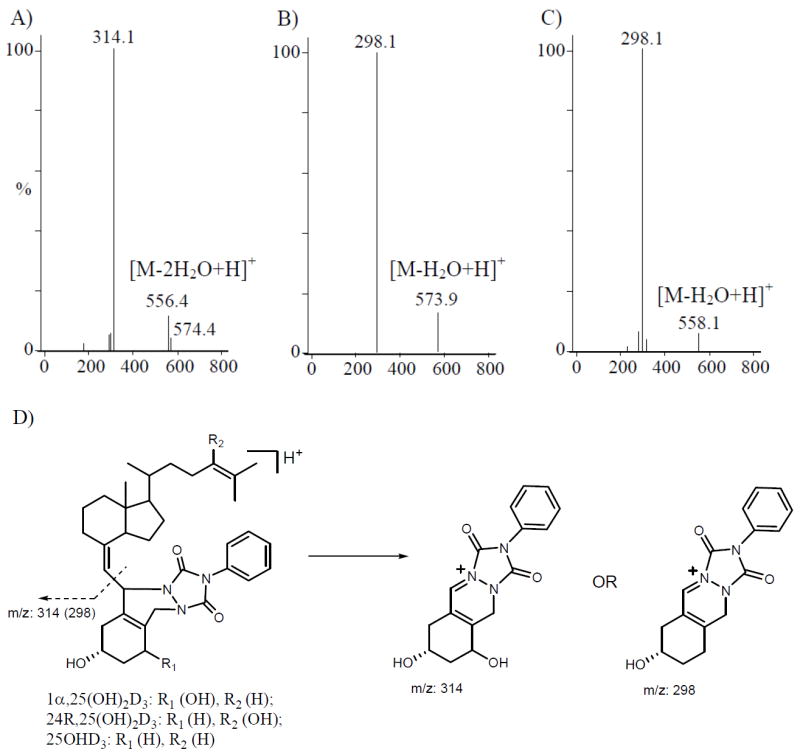
Product ion mass spectra of the derivatized vitamin D3 metabolites. A) 1α,25(OH)2D3; B) 24R,25(OH)2D3; and C) 25OHD3. The product ion spectra were acquired from the dominant [M-H2O+H]+ precursor ion. Proposed fragmentation reaction is shown in figure D. Multiple reaction monitoring (MRM) channel m/z 574 → 314 indicates two hydroxyl groups on the A-ring.
Table 1.
Selected transitions from precursor ions to product ions for quantification.
| PTAD derivatives | Precursor ion (m/z)a | Product ion (m/z) |
|---|---|---|
| 1α,25(OH)2D3 | 574.4 | 314.1 |
| 4β,25(OH)2D3 | 574.4 | 314.1 |
| 24R,25(OH)2D3 | 574.4 | 298.1 |
| 25OHD3 | 558.1 | 298.1 |
| d6-1α,25(OH)2D3 | 580.4 | 314.1 |
| d6-25OHD3 | 564.1 | 298.1 |
[M-H2O+H]+
Derivatization with PTAD produced two epimers, 6S and 6R; as a result, two peaks may be expected for each metabolite in the ion chromatograms. As shown in Figure 3, both 24R,25(OH)2D3 and 25OHD3 contained two peaks, whereas 1α,25(OH)2D3 had only one peak (presumably co-eluting epimers) during chromatography. When two peaks appeared, both major and minor peaks were integrated and combined to minimize the calculation errors, although previous results showed that integration of the major peak was sufficient for quantification [38, 39].
Figure 3.

Representative ion chromatograms of the derivatized vitamin D3 metabolites by LC-MS/MS. The MRM chromatograms were obtained from a standard solution containing d6-1α,25(OH)2D3 (1 ng/mL), 1α,25(OH)2D3 (0.4 ng/mL), 24R,25(OH)2D3 (8 ng/mL), d6-25OHD3 (1 ng/mL) and 25OHD3 (20 ng/mL). The deuterated internal standards of vitamin D3 metabolites were found to elute ~0.1 min earlier than their natural analogs.
Interestingly, a peak interfering with 1α,25(OH)2D3 quantification was present in plasma extracts prepared by both LLE or SPE (Figure 4) and could not be suppressed by adjusting instrumental parameters. Because the interfering peak had the same MRM characteristics to 1α,25(OH)2D3, we speculated that it could also be a dihydroxyvitamin D3 metabolite, with mono-hydroxylation of the A-ring of vitamin D3. The interfering peak co-eluted with 1α,25(OH)2D3 if the mobile phase gradient contained a high percentage of organic solvent, i.e., greater than 70% acetonitrile or 90% methanol. We tested different columns and mobile phase conditions to separate 1α,25(OH)2D3 from the interfering peak. Initially, methanol was used as an organic solvent to give higher signal intensity; however, we substituted it with acetonitrile due to the higher backpressure. Using optimized LC conditions, the interfering peak eluted ~0.3 min earlier than 1α,25(OH)2D3 (Figure 4), which was acceptable for quantification of 1α,25(OH)2D3.
Figure 4.

Chromatographic separation of 1α,25(OH)2D3 from an unknown dihydroxyvitamin D3 metabolite in human plasma. Varying amounts of 1α,25(OH)2D3 were separately spiked into plasma for validation. An arrow indicates the interfering peak with two hydroxyl groups on the A-ring, which could co-elute with 1α,25(OH)2D3 under certain conditions, e.g., 70% acetonitrile.
Method validation
For each metabolite, we plotted the calibration curve as the peak area ratios of each metabolite and its surrogate internal standard versus the corresponding concentrations. Since a deuterated standard was not available for 24R,25(OH)2D3, we used d6-1α,25(OH)2D3 as its internal standard. The interfering metabolite was semi-quantified using the 1α,25(OH)2D3 calibration curve. We obtained the linear range and correlation coefficients using linear regression analysis from the set of six data points in the calibration curves (Table 2). We obtained excellent linearity over the calibration range, and the R2 values for 1α,25(OH)2D3, 24R,25(OH)2D3 and 25OHD3 were 0.999, 0.999 and 0.996, respectively. As shown in Table 2, these metabolites had relatively similar instrumental LOD and LOQ, possibly because of the similarities in structures and ion fragmentation.
We estimated the accuracy and precision of the assay using QC samples at three concentration levels and a plasma sample for which the endogenous levels of the analytes were measured (Table 3). The accuracies were calculated by the equation: mean of determined concentration/nominal concentration. As shown in Table 3, good accuracies (bias < 10%) were observed for all analytes at all three concentrations. The coefficient of variance (% CV), which is used for evaluating precision, ranged from 1.1% to 12.4% (intra-assay) and 1.0% to 13.8% (inter-assay). We also determined the accuracy and precision by adding known amounts of vitamin D3 metabolites to a human plasma sample (Table 4). Because vitamin D3 metabolites are endogenous compounds, the addition of known amounts of metabolites would increase their total plasma levels. As summarized in Table 4, increases were observed in proportion to the amounts added to plasma. The precisions (% CV) of all analytes were < 10% and the accuracies were in the range of 96% to 106%. These data further confirmed that the method was reliable and reproducible for measurement of all targeted analytes in plasma.
Table 3.
Precision and accuracy of vitamin D3 metabolite measurement.
| Compound and concentrations | aIntra-assay (n = 6) | Inter-assay (n = 6) | ||
|---|---|---|---|---|
| bMeasured | c%CV | Measured | %CV | |
| 1α,25(OH)2D3 | ||||
| 50 pg/mL | 48.0 ± 3.3 (96.0%) | 7.0% | 46.4 ± 2.6 (92.8%) | 5.6% |
| 100 pg/mL | 100.3 ± 12.0 (100%) | 11.9% | 102.5 ± 6.8 (103%) | 6.6% |
| 200 pg/mL | 213.1 ± 6.3 (107%) | 2.9% | 206.7 ± 10.6 (103%) | 5.1% |
| native plasma | 85.9 ± 4.6 | 5.3% | 93.4 ± 6.7 | 7.1% |
| 24R,25(OH)2D3 | ||||
| 0.5 ng/mL | 0.50 ± 0.04 (100%) | 8.5% | 0.50 ± 0.07 (100%) | 13.8% |
| 1.0 ng/mL | 1.04 ± 0.07 (104%) | 7.0% | 1.03 ± 0.05 (103%) | 4.6% |
| 2.0 ng/mL | 2.08 ± 0.26 (104%) | 12.4% | 2.07 ± 0.12 (104%) | 5.9% |
| native plasma | 1.19 ± 0.06 | 5.3% | 1.33 ± 0.11 | 8.3% |
| 25OHD3 | ||||
| 10 ng/mL | 9.5 ± 0.2 (95.0%) | 2.1% | 10.0 ± 0.7 (100%) | 7.0% |
| 20.0 ng/mL | 18.8 ± 0.2 (94.0%) | 1.1% | 18.8 ± 0.2 (94.0%) | 1.0% |
| 40.0 ng/mL | 39.8 ± 1.1 (99.5%) | 2.8% | 40.3 ± 0.8 (101%) | 2.1% |
| native plasma | 21.6 ± 0.7 | 3.4% | 23.6 ± 1.1 | 4.7% |
Intra-assay precision was obtained from 6 replicates measured in a single assay.
Inter-assay precision was obtained from 6 assays run over a 10 day period.
Reported as mean ± standard deviation (accuracy).
CV: coefficient of variance is defined as the ratio of the standard deviation to the mean.
Table 4.
Standard addition of vitamin D3 metabolites into human plasma. We extracted spiked human plasma (1 mL) and analyzed the vitamin D3 metabolites by LC-MS/MS. Standard mixtures (10 μL) were spiked into native plasma at various concentrations and methanol (10 μL) was used as vehicle.
| Native plasma | Set 1a | Set 2 | Set 3 | |
|---|---|---|---|---|
| 1α,25(OH)2D3 (pg/mL) | 97 ± 5.7b | 125 ± 3.0 (102%) | 156 ± 6.7 (106%) | 199 ± 9.0 (101%) |
| 24R,25(OH)2D3 (ng/mL) | 1.4 ± 0.04 | 1.9 ± 0.10 (100%) | 2.5 ± 0.14 (104%) | 3.6 ± 0.14 (106%) |
| 25OHD3 (ng/mL) | 24.3 ± 0.43 | 24.4 ± 0.39 (96%) | 27.2 ± 0.33 (101%) | 30.6 ± 0.60 (104%) |
Amounts of 1α,25(OH)2D3, 24R,25(OH)2D3 and 25OHD3:
Set 1: 25 pg, 0.5 ng and 1.25 ng
Set 2: 50 pg, 1.0 ng and 2.50 ng
Set 3: 100 pg, 2.0 ng and 5.00 ng
Concentration was calculated from the standard curve as described in Materials and Methods. Data reported as mean ± standard deviation (accuracy).
Periodate cleavage of plasma extracts
Since the interfering metabolite underwent Diels-Alder derivatization with PTAD and had the same characteristic m/z 314 fragment as 1α,25(OH)2D3, we hypothesized that it was a vitamin D3 metabolite with two hydroxyl groups on the A-ring. A similar observation was reported previously [38], where the authors suggested an endogenous metabolite from epimerization of 1α,25(OH)2D3, 1α,25-dihydroxy-3-epi-vitamin D3 [1α,25(OH)2-3-epi-D3] as a possible candidate. However, 1α,25(OH)2-3-epi-D3 (a gift from Dr. T. Fujishima, Tokushima Bunri University, Japan) did not co-elute with the interfering peak with RT at 12.35 min (Figure 5A). In another experiment, we treated derivatized plasma extracts with sodium periodate, which selectively cleaves C-C bonds with vicinal hydroxyl (or vicinal hydroxyl-ketone) groups. As shown in Figure 5B, upon periodate treatment, the interfering peak disappeared completely, indicating that the putative metabolite contains two vicinal hydroxyl groups. As expected in the control experiments, the 24R,25(OH)2D3 peak was diminished ~95%, but there was no significant change for 1α,25(OH)2D3,. Thus, we speculated this putative vitamin D3 metabolite might be 2,25(OH)2D3 or 4,25(OH)2D3 [42-44]. We performed further experiments for structure elucidation, and the accumulated evidence suggest that it is 4β,25(OH)2D3, and is a metabolite of 25OHD3 produced by cytochrome P450 3A4 (CYP3A4). These findings have been submitted for publication elsewhere [41].
Figure 5.

The MRM (m/z 547 → 314) chromatograms of 1α,25(OH)2-3-epi-D3 and periodate cleavage of the unknown dihydroxyvitamin D3 in human plasma extracts. A) Chromatograms of 1α,25(OH)2D3 and 1α,25(OH)2-3-epi-D3 ; B) Chromatogram of plasma extracts after periodate cleavage. The disappearance of the unknown metabolite peak indicated vicinal hydroxyl groups on the A-ring, subsequently identified as 4β,25(OH)2D3 [41]. As expected, no significant changes was observed with the peak for 1α,25(OH)2D3.
Population levels of vitamin D3 metabolites
Using the newly developed method, we measured plasma concentrations of vitamin D3 metabolites in 25 healthy individuals uniformly recruited from the Seattle, Washington area throughout year (Figure 6). In these healthy subjects, the average (range) plasma concentrations of 25OHD3, 24R,25(OH)2D3, 1α,25(OH)2D3 and 4β,25(OH)2D3 were 25.6 ng/mL (6.9 to 52.6 ng/mL), 2.28 ng/mL (0.13 to 6.19 ng/mL), 61 pg/mL (17 to 101 pg/mL) and 40 pg/mL (2 to 128 pg/mL), respectively. These values are consistent with quantitative data reported previously [38, 40]. Interestingly, both the level of 4β,25(OH)2D3 (R2 = 0.727) and 24R,25(OH)2D3 (R2 = 0. 789) correlated well with the level of 25OHD3. In contrast, the level of 1α,25(OH)2D3 (R2 = 0.455) was only weekly correlated with the level of 25OHD3.
Figure 6.

Plasma concentrations of vitamin D3 metabolites in healthy volunteers (n = 25). Four vitamin D3 metabolites [1α,25(OH)2D3, 4β,25(OH)2D3, 24R,25(OH)2D3, and 25OHD3] were measured by this newly developed LC-MS/MS method. Each data point represents the mean of two determinations.
Conclusions
The accurate measurement of circulating vitamin D metabolites is critical to studies of the metabolic regulation of vitamin D and its impact on health and disease. In this report, we describe a sensitive and more specific method that permits the quantification of major circulating vitamin D3 metabolites in human plasma. Importantly, we found that another dihydroxylvitamin D3 metabolite, 4β,25(OH)2D3, circulates in human plasma at concentrations that could interfere with 1α,25(OH)2D3 measurements. Consistent with our findings, an immunoaffinity extraction method has been developed recently to remove interfering substances in the 1α,25(OH)2D3 quantification prior to LC-MS/MS analysis [45]. In the present study, a complete chromatographic separation of 1α,25(OH)2D3 from 4β,25(OH)2D3 was critical for the quantification of the individual metabolites. The LOQ for 1α,25(OH)2D3 was 25 pg/mL and in some “healthy” subjects, the estimated concentrations of both 1α,25(OH)2D3 and 4β,25(OH)2D3 metabolites were below 25 pg/mL. Thus, for clinical practice, sample enrichment might be required prior to LC-MS/MS analysis to capture accurate concentrations in individuals with apparent vitamin D deficiency, when reporting that the analyte was found at less than the lower detection limit is insufficient.
In general, higher chromatographic resolution and sensitivity with reduction in analysis run time can be achieved with UPLC-MS/MS allowing greater throughput of samples [46]. Recently, two groups reported UPLC-MS/MS methods for the quantification of 1α,25(OH)2D3 in plasma, both of which had a run time one-half that of our newly developed method [38, 39]. However, it appears that 1α,25(OH)2D3 was not fully separated chromatographically from 4β,25(OH)2D3 under those LC conditions, possibly due to the higher percentage of organic solvent in their mobile phases. Indeed, using lower organic composition in the mobile phase and a longer UPLC column, an interfering peak of 1α,25(OH)2D3 was observed [38]. In addition to these two UPLC-based methods, a microflow LC-MS/MS method was used for the quantification of 1α,25(OH)2D3 [39]. Again, a single peak was observed using the same MRM channel (m/z 574 → 314), indicating that 4β,25(OH)2D3 might be co-eluting with 1α,25(OH)2D3. Thus, 1α,25(OH)2D3 concentrations might be over-estimated by incorporation of 4β,25(OH)2D3 into the 1α,25(OH)2D3 quantitative signal under certain LC-MS-based conditions.
We noted marked inter-individual variability in the concentrations of all of the vitamin D3 metabolites measured, including the newly identified 4β,25(OH)2D3 molecule. As indicated by the strong correlation coefficients, much of the variability in 4β,25(OH)2D3 and 24R,25(OH)2D3 concentration could be attributed to differences in the plasma 25OHD3 precursor. However, as the metabolite/parent ratios also varied substantially (not shown), significant differences in either the formation or elimination of the 4β,25(OH)2D3 and 24R,25(OH)2D3 metabolites may be present in vivo. We also observed ethnic and gender differences in the metabolite concentrations or metabolite/parent ratios. Future work is warranted to confirm and explore the biological basis for these differences. Finally, there was more limited variability in plasma 1α,25(OH)2D3 concentrations in these healthy volunteers, perhaps indicative of tighter regulation of 1α,25(OH)2D3.
In summary, we have established a sensitive LC-MS/MS method for quantification of vitamin D3 metabolites. For 1α,25(OH)2D3 quantification, chromatographic separation of 1α,25(OH)2D3 from another dihydroxyvitamin D3 metabolite, 4β,25(OH)2D3 is necessary.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health: R01 GM063666, P01 GM032165, P30 ES07033, and Clinical and Translational Science Award UL1 RR025014 (to YSL). The authors would like to thank Ms. Christine Hoffer for her expert coordination of the healthy volunteer study and Mr. Dustin Ensign for his contributions to some of the early analytical work.
Abbreviation
- CID
collision-induced dissociation
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- HAS
human serum albumin
- LLE
liquid-liquid extraction
- LOD
limit of detection
- LOQ
limit of quantification
- MRM
multiple reaction monitoring
- 1α,25(OH)2D3
1α,25-dihydroxyvitamin D3
- 1α,25(OH)2-3-epi-D3
1α,25-dihydroxy-3-epi-vitamin D3
- 4β,25(OH)2D3
4β,25-dihydroxyvitamin D3
- 24R,25(OH)2D3
24R,25-dihydroxyvitamin D3
- 25OHD3
25-hydroxyvitamin D3
- PTAD
4-phenyl-1,2,4-triazoline-3,5-dione
- QC
quality control
- SPE
solid-phase extraction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mora JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol. 2008;8:685–698. doi: 10.1038/nri2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Plum LA, DeLuca HF. Vitamin D, disease and therapeutic opportunities. Nat Rev Drug Discov. 2010;9:941–955. doi: 10.1038/nrd3318. [DOI] [PubMed] [Google Scholar]
- 3.Zhang R, Naughton DP. Vitamin D in health and disease: current perspectives. Nutr J. 2010;9:65. doi: 10.1186/1475-2891-9-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeLuca HF. The vitamin D story: a collaborative effort of basic science and clinical medicine. The FASEB Journal. 1988;2:224–236. [PubMed] [Google Scholar]
- 5.Holick MF. Vitamin D Deficiency. N Engl J Med. 2007;357:266–281. doi: 10.1056/NEJMra070553. [DOI] [PubMed] [Google Scholar]
- 6.Holick MF. Vitamin D and Sunlight: Strategies for Cancer Prevention and Other Health Benefits. Clin J Am Soc Nephrol. 2008;3:1548–1554. doi: 10.2215/CJN.01350308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohyama Y, Yamasaki T. Eight cytochrome P450s catalyze vitamin D metabolism. Front Biosci. 2004;9:3007–3018. doi: 10.2741/1455. [DOI] [PubMed] [Google Scholar]
- 8.Prosser DE, Jones G. Enzymes involved in the activation and inactivation of vitamin D. Trends Biochem Sci. 2004;29:664–673. doi: 10.1016/j.tibs.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 9.Henry HL, Norman AW. Vitamin D: Metabolism and Biological Actions. Annu Rev Nutr. 1984;4:493–520. doi: 10.1146/annurev.nu.04.070184.002425. [DOI] [PubMed] [Google Scholar]
- 10.Jones G, Strugnell SA, DeLuca HF. Current Understanding of the Molecular Actions of Vitamin D. Physiol Rev. 1998;78:1193–1231. doi: 10.1152/physrev.1998.78.4.1193. [DOI] [PubMed] [Google Scholar]
- 11.Prentice A, Goldberg GR, Schoenmakers I. Vitamin D across the lifecycle: physiology and biomarkers. Am J Clin Nutr. 2008;88:500S–506S. doi: 10.1093/ajcn/88.2.500S. [DOI] [PubMed] [Google Scholar]
- 12.Hollis BW, Horst RL. The assessment of circulating 25(OH)D and 1,25(OH)2D: Where we are and where we are going. J Steroid Biochem Mol Biol. 2007;103:473–476. doi: 10.1016/j.jsbmb.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimada K, Mitamura K, Higashi T. Gas chromatography and high-performance liquid chromatography of natural steroids. J Chromatogr A. 2001;935:141–172. doi: 10.1016/s0021-9673(01)00943-8. [DOI] [PubMed] [Google Scholar]
- 14.Hart GR, Furniss JL, Laurie D, Durham SK. Measurement of vitamin D status: background, clinical use, and methodologies. Clin Lab. 2006;52:335–343. [PubMed] [Google Scholar]
- 15.Glendenning P, Noble JM, Taranto M, Musk AA, McGuiness M, Goldswain PR, Fraser WD, Vasikaran SD. Issues of methodology, standardization and metabolite recognition for 25-hydroxyvitamin D when comparing the DiaSorin radioimmunoassay and the Nichols Advantage automated chemiluminescence protein-binding assay in hip fracture cases. Ann Clin Biochem. 2003;40:546–551. doi: 10.1258/000456303322326470. [DOI] [PubMed] [Google Scholar]
- 16.Hollis BW, Kamerud JQ, Selvaag SR, Lorenz JD, Napoli JL. Determination of vitamin D status by radioimmunoassay with an 125I-labeled tracer. Clin Chem. 1993;39:529–533. [PubMed] [Google Scholar]
- 17.Roth HJ, Schmidt-Gayk H, Weber H, Niederau C. Accuracy and clinical implications of seven 25-hydroxyvitamin D methods compared with liquid chromatography-tandem mass spectrometry as a reference. Ann Clin Biochem. 2008;45:153–159. doi: 10.1258/acb.2007.007091. [DOI] [PubMed] [Google Scholar]
- 18.Carter GD, Jones JC, Berry JL. The anomalous behaviour of exogenous 25-hydroxyvitamin D in competitive binding assays. J Steroid Biochem Mol Biol. 2007;103:480–482. doi: 10.1016/j.jsbmb.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 19.Haddad JG, Chyu KJ. Competitive protein-binding radioassay for 25-hydroxycholecalciferol. J Clin Endocrinol Metab. 1971;33:992–995. doi: 10.1210/jcem-33-6-992. [DOI] [PubMed] [Google Scholar]
- 20.Hamilton JC. A rapid simplified method for plasma 25-hydroxyvitamin D estimation. J Clin Pathol. 1980;33:197–199. doi: 10.1136/jcp.33.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holick MF. Vitamin D status: measurement, interpretation, and clinical application. Ann Epidemiol. 2009;19:73–78. doi: 10.1016/j.annepidem.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de la Hunty A, Wallace AM, Gibson S, Viljakainen H, Lamberg-Allardt C, Ashwell M. UK Food Standards Agency Workshop Consensus Report: the choice of method for measuring 25-hydroxyvitamin D to estimate vitamin D status for the UK National Diet and Nutrition Survey. Br J Nutr. 2010;104:612–619. doi: 10.1017/S000711451000214X. [DOI] [PubMed] [Google Scholar]
- 23.Jones G. Assay of vitamins D2 and D3, and 25-hydroxyvitamins D2 and D3 in human plasma by high-performance liquid chromatography. Clin Chem. 1978;24:287–298. [PubMed] [Google Scholar]
- 24.El-Khoury JM, Reineks EZ, Wang S. Progress of liquid chromatography-mass spectrometry in measurement of vitamin D metabolites and analogues. Clin Biochem. 2010;44:66–76. doi: 10.1016/j.clinbiochem.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 25.Holler U, Quintana AP, Gossl R, Olszewski K, Riss G, Schattner A, Nunes CS. Rapid determination of 25-hydroxy vitamin D3 in swine tissue using an isotope dilution HPLC-MS assay. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:963–968. doi: 10.1016/j.jchromb.2010.02.026. [DOI] [PubMed] [Google Scholar]
- 26.Wallace AM, Gibson S, de la Hunty A, Lamberg-Allardt C, Ashwell M. Measurement of 25-hydroxyvitamin D in the clinical laboratory: current procedures, performance characteristics and limitations. Steroids. 2010;75:477–488. doi: 10.1016/j.steroids.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 27.Herrmann M, Harwood T, Gaston-Parry O, Kouzios D, Wong T, Lih A, Jimenez M, Janu M, Seibel MJ. A new quantitative LC tandem mass spectrometry assay for serum 25-hydroxy vitamin D. Steroids. 2010;75:1106–1112. doi: 10.1016/j.steroids.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 28.Knox S, Harris J, Calton L, Wallace AM. A simple automated solid-phase extraction procedure for measurement of 25-hydroxyvitamin D3 and D2 by liquid chromatography-tandem mass spectrometry. Ann Clin Biochem. 2009;46:226–230. doi: 10.1258/acb.2009.008206. [DOI] [PubMed] [Google Scholar]
- 29.Tai SS, Bedner M, Phinney KW. Development of a candidate reference measurement procedure for the determination of 25-hydroxyvitamin D3 and 25-hydroxyvitamin D2 in human serum using isotope-dilution liquid chromatography-tandem mass spectrometry. Anal Chem. 2010;82:1942–1948. doi: 10.1021/ac9026862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van den Ouweland JM, Beijers AM, Demacker PN, van Daal H. Measurement of 25-OH-vitamin D in human serum using liquid chromatography tandem-mass spectrometry with comparison to radioimmunoassay and automated immunoassay. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:1163–1168. doi: 10.1016/j.jchromb.2010.03.035. [DOI] [PubMed] [Google Scholar]
- 31.Vogeser M. Quantification of circulating 25-hydroxyvitamin D by liquid chromatography-tandem mass spectrometry. J Steroid Biochem Mol Biol. 2010;121:565–573. doi: 10.1016/j.jsbmb.2010.02.025. [DOI] [PubMed] [Google Scholar]
- 32.Casetta B, Jans I, Billen J, Vanderschueren D, Bouillon R. Development of a method for the quantification of 1alpha,25(OH)2-vitamin D3 in serum by liquid chromatography tandem mass spectrometry without derivatization. Eur J Mass Spectrom (Chichester, Eng) 2010;16:81–89. doi: 10.1255/ejms.1024. [DOI] [PubMed] [Google Scholar]
- 33.Kissmeyer AM, Sonne K. Sensitive analysis of 1alpha,25-dihydroxyvitamin D3 in biological fluids by liquid chromatography-tandem mass spectrometry. J Chromatogr A. 2001;935:93–103. doi: 10.1016/s0021-9673(01)00985-2. [DOI] [PubMed] [Google Scholar]
- 34.Higashi T, Shimada K, Toyo’oka T. Advances in determination of vitamin D related compounds in biological samples using liquid chromatography-mass spectrometry: A review. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:1654–1661. doi: 10.1016/j.jchromb.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 35.Higashi T, Awada D, Shimada K. Simultaneous determination of 25-hydroxyvitamin D2 and 25-hydroxyvitamin D3 in human plasma by liquid chromatography-tandem mass spectrometry employing derivatization with a Cookson-type reagent. Biol Pharm Bull. 2001;24:738–743. doi: 10.1248/bpb.24.738. [DOI] [PubMed] [Google Scholar]
- 36.Higashi T, Shimada K. Derivatization of neutral steroids to enhance their detection characteristics in liquid chromatography-mass spectrometry. Anal Bioanal Chem. 2004;378:875–882. doi: 10.1007/s00216-003-2252-z. [DOI] [PubMed] [Google Scholar]
- 37.Higashi T, Yamauchi A, Shimada K. Application of 4-(4-nitrophenyl)-1,2,4-triazoline-3,5-dione to analysis of 25-hydroxyvitamin D3 in human plasma by liquid chromatography/electron capture atmospheric pressure chemical ionization-mass spectrometry. Anal Sci. 2003;19:941–943. doi: 10.2116/analsci.19.941. [DOI] [PubMed] [Google Scholar]
- 38.Aronov PA, Hall LM, Dettmer K, Stephensen CB, Hammock BD. Metabolic profiling of major vitamin D metabolites using Diels-Alder derivatization and ultra-performance liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem. 2008;391:1917–1930. doi: 10.1007/s00216-008-2095-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding S, Schoenmakers I, Jones K, Koulman A, Prentice A, Volmer DA. Quantitative determination of vitamin D metabolites in plasma using UHPLC-MS/MS. Anal Bioanal Chem. 2010;398:779–789. doi: 10.1007/s00216-010-3993-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duan X, Weinstock-Guttman B, Wang H, Bang E, Li J, Ramanathan M, Qu J. Ultrasensitive quantification of serum vitamin D metabolites using selective solid-phase extraction coupled to microflow liquid chromatography and isotope-dilution mass spectrometry. Anal Chem. 2010;82:2488–2497. doi: 10.1021/ac902869y. [DOI] [PubMed] [Google Scholar]
- 41.Wang Z, Senn T, Hashizume T, Lin Y, Thummel K. Novel Pathways of CYP3A-dependent Metabolism of 25-Hydroxyvitamin D3. The FASEB Journal. 2011;25:812.815. [Google Scholar]
- 42.Araya Z, Hosseinpour F, Bodin K, Wikvall K. Metabolism of 25-hydroxyvitamin D3 by microsomal and mitochondrial vitamin D3 25-hydroxylases (CYP2D25 and CYP27A1): a novel reaction by CYP27A1. Biochim Biophys Acta. 2003;1632:40–47. doi: 10.1016/s1388-1981(03)00062-3. [DOI] [PubMed] [Google Scholar]
- 43.Takeda K, Kominato K, Sugita A, Iwasaki Y, Shimazaki M, Shimizu M. Isolation and identification of 2alpha,25-dihydroxyvitamin D3, a new metabolite from Pseudonocardia autotrophica 100U-19 cells incubated with Vitamin D3. Steroids. 2006;71:736–744. doi: 10.1016/j.steroids.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 44.Thierry-Palmer M, Gray TK, Napoli JL. Ring hydroxylation of 25-hydroxycholecalciferol by rat renal microsomes. J Steroid Biochem Mol Biol. 1988;29:623–628. doi: 10.1016/0022-4731(88)90161-6. [DOI] [PubMed] [Google Scholar]
- 45.Yuan C, Kosewick J, He X, Kozak M, Wang S. Sensitive measurement of serum 1α,25-dihydroxyvitamin D by liquid chromatography/tandem mass spectrometry after removing interference with immunoaffinity extraction. Rapid Commun Mass Spectrom. 2011;25:1241–1249. doi: 10.1002/rcm.4988. [DOI] [PubMed] [Google Scholar]
- 46.Xu RN, Fan L, Rieser MJ, El-Shourbagy TA. Recent advances in high-throughput quantitative bioanalysis by LC-MS/MS. J Pharm Biomed Anal. 2007;44:342–355. doi: 10.1016/j.jpba.2007.02.006. [DOI] [PubMed] [Google Scholar]