

Abstract
Background
Studies to dissect the role of calcineurin in pathological cardiac remodeling have relied heavily on murine models, where genetic gain- and loss-of-function manipulations are initiated at or before birth. However, the great majority of clinical cardiac pathology occurs in adults. Yet, nothing is known about the effects of calcineurin when its activation commences in adulthood. Further, despite the fact that ventricular hypertrophy is a well established risk factor for heart failure, the relative pace and progression of these two major phenotypic features of heart disease are unknown.
Methods and Results
We engineered mice harboring in cardiomyocytes a constitutively active calcineurin transgene driven by a tetracycline-responsive promoter element. Expression of the mutant calcineurin transgene was initiated for variable lengths of time to determine the natural history of disease pathogenesis, and to determine when, if ever, these events are reversible. Activation of the calcineurin transgene in adult mice triggered rapid and robust cardiac growth with features characteristic of pathological hypertrophy. Concentric hypertrophy preceded the development of systolic dysfunction, fetal gene activation, fibrosis, and clinical heart failure. Further, cardiac hypertrophy reversed spontaneously when calcineurin activity was turned off, and expression of fetal genes reverted to baseline. Fibrosis, a prominent feature of pathological cardiac remodeling, manifested partial reversibility.
Conclusions
Together, these data establish and define the deleterious effects of calcineurin signaling in adult heart and reveal that calcineurin-dependent hypertrophy with concentric geometry precedes systolic dysfunction and heart failure. Furthermore, these findings demonstrate that during much of the disease process, calcineurin-dependent remodeling remains reversible.
Keywords: heart failure, hypertrophy, remodeling
Introduction
Strong epidemiological evidence links left ventricular hypertrophy with adverse cardiovascular events, including heart failure and death1–4. Consistent with this, current therapies that improve clinical outcomes are often associated with regression of ventricular hypertrophy5–7. However, whereas significant strides have been made recently in elucidating the molecular circuitry governing pathological cardiac remodeling8, few therapies in clinical use target hypertrophic growth mechanisms directly.
Calcineurin is a cytoplasmic protein phosphatase implicated in the pathogenesis of cardiac hypertrophy and heart failure9. Calcineurin dephosphorylates transcription factors of the NFAT (nuclear factor of activated T cells) family, resulting in their translocation to the nucleus and triggering of a complex transcriptional program10. Calcineurin also targets other proteins to activate non-transcriptional signaling cascades. Consistent with widespread involvement in heart disease, studies in animal models have demonstrated a critical role for calcineurin in biomechanical stress-induced hypertrophic remodeling11, 12, and calcineurin activation has been observed in heart failure in humans13–15.
Studies designed to dissect the role of calcineurin signaling in heart disease have relied, by and large, on genetic manipulations in mice. In this context, gain- and loss-of-function manipulations are typically initiated at or before birth. However, recent studies have highlighted the fact that the effects of mechanisms triggered in adulthood oftentimes differ from that seen when the same mechanism is triggered during development, when a host of other events are taking place16–19. Further, the great majority of clinically relevant disease states involving calcineurin occur in adults. However, little is known about the effects of calcineurin activation in adult animal models.
Pathological ventricular remodeling is a dynamic process involving changes in cardiomyocyte size and shape, alterations in gene expression and substrate metabolism, changes in intracellular calcium homeostasis, deposition of excess extracellular matrix, sarcomere recruitment, and more8. In many animal models, pathological stress elicits a hypertrophic growth response that culminates ultimately in contractile dysfunction and heart failure4. In humans, however, where disease-related stress initiates long after cardiac development is complete, this pathogenic progression has been questioned20. Further, the extent to which ventricular hypertrophy is a risk factor for systolic dysfunction may depend on specific phenotypic features, such as whether its geometry is concentric or eccentric3. Given all this, it is important to determine the natural history of load-induced remodeling and to tease out molecular events governing the progression of hypertrophy and failure.
In light of recent advances in our understanding of signaling cascades governing pathological cardiac remodeling21, the hypertrophic growth response per se has emerged has a viable therapeutic target9. This strategy hinges critically, however, on the notion that pathological remodeling is reversible and not just preventable. Yet, little is known at present regarding the extent to which calcineurin-dependent remodeling is reversible. Finally, clinically relevant anti-remodeling therapies, both pharmacological and mechanical, are only modestly efficacious, and hence it is important to tease out the diseased myocyte’s capacity for spontaneous recovery.
Thus, we set out to address three questions: a) what are the effects of calcineurin activation initiated during adulthood; b) is ventricular hypertrophy a requisite precursor of calcineurin-dependent heart failure, and if so, what are its features; and c) is the calcineurin-dependent phenotype reversible in the absence of anti-remodeling therapy? To accomplish this, we engineered mice harboring in cardiomyocytes a constitutively active calcineurin transgene driven by a tetracycline-responsive promoter element. Expression of the mutant calcineurin transgene was initiated in adulthood for variable lengths of time to determine the natural history of disease pathogenesis and associated molecular and functional events, and to identify markers of disease reversibility.
Materials and Methods
A Detailed Methods section is available in the Online Data Supplement.
Animal Care
Transgenic mice that conditionally express a constitutively active calcineurin mutant22 specifically in cardiomyocytes were generated using the α-myosin heavy chain promoter and the tetracycline transactivator off promoter system23,24. Both male and female animals aged 5 to 8 weeks were studied using protocols approved by the animal care and use committee of UT Southwestern Medical Center.
Echocardiography
Echocardiograms were performed using a Sonos 5500 system with a 15 MHz probe, and M-mode and 2D parasternal short axis images were obtained. Left ventricular mass was calculated by the cubed method, as previously described25.
Immunoblot analyses
Immunoblots were performed on protein lysates prepared from left ventricular tissue. All antibodies used are presented in the Data Supplement.
Quantitative real-time RT PCR
RNA was extracted from snap frozen left ventricular tissue and used to prepare cDNA. Real-time PCR was performed using an ABI 7000 system. All primers used are presented in the Data Supplement.
Histology and cardiomyocyte cross-sectional area
Selected hearts were perfusion fixed in paraformaldehyde and subjected to cutting and staining as described previously26. Cardiomyocyte area was measured from hematoxylin and eosin-stained images and from wheat germ agglutinin-stained images. Fibrosis was measured from picrosirius red-stained images. Hydroxyproline analysis was performed based on previously described methods27.
In vivo electrophysiology studies
In vivo electrophysiology studies were performed on sedated mice using a multipolar electrode catheter placed via the right jugular vein as previously described28.
Statistical methods and data handling
All data are presented as mean ± standard deviation (SD). Comparison of data between two groups was performed using the Mann Whitney U test. The authors had full access to, and take full responsibility for, the integrity of these data. All authors have read and agree to the manuscript as written.
Results
To test the effects of calcineurin activation in the adult mammalian heart, we generated transgenic mice that conditionally express a constitutively active calcineurin mutant specifically in cardiomyocytes. We used the tetracycline-inhibited transactivator system under control of the αMHC promoter (αMHC-tTA) to control expression of a constitutively active form of the human calcineurin A subunit (CnA*) (Figure 1A). Several lines of tetO-CnA* mice were generated and then crossed with αMHC-tTA mice. A line of double transgenic mice (tTA/CnA*) was selected for analysis which manifested increased cardiac mass by 8 weeks of age, similar to that previously described for conventional αMHC-calcineurin transgenic mice10. For subsequent experiments, all mice including breeding pairs were administered doxycycline via drinking water so that expression of the CnA* transgene was suppressed in utero and in early post-natal life.
Figure 1. Conditional activation of calcineurin in cardiomyocytes.
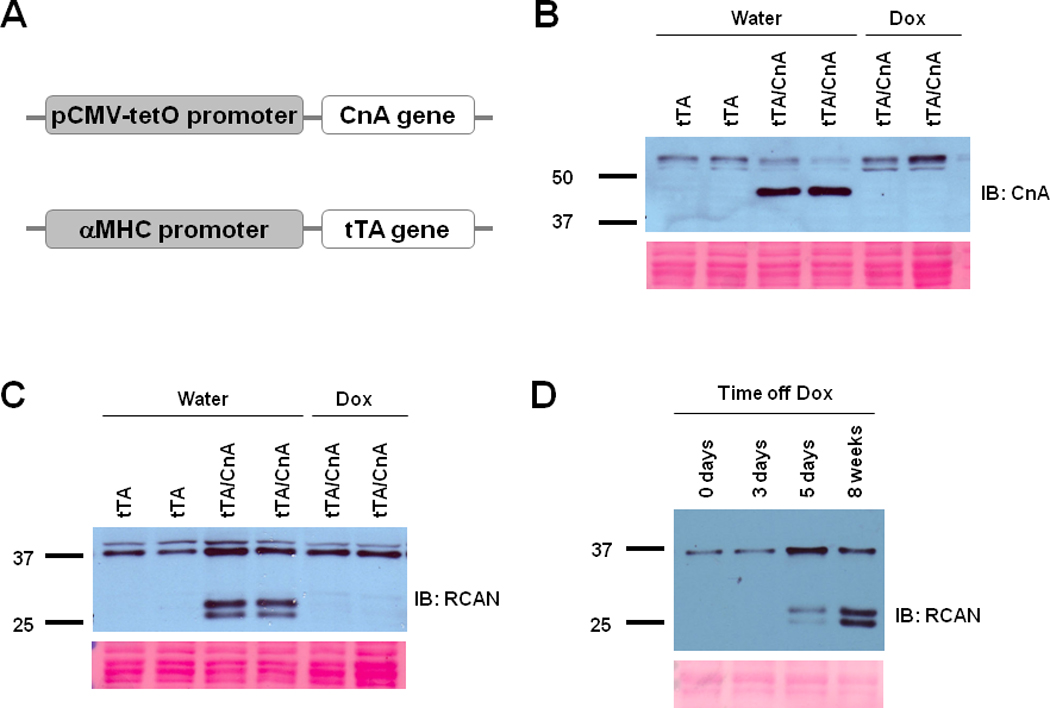
A: Schematic depiction of the transgenic constructs used to engineer double transgenic tTA/CnA* mice. B, C: Representative immunoblots of LV lysates prepared after 8 weeks of treatment with doxycycline (Dox) or water and probed for calcineurin (CnA) (B) (upper bands represent endogenous calcineurin; lower bands represent mutant calcineurin) or RCAN (C) (lower bands represent RCAN1.4 in different phosphorylation states). Ponceau stains show protein loading in each lane. D: Representative immunoblot of LV lysates probed for RCAN from hearts harvested at different times after removal of doxycycline. tTA, single transgenic for αMHC-tTA only; tTA/CnA, double transgenic for both αMHC-tTA and tetO-CnA*.
Doxycycline was withdrawn from several tTA/CnA* mice at age 8 weeks, and hearts were harvested at age 16 weeks. Western blot analysis of protein from ventricular tissue confirmed expression of a truncated calcineurin protein of the expected molecular weight in tTA/CnA* mice (Figure 1B, Supp Figure IA). This protein was not evident in hearts from single transgenic αMHC-tTA mice or from tTA/CnA* mice that continued to receive doxycycline, confirming conditional expression of CnA*.
We investigated a downstream target of calcineurin signaling in order to confirm increased calcineurin activity in tTA/CnA* hearts. The expression of regulator of calcineurin (RCAN1.4) is directly controlled by calcineurin via the NFAT pathway29, 30. We observed a robust increase in RCAN1.4 in hearts of tTA/CnA* mice compared to that of single transgenic αMHC-tTA mice (Figure 1C, Supp Figure IA), occurring within 5 days of doxycycline withdrawal (Figure 1D, Supp Figure IB). This augmentation in RCAN1.4 protein levels was not evident when doxycycline was continued in tTA/CnA* mice (Figure 1C). Thus, doxycycline was effective in suppressing the expression and activity of CnA* in double transgenic tTA/CnA* mice.
Calcineurin activation in the adult heart triggers pathological hypertrophy
To test the effects of prolonged calcineurin activation in adult heart, doxycycline was withdrawn from tTA/CnA* mice at 5 weeks of age, and hearts were harvested at 21 weeks of age (Figure 2A). For comparison, a second group of tTA/CnA* mice was maintained on doxycycline to suppress CnA* throughout the study. First, echocardiograms were performed biweekly to assess serial changes in cardiac size and function. Calcineurin signaling in CnA* ON mice triggered a significant decline in fractional shortening consistent with impaired ventricular systolic function (Figures 2B). Calcineurin activation elicited a 34% increase in LV internal diameter at end-diastole (LVIDd OFF 3.23±0.40 mm, n=10; LVIDd ON 4.34±1.07 mm, n=14, p<0.01), and an even greater increase in LV internal diameter at end-systole (LVIDs OFF 1.04±0.36 mm, LVIDs ON 2.93±1.30, p<0.01, Figures 2B, 2C). The greatest decline in contractile function occurred between 2 to 6 weeks after removing doxycycline. No significant decline in fractional shortening occurred in tTA/CnA* mice that were maintained on doxycycline in the CnA* OFF group.
Figure 2. Hypertrophy and heart failure develop when doxycycline is withheld from tTA/CnA* mice.
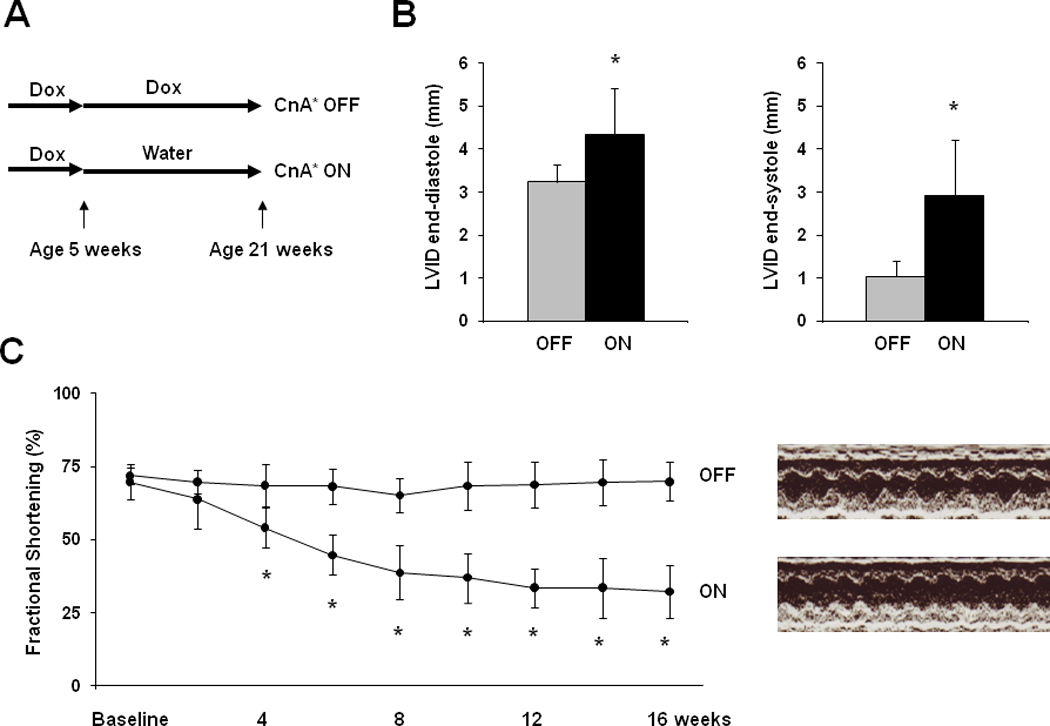
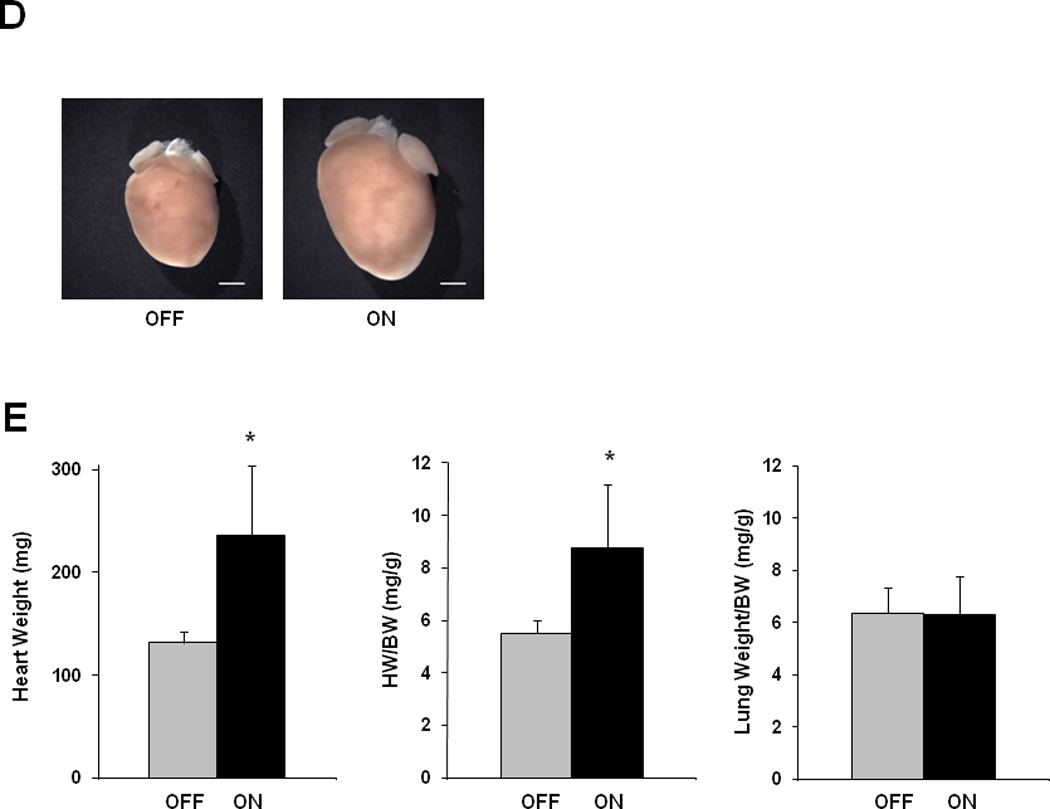
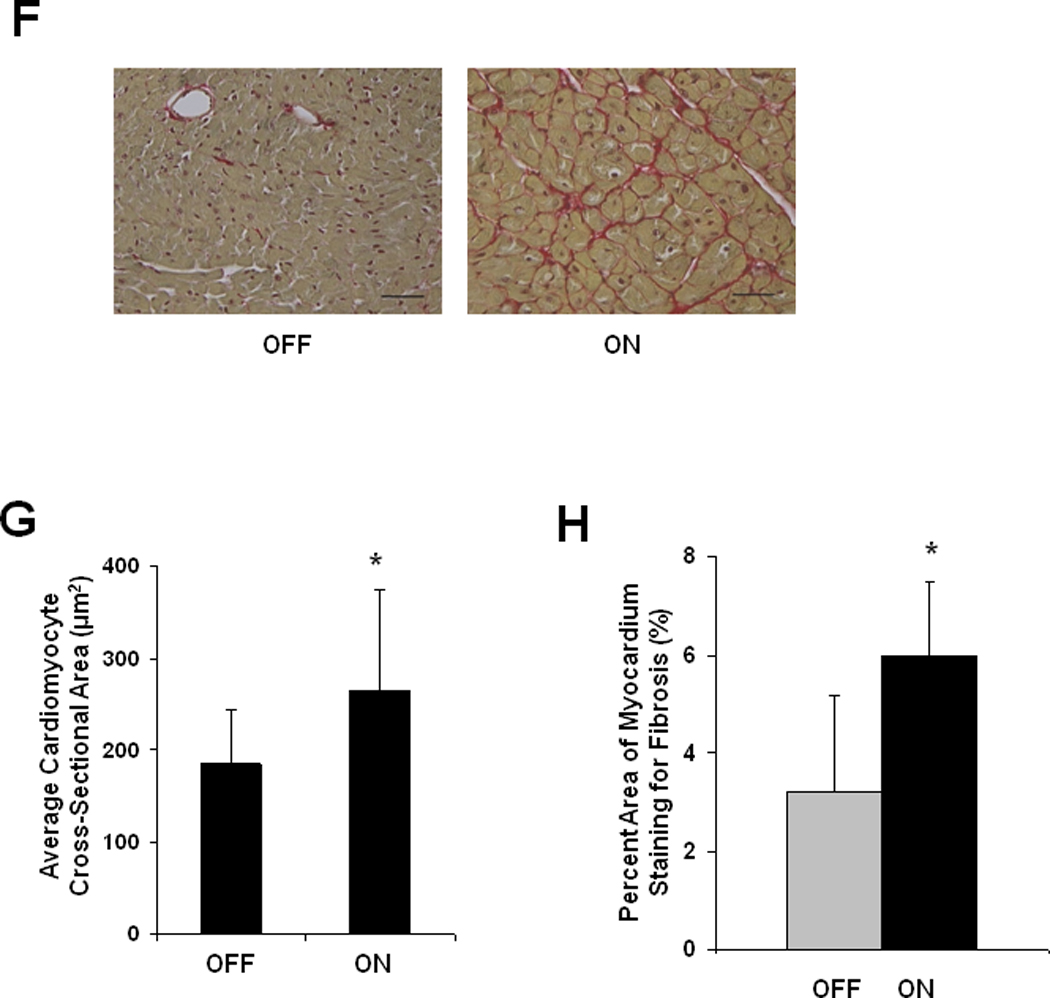
A: Experimental design. Double transgenic tTA/CnA* mice maintained on doxycycline (OFF) were used as controls. B: Left ventricular internal diameter (LVID) at end-diastole and end-systole after 16 weeks of calcineurin activation. C: Mean percent fractional shortening measured from M-mode tracings from mice in the OFF (n=10) and ON (n=14) groups over the course of the experiment. Representative M-mode echocardiographic tracings showing left ventricular mechanical activity at study completion. D: Representative images of hearts at study completion (scale bar = 800 µm). E: Mean absolute heart weight, heart weight normalized to body weight (HW/BW), and lung weight normalized to body weight for mice in the OFF (n=10) and ON (n=14) groups at study completion. F: Representative histological images of picrosirius red staining revealing cardiomyocyte size and fibrosis at study completion. Bar = 80 µm. G: Mean cardiomyocyte cross-sectional area measured from hearts in the OFF and ON groups (4 hearts per group; 60 cells measured per heart; bar = 1 SD). *p<0.01. H. Mean percent area of myocardium staining for fibrosis in the OFF and ON groups (6 images per heart, 6 hearts per group).
Necropsy analysis revealed a significant increase in cardiac mass after 16 weeks of calcineurin activation (Figure 2D, 2E), and three animals (of 17) in the CnA* ON group died before study completion (no deaths of 10 mice occurred in the CnA* OFF group). Lung weights were similar between the two groups, although the two CnA* ON mice with the largest hearts were noted to have lung weights twice that of controls.
Cardiomyocyte hypertrophy was evident upon microscopic examination of sections of ventricular tissue. Cardiomyocyte cross-sectional area increased significantly in CnA* ON mice (266 ± 82 µm2, n=240 cells, 4 hearts) compared to CnA* OFF mice (185 ± 42 µm2, n=240 cells, 4 hearts, p<0.01) (Figure 2F, 2G, Suppl Figure II). We also noted prominent interstitial fibrosis in hearts subjected to 16 weeks of calcineurin activation (Figure 2H). Quantification of fibrosis from histological images revealed more fibrosis in hearts of CnA* ON mice (ON 6.0±1.5%, n=5; OFF 3.2±2.0%, n=6; p=0.02).
We next set out to determine the phenotype of calcineurin-induced ventricular remodeling after just 8 weeks of calcineurin activation (Figure 3A). Single transgenic αMHC-tTA littermates were used as controls, and all mice were subjected to the same schedule of doxycycline. Hearts were harvested 8 weeks after doxycycline removal, a time point associated with significant declines in fractional shortening based on our initial study (Figure 2C). Eight weeks of calcineurin activation in adult hearts triggered profound pathological hypertrophy (Figure 3). Analysis of echocardiographic images revealed significant declines in systolic function (Figure 3B), robust increases in ventricular mass, left ventricular wall thickness, and chamber dimension (Figure 3C). Of note, we observed a subtle increase in cardiac mass in single-transgenic tTA mice compared to wild-type littermates, but there was no significant decline in fractional shortening (Supp Figure III). We, therefore, used single-transgenic tTA mice as controls for comparison to double-transgenic mice. [Head-to-head comparisons of LV mass estimated by echo and measured on necropsy revealed high correlation, Supp Figure IV]
Figure 3. Calcineurin activation in the adult heart triggers pathological hypertrophy.
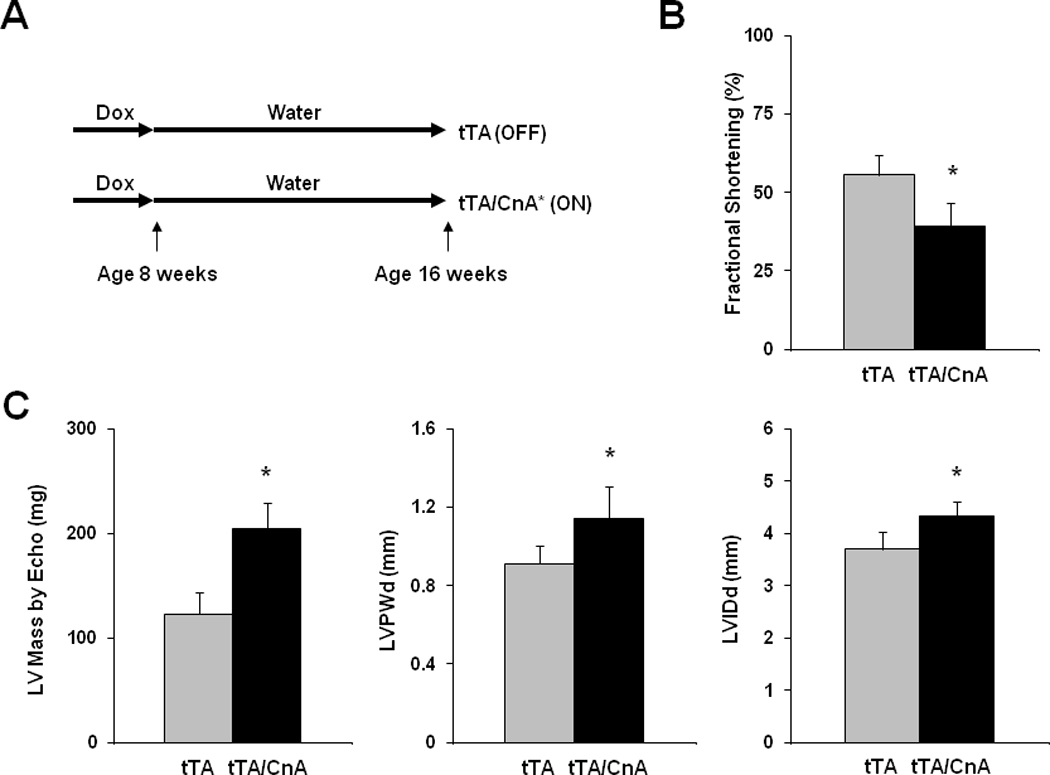
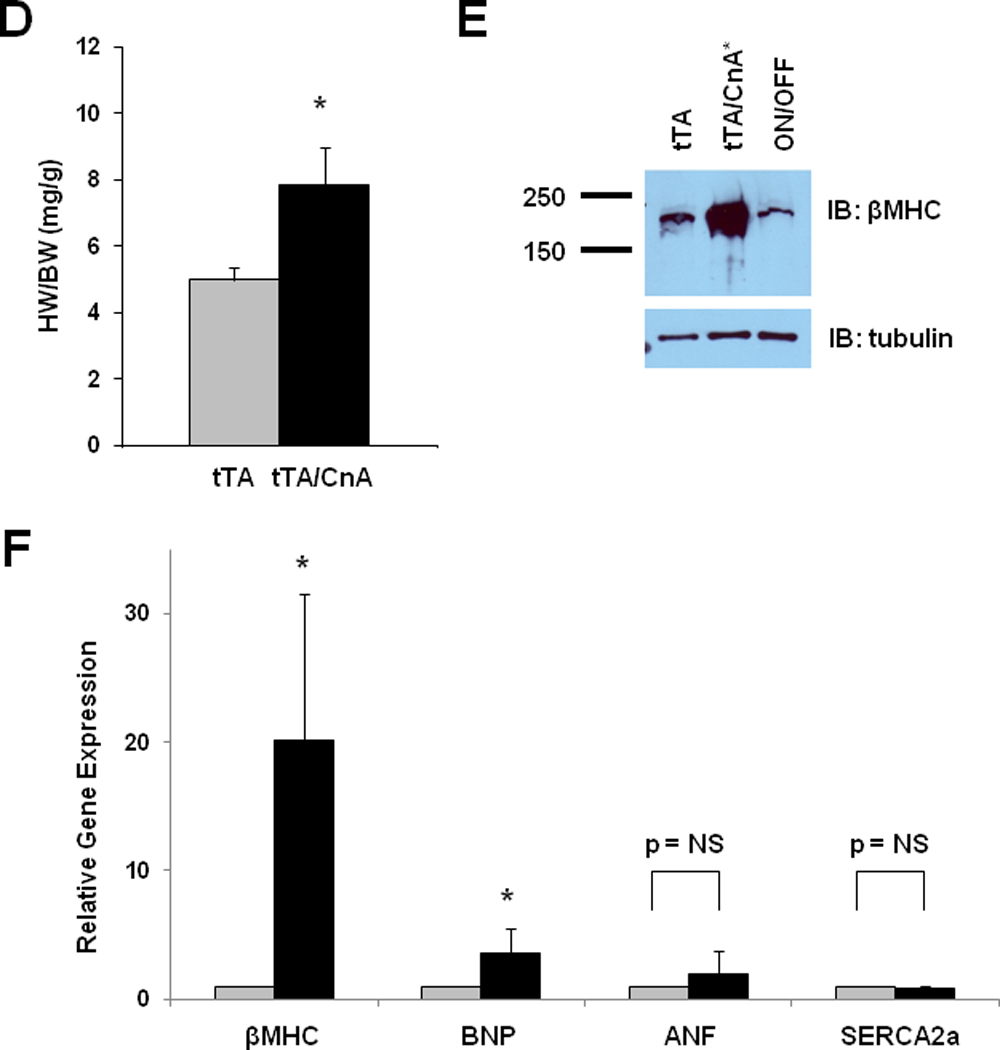
A: Experimental design. Single transgenic tTA mice were used as controls. B: Mean fractional shortening calculated from M-mode tracings for tTA (n=9) and tTA/CnA* (n=11) mice at study completion. C: Mean LV mass, LV posterior wall thickness (LVPWd), and LV internal diameter in diastole (LVIDd) measured from 2D echocardiographic images at study completion. D: Mean heart weight normalized to body weight (HW/BW) measured at necropsy. E: Representative immunoblot of LV lysates from tTA, tTA/CnA*, and ON/OFF hearts probed for βMHC. F: Mean mRNA levels of βMHC, BNP, ANF, and SERCA2a determined by real-time RT-PCR from RNA isolated from tTA (gray bars; n=6) and tTA/CnA* (black bars, n=8) hearts. mRNA levels are calculated relative to cyclophilin mRNA and normalized to control levels. *p < 0.05 ON: double-transgenic tTA/CnA mice; OFF: single transgenic tTA mice. ON/OFF: double-transgenic tTA/CnA mice after doxycycline was withdrawn for 8 weeks and then restored for 8 weeks.
Post-mortem analysis revealed consistent results; heart weight:body weight ratios increased 48% (p<0.05) in tTA/CnA* mice compared to hearts from control mice (Figure 3D). To evaluate for activation of the fetal gene program, a marker of pathological remodeling in heart, we extracted protein and RNA from ventricular tissue 8 weeks after removing doxycycline. Calcineurin signaling triggered dramatic increases in β myosin heavy chain (βMHC) protein and mRNA (Figure 3E, 3F, Supp Figure IC). There was also a significant increase in BNP expression, but no statistically significant changes in ANF or SERCA2a expression were observed.
Calcineurin-induced ventricular hypertrophy precedes systolic dysfunction
In animal models where calcineurin is activated early in life, hypertrophic growth of the heart precedes contractile dysfunction and heart failure. In humans, however, where disease-related stress initiates long after cardiac development is complete, this pathogenic progression has been questioned20. Does ventricular hypertrophy progress routinely to heart failure, or does ventricular hypertrophy develop secondarily, as a consequence of systolic dysfunction? Further, the extent to which ventricular hypertrophy is a risk factor for systolic dysfunction may depend on specific phenotypic features, such as whether it is concentric or eccentric3. To test these important questions, we studied 8-week old male mice with serial echocardiograms performed biweekly during calcineurin activation. First, we measured left ventricular mass and wall thickness by echocardiogram, observing robust increases within two weeks (Figure 4A, 4B). Next, we evaluated ventricular size and performance. Interestingly, there was no significant decrease in fractional shortening at two weeks despite the development of ventricular hypertrophy (Figure 4C). Fractional shortening was significantly decreased by four weeks, but left ventricular chamber dimension remained unchanged until 8 weeks (Figure 4D). Thus, calcineurin activation in adult mice triggered ventricular hypertrophy followed by systolic dysfunction, and left ventricular chamber dilation occurred last. There was no significant difference in heart rates in these mice at the time the echocardiograms were performed (Supp Figure V).
Figure 4. Calcineurin-induced ventricular hypertrophy precedes systolic dysfunction, and fetal gene activation is delayed after calcineurin activation.
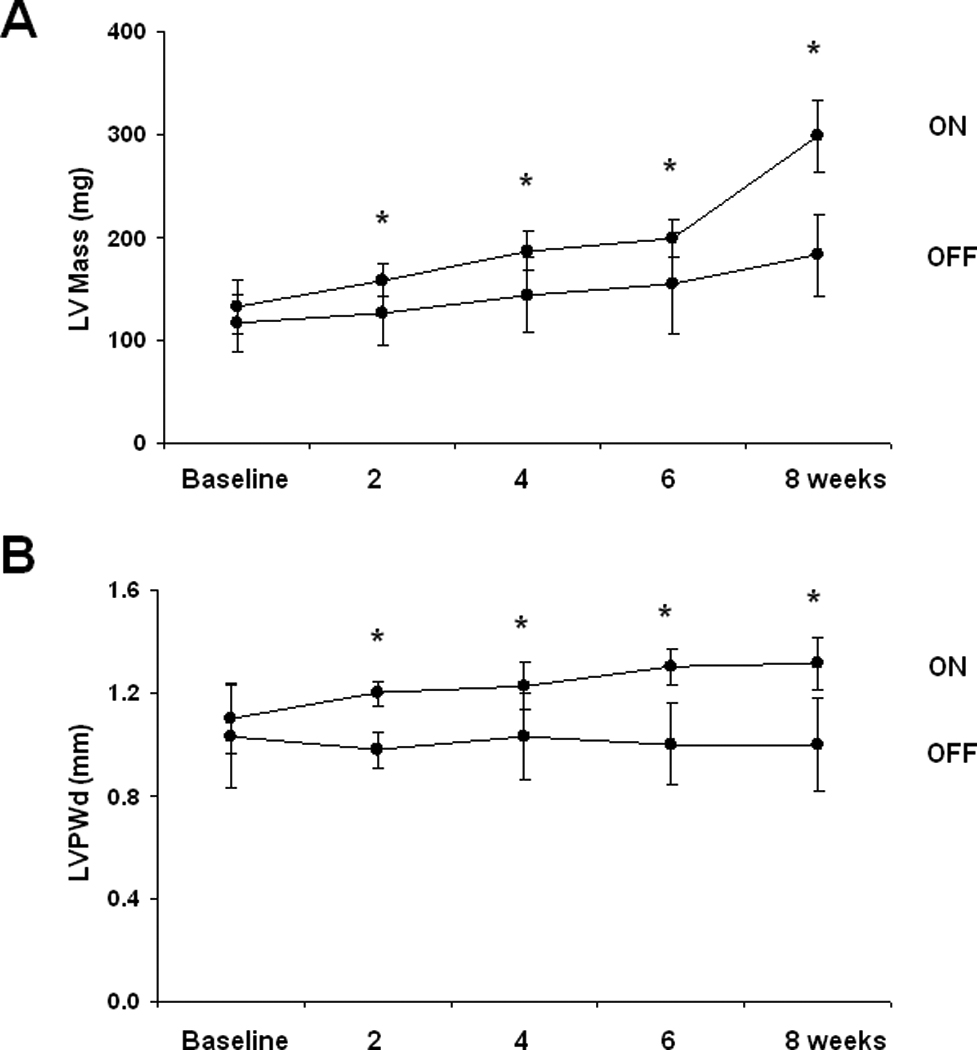
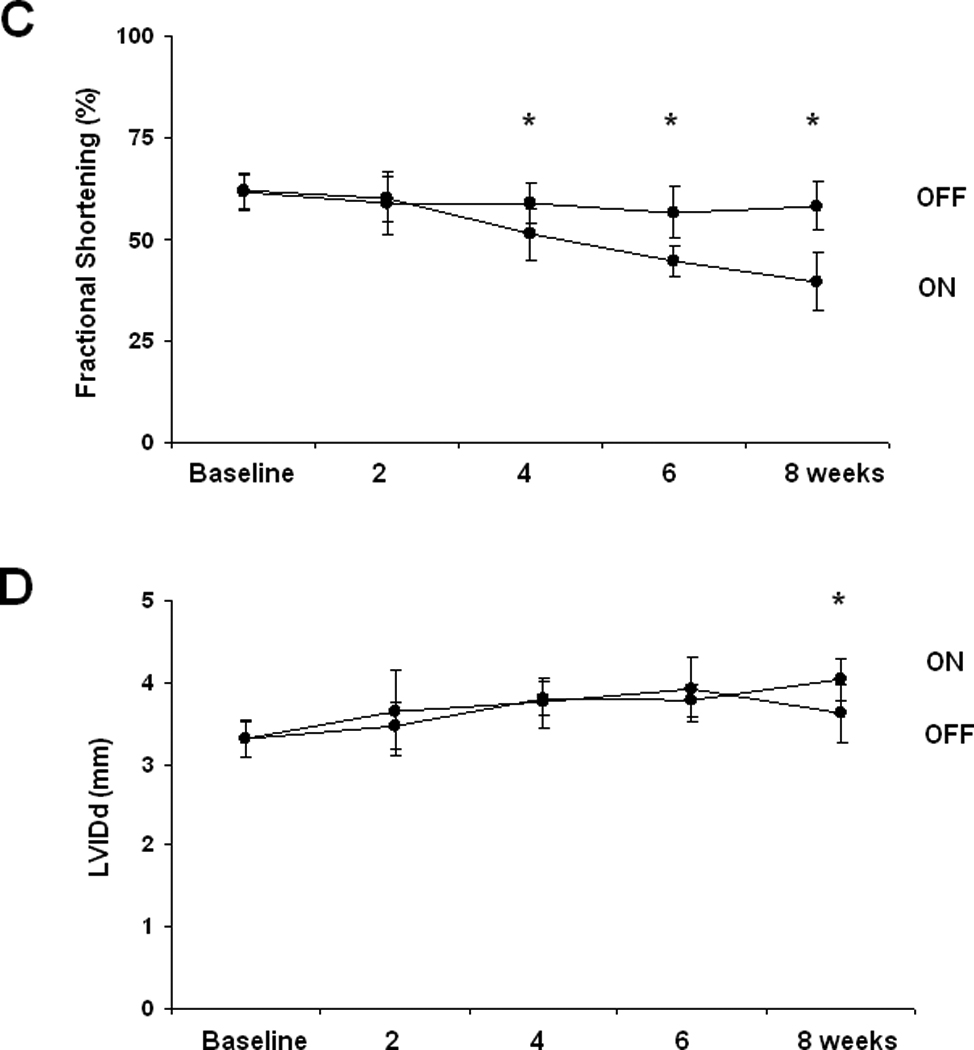
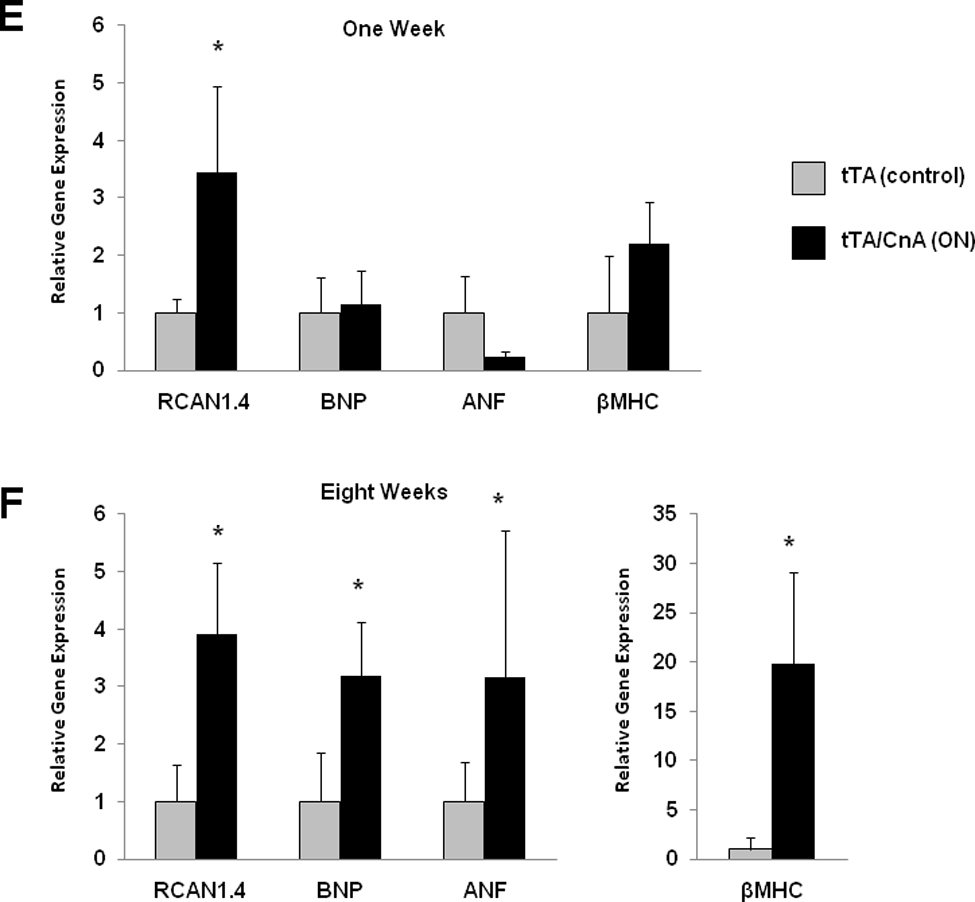
A: Mean LV mass calculated from serial 2D echocardiographic images of tTA (n=9) and tTA/CnA* (n=9) mice recorded after removing doxycycline. B: Mean LV posterior wall thickness at end-diastole (LVPWd) by serial echocardiography after removing doxycycline. C: Mean percent fractional shortening from serial M-mode tracings obtained after removing doxycycline. D: Mean LV internal diameter in diastole (LVIDd) measured from serial 2D echocardiographic images obtained after removing doxycycline. E,F: Quantitative real-time RT-PCR measurements of mRNA levels of indicated genes from RNA isolated from selected hearts at (E) one week and (F) eight weeks after removing doxycycline. mRNA levels are calculated relative to cyclophilin mRNA and normalized to control levels. *p < 0.01. ON: double-transgenic tTA/CNA mice; OFF: single transgenic tTA mice.
In order to evaluate the relationship between gene expression and ventricular remodeling, we collected RNA from mice at one week and eight weeks after removing doxycycline. Just one week after removing doxycycline, we observed a robust increase in RCAN1.4 expression consistent with activation of calcineurin (Figure 4E). Immunoblots for RCAN1.4 also confirmed robust calcineurin activation at one week (data not shown). There was, however, no significant change in expression of fetal gene markers at this early time point. After eight weeks of calcineurin activation, however, βMHC and BNP expression had increased significantly (Figure 4F). These data, then, demonstrate that calcineurin activation alone does not provoke an immediate increase in βMHC or BNP expression. Rather, calcineurin activation may trigger a cascade of molecular signaling and remodeling events that eventually results in increased expression of fetal gene markers.
Adverse remodeling regresses after activated calcineurin is turned off
Adults with heart disease often present to medical attention with significant hypertrophy and/or systolic dysfunction. However, the extent to which calcineurin-dependent adverse remodeling is reversible in the absence of therapy is unknown. To test this, doxycycline was withdrawn from a group of tTA/CnA* mice for 8 weeks in order to activate calcineurin signaling; doxycycline was subsequently restored for an additional 8 weeks (Figure 5A). In these ON/OFF mice, ventricular function declined during the first 8 weeks similar to that of the ON group of mice (Figure 5B). Then, after doxycycline was restored, ventricular function began to improve, reaching statistical significance at 10 weeks (2 weeks after restarting doxycycline) (Figure 5B). This recovery of ventricular function occurred without the assistance of pharmacological therapies for heart failure. At study completion, the average cardiac mass for mice in the ON/OFF group was significantly less than that of mice in the ON group subjected to 16 weeks of persistent CnA* expression (Figure 5C). There was variability in the degree of reverse remodeling, but no specific parameters of ventricular hypertrophy or chamber dimension were predictive of the extent of reverse remodeling (Online Supplement, Supp Fig VI, Supp Fig VII).
Figure 5. Impaired cardiac function recovers after activated calcineurin is extinguished.
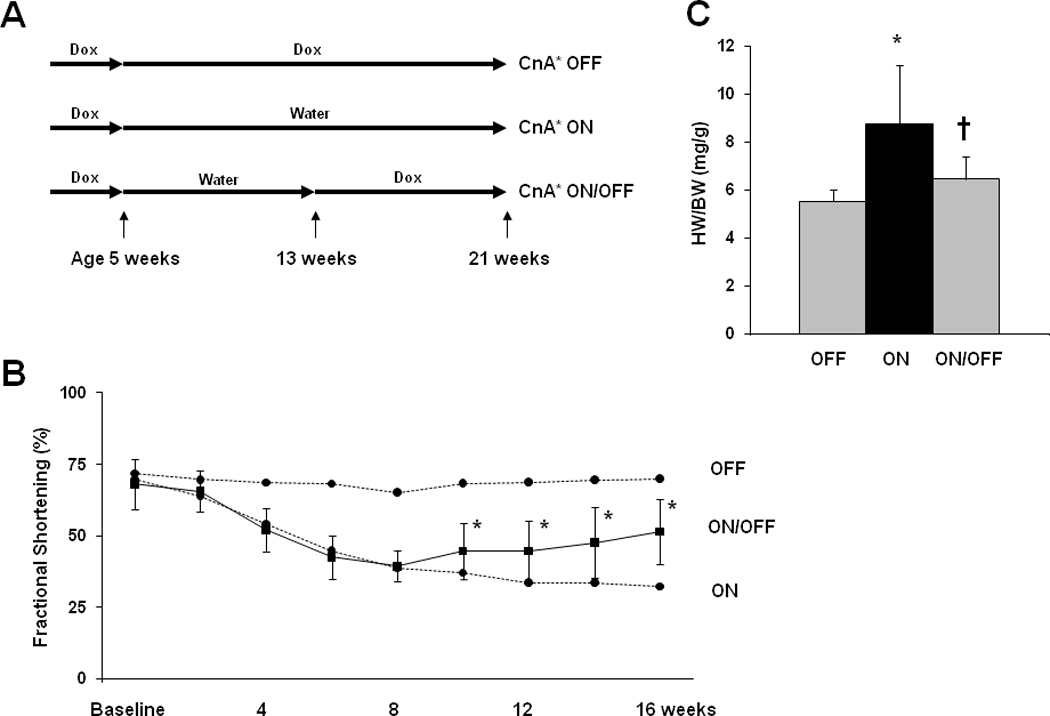
A: Experimental design. Double transgenic mice maintained on doxycycline were used as controls. B: Mean percent fractional shortening measured from serial M-mode tracings of tTA/CnA* mice in the ON/OFF group (n=16). Dotted lines represent mean percent fractional shortening in the OFF (n=10) and ON groups (n=14). *p < 0.01. C: Mean HW/BW at study completion. *p < 0.01 for ON vs OFF; † p < 0.01 for ON/OFF vs ON.
To evaluate the kinetics of extracellular matrix deposition during the course of calcineurin-dependent ventricular remodeling, we quantified ventricular collagen accumulation by hydroxyproline assay. Collagen content within the LV did not increase significantly after 8 weeks of calcineurin activation (ON 0.9±0.2 mg/g, n=3; OFF 0.9±0.2, n=3; p=NS) despite significant ventricular dilatation and declines in systolic function (Figure 5). By contrast, after 16 weeks of calcineurin signaling, collagen content was significantly increased both by hydroxyproline assay (ON 1.6±0.1 mg/g, n=2; OFF 0.80±0.05, n=2, p<0.05) and analysis of picrosirius red-stained images (ON 6.0±1.5%, n=5; OFF 3.2±2.0%, n=6; p=0.02). Collagen content in hearts of ON/OFF mice was similar to control levels (ON/OFF 4.0±2.0%, n=6). Surprisingly, we were unable to induce ventricular tachyarrhythmia in ON mice despite the presence of significant interstitial fibrosis (Online Supplement, Supp Fig VIII). In aggregate, these findings suggest that ventricular fibrosis is a relatively late-arriving feature of the adverse remodeling phenotype, developing after both ventricular hypertrophy and altered contractile function are present.
From these data, it is unclear whether cardiac mass decreased when calcineurin signaling was extinguished or whether hearts in ON/OFF mice simply stopped growing when CnA* was turned off. To test this, we performed two additional, parallel experiments on tTA/CnA* mice. For study A, CnA* was activated at age 8 weeks, and mice were sacrificed at age 16 weeks. For study B, CnA* was also activated at age 8 weeks, echocardiograms were performed at age 16 weeks to confirm depressed ventricular function, and CnA* was suppressed until age 24 weeks at which time mice were sacrificed. As in the previous experiments, echocardiography confirmed recovery of ventricular function and a decrease in LV mass when CnA* was turned off in Study B (Figure 6A, 6B). Analysis of protein from left ventricular tissue confirmed robust activation of calcineurin in tTA/CnA* hearts at the time of sacrifice in study A (Figure 6C). By contrast, calcineurin activity was not elevated in tTA/CnA* hearts at study completion in study B, confirming successful suppression of CnA* during the second half of the experiment. Importantly, heart weights at the completion of study B were significantly lower than those at the completion of study A, despite having been subjected to the same 8 weeks of CnA* activation (Figure 6D).
Figure 6. Pathological hypertrophy regresses after activated calcineurin is extinguished.
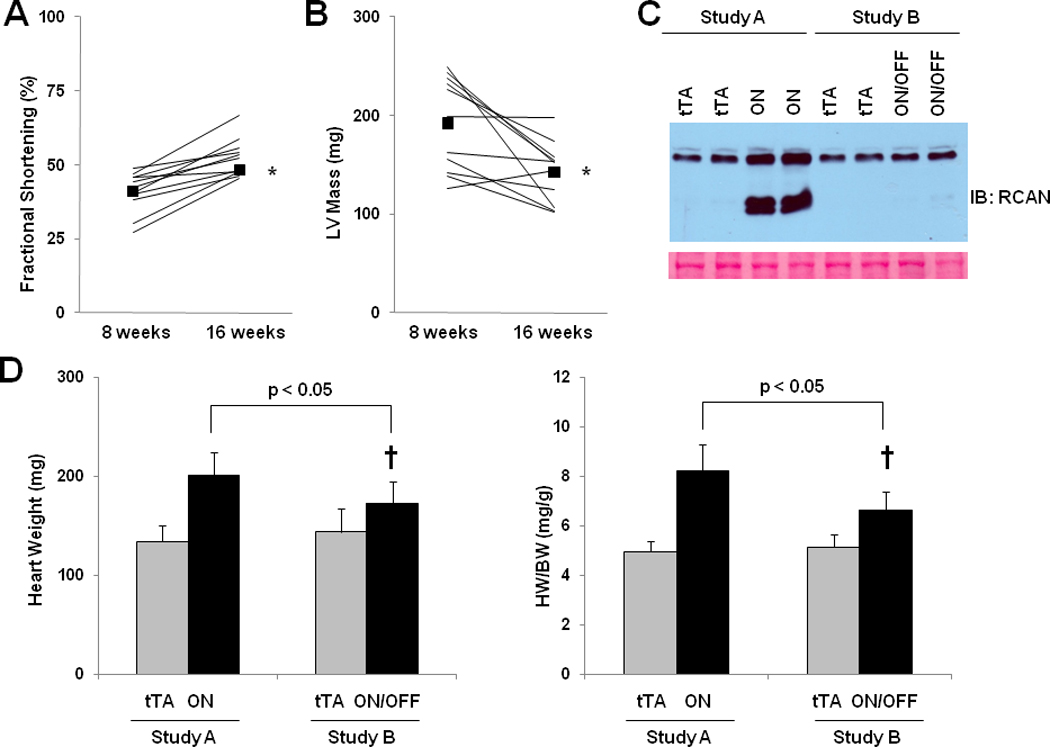
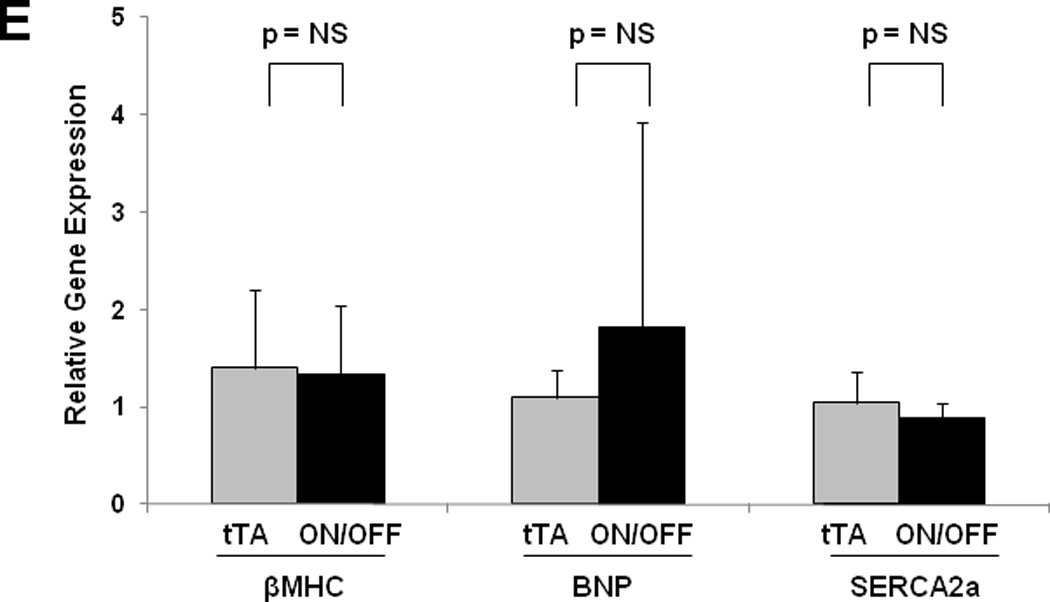
Fractional shortening (A) and LV mass by echocardiography (B) for ON/OFF mice (n=11) before and after restarting doxycycline. Squares represent means. C: Representative immunoblot of LV lysates probed for RCAN from treatment groups as listed. Ponceau stain shows protein loading for each lane. D: Mean absolute heart weight and HW/BW for OFF (n=9) and ON (n=11) mice in study A and OFF (n=4) and ON/OFF (n=6) mice in study B. † p < 0.05 for ON/OFF (study B) vs. ON (study A) E: Mean mRNA levels of βMHC, BNP, and SERCA2a determined by real-time RT-PCR from RNA isolated from OFF (gray bars; n=4) and ON/OFF (black bars, n=6) hearts. mRNA levels are calculated relative to cyclophilin mRNA and normalized to control levels. F: Mean mRNA levels of MuRF3 determined by real-time RT-PCR from RNA isolated from OFF, ON, and ON/OFF hearts at 8 weeks (after removing doxycycline) or 16 weeks (after removing and then restarting doxycycline). ON: tTA/CnA* mice with Dox removed for 8 weeks; ON/OFF: tTA/CnA* mice with Dox removed for 8 weeks then restarted for 8 weeks; OFF: single transgenic tTA mice served as controls.
Activation of the genes coding for βMHC and BNP was detected after 8 weeks of calcineurin activation (Figure 3E). After 8 weeks of recovery in study B, βMHC and BNP mRNA returned to control levels (Figure 6E). Interestingly, those mice with limited recovery of fractional shortening (less than 50% at after 8 weeks of recovery) also had normal expression of βMHC (included in Figure 6E). Conversely, BNP expression remained elevated in some but not all mice with limited recovery.. Together, these data lend strong support to the concept that calcineurin-dependent remodeling of the heart can be reversed even without anti-remodeling therapy.
Discussion
In recent years, calcineurin has emerged as a major mechanism of pathological cardiac remodeling, active in numerous forms of heart disease. As such, this molecule has generated much interest as a target of therapy. Here, we addressed several questions relevant to the therapeutic targeting of calcineurin in heart disease: what are the effects of calcineurin activation in the fully developed adult heart; does calcineurin-dependent hypertrophy precede contractile dysfunction, and, if so, what are its phenotypic features; to what extent are these phenotypes reversible; can reversal be accomplished in the absence of therapy? To accomplish this, we engineered a mouse model that provides high fidelity, spatiotemporal control of calcineurin activation. From these experiments, we report the following significant findings: a) calcineurin activation in adulthood triggers robust pathological remodeling; b) ventricular hypertrophy, concentric in nature, precedes systolic dysfunction and tissue fibrosis; c) ventricular fibrosis develops after both hypertrophy and contractile dysfunction have arisen; d) hypertrophic growth, and contractile dysfunction are each partially and spontaneously reversible. Together, these findings shed new light on the adverse remodeling response elicited by calcineurin and raise yet further the prospects of targeting this molecule for therapeutic gain.
Calcineurin signaling is maladaptive in adult heart
Heart disease is the leading cause of morbidity and mortality in the industrialized world, contributing importantly to ever-expanding health care expenditures31. The pathogenesis of many forms of heart disease involves a set of complex remodeling processes, many of which are independent risk factors for adverse clinical events8, 32. For example, the presence of left ventricular hypertrophy in patients with hypertension is associated with substantially increased morbidity and mortality1. Also, a decline in ejection fraction after myocardial infarction is a major predictor of increased risk of developing heart failure or sudden death33. In many of these events, persistent or excessive activation of the calcineurin signaling pathway is thought to play a significant role21.
Previous models of calcineurin activation in neonatal murine hearts demonstrated an almost 3-fold increase in cardiac mass at only 3 to 12 weeks of age10. Working with adult myocytes, we observed significantly less hypertrophy despite the presence of robust calcineurin activation for 16 weeks. In addition, adult mice manifested less than 10% mortality at 16 weeks, which contrasts with the greater than 50% mortality at 16 weeks observed when calcineurin is activated in neonatal mice34. Together, these findings are consistent with a model where pathological signaling events activated early in life interact with developmental pathways, often compounding the resulting adverse phenotype.
Our model of conditional activation of calcineurin provided an opportunity to investigate the time course of events that occur with calcineurin signaling. We found that ventricular hypertrophy is an early event in the progression of calcineurin-induced heart failure. Left ventricular mass, measured noninvasively by echocardiography, was significantly increased after just 2 weeks of calcineurin activation, while left ventricular cavity size and ejection fraction remained normal. Conversely, we found that tissue fibrosis and fetal gene activation are late-appearing aspects of the phenotype, emerging only after significant contractile dysfunction has developed. A dramatic increase in βMHC expression was apparent after several weeks of calcineurin activation. Despite an increase in RCAN1.4 during the first week, confirming calcineurin activation, we did not observe a significant increase in βMHC at this early time point. This suggests that calcineurin triggers a cascade of signaling events that later leads to the rise in βMHC. Yet, when calcineurin was turned off, βMHC returned to control levels. The contribution of βMHC expression to systolic dysfunction remains uncertain.
These findings lend further support to the notion that calcineurin-induced hypertrophy is maladaptive, as a decline in systolic function ultimately developed in these mice without any additional stressors, such as myocardial infarction or pressure overload. Furthermore, these data are consistent with previous findings that inhibition of calcineurin-dependent signaling in the setting of pressure overload blunts hypertrophy without adverse effects on cardiac function or clinical events9, 35.
Calcineurin as a therapeutic target
While preventive strategies will undoubtedly remain cornerstones of therapy, many patients do not present to clinical attention until after significant structural heart disease has already developed. It is therefore critical to parse the various pathological elements and determine and quantify their potential for reversal. Some, such as myocyte death, have essentially no potential for recovery. Others, however, such as cellular hypertrophy, contractile dysfunction, and fibrosis hold potential for significant reversibility. Prior to this study, however, nothing was known about the kinetics of development of the multiple phenotypes elicited by calcineurin, their emergence in adult heart, and their potential for reversal.
Both pharmacological and device-based therapies improve clinical outcomes in association with reverse remodeling5, 36, 37, and calcineurin signaling mechanisms may prove to be new targets for recovery of cardiac function. For example, calcineurin suppression by systemic administration of cyclosporine A is capable of reversing cardiac hypertrophy38, although cyclosporine A may trigger hypertrophy in some models of heart disease39. Rapamycin treatment will reverse hypertrophy induced by pressure overload40, 41 and in AKT transgenic mice42. Our results here extend these observations by demonstrating the reversibility of some of the effects of calcineurin signaling. Further, that this reversal occurs when myocyte-specific calcineurin activity is suppressed points to a cell-autonomous mechanism of reverse remodeling.
Variable degrees of reverse remodeling were observed in ON/OFF mice, but left ventricular geometry did not emerge as a robust predictor of the potential for recovery of function; mice with the greatest LV dilation or lowest percent fractional shortening following 8 weeks of calcineurin activation sometimes demonstrated significant reverse remodeling when calcineurin activation was turned off. Thus, other factors may mark, or even determine, the potential for recovery of ventricular function. Interstitial fibrosis emerged as a late manifestation of calcineurin-triggered remodeling, possibly the result of activation of secondary signaling cascades. Excessive fibrosis in some hearts may have limited reverse remodeling when calcineurin signaling was extinguished.
A major objective of our study was to determine whether hypertrophy was reversible if the underlying cause were removed. As some evidence suggests that the capacity for cardiac plasticity and repair declines age, we chose to study young adult mice in an effort to obviate confounding, age-dependent changes in plasticity. In other words, we set out specifically to study young adults – with presumably maximal capacity for reversibility – in order to determine whether such reversibility is possible in the setting of a robust signaling cascade (viz. calcineurin). Moving forward, having established for the first time that such reversibility of remodeling is, indeed, possible, it will be of interest to test for age-dependent declines in cardiac plasticity.
Increased calcineurin activity has been reported in the hearts of patients with multiple forms of structural heart disease13–15. Further, inhibition of calcineurin via either pharmacological or transgenic strategies blunts hypertrophic growth in a variety of animal models of heart disease11, 12, 35, 43. Importantly, suppression of hypertrophic growth by calcineurin inhibition is not associated with impairment of cardiac function or increased mortality9. Consistent with this, data reported here demonstrate that calcineurin-induced cardiac hypertrophy results, in fact, in impaired ventricular function. These findings, then, add to a growing literature implicating the hypertrophic growth response per se as a maladaptive response to stress and a viable target for therapeutic intervention9.
Conclusion
Calcineurin activation in the adult heart triggers pathological remodeling which is, at least in part, reversible. Whereas certain forms of hypertrophic growth are thought to represent compensatory responses to environmental stress, calcineurin-induced remodeling appears to be uniquely maladaptive, progressing ultimately to systolic dysfunction, heart failure, and premature mortality. This progression, however, is reversible, suggesting that novel calcineurin-targeting therapies may prove effective in further improving outcomes and quality of life for patients with heart disease.
Novelty and Significance.
What is Known?
Left ventricular hypertrophy is associated with adverse cardiovascular events, including heart failure and death.
Calcineurin is a cytoplasmic protein phosphatase implicated in the pathogenesis of cardiac hypertrophy.
What New Information Does This Article Contribute?
Calcineurin signaling in the adult heart triggers ventricular hypertrophy with markers of pathological remodeling.
Calcineurin-induced ventricular hypertrophy precedes the development of systolic dysfunction and heart failure.
Calcineurin-induced cardiac hypertrophy reverses when calcineurin signaling is turned off.
Fetal gene expression and ventricular fibrosis, each a late manifestation of pathological remodeling, manifest significant reversibility.
Strong epidemiological evidence links left ventricular hypertrophy with adverse cardiovascular events, and heart failure therapies that improve clinical outcomes are often associated with regression of hypertrophy. Calcineurin may play a role in the pathogenesis of hypertrophic heart disease, yet little is known regarding the mechanisms of calcineurin-induced ventricular hypertrophy in the adult heart. Data reported here demonstrate that calcineurin signaling in adult cardiomyocytes triggers pathological ventricular hypertrophy and heart failure. Hypertrophy, heart failure, and other aspects of the pathological remodeling response reverse when calcineurin activation is eliminated.
This study is significant for defining the time course and natural history of remodeling events in adult heart triggered by calcineurin signaling. Ventricular hypertrophy precedes the development of systolic dysfunction and heart failure. A robust increase in beta myosin heavy chain expression occurs, but this is not apparent during the first week of calcineurin signaling. Beta myosin heavy chain expression returns to baseline levels as calcineurin-induced hypertrophy reverses. Future studies are warranted to investigate calcineurin and its downstream effectors as therapeutic targets to prevent, and possibly reverse, relevant features of the pathologically remodeled ventricle.
Supplementary Material
Acknowledgements
We thank Dr. Rhonda Bassel-Duby for critical support and Ms. Beverley Huet for expert assistance with the statistical analyses.
Source of Funding
This work was supported by grants from the NIH (HL-075173, JAH; HL-080144, JAH; HL-090842, JAH; HL-072016, BAR; HL-097768, BAR), AHA (0640084N, JAH; 0655202Y, BAR), ADA (7-08-MN-21-ADA, JAH), Deutsche Forschngsgemeinschaft (BA 2258/1-1, JB), and the AHA-Jon Holden DeHaan Foundation (0970518N, JAH).
Non-standard Abbreviations and Acronyms
- NFAT
nuclear factor of activated T cells
- αMHC
alpha myosin heavy chain
- βMHC
beta myosin heavy chain
- tTA
tetracycline transactivator
- CnA
calcineurin A
- RCAN
regulator of calcineurin
- LVIDd
left ventricular internal diameter at end-diastole
- LVIDs
left ventricular internal diameter at end-systole
- LVPWd
left ventricular posterior wall thickness at end-diastole
- IVSd
interventricular septal thickness at end-diastole
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest Disclosures
None
References
- 1.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the framingham heart study. N Engl J Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 2.Levy D, Larson MG, Vasan RS, Kannel WB, Ho KK. The progression from hypertension to congestive heart failure. JAMA. 1996;275:1557–1562. [PubMed] [Google Scholar]
- 3.Drazner MH, Rame JE, Marino EK, Gottdiener JS, Kitzman DW, Gardin JM, Manolio TA, Dries DL, Siscovick DS. Increased left ventricular mass is a risk factor for the development of a depressed left ventricular ejection fraction within five years: The cardiovascular health study. J Am Coll Cardiol. 2004;43:2207–2215. doi: 10.1016/j.jacc.2003.11.064. [DOI] [PubMed] [Google Scholar]
- 4.Berenji K, Drazner MH, Rothermel BA, Hill JA. Does load-induced ventricular hypertrophy progress to systolic heart failure? Am J Physiol Heart Circ Physiol. 2005;289:H8–H16. doi: 10.1152/ajpheart.01303.2004. [DOI] [PubMed] [Google Scholar]
- 5.Groenning BA, Nilsson JC, Sondergaard L, Fritz-Hansen T, Larsson HB, Hildebrandt PR. Antiremodeling effects on the left ventricle during beta-blockade with metoprolol in the treatment of chronic heart failure. J Am Coll Cardiol. 2000;36:2072–2080. doi: 10.1016/s0735-1097(00)01006-8. [DOI] [PubMed] [Google Scholar]
- 6.Okin PM, Devereux RB, Jern S, Kjeldsen SE, Julius S, Nieminen MS, Snapinn S, Harris KE, Aurup P, Edelman JM, Wedel H, Lindholm LH, Dahlof B. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive treatment and the prediction of major cardiovascular events. Jama. 2004;292:2343–2349. doi: 10.1001/jama.292.19.2343. [DOI] [PubMed] [Google Scholar]
- 7.Devereux RB, Wachtell K, Gerdts E, Boman K, Nieminen MS, Papademetriou V, Rokkedal J, Harris K, Aurup P, Dahlof B. Prognostic significance of left ventricular mass change during treatment of hypertension. Jama. 2004;292:2350–2356. doi: 10.1001/jama.292.19.2350. [DOI] [PubMed] [Google Scholar]
- 8.Hill JA, Olson EN. Cardiac plasticity. New Engl J Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 9.Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: A new therapeutic target? Circulation. 2004;109:1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- 10.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sussman MA, Lim HW, Gude N, Taigen T, Olson EN, Robbins J, Colbert MC, Gualberto A, Wieczorek DF, Molkentin JD. Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science. 1998;281:1690–1693. doi: 10.1126/science.281.5383.1690. [DOI] [PubMed] [Google Scholar]
- 12.Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, Kerber RE, Weiss RM. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2863–2869. doi: 10.1161/01.cir.101.24.2863. [DOI] [PubMed] [Google Scholar]
- 13.Lim HW, Molkentin JD. Calcineurin and human heart failure. Nature Medicine. 1999;5:246–247. doi: 10.1038/6430. [DOI] [PubMed] [Google Scholar]
- 14.Diedrichs H, Chi M, Boelck B, Mehlhorm U, Schwinger RH. Increased regulatory activity of the calcineurin/nfat pathway in human heart failure. Eur J Heart Fail. 2004;6:3–9. doi: 10.1016/j.ejheart.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 15.Diedrichs H, Hagemeister J, Chi M, Boelck B, Muller-Ehmsen J, Schneider CA. Activation of the calcineurin/nfat signalling cascade starts early in human hypertrophic myocardium. J Int Med Res. 2007;35:803–818. doi: 10.1177/147323000703500609. [DOI] [PubMed] [Google Scholar]
- 16.Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, Saffitz J, Chien K, Xiao RP, Kass DA, Wang Y. The in vivo role of p38 map kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci U S A. 2001;98:12283–12288. doi: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Syed F, Odley A, Hahn HS, Brunskill EW, Lynch RA, Marreez Y, Sanbe A, Robbins J, Dorn GW., 2nd Physiological growth synergizes with pathological genes in experimental cardiomyopathy. Circ Res. 2004;95:1200–1206. doi: 10.1161/01.RES.0000150366.08972.7f. [DOI] [PubMed] [Google Scholar]
- 18.Martindale JJ, Wall JA, Martinez-Longoria DM, Aryal P, Rockman HA, Guo Y, Bolli R, Glembotski CC. Overexpression of mitogen-activated protein kinase kinase 6 in the heart improves functional recovery from ischemia in vitro and protects against myocardial infarction in vivo. J Biol Chem. 2005;280:669–676. doi: 10.1074/jbc.M406690200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Streicher JM, Ren S, Herschman H, Wang Y. Mapk-activated protein kinase-2 in cardiac hypertrophy and cyclooxygenase-2 regulation in heart. Circ Res. 106:1434–1443. doi: 10.1161/CIRCRESAHA.109.213199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drazner MH. The transition from hypertrophy to failure: How certain are we? Circulation. 2005;112:936–938. doi: 10.1161/CIRCULATIONAHA.105.558734. [DOI] [PubMed] [Google Scholar]
- 21.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 22.O'Keefe SJ, Tamura J, Kincaid RL, Tocci MJ, O'Neill EA. Fk-506- and csa-sensitive activation of the interleukin-2 promoter by calcineurin. Nature. 1992;357:692–694. doi: 10.1038/357692a0. [DOI] [PubMed] [Google Scholar]
- 23.Zhu Z, Zheng T, Lee CG, Homer RJ, Elias JA. Tetracycline-controlled transcriptional regulation systems: Advances and application in transgenic animal modeling. Semin Cell Dev Biol. 2002;13:121–128. doi: 10.1016/s1084-9521(02)00018-6. [DOI] [PubMed] [Google Scholar]
- 24.McCloskey DT, Turnbull L, Swigart PM, Zambon AC, Turcato S, Joho S, Grossman W, Conklin BR, Simpson PC, Baker AJ. Cardiac transgenesis with the tetracycline transactivator changes myocardial function and gene expression. Physiol Genomics. 2005;22:118–126. doi: 10.1152/physiolgenomics.00016.2005. [DOI] [PubMed] [Google Scholar]
- 25.Collins KA, Korcarz CE, Shroff SG, Bednarz JE, Fentzke RC, Lin H, Leiden JM, Lang RM. Accuracy of echocardiographic estimates of left ventricular mass in mice. Am J Physiol Heart Circ Physiol. 2001;280:H1954–H1962. doi: 10.1152/ajpheart.2001.280.5.H1954. [DOI] [PubMed] [Google Scholar]
- 26.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woessner JF., Jr The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch Biochem Biophys. 1961;93:440–447. doi: 10.1016/0003-9861(61)90291-0. [DOI] [PubMed] [Google Scholar]
- 28.Massare J, Berry JM, Luo X, Rob F, Johnstone JL, Shelton JM, Bassel-Duby R, Hill JA, Naseem RH. Diminished cardiac fibrosis in heart failure is associated with altered ventricular arrhythmia phenotype. J Cardiovasc Electrophysiol. 2010;21:1031–1037. doi: 10.1111/j.1540-8167.2010.01736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Rothermel B, Vega RB, Frey N, McKinsey TA, Olson EN, Bassel-Duby R, Williams RS. Independent signals control expression of the calcineurin inhibitory proteins mcip1 and mcip2 in striated muscles. Circ Res. 2000;87:E61–E68. doi: 10.1161/01.res.87.12.e61. [DOI] [PubMed] [Google Scholar]
- 30.Rothermel BA, Vega RB, Williams RS. The role of modulatory calcineurin-interacting proteins in calcineurin signaling. Trends Cardiovasc Med. 2003;13:15–21. doi: 10.1016/s1050-1738(02)00188-3. [DOI] [PubMed] [Google Scholar]
- 31.Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Greenlund KJ, Hailpern SM, Heit JA, Michael Ho P, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, McDermott MM, Meigs JB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Rosamond WD, Sorlie PD, Stafford RS, Turan TN, Turner MB, Wong ND, Wylie-Rosett J. Executive summary: Heart disease and stroke statistics--2011 update: A report from the american heart association. Circulation. 2011;123:459–463. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. Controversies in ventricular remodelling. Lancet. 2006;367:356–367. doi: 10.1016/S0140-6736(06)68074-4. [DOI] [PubMed] [Google Scholar]
- 33.Solomon SD, Zelenkofske S, McMurray JJ, Finn PV, Velazquez E, Ertl G, Harsanyi A, Rouleau JL, Maggioni A, Kober L, White H, Van de Werf F, Pieper K, Califf RM, Pfeffer MA. Sudden death in patients with myocardial infarction and left ventricular dysfunction, heart failure, or both. N Engl J Med. 2005;352:2581–2588. doi: 10.1056/NEJMoa043938. [DOI] [PubMed] [Google Scholar]
- 34.Dong D, Duan Y, Guo J, Roach DE, Swirp SL, Wang L, Lees-Miller JP, Sheldon RS, Molkentin JD, Duff HJ. Overexpression of calcineurin in mouse causes sudden cardiac death associated with decreased density of k+ channels. Cardiovasc Res. 2003;57:320–332. doi: 10.1016/s0008-6363(02)00661-2. [DOI] [PubMed] [Google Scholar]
- 35.Hill JA, Rothermel B, Yoo KD, Cabuay B, Demetroulis E, Weiss RM, Kutschke W, Bassel-Duby R, Williams RS. Targeted inhibition of calcineurin in pressure-overload cardiac hypertrophy. Preservation of systolic function. J Biol Chem. 2002;277:10251–10255. doi: 10.1074/jbc.M110722200. [DOI] [PubMed] [Google Scholar]
- 36.Devereux RB, Wachtell K, Gerdts E, Boman K, Nieminen MS, Papademetriou V, Rokkedal J, Harris K, Aurup P, Dahlof B. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA. 2004;292:2350–2356. doi: 10.1001/jama.292.19.2350. [DOI] [PubMed] [Google Scholar]
- 37.Cleland JG, Daubert JC, Erdmann E, Freemantle N, Gras D, Kappenberger L, Tavazzi L. The effect of cardiac resynchronization on morbidity and mortality in heart failure. N Engl J Med. 2005;352:1539–1549. doi: 10.1056/NEJMoa050496. [DOI] [PubMed] [Google Scholar]
- 38.Lim HW, De Windt LJ, Mante J, Kimball TR, Witt SA, Sussman MA, Molkentin JD. Reversal of cardiac hypertrophy in transgenic disease models by calcineurin inhibition. J Mol Cell Cardiol. 2000;32:697–709. doi: 10.1006/jmcc.2000.1113. [DOI] [PubMed] [Google Scholar]
- 39.Fatkin D, McConnell BK, Mudd JO, Semsarian C, Moskowitz IG, Schoen FJ, Giewat M, Seidman CE, Seidman JG. An abnormal ca(2+) response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy. J Clin Invest. 2000;106:1351–1359. doi: 10.1172/JCI11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, Izumo S. Inhibition of mtor signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050–3055. doi: 10.1161/01.CIR.0000130641.08705.45. [DOI] [PubMed] [Google Scholar]
- 41.Gao XM, Wong G, Wang B, Kiriazis H, Moore XL, Su YD, Dart A, Du XJ. Inhibition of mtor reduces chronic pressure-overload cardiac hypertrophy and fibrosis. J Hypertens. 2006;24:1663–1670. doi: 10.1097/01.hjh.0000239304.01496.83. [DOI] [PubMed] [Google Scholar]
- 42.Ha T, Li Y, Gao X, McMullen JR, Shioi T, Izumo S, Kelley JL, Zhao A, Haddad GE, Williams DL, Browder IW, Kao RL, Li C. Attenuation of cardiac hypertrophy by inhibiting both mtor and nfkappab activation in vivo. Free Radic Biol Med. 2005;39:1570–1580. doi: 10.1016/j.freeradbiomed.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 43.van Rooij E, Doevendans PA, Crijns HJ, Heeneman S, Lips DJ, van Bilsen M, Williams RS, Olson EN, Bassel-Duby R, Rothermel BA, De Windt LJ. Mcip1 overexpression suppresses left ventricular remodeling and sustains cardiac function after myocardial infarction. Circ Res. 2004;94:e18–e26. doi: 10.1161/01.RES.0000118597.54416.00. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.