

Abstract
Studies have linked sex-biased neurodevelopmental disorders, including autism and schizophrenia, with fetal antecedents such as prenatal stress. Further, these outcomes can persist into subsequent generations, raising the possibility that aspects of heritability in these diseases involve epigenetic mechanisms. Utilizing a mouse model in which we previously identified a period in early gestation when stress results in dysmasculinized and stress-sensitive male offspring, we have examined programming effects in second-generation offspring of prenatally stressed (F2-S) or control (F2-C) sires. Examination of gene expression patterns during the perinatal sensitive period, when organizational gonadal hormones establish the sexually dimorphic brain, confirmed dysmasculinization in F2-S males, where genes important in neurodevelopment showed a female-like pattern. Analyses of the epigenomic miRNA environment detected significant reductions in miR-322, miR-574, and miR-873 in the F2-S male brain, levels that were again more similar to those of control females. Increased expression of a common gene target for these three miRNAs, β-glycan, was confirmed in these males. These developmental effects were associated with the transmission of a stress-sensitive phenotype and shortened anogenital distance in adult F2-S males. As confirmation that the miRNA environment is responsive to organizational testosterone, neonatal males administered the aromatase inhibitor formestane exhibited dramatic changes in brain miRNA patterns, suggesting that miRNAs may serve a previously unappreciated role in organizing the sexually dimorphic brain. Overall, these data support the existence of a sensitive period of early gestation when epigenetic programming of the male germline can occur, permitting transmission of specific phenotypes into subsequent generations.
Introduction
Epidemiological studies have linked prenatal stress to increases in the incidence of neurodevelopmental disorders, including schizophrenia and autism spectrum disorders, associations that are often sex dependent (Huttunen and Niskanen, 1978; van Os and Selten, 1998; Khashan et al., 2008; Kinney et al., 2008). These diseases often display sex differences in prevalence, presentation, or therapeutic outcomes (Bale et al., 2010). While many factors likely contribute to these differences, sex-specific responses to fetal antecedents are likely involved (Weinstock, 2007).
We have previously identified early gestation as a specific window of sensitivity during which male mice were susceptible to the programming effects of maternal stress. These males exhibited physiological and behavioral stress sensitivity and cognitive deficits, endophenotypes associated with human neuropsychiatric disease. In addition, these changes reduced or disrupted established sex differences by dysmasculinizing male offspring measures of stress responsivity (Mueller and Bale, 2007, 2008). Similar disruptions of sex differences in behavior, morphology, and gene expression profiles have previously been reported in studies using prenatal stress paradigms across multiple species (Ward, 1972; Meisel et al., 1979; Reznikov et al., 1999; Kapoor and Matthews, 2005; Biala et al., 2010). The organizational/activational hypothesis of brain development suggests that a surge of gonadal hormones organize the brain in a sexually dimorphic manner during the perinatal sensitive period. Then in adulthood, gonadal hormones can activate this organized neurocircuitry to express appropriate sex-specific behavioral phenotypes, including stress axis responsivity (Phoenix et al., 1959; Arnold and Gorski, 1984; Seale et al., 2005; Bingham and Viau, 2008). The disruption of sex differences identified in our model suggests that early prenatal stress alters the trajectory of neurodevelopment during the perinatal period.
Fetal antecedents likely contribute to adult disease through programming changes in the epigenome. Examples of this phenomenon are emerging in human studies. For example, infants with prenatal exposure to maternal depression or anxious mood exhibited increased glucocorticoid methylation, which was associated with a heightened cortisol response to a mild stressor (Oberlander et al., 2008). Such programming effects may transmit to subsequent generations, predisposing offspring to disease. Animal models have clearly established a role for epigenetics in transgenerational phenotypic inheritance following exposure to environmental factors such as maternal stress, diet, and endocrine disruptors (Dunn and Bale, 2009; Franklin et al., 2010; Guerrero-Bosagna et al., 2010; Skinner, 2011). Importantly, in rodent models, inheritance of a phenotype through the paternal lineage excludes confounding effects of the maternal intrauterine environment and postnatal rearing behavior, and indicates gametic epigenetic transmission (Youngson and Whitelaw, 2008). These phenomena raise the interesting possibility that the heritability observed in neurodevelopmental diseases could be the product of both classic genetic and non-Mendelian, or epigenetic, mechanisms (Maher, 2008; Slatkin, 2009). Therefore, we used our mouse model to determine epigenetic programming effects and the transmission of a dysmasculinized phenotype to second-generation (F2) male offspring via the paternal lineage, eventually focusing mechanistically on analysis of the miRNA environment as a form of transcriptional regulation with broad potential to impact developmental processes.
Materials and Methods
Animals
All dams bred for gestational stress studies were virgin, experimentally naive C57BL/6:129 F1 hybrid 5-week-old mice purchased from The Jackson Laboratory. Offspring from these breedings were used to generate the second-generation (F2) litters for transgenerational studies. Justification for using a hybrid background strain in these studies is related to stress responsivity phenotypes and physiology. C57BL/6 are extremely low-stress responders and display low levels of maternal care, making them poor choices for studies focusing on neurodevelopment. While 129 mice are great stress responders and show high levels of quality maternal care, they frequently lack a fully formed corpus callosum and are poor performers in behavioral tests, especially learning and memory tasks. However, the combination of these two strains produces a hybrid vigor that has served our research well with predictable stress responses, behavioral outcomes, and sex differences in stress physiology and behavioral tests (Mueller and Bale, 2006, 2007, 2008). Pregnancy was established by confirmation of a copulation plug (checked for each morning between 7:00 and 8:00 A.M.). Presence of a copulation plug denoted experimental day 1 for early prenatal stress exposure. The pregnant female was individually housed, given a cotton nestlet, and randomly assigned to a stress treatment or control group. Food (Purina Rodent Chow; 28.1% protein, 59.8% carbohydrate, 12.1% fat) and water was provided ad libitum throughout the study. All studies were performed according to experimental protocols approved by the University of Pennsylvania Institutional Animal Care and Use Committee, and all procedures were conducted in accordance with institutional guidelines.
Early prenatal stress
Administration of chronic variable stress was performed as described previously (Mueller and Bale, 2006). Briefly, pregnant dams were randomly assigned to either an experimental treatment group to receive chronic variable stress during gestation days 1–7, or to a control nonstressed treatment group. Pregnant dams assigned to the stress group experienced a different stressor on each of the seven days. Stressors included: 36 h constant light, 15 min of fox odor exposure (1:10,000 2,4,5-trimethylthiazole, Acros Organics), novel objects in cage overnight (eight marbles of similar size and color), 5 min restraint stress in a 50 ml conical tube, novel white noise overnight (Sleep machine, Brookstone), multiple cage changes, and water saturated bedding overnight. These mild stressors were selected to be nonhabituating and to not induce pain. We previously demonstrated that this chronic variable stress paradigm does not affect maternal food or water intake, weight gain, gestation length, litter size, or postpartum maternal behaviors (Mueller and Bale, 2006).
Offspring
To determine whether the dysmasculinized stress-sensitive phenotype identified in first-generation (F1) prenatally stressed males could be transmitted through the paternal lineage to second-generation (F2) offspring, F1 control or prenatally stressed males were bred with F1 control females to generate F2 control litters (F2-C n = 9 litters, n = 69 total animals) or F2 stress litters (F2-S n = 9 litters, n = 59 total animals). Molecular, physiological, morphological, and behavioral correlates of the dysmasculinized stress-sensitive phenotype were then tested in these F2 offspring. All experimental n values described throughout the manuscript refer to litter n values.
Postnatal day 1 brain expression analysis
To explore mechanisms through which the F1 dysmasculinized stress-sensitive phenotype may be programmed in the F2 generation, we examined whole-brain gene expression in male and female neonates at birth, a period critical for the organization of the sexually dimorphic brain by gonadal hormones. One male and one female neonate per litter were killed on the day of parturition. Whole brains were dissected, frozen in liquid nitrogen, and stored at −80°C before assay. Neonate sex was confirmed by SRY genotyping of tail genomic DNA as previously described (Mueller and Bale, 2008). Whole brains were sonicated in TRIzol reagent (Invitrogen) and total RNA was isolated according to manufacturer's protocol.
Custom Taqman qRT-PCR Array.
Two hundred fifty nanograms of total RNA from postnatal day 1 (PN1) brains was reverse transcribed to cDNA using the High-Capacity cDNA reverse transcriptase kit (Applied Biosystems). Expression levels of 93 genes important in neurodevelopment were determined using a custom Taqman array (F2-C♂ n = 4, F2-S♂ n = 3, and F2-C♀ n = 4) (Applied Biosystems). Analysis was performed using the comparative Ct method. Eighteen svedberg rRNA was used as an endogenous loading control. Expression levels of each sample were normalized to the average F2-C♂ expression level.
miRNA Taqman qRT-PCR Array.
Five hundred nanograms of total RNA from PN1 brain was reverse transcribed to cDNA using Megaplex RT pool A primers and Multiscribe reverse transcriptase (Applied Biosystems). Expression levels of 239 miRNAs were determined using the Taqman Array MicroRNA card A Array (F2-C♂ n = 5, F2-S♂ n = 3, and F2-C♀ n = 5) (Applied Biosystems). Analysis was performed using the comparative Ct method. For each sample, the average of the Ct values of sno135 and sno202 was used as an endogenous loading control. Expression levels of each sample were normalized to the average F2-C♂ expression level.
qRT-PCR of predicted miRNA target transcripts.
Predicted miRNA targets were identified using the web-based algorithm miRDB (Wang, 2008; Wang and El Naqa, 2008). Expression of mRNAs that were shared predicted targets of miRNAs identified as significantly changed by early prenatal stress, and with potential relevance to disease mechanisms, were assayed by Taqman qRT-PCR (Applied Biosystems). GAPDH was used as an endogenous loading control. Samples were run in triplicate, and expression was determined using the comparative Ct method. Expression levels of each sample were normalized to the average F2-C♂ expression level.
F2 physiological and behavioral measures
For each test, one female and one male per litter were examined to avoid litter effects. An investigator blind to animal treatment group and sex performed scoring of behavioral tests.
Tail suspension test.
The tail suspension test (TST) was performed as previously described to examine active versus passive stress coping strategies (Steru et al., 1985). Mice were secured to a rod by adhesive tape placed ∼1 cm from the tip of the tail, and suspended 50 cm from the bench-top in a visually isolated area (F2-C♂ n = 7, F2-S♂ n = 7, F2-C♀ n = 8, and F2-S♀ n = 6). Immobility time, defined as the absence of all movement except for whisker movement or respiration, was scored for the 6 min test session.
Barnes maze.
As a test with predictable sex differences in outcome measures of learning and memory, we conducted the Barnes maze (F2-C♂ n = 7, F2-S♂ n = 7, F2-C♀ n = 9, and F2-C♀ n = 9) as previously described (Mueller and Bale, 2007). Briefly, the maze consists of a black circular disk (90 cm in diameter) with 24 holes evenly spaced around its perimeter. An escape box is located under one of the holes. The location of the escape box remains constant throughout training. The disk is elevated 70 cm above the floor and situated in a room with white walls containing three distinct visual cues around the perimeter of the maze. All mice were trained on the maze 2 trials/d for 3 d. Trials within each day were separated by 4 h. To begin each trial, the mouse was placed under a glass beaker in the center of the maze for 15 s before trial start. Latency to identify the target escape box was determined. Each trial was terminated when the mouse located and entered the target escape box or after 4 min elapsed. If the mouse did not successfully locate the target box, the investigator guided the mouse to the target and a latency to target of 240 s was assigned.
HPA response to restraint stress.
The HPA axis response to acute stress was measured by exposing adult F2 mice to a 15 min restraint in a 50 ml conical tube (F2-C♂ n = 7, F2-S♂ n = 7, F2-C♀ n = 7, and F2-C♀ n = 9). Testing occurred 2–5 h following lights on. Blood samples were collected from a tail nick at four time points: (1) time 0, immediately upon removal from the cage, (2) time 15, immediately after the restraint stress, (3) time 30, following 15 min recovery in the home cage, and (4) time 90, following 75 min recovery in the home cage. Samples were collected into EDTA-treated tubes, centrifuged, and plasma was stored at −80°C until corticosterone levels were measured by radioimmunoassay (MP Biomedicals). The minimum detection limit of the assay was 7.7 ng/ml, and the intra-assay coefficient of variation was 7.1%.
Morphology.
One experimentally naive male and female per litter were killed as adults (F2-C♂ n = 7, F2-S♂ n = 7, and F2-C♀ n = 9). Anogenital distances and left testis weights were obtained in males.
Effects of an aromatase inhibitor on the neonatal brain miRNA environment
Formestane administration.
To examine the influence of organizational estradiol on the neonatal brain miRNA environment, a separate cohort of C57BL/6:129 control pups were treated with an aromatase inhibitor or vehicle on the morning following parturition. Male pups were randomly assigned to receive 20 μg of formestane (Sigma-Aldrich) in 20 μl of sesame oil with 10% ethanol (n = 7 from 6 litters) or vehicle injections (n = 7 from 6 litters). This dose, after adapting for differences in rat versus mouse neonate weight, was previously shown to reduce male hypothalamic estrogen to female levels (Amateau et al., 2004). All female pups received vehicle injections (n = 8 from 5 litters). Injections were administered subcutaneously between the shoulders, and the injection site was treated with New Skin liquid bandage to prevent leakage.
miRNA expression analysis.
Pups were killed 24 h after treatment (PN2). Whole brains were dissected, frozen in liquid nitrogen, and stored at −80°C before assay. Neonate sex was confirmed by SRY genotyping of tail genomic DNA as previously described (Mueller and Bale, 2008). Whole brains were sonicated in TRIzol reagent (Invitrogen) and total RNA was isolated according to the manufacturer's protocol.
Five hundred nanograms of total RNA from PN2 brain was reverse transcribed to cDNA using Megaplex RT pool A primers and Multiscribe reverse transcriptase (Applied Biosystems). Expression levels of 239 miRNAs were determined using the Taqman Array MicroRNA card A Array (Applied Biosystems). Analysis was performed using the comparative Ct method. For each sample, the average of the Ct values of sno135 and sno202 was used as an endogenous loading control. Expression levels of each sample were normalized to the average expression level of vehicle-treated males.
Statistics
An investigator blind to animal treatment group and sex conducted all studies and analyses. To control for litter effects in prenatal manipulations, only one male or female from a litter was used for a test or assay. We tested the null hypothesis that early prenatal stress exposed males could not transmit their dysmasculinized stress-sensitive phenotype to their second-generation offspring. Morphological measures and immobility in the TST were analyzed by one-tailed Student's t test, testing for effects identified in early prenatal stress exposed (F1) sires. Barnes maze performance and corticosterone response to restraint was analyzed by one-way ANOVA for paternal (F1) prenatal stress exposure, and trial or time as repeated measure, respectively. miRNA and gene target expression in F2-C♂, F2-S♂, and F2-C♀ PN1 whole brains was analyzed by one-way ANOVA, using Student's t tests for post hoc analysis. miRNA expression data in control or formestane-treated brains were analyzed by hierarchical clustering of samples, using Pearson correlation as a metric. Treatment effects on individual miRNAs were identified by two-tailed Student's t tests. Heat maps and hierarchical clustering was performed using MultiExperiment Viewer (TM4.org). All other statistics were performed using JMP8 (SAS).
Results
F2 gene expression analysis during the perinatal sensitive period
F2 PN1 brain gene expression analysis
To explore mechanisms through which the F1 dysmasculinized stress-sensitive phenotype may be programmed in the F2 generation, we examined brain gene expression in male and female neonates at birth, a period critical for the organization of the sexually dimorphic brain by gonadal hormones. Expression data for individual genes are presented in Table 1. These data are also displayed in a heat map to highlight the broad similarities in gene expression that exist between F2-S♂ and F2-C♀ relative to F2-C♂ (Fig. 1A). Mean F2-S♂ expression of the 17 genes that display significant basal sex differences in PN1 whole brain are plotted on a continuum between average F2-C♂ and F2-C♀ expression (Fig. 1B). In F2-S♂, 13 of these 17 genes displayed expression levels closer to F2-C♀ than to F2-C♂ levels.
Table 1.
PN1 brain gene expression assayed by Custom Taqman qRT-PCR Array for genes important in neurodevelopment
| Gene | F2-C♂ | F2-C♀ | p value (t test) | Gene | F2-C♂ | F2-C♀ | p value (t test) |
|---|---|---|---|---|---|---|---|
| Bax | 1 ± 0.06 | 1.2 ± 0.07 | 0.07 | Hsp90aa1 | 1 ± 0.13 | 1.3 ± 0.16 | 0.24 |
| Bcl2l1 | 1 ± 0.02 | 1.3 ± 0.09 | 0.04* | Hspb1 | 1 ± 0.09 | 0.9 ± 0.1 | 0.61 |
| Bcl2 | 1 ± 0.08 | 1.3 ± 0.09 | 0.05* | Htr1a | 1 ± 0.1 | 1.2 ± 0.12 | 0.27 |
| Bdnf | 1 ± 0.15 | 1.5 ± 0.14 | 0.07 | Htr2b | 1 ± 0.47 | 2.1 ± 0.45 | 0.14 |
| Casp2 | 1 ± 0.07 | 1.1 ± 0.03 | 0.17 | Htr2c | 1 ± 0.19 | 1.1 ± 0.14 | 0.68 |
| Casp3 | 1 ± 0.06 | 1.2 ± 0.07 | 0.06 | Il6 | 1 ± 0.2 | 1 ± 0.28 | 0.95 |
| Casp7 | 1 ± 0.13 | 1.5 ± 0.19 | 0.12 | Il6ra | 1 ± 0.12 | 1.5 ± 0.19 | 0.12 |
| Casp8 | 1 ± 0.09 | 1.2 ± 0.1 | 0.16 | Il6st | 1 ± 0.003 | 1.3 ± 0.15 | 0.13 |
| Cd40 | 1 ± 0.1 | 1.2 ± 0.13 | 0.34 | Kcnj9 | 1 ± 0.18 | 1.5 ± 0.29 | 0.2 |
| Chrm1 | 1 ± 0.09 | 1.3 ± 0.11 | 0.05* | Mbd2 | 1 ± 0.13 | 1.3 ± 0.21 | 0.24 |
| Chrm4 | 1 ± 0.12 | 1.4 ± 0.2 | 0.18 | Mbd3 | 1 ± 0.04 | 1.2 ± 0.11 | 0.19 |
| Chrna4 | 1 ± 0.03 | 1.2 ± 0.09 | 0.04* | Mecp2 | 1 ± 0.02 | 1.2 ± 0.09 | 0.06 |
| Chrna7 | 1 ± 0.05 | 1.2 ± 0.07 | 0.06 | Mtap2 | 1 ± 0.11 | 1.2 ± 0.07 | 0.22 |
| Chrnb2 | 1 ± 0.04 | 1.3 ± 0.06 | 0.01* | Ncor1 | 1 ± 0.02 | 1.2 ± 0.09 | 0.07 |
| Cntf | 1 ± 0.14 | 1.2 ± 0.13 | 0.45 | Ncor2 | 1 ± 0.06 | 1.2 ± 0.11 | 0.17 |
| Creb1 | 1 ± 0.11 | 1.4 ± 0.09 | 0.03* | Nefh | 1 ± 0.16 | 1.1 ± 0.18 | 0.85 |
| Crebbp | 1 ± 0.07 | 1.3 ± 0.08 | 0.06 | Nefl | 1 ± 0.13 | 1.1 ± 0.1 | 0.73 |
| Crfbp | 1 ± 0.06 | 1.2 ± 0.17 | 0.3 | Ngf | 1 ± 0.09 | 1.2 ± 0.14 | 0.32 |
| Crf | 1 ± 0.23 | 1.5 ± 0.27 | 0.24 | Ngfrap1 | 1 ± 0.09 | 1.3 ± 0.14 | 0.12 |
| Crfr1 | 1 ± 0.13 | 1.4 ± 0.09 | 0.04* | Ngfr | 1 ± 0.08 | 0.9 ± 0.09 | 0.37 |
| Crfr2 | 1 ± 0.22 | 0.6 ± 0.16 | 0.16 | Nr3c1 | 1 ± 0.08 | 1.1 ± 0.12 | 0.44 |
| Disc1 | 1 ± 0.1 | 1.1 ± 0.11 | 0.51 | Nrg1 | 1 ± 0.05 | 1.2 ± 0.1 | 0.09 |
| Dnmt1 | 1 ± 0.04 | 1.1 ± 0.08 | 0.19 | Nrg4 | 1 ± 0.41 | 1 ± 0.54 | 0.99 |
| Dnmt3a | 1 ± 0.07 | 1.2 ± 0.07 | 0.14 | Ntf3 | 1 ± 0.08 | 1.2 ± 0.13 | 0.26 |
| Dnmt3b | 1 ± 0.17 | 1.5 ± 0.14 | 0.06 | Ntf5 | 1 ± 0.29 | 0.8 ± 0.27 | 0.71 |
| Ep300 | 1 ± 0.08 | 1.5 ± 0.17 | 0.07 | Ntrk1 | 1 ± 0.07 | 1.1 ± 0.18 | 0.54 |
| Fos | 1 ± 0.09 | 1.2 ± 0.13 | 0.32 | Ntrk2 | 1 ± 0.04 | 1.3 ± 0.04 | 0.004* |
| Gabbr2 | 1 ± 0.04 | 1.4 ± 0.15 | 0.07 | Ntrk3 | 1 ± 0.004 | 1.2 ± 0.06 | 0.08 |
| Gabra2 | 1 ± 0.2 | 0.8 ± 0.15 | 0.47 | Sin3a | 1 ± 0.05 | 1.1 ± 0.04 | 0.19 |
| Gabra3 | 1 ± 0.08 | 1.3 ± 0.1 | 0.05* | Slc1a2 | 1 ± 0.12 | 1.2 ± 0.15 | 0.34 |
| Gabrb3 | 1 ± 0.08 | 1.5 ± 0.13 | 0.03* | Slc1a3 | 1 ± 0.06 | 1.2 ± 0.07 | 0.1 |
| Gad1 | 1 ± 0.08 | 1.3 ± 0.15 | 0.14 | Slc6a1 | 1 ± 0.45 | 1.9 ± 0.58 | 0.26 |
| Gad2 | 1 ± 0.04 | 1.2 ± 0.11 | 0.15 | Slc6a2 | 1 ± 0.1 | 1.4 ± 0.29 | 0.36 |
| Gdnf | 1 ± 0.12 | 1.3 ± 0.1 | 0.11 | Slc6a3 | 1 ± 0.05 | 1.5 ± 0.17 | 0.03* |
| Gfap | 1 ± 0.13 | 1.5 ± 0.27 | 0.14 | Slc6a4 | 1 ± 0.19 | 1.4 ± 0.15 | 0.16 |
| Gfra1 | 1 ± 0.09 | 1.3 ± 0.14 | 0.19 | Stat1 | 1 ± 0.08 | 1.2 ± 0.09 | 0.18 |
| Gfra2 | 1 ± 0.11 | 1.2 ± 0.14 | 0.24 | Stat2 | 1 ± 0.08 | 1.3 ± 0.07 | 0.04* |
| Gfra3 | 1 ± 0.34 | 1.5 ± 0.2 | 0.23 | Stat3 | 1 ± 0.04 | 1.2 ± 0.11 | 0.16 |
| Gmfb | 1 ± 0.07 | 1.2 ± 0.1 | 0.18 | Stat4 | 1 ± 0.19 | 1.8 ± 0.26 | 0.09 |
| Gmfg | 1 ± 0.06 | 0.8 ± 0.21 | 0.52 | Tgfa | 1 ± 0.03 | 1.4 ± 0.12 | 0.03* |
| Hdac1 | 1 ± 0.04 | 1.3 ± 0.09 | 0.03* | Tgfb1i1 | 1 ± 0.12 | 1.3 ± 0.18 | 0.28 |
| Hdac2 | 1 ± 0.03 | 1.1 ± 0.04 | 0.1 | Tgfb1 | 1 ± 0.06 | 1.4 ± 0.16 | 0.08 |
| Hdac3 | 1 ± 0.04 | 1.2 ± 0.07 | 0.04* | Tnf | 1 ± 0.39 | 0.9 ± 0.3 | 0.8 |
| Hells | 1 ± 0.17 | 1.3 ± 0.09 | 0.25 | Tph2 | 1 ± 0.49 | 1.7 ± 0.41 | 0.31 |
| Hif1a | 1 ± 0.07 | 1.2 ± 0.1 | 0.14 | Ucn | 1 ± 0.25 | 0.9 ± 0.21 | 0.85 |
| Hif3a | 1 ± 0.24 | 1 ± 0.26 | 0.98 | Vegfa | 1 ± 0.09 | 1.4 ± 0.06 | 0.01* |
| Hsp90aa1 | 1 ± 0.13 | 1.3 ± 0.16 | 0.24 | Zfp110 | 1 ± 0.05 | 1.2 ± 0.05 | 0.03* |
Bold and asterisks indicate significance (p < 0.05).
Figure 1.

Second-generation males from the paternal stress lineage (F2-S) show dysmasculinized brain gene expression and miRNA expression patterns on PN1. A, Heat map illustration of custom Taqman qRT-PCR Array results demonstrating a broad shift in gene expression in the PN1 brain of F2-S male mice from a male-typical (F2-C♂) to a more female-typical (F2-C♀) pattern. B, Statistical analyses for sex differences detected 17 genes in the PN1 brain from our custom Taqman Array. In F2-S male PN1 brains, 13 of these 17 genes displayed expression levels closer to F2-C females than to F2-C male levels. C, As F2-S males show a reduced organizational masculinization, we examined gene expression for central estrogen programming targets: aromatase, and estrogen receptor α (ERα) and β (ERβ). ERβ was significantly increased in the F2-S male PN1 brain compared to F2-C male. D, Examination of the miRNA environment in F2 PN1 brain was examined using a miRNA Array. miR-322, miR-574–3p, and miR-873 expression were dysmasculinized in F2-S male mice. A single predicted shared gene target of these three miRs, β-glycan (TGFβr3), was identified by the database miRDB.org and examined in F2 PN1 brain. Where we found a reduction in miR expression in F2-S male mice, we detected an expected increase in expression of β-glycan. All data are mean per group ± SEM, n = 3–5 litters/group, *p < 0.05.
To identify potential mediators of the program of dysmasculinized gene expression, we assayed the F2 PN1 brain expression of aromatase, ERα, and ERβ, known effectors of masculinization during the perinatal sensitive period (Fig. 1C). While there was no significant effect of group on aromatase (F(2,10) = 0.69; p = 0.70) or ERα levels (F(2,10) = 1.51; p = 0.27), ERβ expression was elevated in F2-S♂ compared to F2-C♂ (F(2,10) = 4.23; p = 0.05).
F2 postnatal day 1 (PN1) brain expression of miRNA and predicted targets
To identify potential alternative mediators of a program of dysmasculinized gene expression in F2-S males, the F2 PN1 brain miRNA environment was assayed. miRNAs with expression levels that displayed a statistically significant effect of group are displayed in Table 2. The expression of three of these miRNAs (miR-322, miR-574–3p, and miR-873) appeared dysmasculinized in F2-S♂ (Fig. 1D).
Table 2.
PN1 brain expression of statistically significant miRNAs assayed by Taqman qRT-PCR Array
| miRNA | F2-C♂ | F2-S♂ | F2-C♀ | ANOVA |
|---|---|---|---|---|
| miR-322 | 1 ± 0.05A | 0.71 ± 0.06B | 0.81 ± 0.08AB | F(2,10) = 4.61; p = 0.04 |
| miR-574-3p | 1 ± 0.08A | 0.81 ± 0.06AB | 0.7 ± 0.06B | F(2,8) = 4.48; p = 0.05 |
| miR-873 | 1 ± 0.26A | 0.33 ± 0.1AB | 0.14 ± 0.06B | F(2,7) = 5.0; p = 0.04 |
| miR-302b | 1 ± 0.18A | 2.23 ± 0.28B | 1.07 ± 0.23A | F(2,8) = 5.22; p = 0.04 |
| miR-28 | 1 ± 0.11A | 1.43 ± 0.08B | 1.08 ± 0.08A | F(2,8) = 4.43; p = 0.04 |
| miR-216b | 1 ± 0.11A | 1.11 ± 0.15A | 0.38 ± 0.04B | F(2,10) = 16.51; p = 0.0007 |
| miR-532-5p | 1 ± 0.05A | 0.97 ± 0.02AB | 0.83 ± 0.04B | F(2,9) = 4.66; p = 0.04 |
Superscript indicates post hoc analysis
To determine whether miRNA changes in F2 PN1 brains were associated with altered expression of target transcripts, expression of genes that were shared predicted targets of miRNAs identified as significantly changed by early prenatal stress, and with potential relevance to disease mechanisms, were assayed by Taqman qRT-PCR. β-Glycan was the only predicted target of all three dysmasculinized miRNAs (miR-322, miR-574–3p, and miR-873). There was significantly greater expression of β-glycan in F2-S♂ than in F2-C♂ (F(2,10) = 4.99; p = 0.03) (Fig. 1D). There was also significantly greater expression of Reep3, the shared predicted target of miR-302b and miR-28, in F2-S♂ (1.23 ± 0.11) compared to F2-C♀ (0.91 ± 0.06) (F(2,10) = 4.64; p = 0.04). There were no significant differences in expression of the additional predicted targets of miR-322 and miR-873, Plxna2 (F(2,10) = 0.72; p = 0.51) and Prkar2a (F(2,10) = 0.23; p = 0.23), or of the predicted targets of miR-28 and miR-302b, Unk (F(2,10) = 0.37; p = 0.70) and Hif1an (F(2,10) = 0.68; p = 0.53).
Analysis of adult F2 behavior and physiology
F2 adult male morphology
To assess the degree to which paternal (F1) prenatal stress exposure affects morphological measures of masculinization, we examined male anogenital distances and testis weights (Fig. 2A,B). F2-S♂ had reduced anogenital distances (one-tail t = 2.7; p = 0.01) and reduced testis weights (one-tail t = 1.97; p = 0.04).
Figure 2.

Analyses of physiological and behavioral measures in adult second-generation males from the paternal stress lineage (F2-S♂) show a similar dysmasculinized physiology and stress-sensitive phenotype as their sires. As further evidence of dysmasculinization programmed during the perinatal period by testosterone, adult F2-S males showed a significant reduction in both anogenital distance lengths (A) and testis weights (B) compared to control males (F2-C♂). Behaviorally, while not all aspects of the first-generation stress-sensitive phenotype were transmitted along the paternal lineage to second-generation (F2) male offspring, we did detect increased immobility in a tail suspension test in F2-S male mice (C). Similar to first-generation findings, no further increase in immobility was observed in F2-S female offspring (F2-S♀) compared to control females (F2-C♀). E, While there was no statistically significant effect of F2-S in latency to locate the target in the Barnes maze spatial learning task, the direction of effect in F2-S males was similar to that previously reported for first-generation offspring. F, No differences were detected in latencies in F2 control and stress females. No main effects of F2-S in either male (G) or female (H) offspring were detected for corticosterone levels in response to an acute restraint stress. All data are mean per group ± SEM, n = 6–9 litters/group, *p < 0.05.
F2 adult behavior
To determine whether prenatal stressed males (F1) could transmit their dysmasculinized stress-sensitive phenotype to F2 offspring, we examined F2 adult performance in the TST and Barnes maze. F2-S♂ spent significantly more time immobile than F2-C♂ (one-tailed t(12) = 1.85, p = 0.04) (Fig. 2C). There was no corresponding increase in F2-S♀ immobility relative to F2-C♀ (one-tailed t(12) = 0.17, p = 0.44) (Fig. 2D). Analyzing Barnes maze performance, there was no statistically significant between-subjects effect of paternal (F1) prenatal stress exposure in males (F(1,9) = 0.09; p = 0.40). As expected, there was a significant within subjects effect of time (F(5,5) = 6.91; p = 0.03) (Fig. 2E). In females, there was also no significant between-subjects effect of paternal (F1) prenatal stress exposure (F(1,11) = 0.03; p = 0.57), and again there was a significant within-subjects effect of time (F(5,7) = 4.34; p = 0.02) (Fig. 2F).
F2 adult HPA stress axis
To examine the impact of paternal (F1) prenatal stress exposure on F2 offspring HPA axis sensitivity, we examined corticosterone levels in response to a 15 min restraint stress. In males, there was no significant between-subjects effect of paternal prenatal stress exposure on corticosterone levels (F(1,12) = 0.96; p = 0.35), but there was a significant within-subjects effect of time (F(3,10) = 51.0; p < 0.0001) (Fig. 2G). In females, as in males, there was no significant between-subjects effect of paternal prenatal stress exposure on corticosterone response (F(1,14) = 0.89; p = 0.36), though there was a significant within-subjects effect of time (F(3,12) = 33.5; p < 0.0001) (Fig. 2H).
Effects of formestane treatment on the neonatal miRNA environment
To determine the role of organizational gonadal hormones in the regulation of the neonatal brain miRNA environment, we assayed changes in brain miRNA complement 24 h following a PN1 injection of the aromatase inhibitor, formestane. These data are displayed in a heat map (Fig. 3). Hierarchical clustering analysis using Pearson correlation as a metric successfully segregated male vehicle samples from formestane-treated male and female vehicle samples, while it was unable to distinguish between formestane-treated male and vehicle-treated female samples (Fig. 3). Formestane significantly increased expression of miR-143 (t(12) = 3.0, p = 0.02), miR-152 (t(12) = 3.69, p = 0.005), miR-18a (t(12) = 2.35, p = 0.04), miR-298 (t(12) = 2.24, p = 0.05), miR-301b (t(12) = 2.29, p = 0.04), miR-34a (t(12) = 2.65, p = 0.03), miR-362–3p (t(12) = 2.79, p = 0.02), miR-365 (t(12) = 2.75, p = 0.02), miR-384–3p (t(12) = 3.34, p = 0.007), miR-448 (t(12) = 2.47, p = 0.03), miR-451 (t(12) = 2.57, p = 0.03), and miR-674 (t(12) = 2.38, p = 0.04). Formestane significantly reduced expression of miR-133b (t(12) = 4.16, p = 0.002), miR-15a (t(12) = 2.23, p = 0.05), miR-467c (t(12) = 2.47, p = 0.03), and miR-671–3p (t(12) = 2.54, p = 0.04).
Figure 3.
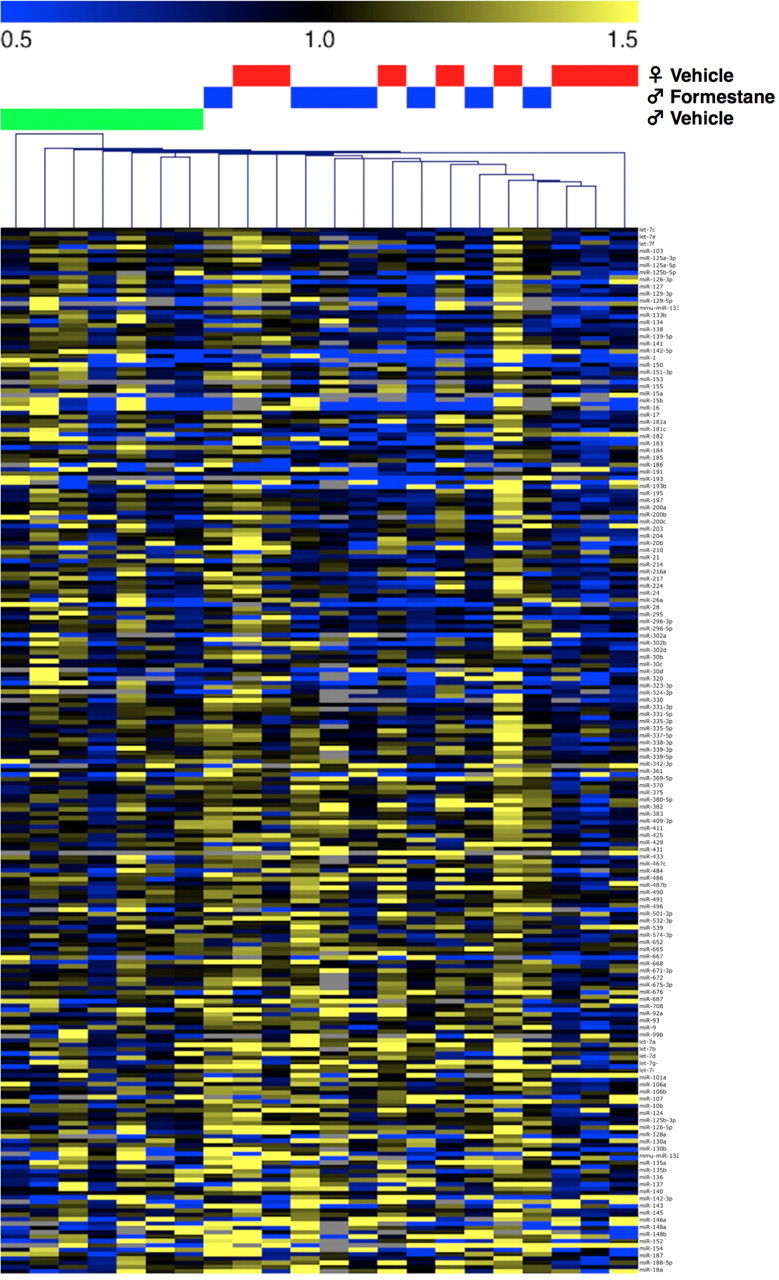
Aromatase inhibition dramatically dysmasculinizes the neonatal brain miRNA environment during the perinatal sensitive period. Administration of a single injection of the aromatase inhibitor, formestane (20 μg), on PN1 produced a profound effect on the brain miRNA environment as analyzed by miRNA Taqman qRT-PCR Array. Pearson Correlational Hierarchical Clustering analysis of miRNA expression patterns was unable to distinguish between control female and formestane-treated males, while completely segregating control male samples from these groups, supporting a novel role of miRNA in organizing the sexually dimorphic brain. All data are mean per group ± SEM, n = 7–8 litters/group.
Discussion
Epidemiological studies have linked sex-biased neurodevelopmental disorders, including autism and schizophrenia, with prenatal stress (Huttunen and Niskanen, 1978; van Os and Selten, 1998; Khashan et al., 2008; Kinney et al., 2008). Animal models of prenatal and postnatal stress have provided insight into sensitive periods and sex-specific vulnerabilities related to neurodevelopmental disorder etiology (Champagne and Meaney, 2007; Mueller and Bale, 2007; Kapoor and Matthews, 2008; Mueller and Bale, 2008; Cottrell and Seckl, 2009; Biala et al., 2010; Eiland and McEwen, 2010; Ivy et al., 2010; Korosi et al., 2010). We previously identified early gestation as a period sensitive to the sex-specific programming effects of prenatal stress in which male offspring showed a dysmasculinized phenotype in behavioral and physiological stress measures as adults (Mueller and Bale, 2007, 2008). As certain disease outcomes persist into subsequent generations, we examined the paternal transmission and programming of the prenatal stress induced dysmasculinized phenotype in second-generation (F2) offspring.
F2 brain gene expression was examined during the perinatal sensitive period to identify mechanisms of a disruption in masculinization in the F2 male brain. This period is critical for the organization of the sexually dimorphic brain by gonadal hormones. Using a custom Taqman qRT-PCR Array for genes involved in neurodevelopment, we observed a broad shift in expression from a male-typical to a more female-typical pattern in the F2 male offspring of prenatally stressed sires (F2-S). In F2-S male PN1 brains, 13 of 17 genes with statistical sex differences displayed expression levels closer to F2-C females than to F2 control (F2-C) male levels. These data correlate with diminished sex differences previously reported in adult hippocampal gene expression of prenatally stressed rats, supporting the hypothesis that disrupted masculinization during the perinatal sensitive period may be a mechanism through which paternal (F1) prenatal stress exposure impacts F2 offspring development (Biala et al., 2010).
Sex differences in gene expression result from combinations of chromosomal and hormonal effects. The male brain is organized in a sex-specific manner by a surge of testosterone during the perinatal sensitive period (Phoenix et al., 1959; McCarthy et al., 2009a). Testosterone is converted to estradiol by a neuronal-specific aromatase where it alters gene expression to masculinize and defeminize neurocircuitry through the estrogen receptors ERα and ERβ. We examined the expression of these primary effectors to determine whether their dysregulation was associated with the broad shift in gene expression observed in F2-S males. While aromatase expression was unchanged, both ERα and ERβ appeared upregulated, an effect suggestive of reduced ligand availability supporting a hypothesis for decreased perinatal testosterone in F2-S males. To identify potential alternative mediators of the dysmasculinized gene expression in F2-S males, we examined the PN1 brain miRNA environment. miRNAs are small non-protein-coding RNAs involved in the posttranscriptional regulation of genes (Bartel, 2009). Interestingly, a single miRNA may interact with up to a hundred target transcripts, potentially regulating critical gene families involved in early neurodevelopment. We identified 3 miRNAs whose expression appeared dysmasculinized in F2-S males, and 2 miRNAs that showed a significant effect of paternal (F1) prenatal stress. Several of these miRNAs have known functions in peripheral tissues (Caruso et al., 2010; Ghosh et al., 2010; Qin et al., 2010). To determine whether these changes had functional consequences on gene expression, we identified predicted gene targets using the web-based algorithm miRDB (Wang, 2008; Wang and El Naqa, 2008). Only one gene, β-glycan (TGFβr3), was a shared predicted target of all three dysmasculinized miRNAs. As would be predicted based on the reduced expression of miR-322, miR-574-3p, and miR-873, β-glycan expression was significantly increased in the F2-S male PN1 brain. Beta-glycan is a member of the TGFβ superfamily expressed in adult brain, pituitary, and gonadal tissues where it acts as an accessory protein, binding other TGFβ isoforms, such as inhibin A, and increasing their receptor affinity (Lewis et al., 2000; MacConell et al., 2002). Interestingly, in pituitary gonadotrophs and gonadal Leydig or theca cells, β-glycan is involved in regulating the release of gonadal hormones (MacConell et al., 2002; Chapman and Woodruff, 2003; Wiater et al., 2009). As a role for β-glycan in neurodevelopment has not been identified, our data suggest that it may serve an unappreciated role in the organization of the sexually dimorphic brain.
As an additional physiological marker programmed by perinatal testosterone, adult male anogenital distances were measured (Scott et al., 2008). As predicted, F2-S males showed a significantly reduced anogenital distance and adult testis weights, supportive of decreased testosterone exposure during the perinatal sensitive period. Interestingly, studies examining prenatal stress during late pregnancy have also reported decreased perinatal testosterone, adult anogenital distance, and testis weight in rats (Dahlöf et al., 1978; Ward and Weisz, 1980). It is important to note that we are examining these measures in F2 animals that were not themselves exposed to any prenatal manipulation. Thus, these data suggest that reduced exposure to organizational hormones during the perinatal sensitive period is a common marker of gestational stress experience, and that this effect can be transmitted along the paternal lineage to F2 male offspring. Of translational importance, male schizophrenics have been reported to display reduced circulating testosterone and disruptions in brain masculinization (Gur et al., 2004; Goldstein et al., 2007). Further, boys with prepsychotic prodromal symptoms had significantly lower testosterone levels during adolescents, a period of increased psychotic disorder onset (van Rijn et al., 2011).
To examine F2-S male adult dysmasculinized and stress-sensitive phenotypes, we measured their stress responsivity in the tail suspension test, Barnes maze, and HPA stress axis. These tests were selected as they measure predictable sex differences in stress-provoking environments, and performance in these tests was previously found to be significantly dysmasculinized in F1 prenatally stressed males (Mueller and Bale, 2007, 2008). In the tail suspension test, F2-S males spent significantly more time immobile than F2-C males. No effect of paternal prenatal stress was detected in females. These results are similar to those reported in the first generation (Mueller and Bale, 2008). While there was not a statistically significant effect of F2-S on overall performance in the Barnes maze or HPA axis sensitivity, males did show a general trend for a pattern of stress responsivity similar to that identified in F1 prenatally stressed males (Mueller and Bale, 2008). Thus, it appears that aspects of the adult dysmasculinized stress-sensitive phenotype were transmitted from F1 prenatally stressed sires to their F2 male offspring. It is also possible that increased numbers of litters may have provided sufficient statistical power to identify significant effects in additional stress tests.
As our data point to a likely reduction in testosterone-mediated developmental organization in F2-S male brains, we hypothesized that miRNAs in the brain are responsive to organizational gonadal hormones. Therefore, in a subsequent study examining the influences of estradiol on the neonatal brain miRNA environment, we administered the aromatase inhibitor, formestane, to PN1 male neonates. miRNA expression was then assayed using a miRNA Taqman qRT-PCR Array. Aromatase inhibition dramatically dysmasculinized the brain miRNA environment where statistical hierarchical clustering was unable to distinguish between formestane-treated males and control females based on miRNA expression patterns, while completely segregating control males from these groups. Thus, these data confirm the dynamic response of the miRNA environment during this critical window. Gonadal hormones have previously been shown to regulate miRNAs in peripheral target tissues (Klinge, 2009; Delić et al., 2010; Narayanan et al., 2010). However, our data appear to demonstrate a novel impact of organizational hormones on brain miRNA expression during the perinatal sensitive period. Epigenetic mechanisms have been attributed to gonadal hormone status and shown to influence brain sexual differentiation and may intersect with miRNAs to program the sexually dimorphic brain (McCarthy et al., 2009b; Auger and Auger, 2011; Auger et al., 2011).
Our studies provide intriguing evidence for the paternal transmission of prenatal stress effects on neurodevelopmental processes including programming of the miRNA environment and adult stress responsivity. Transmission through the paternal lineage excludes confounds associated with maternal transmission, such as the intrauterine environment or maternal behaviors, and implicates gametic epigenetic mechanisms (Youngson and Whitelaw, 2008). However, we cannot completely discount paternal experience effects that, while unlikely, could occur during the brief time the males are in the cage with females where stress-sensitive F1 males may impart some aspect of their behavior upon the pregnant dam. A recent report examining a postnatal stress model has also demonstrated the ability of early-life maternal separation to alter adult behavior and methylation patterns of several genes in the germ line of male mice, with effects persisting into second-generation offspring (Franklin et al., 2010). In addition, the dysmasculinization we observed in F2-S male offspring importantly points to a developmental window of susceptibility during which the programming effects of early prenatal stress exposure may manifest. As such, identifying developmental processes affected during this window, such as the dynamic changes in miRNAs detected, may lead to critical therapeutic targets or biomarkers predictive for neurodevelopmental diseases, particularly in at-risk pregnancies. Overall, these data support an early gestational period vulnerable to prenatal stress epigenetic programming of the male germline, permitting paternal transmission into subsequent generations.
Footnotes
This work was supported by NIH Grants MH087597 and MH091258. We thank A. Scott, D. Fisher, and C. Taylor for technical assistance, A. Morgan for graphics support, and G. Dunn for scientific discussion.
References
- Amateau SK, Alt JJ, Stamps CL, McCarthy MM. Brain estradiol content in newborn rats: sex differences, regional heterogeneity, and possible de novo synthesis by the female telencephalon. Endocrinology. 2004;145:2906–2917. doi: 10.1210/en.2003-1363. [DOI] [PubMed] [Google Scholar]
- Arnold AP, Gorski RA. Gonadal steroid induction of structural sex differences in the central nervous system. Annu Rev Neurosci. 1984;7:413–442. doi: 10.1146/annurev.ne.07.030184.002213. [DOI] [PubMed] [Google Scholar]
- Auger AP, Auger CJ. Epigenetic turn ons and turn offs: chromatin reorganization and brain differentiation. Endocrinology. 2011;152:349–353. doi: 10.1210/en.2010-0793. [DOI] [PubMed] [Google Scholar]
- Auger CJ, Coss D, Auger AP, Forbes-Lorman RM. Epigenetic control of vasopressin expression is maintained by steroid hormones in the adult male rat brain. Proc Natl Acad Sci U S A. 2011;108:4242–4247. doi: 10.1073/pnas.1100314108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale TL, Baram TZ, Brown AS, Goldstein JM, Insel TR, McCarthy MM, Nemeroff CB, Reyes TM, Simerly RB, Susser ES, Nestler EJ. Early life programming and neurodevelopmental disorders. Biol Psychiatry. 2010;68:314–319. doi: 10.1016/j.biopsych.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biala YN, Bogoch Y, Bejar C, Linial M, Weinstock M. Prenatal stress diminishes gender differences in behavior and in expression of hippocampal synaptic genes and proteins in rats. Hippocampus. 2010 doi: 10.1002/hipo.20825. [DOI] [PubMed] [Google Scholar]
- Bingham B, Viau V. Neonatal gonadectomy and adult testosterone replacement suggest an involvement of limbic arginine vasopressin and androgen receptors in the organization of the hypothalamic-pituitary-adrenal axis. Endocrinology. 2008;149:3581–3591. doi: 10.1210/en.2007-1796. [DOI] [PubMed] [Google Scholar]
- Caruso P, MacLean MR, Khanin R, McClure J, Soon E, Southgate M, MacDonald RA, Greig JA, Robertson KE, Masson R, Denby L, Dempsie Y, Long L, Morrell NW, Baker AH. Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler Thromb Vasc Biol. 2010;30:716–723. doi: 10.1161/ATVBAHA.109.202028. [DOI] [PubMed] [Google Scholar]
- Champagne FA, Meaney MJ. Transgenerational effects of social environment on variations in maternal care and behavioral response to novelty. Behav Neurosci. 2007;121:1353–1363. doi: 10.1037/0735-7044.121.6.1353. [DOI] [PubMed] [Google Scholar]
- Chapman SC, Woodruff TK. Betaglycan localization in the female rat pituitary: implications for the regulation of follicle-stimulating hormone by inhibin. Endocrinology. 2003;144:5640–5649. doi: 10.1210/en.2003-0670. [DOI] [PubMed] [Google Scholar]
- Cottrell EC, Seckl JR. Prenatal stress, glucocorticoids and the programming of adult disease. Front Behav Neurosci. 2009;3:19. doi: 10.3389/neuro.08.019.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlöf LG, Hård E, Larsson K. Influence of maternal stress on the development of the fetal genital system. Physiol Behav. 1978;20:193–195. doi: 10.1016/0031-9384(78)90072-0. [DOI] [PubMed] [Google Scholar]
- Delić D, Grosser C, Dkhil M, Al-Quraishy S, Wunderlich F. Testosterone-induced upregulation of miRNAs in the female mouse liver. Steroids. 2010;75:998–1004. doi: 10.1016/j.steroids.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Dunn GA, Bale TL. Maternal high-fat diet promotes body length increases and insulin insensitivity in second-generation mice. Endocrinology. 2009;150:4999–5009. doi: 10.1210/en.2009-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiland L, McEwen BS. Early life stress followed by subsequent adult chronic stress potentiates anxiety and blunts hippocampal structural remodeling. Hippocampus. 2010 doi: 10.1002/hipo.20862. [DOI] [PubMed] [Google Scholar]
- Franklin TB, Russig H, Weiss IC, Gräff J, Linder N, Michalon A, Vizi S, Mansuy IM. Epigenetic transmission of the impact of early stress across generations. Biol Psychiatry. 2010;68:408–415. doi: 10.1016/j.biopsych.2010.05.036. [DOI] [PubMed] [Google Scholar]
- Ghosh G, Subramanian IV, Adhikari N, Zhang X, Joshi HP, Basi D, Chandrashekhar YS, Hall JL, Roy S, Zeng Y, Ramakrishnan S. Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-alpha isoforms and promotes angiogenesis. J Clin Invest. 2010;120:4141–4154. doi: 10.1172/JCI42980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JM, Seidman LJ, Makris N, Ahern T, O'Brien LM, Caviness VS, Jr, Kennedy DN, Faraone SV, Tsuang MT. Hypothalamic abnormalities in schizophrenia: sex effects and genetic vulnerability. Biol Psychiatry. 2007;61:935–945. doi: 10.1016/j.biopsych.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Guerrero-Bosagna C, Settles M, Lucker B, Skinner MK. Epigenetic transgenerational actions of vinclozolin on promoter regions of the sperm epigenome. PLoS ONE. 2010;5:e13100. doi: 10.1371/journal.pone.0013100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur RE, Kohler C, Turetsky BI, Siegel SJ, Kanes SJ, Bilker WB, Brennan AR, Gur RC. A sexually dimorphic ratio of orbitofrontal to amygdala volume is altered in schizophrenia. Biol Psychiatry. 2004;55:512–517. doi: 10.1016/j.biopsych.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Huttunen MO, Niskanen P. Prenatal loss of father and psychiatric disorders. Arch Gen Psychiatry. 1978;35:429–431. doi: 10.1001/archpsyc.1978.01770280039004. [DOI] [PubMed] [Google Scholar]
- Ivy AS, Rex CS, Chen Y, Dubé C, Maras PM, Grigoriadis DE, Gall CM, Lynch G, Baram TZ. Hippocampal dysfunction and cognitive impairments provoked by chronic early-life stress involve excessive activation of CRH receptors. J Neurosci. 2010;30:13005–13015. doi: 10.1523/JNEUROSCI.1784-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Matthews SG. Short periods of prenatal stress affect growth, behaviour and hypothalamo-pituitary-adrenal axis activity in male guinea pig offspring. J Physiol. 2005;566:967–977. doi: 10.1113/jphysiol.2005.090191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Matthews SG. Prenatal stress modifies behavior and hypothalamic-pituitary-adrenal function in female guinea pig offspring: effects of timing of prenatal stress and stage of reproductive cycle. Endocrinology. 2008;149:6406–6415. doi: 10.1210/en.2008-0347. [DOI] [PubMed] [Google Scholar]
- Khashan AS, Abel KM, McNamee R, Pedersen MG, Webb RT, Baker PN, Kenny LC, Mortensen PB. Higher risk of offspring schizophrenia following antenatal maternal exposure to severe adverse life events. Arch Gen Psychiatry. 2008;65:146–152. doi: 10.1001/archgenpsychiatry.2007.20. [DOI] [PubMed] [Google Scholar]
- Kinney DK, Miller AM, Crowley DJ, Huang E, Gerber E. Autism prevalence following prenatal exposure to hurricanes and tropical storms in Louisiana. J Autism Dev Disord. 2008;38:481–488. doi: 10.1007/s10803-007-0414-0. [DOI] [PubMed] [Google Scholar]
- Klinge CM. Estrogen regulation of MicroRNA expression. Curr Genomics. 2009;10:169–183. doi: 10.2174/138920209788185289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korosi A, Shanabrough M, McClelland S, Liu ZW, Borok E, Gao XB, Horvath TL, Baram TZ. Early-life experience reduces excitation to stress-responsive hypothalamic neurons and reprograms the expression of corticotropin-releasing hormone. J Neurosci. 2010;30:703–713. doi: 10.1523/JNEUROSCI.4214-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis KA, Gray PC, Blount AL, MacConell LA, Wiater E, Bilezikjian LM, Vale W. Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature. 2000;404:411–414. doi: 10.1038/35006129. [DOI] [PubMed] [Google Scholar]
- MacConell LA, Leal AM, Vale WW. The distribution of betaglycan protein and mRNA in rat brain, pituitary, and gonads: implications for a role for betaglycan in inhibin-mediated reproductive functions. Endocrinology. 2002;143:1066–1075. doi: 10.1210/endo.143.3.8707. [DOI] [PubMed] [Google Scholar]
- Maher B. Personal genomes: the case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- McCarthy MM, Wright CL, Schwarz JM. New tricks by an old dogma: mechanisms of the organizational/activational hypothesis of steroid-mediated sexual differentiation of brain and behavior. Horm Behav. 2009a;55:655–665. doi: 10.1016/j.yhbeh.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MM, Auger AP, Bale TL, De Vries GJ, Dunn GA, Forger NG, Murray EK, Nugent BM, Schwarz JM, Wilson ME. The epigenetics of sex differences in the brain. J Neurosci. 2009b;29:12815–12823. doi: 10.1523/JNEUROSCI.3331-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RL, Dohanich GP, Ward IL. Effects of prenatal stress on avoidance acquisition, open-field performance and lordotic behavior in male rats. Physiol Behav. 1979;22:527–530. doi: 10.1016/0031-9384(79)90020-9. [DOI] [PubMed] [Google Scholar]
- Mueller BR, Bale TL. Impact of prenatal stress on long term body weight is dependent on timing and maternal sensitivity. Physiol Behav. 2006;88:605–614. doi: 10.1016/j.physbeh.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Mueller BR, Bale TL. Early prenatal stress impact on coping strategies and learning performance is sex dependent. Physiol Behav. 2007;91:55–65. doi: 10.1016/j.physbeh.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Mueller BR, Bale TL. Sex-specific programming of offspring emotionality after stress early in pregnancy. J Neurosci. 2008;28:9055–9065. doi: 10.1523/JNEUROSCI.1424-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R, Jiang J, Gusev Y, Jones A, Kearbey JD, Miller DD, Schmittgen TD, Dalton JT. MicroRNAs are mediators of androgen action in prostate and muscle. PLoS ONE. 2010;5:e13637. doi: 10.1371/journal.pone.0013637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3:97–106. doi: 10.4161/epi.3.2.6034. [DOI] [PubMed] [Google Scholar]
- Phoenix CH, Goy RW, Gerall AA, Young WC. Organizing action of prenatally administered testosterone propionate on the tissues mediating mating behavior in the female guinea pig. Endocrinology. 1959;65:369–382. doi: 10.1210/endo-65-3-369. [DOI] [PubMed] [Google Scholar]
- Qin L, Chen Y, Niu Y, Chen W, Wang Q, Xiao S, Li A, Xie Y, Li J, Zhao X, He Z, Mo D. A deep investigation into the adipogenesis mechanism: profile of microRNAs regulating adipogenesis by modulating the canonical Wnt/beta-catenin signaling pathway. BMC Genomics. 2010;11:320. doi: 10.1186/1471-2164-11-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznikov AG, Nosenko ND, Tarasenko LV. Prenatal stress and glucocorticoid effects on the developing gender-related brain. J Steroid Biochem Mol Biol. 1999;69:109–115. doi: 10.1016/s0960-0760(98)00151-4. [DOI] [PubMed] [Google Scholar]
- Scott HM, Hutchison GR, Jobling MS, McKinnell C, Drake AJ, Sharpe RM. Relationship between androgen action in the “male programming window,” fetal sertoli cell number, and adult testis size in the rat. Endocrinology. 2008;149:5280–5287. doi: 10.1210/en.2008-0413. [DOI] [PubMed] [Google Scholar]
- Seale JV, Wood SA, Atkinson HC, Lightman SL, Harbuz MS. Organizational role for testosterone and estrogen on adult hypothalamic-pituitary-adrenal axis activity in the male rat. Endocrinology. 2005;146:1973–1982. doi: 10.1210/en.2004-1201. [DOI] [PubMed] [Google Scholar]
- Skinner MK. Role of epigenetics in developmental biology and transgenerational inheritance. Birth Defects Res C Embryo Today. 2011;93:51–55. doi: 10.1002/bdrc.20199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M. Epigenetic inheritance and the missing heritability problem. Genetics. 2009;182:845–850. doi: 10.1534/genetics.109.102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steru L, Chermat R, Thierry B, Simon P. The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology (Berl) 1985;85:367–370. doi: 10.1007/BF00428203. [DOI] [PubMed] [Google Scholar]
- van Os J, Selten JP. Prenatal exposure to maternal stress and subsequent schizophrenia. The May 1940 invasion of The Netherlands. Br J Psychiatry. 1998;172:324–326. doi: 10.1192/bjp.172.4.324. [DOI] [PubMed] [Google Scholar]
- van Rijn S, Aleman A, de Sonneville L, Sprong M, Ziermans T, Schothorst P, van Engeland H, Swaab H. Neuroendocrine markers of high risk for psychosis: salivary testosterone in adolescent boys with prodromal symptoms. Psychol Med. 2011 doi: 10.1017/S0033291710002576. [DOI] [PubMed] [Google Scholar]
- Wang X. miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA. 2008;14:1012–1017. doi: 10.1261/rna.965408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, El Naqa IM. Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics. 2008;24:325–332. doi: 10.1093/bioinformatics/btm595. [DOI] [PubMed] [Google Scholar]
- Ward IL. Prenatal stress feminizes and demasculinizes the behavior of males. Science. 1972;175:82–84. doi: 10.1126/science.175.4017.82. [DOI] [PubMed] [Google Scholar]
- Ward IL, Weisz J. Maternal stress alters plasma testosterone in fetal males. Science. 1980;207:328–329. doi: 10.1126/science.7188648. [DOI] [PubMed] [Google Scholar]
- Weinstock M. Gender differences in the effects of prenatal stress on brain development and behaviour. Neurochem Res. 2007;32:1730–1740. doi: 10.1007/s11064-007-9339-4. [DOI] [PubMed] [Google Scholar]
- Wiater E, Lewis KA, Donaldson C, Vaughan J, Bilezikjian L, Vale W. Endogenous betaglycan is essential for high-potency inhibin antagonism in gonadotropes. Mol Endocrinol. 2009;23:1033–1042. doi: 10.1210/me.2009-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngson NA, Whitelaw E. Transgenerational epigenetic effects. Annu Rev Genomics Hum Genet. 2008;9:233–257. doi: 10.1146/annurev.genom.9.081307.164445. [DOI] [PubMed] [Google Scholar]