

Abstract
Biogenesis of a specialized organelle that supports intracellular replication of Legionella pneumophila involves the fusion of secretory vesicles exiting the endoplasmic reticulum (ER) with phagosomes containing this bacterial pathogen. Here, we investigated host plasma membrane SNARE proteins to determine whether they play a role in trafficking of vacuoles containing L. pneumophila. Depletion of plasma membrane syntaxins by RNA interference resulted in delayed acquisition of the resident ER protein calnexin and enhanced retention of Rab1 on phagosomes containing virulent L. pneumophila, suggesting that these SNARE proteins are involved in vacuole biogenesis. Plasma membrane-localized SNARE proteins syntaxin 2, syntaxin 3, syntaxin4 and SNAP23 localized to vacuoles containing L. pneumophila. The ER-localized SNARE protein Sec22b was found to interact with plasma membrane SNAREs on vacuoles containing virulent L. pneumophila, but not on vacuoles containing avirulent mutants of L. pneumophila. The addition of α-SNAP and N-ethylmaleimide-sensitive factor (NSF) to the plasma membrane SNARE complexes formed by virulent L. pneumophila resulted in the dissociation of Sec22b, indicating functional pairing between these SNAREs. Thus, L. pneumophila stimulates the non-canonical pairing of plasma membrane t-SNAREs with the v-SNARE Sec22b to promote fusion of the phagosome with ER-derived vesicles. The mechanism by which L. pneumophila promotes pairing of plasma membrane syntaxins and Sec22b could provide unique insight into how the secretory vesicles could provide an additional membrane reserve subverted during phagosome maturation.
Keywords: endoplasmic reticulum, Dot/Icm system, Legionella pneumophila, membrane fusion, Sec22b, SNARE, syntaxin, vacuole
Many intracellular pathogens have the capacity to manipulate the transport and fusion of the membrane-bound vacuoles in which they reside (1). Modulating vacuole transport is critical for pathogen survival and replication within a eukaryotic host cell. To understand how bacteria can influence transport of the vacuoles they occupy, we have been investigating biogenesis of vacuoles containing Legionella pneumophila. After being internalized by a phagocytic host cell, vacuoles containing L. pneumophila have the ability to intercept early secretory vesicles transiting between the endoplasmic reticulum (ER) and Golgi (2–5). These early secretory vesicles fuse with vacuoles containing L. pneumophila and convert a vacuole derived from the plasma membrane into a compartment into a specialized organelle that has similarities to an ER-Golgi intermediate compartment (6–8).
The processes by which L. pneumophila promotes the association and fusion of ER-derived vesicles with the vacuole require a bacterial secretion system called Dot/Icm (7,8), which is a device that delivers bacterial effector proteins into the host cytosol (9). Mutant L. pneumophila with a defective Dot/Icm system resides in a more conventional phagosome that fuses with early and late endosomes (10). Effector proteins translocated into host cells by the Dot/Icm system include an Arfguanine nucleotide exchange factor (GEF) called RalF (11), a Rab1 GEF called DrrA (also known as SidM) (12,13)and a Rab1 GTPase-activating protein (GAP) called LepB (14). The ability of these bacterial effector proteins to target specific host factors that regulate transport and fusion of early secretory organelles highlights the sophisticated strategies this pathogen has evolved for manipulating membrane transport proteins in the host secretory pathway.
Soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor (SNARE) proteins are critical components of the membrane fusion machinery (15). Intracellular membrane fusion events between donor compartment-derived vesicles and acceptor membranes are mediated by the interaction of a v-SNARE on the vesicle membrane with t-SNARE complex on the opposing membrane of the target organelle (16–19). In SNARE-mediated membrane fusion events, the v-SNARE provides a helix that pairs with three helices provided by the t-SNARE complex, which generates a four-helix bundle called a trans-SNARE complex. Formation of a trans-SNARE complex facilitates membrane fusion by bringing the two opposing membranes in close proximity (20,21). After membrane fusion, the cis-SNARE complex is disassembled, which facilitates the recycling of the t-SNARE complex to donor membranes. SNARE disassembly is catalyzed by soluble N-ethylmaleimide-sensitive factor attachment protein (α-SNAP) and NSF (22,23).
ER-derived vesicles display a v-SNARE called Sec22b that forms a complex with the Golgi-localized t-SNARE complex consisting of the proteins membrin, rBet1 and syntaxin 5 (Stx5), which promote membrane fusion (24,25). Vacuoles containing wild-type L. pneumophila acquire Sec22b, but vacuoles containing L. pneumophila mutants with a defective Dot/Icm secretion system do not acquire Sec22b (4,5). These results are consistent with the Dot/Icm system promoting the recruitment and fusion of ER-derived vesicles with vacuoles containing L. pneumophila. It was also observed that vacuoles containing wild-type L. pneumophila that acquire Sec22b do not contain detectable amounts of the cognate t-SNARE consisting of the proteins rBet1, Stx5 and membrin (4,5), raising the question of whether a t-SNARE is present on vacuoles containing L. pneumophila that can be recognized by Sec22b.
Biochemical studies using purified SNARE proteins from Saccharomyces cerevisiae found that the plasma membrane t-SNARE consisting of the proteins Sso1 and Sec9c could promote fusion of vesicles containing the ER-localized v-SNARE Sec22 (18). It has been suggested that mammalian macrophages possess a membrane fusion pathway that allows phagosomes containing latex bead particles or microbes to acquire membranes derived from the ER (26). However, fusion of ER-derived vesicles with conventional phagosomes has been difficult to study, suggesting this pathway might be tightly controlled. Here, we provide evidence in support of this hypothesis by showing that L. pneumophila infection stimulates an endogenous pathway that promotes the fusion of ER-derived vesicles with phagosomes by enhancing interactions between Sec22b and plasma membrane-localized t-SNAREs.
Results
Plasma membrane syntaxins are involved in biogenesis of Legionella pneumophila-containing vacuoles
To determine whether host plasma membrane SNAREs might be involved in the biogenesis of the L. pneumophila-containing vacuole, RNA interference (RNAi) was used to deplete syntaxin 2 (Stx2), syntaxin 3 (Stx3) and syntaxin 4 (Stx4), which comprise the non-neuronal syntaxins localized to the plasma membrane in mammalian cells (27). To facilitate these RNAi studies, we established a HEK293 cell line that produces the FcγRII protein, enabling these cells to efficiently internalize immunoglobulin G (IgG)-opsonized L. pneumophila (Figure S1). Silencing by RNAi reduced the levels of syntaxins when the cells were treated singularly and in combination (Figure 1A). Immunofluorescence micrographs showed a drop in the staining intensity of plasma membrane syntaxins after RNAi treatment, whereas the staining intensity of the plasma membrane-localized FcγRII protein expressed in these cells was not affected (Figure 1B).
Figure 1.
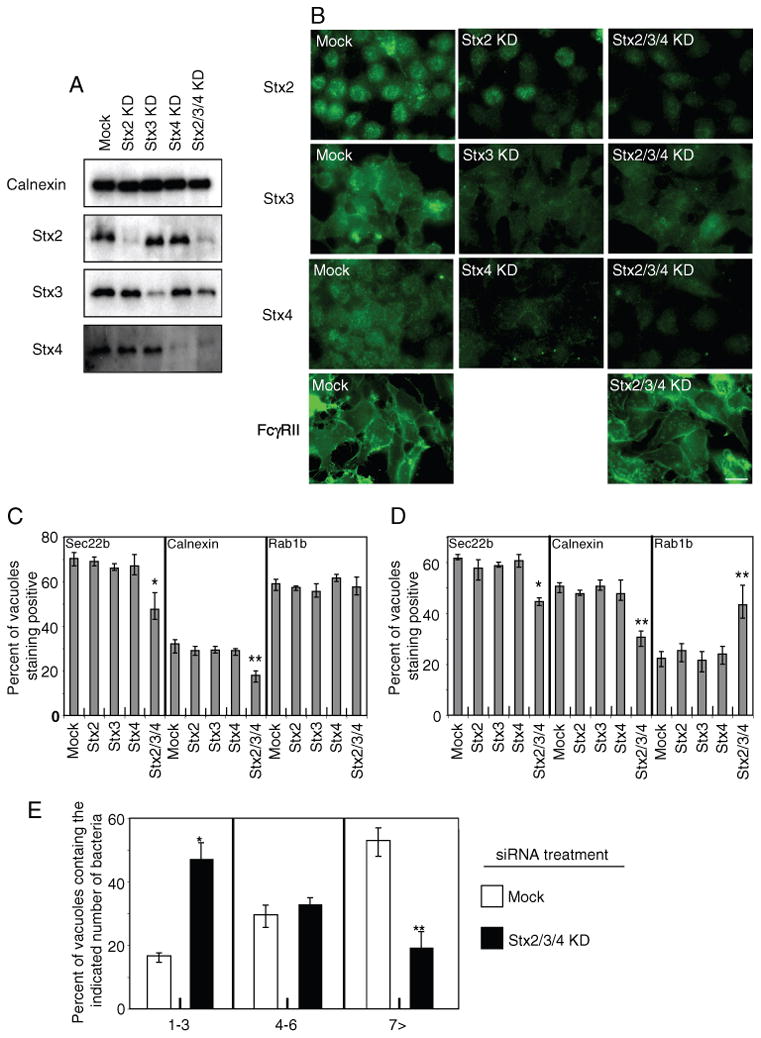
Dynamics of Sec22b, calnexin and Rab1b recruitment to vacuoles containing L. pneumophila after silencing of plasma membrane syntaxins. A) Cell lysates were obtained from HEK293-FcγRII cells transfected for 72 h with the siRNA indicated (top), and immunoblot analysis was used to measure the levels of the proteins indicated (left). Calnexin levels were used to assess equal protein loading. B) HEK293-FcγRII cells were treated with the indicated siRNA or mock treated as indicated in the upper left corner of each image panel. Cells were fixed and stained with antibodies specific for the proteins indicated on the left of each row. FcγRII staining was conducted to determine if the triple syntaxin knockdown (Stx2/3/4 KD) affected the localization of this plasma membrane protein. Scale bar = 5 μm. C and D) HEK293-FcγRII cells were transfected with siRNA targeting the indicated syntaxins (Stx2, Stx3, Stx4, Stx2/3/4) or treated with control siRNA (mock) as indicated at the bottom of the graph. Cells were fixed either 1 (C) or 4 h (D) after infection with wild-type L. pneumophila, and stained with antibodies to either Sec22b, calnexin or Rab1b as indicated in each panel. Colocalization of Sec22b, calnexin and Rab1b was assessed by fluorescence microscopy and the data are presented as the percent of vacuoles that were positive for each marker. Data are the mean ± SEM of three independent experiments in which 100 vacuoles were scored [*p < 0.02 and **p < 0.04 (C), *p < 0.002 and **p < 0.001 (D), as compared with mock-treated]. E) HEK293-FcγRII cells were transfected with an siRNA pool to silence the production of the plasma membrane-localized syntaxins (Stx2/3/4; black bars) or treated with control siRNA (mock; white bars) as indicated. Cells were fixed 8 h after infection with wild-type L. pneumophila. Intracellular replication of L. pneumophila was assessed by counting the number of bacteria residing in a single vacuole in the infected cells. The data are presented as the percent of vacuoles in mock-transfected cells (white bars) and Stx2/3/4 siRNA-transfected cells (black bars) that contained 1–3 bacteria, 4–6 bacteria or >7 bacteria. Data are the mean ± SEM of three independent experiments in which 100–120 vacuoles were scored (*p < 0.009 and **p < 0.02, as compared with mock-treated).
Vacuoles containing L. pneumophila mature into organelles that support bacterial replication through a biphasic process. After uptake, vacuoles containing L. pneumophila first interact with secretory vesicles derived from the ER, which correlates with the acquisition of Rab1 and the transmembrane protein Sec22b (4,5). After fusion with ER-derived vesicles, vacuoles containing L. pneumophila interact with the ER network and acquire resident ER proteins such as calnexin and glucose 6-phosphatase, which correlates with Rab1 cycling off the vacuole at this stage (5,7,8). To determine whether plasma membrane syntaxins were involved in maturation of vacuoles containing L. pneumophila, the kinetics of Rab1, Sec22b and calnexin association with vacuoles was assessed after syntaxin production was silenced by RNAi.
No significant difference in the percent of vacuoles staining positive for Sec22b, Rab1 and calnexin was observed at 1 and 4 h after infection by L. pneumophila when control cells were compared with cells in which a single plasma membrane-localized syntaxin had been silenced (Figure 1C, D). Thus, elimination of a single plasma membrane syntaxin did not abrogate trafficking of L. pneumophila to a measurable degree. When all three syntaxins were silenced, however, the kinetics of Sec22b and calnexin recruitment to vacuoles containing L. pneumophila was slower and Rab1 did not cycle off vacuoles as quickly as in control cells (Figure 1C, D). Silencing of Sec22b by RNAi resulted in a similar phenotype (Figure S2), consistent with plasma membrane syntaxins functioning in association with Sec22b during L. pneumophila infection. Importantly, a defect in the intracellular replication of L. pneumophila was observed in cells where the three plasma membrane-localized syntaxins were silenced, and this defect was similar to that observed in cells where Sec22b was silenced (Figure 1E). These data suggest that formation of the vacuole in which L. pneumophila replicates involves fusion events mediated by plasma membrane-localized syntaxins contained on the early vacuole and Sec22b on the ER-derived vesicles recruited to the vacuole after infection.
Plasma membrane t-SNAREs are present on vacuoles containing Legionella pneumophila
To further investigate the role of plasma membrane t-SNAREs in the biogenesis of vacuoles containing L. pneumophila, we examined the localization of endogenous t-SNARE proteins during infection. In non-neuronal cells, a plasma membrane t-SNARE complex typically consists of a transmembrane syntaxin protein associated with the peripheral membrane protein SNAP23. To create the four-helix trans-SNARE complex that promotes vesicle fusion with target membranes, the syntaxin protein contributes one helix, SNAP23 contributes two helices and the v-SNARE contributes one helix (15). As shown in Figure 2A, endogenous Stx2, Stx3, Stx4 and SNAP23 were all present on vacuoles containing L. pneumophila in the macrophage-like cell line RAW264.7. Consistent with the early phagosome containing L. pneumophila being derived from the plasma membrane, there was no difference observed in the localization of these SNARE proteins on vacuoles containing wild-type L. pneumophila compared with a type IV secretion system-deficient dotA mutant of L. pneumophila that is unable to modulate trafficking of the vacuole (Figure 2B, C). To determine whether localization of plasma membrane t-SNAREs was a property specific for macrophage-like cells, we examined localization of these SNAREs after uptake of opsonized L. pneumophila into HEK293 cells stably producing the FcγRII protein. Similar to what was observed in RAW 264.7 macrophage-like cells, endogenous t-SNARE proteins produced by the HEK293-FcγRII cells were present on vacuoles containing L. pneumophila (Figure 3). Thus, the L. pneumophila Dot/Icm system does not exclude the localization of plasma membrane-localized t-SNARE complexes during phagosome formation and these t-SNARE molecules are present on vacuoles containing L. pneumophila in both professional phagocytes and cells that are normally non-phagocytic.
Figure 2.
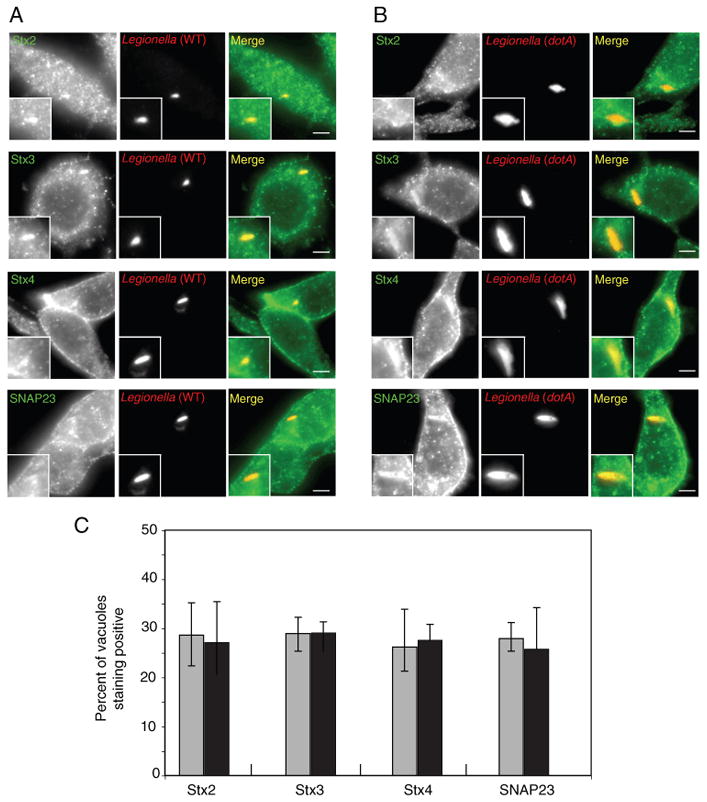
Localization of endogenous plasma membrane SNAREs to vacuoles containing L. pneumophila in RAW 264.7 cells. A and B) RAW 264.7 macrophage-like cells were infected with dsRed-expressing wild-type L. pneumophila (A) or the dotA mutant (B). At 1 h after infection, the cells were fixed and stained with antibodies specific for the proteins listed on the left (Stx2, Stx3, Stx4 or SNAP23). The insert contains a magnified image of the vacuole from each panel. Bar = 1 μm. C) Localization of the SNAREs indicated at the bottom of the graph (Stx2, Stx3, Stx4, SNAP23) was assessed for vacuoles containing either wild-type L. pneumophila (gray bars) or a dotA mutant (black bars) at 1 h after infection. Values are the mean ± SEM of three independent experiments in which 70 vacuoles were scored.
Figure 3.
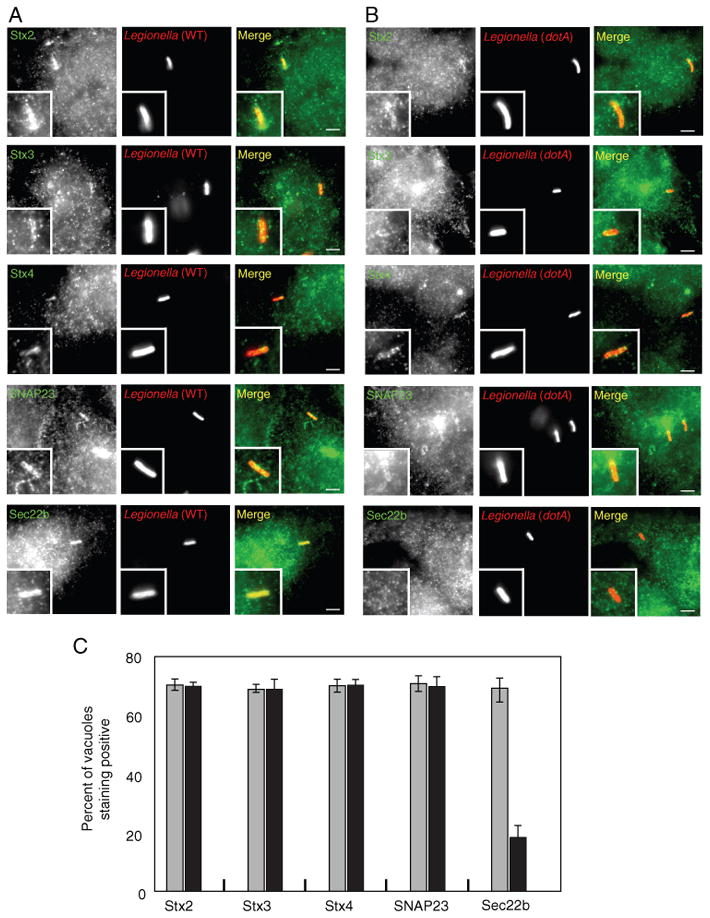
Localization of endogenous plasma membrane SNAREs to vacuoles containing L. pneumophila in HEK293-FcγRII cells. A and B) HEK293-FcγRII cells were infected with dsRed-expressing wild-type L. pneumophila (A) or the dotA mutant (B). At 1 h after infection, the cells were fixed and stained with antibodies specific for the protein listed on the left (Stx2, Stx3, Stx4, SNAP23 or Sec22b). The insert contains a magnified image of the vacuole from each panel. Bar = 1 μm. C) Localization of the protein indicated at the bottom of the graph (Stx2, Stx3, Stx4, SNAP23, Sec22b) was assessed for vacuoles containing either wild-type L. pneumophila (gray bars) or a dotA mutant (black bars) at 1 h after infection. Values are the mean ± SEM of three independent experiments in which 70 vacuoles were scored.
The Dot/Icm system modulates v-SNARE recruitment to vacuoles containing Legionella pneumophila
Plasma membrane t-SNAREs play a role in the fusion of intracellular vesicles with the plasma membrane and also in the fusion of plasma membrane-derived vesicles with endosomes (28,29). It has previously been shown that VAMP7 (TI-VAMP) is a v-SNARE localized to late endosomes. VAMP7 can pair with t-SNAREs consisting of a plasma membrane syntaxin and SNAP23 and this association promotes fusion of late endosomes with target membranes (30,31). Importantly, VAMP7 is involved in the fusion of phagosomes with late endosomes (30). HEK293-FcγRII cells stably producing a 3x-FLAG-tagged VAMP7 protein were used to assess whether plasma membrane-localized t-SNAREs on vacuoles containing L. pneumophila generate a functional trans-SNARE complex with vesicles displaying the v-SNARE VAMP7. For comparative analysis, these experiments were done in parallel with HEK293-FcγRII cells stably producing a 3x-FLAG-tagged version of the v-SNARE protein Sec22b.
Staining by immunofluorescence indicated that the two cell lines expressed 3x-FLAG-Sec22b and 3x-FLAG-VAMP7 at similar levels. In addition, FcγRII expression in the two cell lines was not affected during selection of cells stably producing the 3x-FLAG-tagged v-SNARE proteins (Figure 4A). When these cells were infected for 1 h with wild-type L. pneumophila, the majority of vacuoles stained positive for 3x-FLAG-Sec22b, whereas 3x-FLAG-VAMP7 staining was not observed on most vacuoles containing wild-type L. pneumophila (Figure 4B; left three columns). Cells were then infected with a dotA mutant to determine whether the absence of 3x-FLAG-VAMP7 staining on the vacuoles could be the result of L. pneumophila actively modulating SNARE interactions. On vacuoles containing dotA mutant L. pneumophila an opposite trend was observed, with the majority of vacuoles staining positive for 3x-FLAG-VAMP7 and very few vacuoles staining positive for 3x-FLAG-Sec22b (Figure 4B; right three columns). Similar results were obtained following infection of RAW264.7 macrophage-like cells and examining the localization of Sec22b and VAMP7 expressed endogenously (Figure 5). These results suggest that L. pneumophila Dot/Icm system modulates trafficking in part by altering the preferred utilization of VAMP7 as a v-SNARE for interactions with t-SNAREs on the plasma membrane-derived vacuole.
Figure 4.
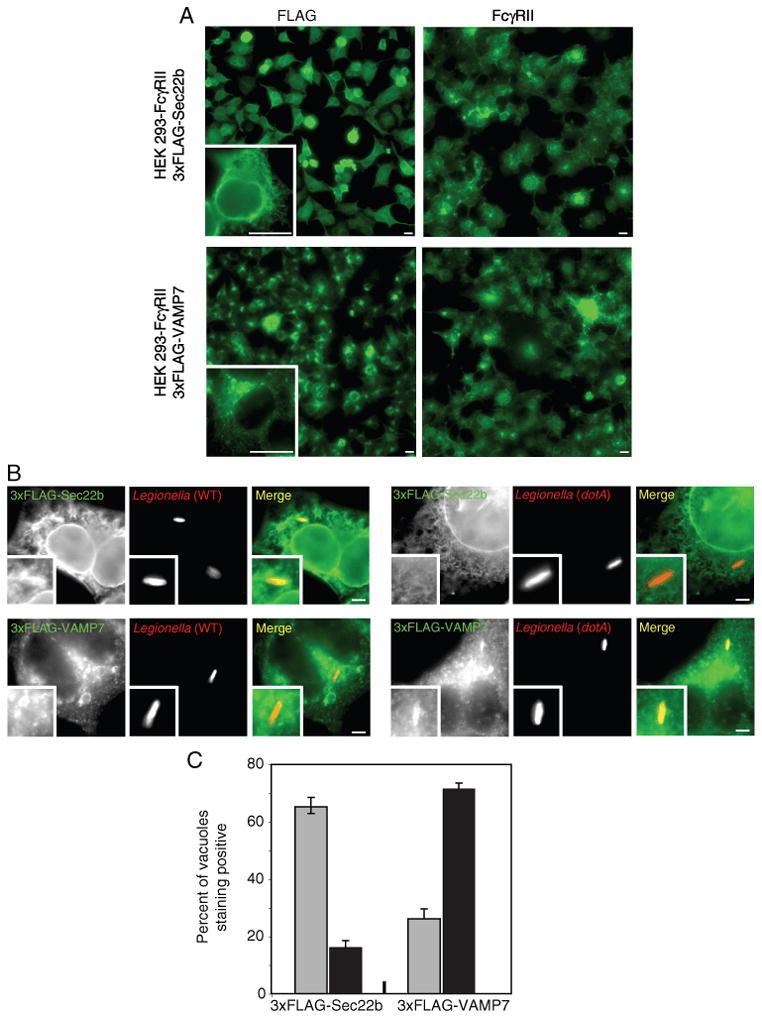
Differential localization of FLAG-tagged VAMP7 and Sec22b to vacuoles containing L. pneumophila in HEK293-FcγRII cells. A) Fluorescence micrographs of stable HEK293-FcγRII cell lines producing either 3x-FLAG-Sec22b (top panels) or 3x-FLAG-VAMP7 (bottom panels). Cells were stained with an antibody specific for FLAG (left panels) or an antibody specific for FcγRII (right panels). A higher magnification of a representative cell stained with the anti-FLAG antibody is shown in the insert. Bar = 5 μm. B) Fluorescence micrographs of stable HEK293-FcγRII cell lines producing either 3x-FLAG-Sec22b (top panels) or 3x-FLAG-VAMP7 (bottom panels) with either wild-type L. pneumophila (left series) or a dotA mutant (right series) at 1 h after infection. Anti-FLAG staining is shown in green and the L. pneumophila staining is shown in red in the merged panels. The insert contains a magnified image of the vacuole from each panel. Bar = 1 μm. C) Localization of 3x-FLAG-Sec22b and 3x-FLAG-VAMP7 was assessed for vacuoles containing either wild-type L. pneumophila (gray bars) or a dotA mutant (black bars) at 1 h after infection. Values are the mean ± SEM of three independent experiments in which 100 vacuoles were scored.
Figure 5.
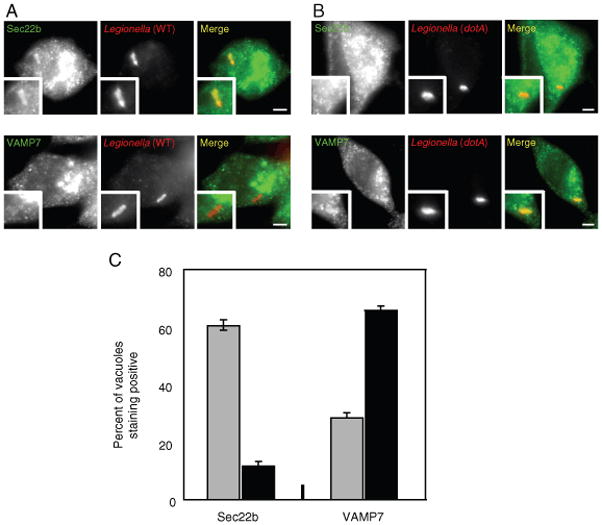
Differential localization of endogenous VAMP7 and Sec22b to vacuoles containing L. pneumophila in RAW 264.7 cells. A and B) Fluorescence micrographs of RAW 264.7 macrophage-like cells stained with an antibody specific for Sec22b (top panels) or VAMP7 (bottom panels) with either wild-type L. pneumophila (A) or a dotA mutant (B) at 1 h after infection. The insert contains a magnified image of the vacuole from each panel. Bar = 1 μm. C) Localization of Sec22b and VAMP7 was assessed for vacuoles containing either wild-type L. pneumophila (gray bars) or a dotA mutant (black bars) at 1 h after infection. Values are the mean ± SEM of three independent experiments in which 100 vacuoles were scored.
The Dot/Icm system stimulates functional interactions between Sec22b and plasma membrane-localized syntaxins
Although the immunofluorescence localization studies were consistent with L. pneumophila altering v-SNARE utilization, whether Sec22b was functioning as a v-SNARE in combination with a plasma membrane-localized t-SNARE remained to be determined. To address this question, we investigated whether L. pneumophila infection stimulated the formation of a SNAREpin assembly consisting of Sec22b paired with a plasma membrane-associated t-SNARE complex. Protein complexes were precipitated from HEK293-FcγRII cells stably producing 3x-FLAG-Sec22b after infection with wild-type or dotA mutant L. pneumophila. As expected, an interaction between 3x-FLAG-Sec22b and Stx5 was detected in cells regardless of infection conditions (Figure 6A). There was no detectable immunoprecipitation of calnexin, which indicates that the precipitates are free of non-interacting components of the ER vesicles containing 3x-FLAG-Sec22b (Figure 6A). When the 3x-FLAG-Sec22b complexes were probed for components of the plasma membrane-localized t-SNARE complexes, infection by wild-type L. pneumophila was found to enhance complex formation between Sec22b and plasma membrane syntaxins (Figure 6A). Infection by dotA mutant L. pneumophila did not enhance interactions between 3x-FLAG-Sec22b and plasma membrane syntaxins (Figure 6A). An interaction between Sec22b and endosomal Stx7 was not detected in cells infected with wild-type L. pneumophila or the dotA mutant (Figure 6A). In two independent experiments where immunoprecipitation of syntaxin proteins with Sec22b was measured by densitometry, it was determined that infection by wild-type L. pneumophila significantly enhanced the association of plasma membrane syntaxins and Sec22b (Figure S3). In addition, it was found that the association between plasma membrane syntaxins and Sec22b increased proportionally with an increase in the multiplicity of infection (MOI) by wild-type L. pneumophila (Figure S4). Consistent with the hypothesis that SNAP23 functions in association with syntaxins to create a t-SNARE complex, coprecipitation of SNAP23 with Sec22b was observed in L. pneumophila-infected cells. An association between SNAP23and Sec22b was also detected in uninfected cells, which was expected based on previous studies showing that Sec22b forms a SNARE complex with ER-localized Stx18 and SNAP23 (32).
Figure 6.
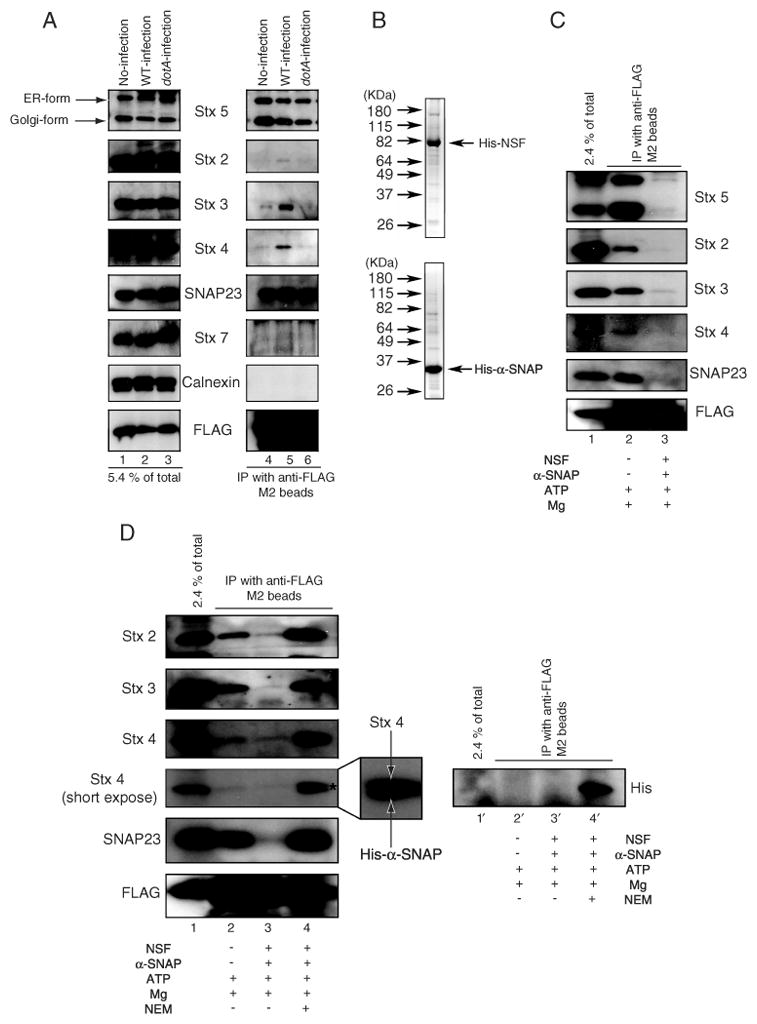
Sec22b forms a complex with plasma membrane t-SNAREs upon infection with wild-type L. pneumophila. A) HEK293-FcγRII 3x-FLAG-Sec22b cells were infected without (lanes 1 and 4) or with wild-type L. pneumophila (lanes 2 and 5) or dotA mutant (lanes 3 and 6). Sec22b was immunoprecipitated with anti-FLAG M2 beads from lysates prepared 1 h after infection. A portion of the cell lysate (5.4% of total; left lanes) and the immunoprecipitated proteins (IP with anti-FLAG M2 beads; right lanes) were separated by SDS PAGE and the amounts of the indicated proteins (Stx5, Stx2, Stx3, Stx4, SNAP23, calnexin, FLAG-Sec22b) were determined by immunoblot analysis. B) Recombinant His6-NSF and His6-α-SNAP were separated by SDS PAGE and stained with coomasie blue. Indicated on the left are the positions of weight markers. C) HEK293-FcγRII 3x-FLAG-Sec22b cells were infected with wild-type L. pneumophila. Sec22b was immunoprecipitated with anti-FLAG M2 beads from lysates prepared 1 h after infection. As indicated in the bottom legend, cell lysates were treated with 0.5 mm ATP and 8 mm MgCl2 (lane 2) or 0.5 mm ATP, 8 mm MgCl2, 10 μg/mL His6-NSF and 5 μg/mL His6-α-SNAP (lane 3) at 16°C for 1 h prior to immunoprecipitation. A portion of the cell lysate (2.4% of total; lane 1) and the immunoprecipitated proteins (IP with anti-FLAG M2 beads; lanes 2 and 3) were separated by SDS PAGE and the amounts of the indicated proteins (Stx5, Stx2, Stx3, Stx4, SNAP23, FLAG-Sec22b) were determined by immunoblot analysis. D) HEK293-FcγRII 3x-FLAG-Sec22b cells were infected with wild-type L. pneumophila. Sec22b was immunoprecipitated with anti-FLAG M2 beads from lysates prepared 1h after infection. As indicated in the bottom legend, cell lysates were treated with 0.5 mm ATP and 8 mm MgCl2(lanes 2 and 2′ ), 0.5 mm ATP, 8 mm MgCl2, 10 μg/mL His6-NSF and 5 μg/mL His6-α-SNAP (lanes 3 and 3′) or 0.5 mm ATP, 8 mm MgCl2, 10 μg/mL NEM-pretreated His6-NSF and 5 μg/mL His6-α-SNAP (lanes 4 and 4′) at 16°C for 1 h. After incubation, the samples were immunoprecipitated with anti-FLAG M2 beads. A portion of the cell lysate (2.4% of total; lanes 1 and 1′) and the immunoprecipitated proteins (IP with anti-FLAG M2 beads) were separated by SDS PAGE and the amounts of the indicated proteins (Stx2, Stx3, Stx4, SNAP23, FLAG-Sec22b) were determined by immunoblot analysis in lanes 1–4. Asterisk and magnification of the Stx4 band in lane 4 shows the presence of His6-α-SNAP, which was detected by the anti-Stx4 antibody. An anti-His immunoblot shown on the right (lanes 1′ – 4′) confirms His6-α-SNAP was immunoprecipitated with FLAG-Sec22b in the NEM-treated NSF-containing sample (lane 4′).
To determine whether the complex formed between 3x-FLAG-Sec22b and plasma membrane-localized syntaxins represents a functional SNAREpin assembly, we asked whether this complex was disassembled upon the addition of recombinant His6-tagged NSF and α-SNAP purified from Escherichia coli (Figure 6B). Stx5 was dissociated from 3x-FLAG-Sec22bupon incubation of cell lysates with recombinant NSF and α-SNAP, validating that these recombinant proteins were active (Figure 6C; lane 3). Importantly, plasma membrane syntaxins were dissociated from 3x-FLAG-Sec22b when extracts from cells infected with wild-type L. pneumophila were incubated with recombinant NSF and α-SNAP (Figure 6C; lane 3).
N-ethylmaleimide (NEM) treatment was used to inactivate the NSF protein before incubation with cell lysates to validate that the SNARE dissociation observed was mediated specifically by NSF (33). NEM-treated NSF was unable to dissociate plasma membrane-localized syntaxins and actually enhanced these interactions (Figure 6D; cf. lanes 2 and 4). This result was not unexpected because α-SNAP has been shown to bind and stabilize a functional SNARE complex prior to disassembly by NSF. As shown in the enlarged region of lane 4 of the Stx4 blot in Figure 6D, a doublet consisting of Stx4 and α-SNAP was detected in the sample treated with NEM. This cross-reactivity resulted from the polyclonal anti-Stx4 antibody being generated against a recombinant His-tagged Stx4 derivative, which is why the anti-Stx4 antibody recognized both proteins. The association of α-SNAP with a 3x-FLAG-Sec22b complex in the NEM-treated samples was confirmed by stripping the blot and reprobing with a monoclonal anti-His antibody (Figure 6D; lane 4′). These observations confirm that the L. pneumophila Dot/Icm system modulates SNARE usage and stimulates functional interactions between the v-SNARE Sec22b and plasma membrane-localized t-SNAREs.
Discussion
Bacterial pathogens are known to exploit eukaryotic proteins to promote their uptake and replication inside of host cells. Because of their ability to strongly stimulate specific host pathways, pathogens are valuable tools for uncovering normal cellular processes that are difficult to investigate, usually because the regulatory processes that govern critical biochemical activities are not understood. In this study, we showed that L. pneumophila promotes functional associations between host plasma membrane syntaxin proteins and the SNARE protein Sec22b localized to ER-derived vesicles. These data indicate that one of the pathways used by this pathogen to remodel the vacuole in which it resides involves the subversion of the host SNARE machinery.
These data could provide mechanistic insight into a pathway described previously that involves the direct fusion of ER-derived membranes with phagosomes (26,34). This process has been called ER-mediated phagocytosis, although it may occur primarily after a phagosome has been formed. It has been suggested that fusion of ER-derived vesicles with phagosomes could facilitate membrane exchange to protect the integrity of the cell by preventing the rapid depletion of lipid and protein molecules that comprise the plasma membrane. The importance of ER-mediated phagocytosis has been questioned, however, based on the inability of several groups using independent methods to validate that ER-derived membranes contribute significantly to phagosome biogenesis (35,36). The sensitivity of different visualization techniques employed by these independent groups may explain different conclusions concerning the significance of the association of the ER with phagosomes. Most likely, what these data indicate is that ER fusion with phagosomes is a tightly regulated process that is not stimulated to an appreciable level in most standard assays that examine the uptake and trafficking of inert particles or organisms that reside initially in canonical early phagosomes, which would explain these seemingly contradictory results obtained by independent groups.
Our data indicate that L. pneumophila activates a vesicular transport pathway that recruits ER-derived membranes to the phagosome. We show that this pathway involves the utilization of host plasma membrane syntaxins and SNAP23 as components of a t-SNARE complex that associates with Sec22b on the membrane of ER-derived vesicles. Consistent with data indicating fusion of ER-derived vesicles with phagosomes is normally difficult to observe, we found no evidence of plasma membrane syntaxins being associated with Sec22b when cells were infected with mutant L. pneumophila deficient in the Dot/Icm secretion system, indicating that during normal endocytic maturation plasma membrane syntaxins function in association with endosomal v-SNAREs, such as VAMP7. By contrast, wild-type L. pneumophila stimulated the association of Sec22b with plasma membrane syntaxins.
The association of Sec22b with plasma membrane syntaxins had not been observed before in a biological system, however, the pioneering work on biochemical interactions between yeast SNAREs that were sufficient to promote membrane fusion suggested such interactions were possible (18). The yeast homolog Sec22p was shown to form a functional trans-SNARE complex with the plasma membrane syntaxin Sso1 and the SNAP23 homolog Sec9c in an in vitro liposome fusion assay (18). Initially, it was suggested that the interaction between Sec22b and Sso1/Sec9c would not occur in a cellular context, because in the canonical secretory pathway ER-derived vesicles are restricted from associating with the plasma membrane. However, the finding that ER components can be detected on phagosomes suggested that interactions between the ER and plasma membrane SNAREs may not be restricted. It was also shown that inhibition of Sec22b function had a detrimental effect on the phagocytic capacity of macrophages, suggesting that Sec22b may play an important role in this cellular process (37). Thus, both biochemical and cellular studies suggest that an endogenous pathway could promote the fusion of ER-derived vesicles with phagosomes containing plasma membrane-localized SNAREs. Accordingly, our data suggest that L. pneumophila may have evolved mechanisms to subvert an endogenous pathway to facilitate fusion of ER-derived vesicles with the vacuole in which it resides.
These data suggest that Sec22b is a multifunctional v-SNARE that is involved in several different membrane fusion pathways. Understanding how cells regulate the association of Sec22b with other SNAREs should provide important insight into how processes that involve fusion of ER-derived vesicles with phagosomes are regulated. Our data indicate that L. pneumophila directs the pairing of Sec22b with plasma membrane-localized syntaxins by a process that requires a functional Dot/Icm secretion system. This implies that there are substrates translocated by the Dot/Icm system that can stimulate the pathway directing the association of plasma membrane syntaxins and Sec22b. There are more than 150 different proteins translocated into eukaryotic cells by the Dot/Icm system (9). Functional redundancy among this large repertoire of effectors provides the organism with multiple means to subvert ER-derived vesicles. At present, we have not identified a strain deficient in an L. pneumophila effector that has lost the ability to direct the association of plasma membrane syntaxins with Sec22b (data not shown). A recent study has suggested that the L. pneumophila protein IcmG/DotF might play a possible role in modulating SNARE interactions; however, there is no evidence currently that IcmG/DotF is delivered across the vacuole membrane into the cytosol during infection and assessing the role of IcmG/DotF during infection is further complicated by the fact that this protein is an important component of the Dot/Icm secretion apparatus (38). This might indicate that there are several effectors with a biochemical activity that is sufficient to stimulate the association of Sec22b with plasma membrane syntaxins. Alternatively, there could be more than one mechanism by which L. pneumophila is able to stimulate this pathway, and each alone is sufficient. Thus, future studies aimed at the identification of L. pneumophila effectors that can promote interactions between Sec22b and plasma membrane syntaxins should reveal important regulatory processes that govern the usage of Sec22b as a v-SNARE molecule.
Materials and Methods
Rabbit polyclonal antibodies to Stx2, Stx4 and Stx7 were purchased from Synaptic Systems. Rabbit polyclonal antibodies to SNAP23 and calnexin were obtained from Abcam and Stress Gen, respectively. A goat polyclonal antibody to Stx3 and a rabbit polyclonal antibody to Stx5 and Rab1b were purchased from Santa Cruz. Monoclonal antibody to VAMP7 was obtained from Novus Biologicals. Monoclonal antibodies to FLAG and His were purchased from Sigma-Aldrich. A rabbit polyclonal antibody to Sec22b and rabbit polyclonal and mouse polyclonal antibodies to L. pneumophila were generated for this study.
Bacterial strains and cell culture
Growth of L. pneumophila strains (wild-type; Lp01 and dotA mutant CR58) and host cell infection conditions have been described previously (11,39,40). RAW 264.7 macrophages and HEK293 cells were maintained in RPMI-1640 medium (GIBCO BRL) supplemented with 10% FBS and DMEM supplemented with 10% FBS, respectively. Mammalian cells were cultured at 37°C in 5% CO2.
Stable cell lines
To construct an expression vector that produces FcγRII, cDNA was amplified by polymerase chain reaction (PCR) from a plasmid described previously (41). The FcγRII cDNA was inserted into the HindIII/EcoRV sites of pcDNA 3.1/Hygro (Clontech). Plasmid DNA was tranfected into HEK293 cells and the cells expressing FcγRII (293 FcγRII cells) were selected by culturing in DMEM supplemented with 10% FBS in the presence of 200 μg/mL Hygromycin (Sigma-Aldrich).
cDNAs of mouse Sec22b and rat VAMP7 were generated by PCR from myc-Sec22b (24) and YFP-VAMP7 (31), respectively. The cDNAs were inserted into the 3x-FLAG sequence-inserted pcDNA 4TO (14). To establish the 293 FcγRII cells that stably express 3x-FLAG-Sec22b and -VAMP7, 293 FcγRII cells were transfected with 3x-FLAG-Sec22b or -VAMP7. Drug-resistant clones were selected by culturing them in DMEM supplemented with 10% FBS and 200 μg/mL Hygromycin in the presence of 400 μg/mL Zeocin (Invitrogen).
Immunofluorescence microscopy
Legionella pneumophila infection of mammalian cells was described previously (11,39,40). To examine Legionella containing vacuole (LCV) localization of endogenous plasma membrane t-SNARE molecules in RAW 264.7 macrophages, cells were infected with dsRed-expressing wild-type L. pneumophila or dotA mutant at an MOI of one for 1 h. Cells were fixed for 5 min in ice-cold methanol, and blocked with 2% BSA in PBS. Samples were stained with antibodies to Stx2, Stx3, Stx4 or SNAP23 and Alexa-488-labeled secondary antibody (Invitrogen). To examine LCV localization of endogenous Sec22b and VAMP7 in RAW 264.7 macrophages, cells were infected as described earlier. After infection, cells were fixed in 4% paraformaldehyde (PFA) in PBS for 15 min at room temperature. Samples were stained with rabbit or mouse anti-L. pneumophila antibodies to detect the extracellular bacteria before permeabilization. After staining extracellular bacteria, cells were permeabilized with ice-cold methanol, blocked and stained with antibodies to Sec22b or VAMP7, and Alexa-488 anti-rabbit or mouse IgG secondary Abs to detect Sec22b or VAMP7, and Alexa-350 anti-rabbit or anti-mouse IgG secondary antibodies to identify the extracellular bacteria. To examine endogenous plasma membrane t-SNARE molecules and Sec22b in HEK293-FcγRII cells and 3x-FLAG-Sec22b and 3x-FLAG VAMP7 in HEK293-FcγRII stable cell lines, L. pneumophila was opsonized with rabbit or mouse anti-L. pneumophila antibodies (1:1000), and cells were infected at an MOI of one for 1 h. The same fixation and staining procedure used for staining of Sec22b/VAMP7 in RAW 264.7 macrophages was used to detect the target proteins and extra/intracellular bacteria.
Preparation of lysates from Legionella pneumophila-infected cells
To prepare lysates from L. pneumophila-infected HEK293-FcγRII 3x-FLAG-Sec22b cells for immunoprecipitation, the cells were plated in 6-well tissue culture dishes at a density of 1 × 106 cells/well. After an overnight incubation, cells were infected without or with wild-type L. pneumophila or dotA mutant that were opsonized with rabbit anti-L. pneumophila antibody (1:1000 dilution) at an MOI of 20. After 1 h of infection, cells were collected by centrifugation at 1000 ×g for 10 min. Cell pellets were washed once in PBS and resuspended in lysis buffer [150 mm KCl,20 mm HEPES KOH (pH 7.2), 2 mm ethylenediaminetetraacetic acid (EDTA), 0.5 mg/mL leupeptin, 2 mm pepstatin A, 1 mm phenylmethylsulfonyl fluoride, 1 mm dithiothreitol and 1% Triton-X-100]. The lysates were centrifuged in a microcentrifuge at 10,000 × g for10 min. All collection and preparation steps were conducted at 4°C.
Purification of His6-NSF and α-SNAP
Full-length cDNA encoding human NSF and α-SNAP were amplified by PCR from plasmid DNA (Open Biosystems) and inserted into the pQE-30 vector. Escherichia coli XLI-Blue cells transformed with vectors producing the indicated proteins were grown at 37°C in Luria broth broth containing appropriate antibiotic to an optical density (OD ) > 0.5. Isopropylthiogalactopyranoside was added to the medium at final concentration of 0.5 mm and samples were incubated for 3 h at 37°C. The cells were harvested by centrifugation, and the His-tagged proteins were purified as described previously (19,42). Purified proteins were stored at −70°C until use.
RNA interference
RNA duplexes targeting Stx2, Stx3, Stx4 and Sec22b were purchased from Dharmacon as siRNAplusSmart Pools. The transfection of RNA duplexes into HEK293-FcγRII cells was conducted using Lipofectamine 2000 (Invitrogen) according to manufacturer’s directions. The final concentration of RNA duplexes was 50 nm. Knockdown efficiency was determined 72 h post-transfection by immunoblot, immunofluorescence and infection experiments. For infection studies, mock-or siRNA-treated cells were infected with opsonized L. pneumophila, then processes as described above for the stable cell lines.
Supplementary Material
Establishment of the HEK293-FcγRII cell line. A) HEK293 cells transfected without (top panel) or with (middle panel) plasmid for pcDNA-FcγRII and drug-screened HEK293-FcγRII cells (bottom panel) were fixed and stained with anti-FcγRII antibody and 4, 6-diamidino-2-phenylindole (DAPI). All images were obtained with same excitation intensity. B)HEK293-FcγRII cells were infected with opsonized (anti-rabbit Legionella antibody) wild-type L. pneumophila (left column) or the dotA mutant (right column). At 1 (top and second rows) or 8 h (third and bottom rows) after infection, cells were fixed and stained with fluorescein-labeled anti-rabbit IgG secondary antibody to detect the bacteria.
Sec22b is important for maturation of the L.pneumophila-containing vacuole. A) To assess the efficiency of Sec22b silencing, cell lysates were obtained from HEK293-FcγRII cells transfected for 72 h with the siRNA indicated (top), and immunoblot analysis was used to measure the levels of the proteins indicated (right). Calnexin, Stx3 and SNAP 23 levels were used to assess equal protein loading. B) HEK293-FcγRII cells were treated with the Sec22b siRNA or mock treated at 72 h. After treatment, cells were fixed and stained with antibodies specific for Sec22b. Images show decreased Sec22b staining after silencing. Scale bar = 5 μm. C and D) HEK293-FcγRII cells were transfected with siRNA targeting Sec22b or treated with control siRNA (mock) as indicated at the bottom of the graph. Cells were fixed either 1 (C) or 4 h (D) after infection with wild-type L. pneumophila, and stained with antibodies to either calnexinor Rab1b as indicated in each panel. Colocalization of calnexin and Rab1b was assessed by fluorescence microscopy, and the data are presented as the percent of vacuoles that were positive for each marker. Data are the mean ± SEM of three independent experiments in which 100 vacuoles were scored [*p < 0.005 (C), *p < 0.004 and **p < 0.007 (D), as compared with mock-treated].
Quantitative analysis of Sec22b complexes containing plasma membrane-localized syntaxins. A) HEK293-FcγRII 3x-FLAG-Sec22b cells were infected without or with wild-type L. pneumophila or the dotA mutant. Sec22b was immunoprecipitated with anti-FLAG M2 beads from lysates prepared 1 h after infection. A portion of the cell lysate (3% of total; top rows) and the immunoprecipitated proteins (IP with anti-FLAG M2 beads; bottom rows) were separated by SDS PAGE and the amounts of the indicated proteins (Stx2, Stx3, Stx4, SNAP23, FLAG-Sec22b) were determined by immunoblot analysis. After detection, ImageJ was used to measure the band intensity of the input and that of the immunoprecipated proteins. The calculation for the amount of total protein was determined by multiplying the intensity of the band in the cell lysate by 33.33. The calculation for the percentage of total protein in the immunoprecipitated fraction was determined by dividing the band intensity of the immunoprecipated protein by calculated value for the total amount of protein in the lysate (percent of total values shown below each lane). The results from two independent experiments are shown. B) To measure the amount of Stx3 in the total cell lysate and the amount of Stx3 associated with Sec22b, cell infection and immunoprecipitations were done as described in Figure S3A. To generate a standard curve to calculate Stx3 amounts, 25, 50 or 100 ng of purified recombinant MBP-Stx3 ΔTM was loaded in duplicate and detected by immunoblot analysis with the anti-Stx3 antibody. In parallel, 0.75, 1.5 and 3% of input lysate from uninfected, wild-type or dotA-infected cells was loaded and 10 and 80% of the FLAG-tagged immunoprecipitated Sec22b was included on the blots. In addition, 1.5% of input of uninfected, wild-type or dotA-infected cell lysate were loaded with 10% of FLAG-tagged immunoprecipitated Sec22b and blotted with anti-FLAG antibody to control for equal loading. ImageJ was used to determine the band intensity of each lane. A standard curve was generated for the MBP-Stx3 ΔTM amounts and the calculated band intensities. The band intensities of Stx3 in each cell lysate and in the corresponding immunoprecipitates were used to determine protein amounts by plotting these values on the standard curve. Images shown were used to determine protein amounts and were generated from the same exposure on a single sheet of film.
The amount of plasma membrane t-SNAREs that coprecipitate with Sec22b increases with an increase in the multiplicity of infection by wild-type L. pneumophila. HEK293-FcγRII 3x-FLAG-Sec22b cells were infected without or with wild-type L. pneumophila at an MOI of 20 and 80. Sec22b was immunoprecipitated with anti-FLAG M2 beads from lysates prepared 1 h after infection. A portion of the cell lysate (5% of total; left lanes) and the immunoprecipitated proteins (IP with anti-FLAG M2 beads; right lanes) were separated by SDS PAGE and the amounts of the indicated proteins (Stx2, Stx3, Stx4, SNAP23, FLAG-Sec22b) were determined by immunoblot analysis.
Acknowledgments
We thank Drs Norma Andrews and Jesse Hay for providing plasmids encoding VAMP7 and Sec22b, respectively. We also thank Dr Shaeri Mukherjee for technical assistance. This work was supported by NIH grant R01AI041699 to C. R. R.
References
- 1.Kumar Y, Valdivia RH. Leading a sheltered life: intracellular pathogens and maintenance of vacuolar compartments. Cell Host Microbe. 2009;5:593–601. doi: 10.1016/j.chom.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Swanson MS, Isberg RR. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect Immun. 1995;63:3609–3620. doi: 10.1128/iai.63.9.3609-3620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kagan JC, Roy CR. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat Cell Biol. 2002;4:945–954. doi: 10.1038/ncb883. [DOI] [PubMed] [Google Scholar]
- 4.Derre I, Isberg RR. Legionella pneumophila replication vacuole formation involves rapid recruitment of proteins of the early secretory system. Infect Immun. 2004;72:3048–3053. doi: 10.1128/IAI.72.5.3048-3053.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kagan JC, Stein MP, Pypaert M, Roy CR. Legionella subvert the functions of Rab1 and Sec22b to create a replicative organelle. J Exp Med. 2004;199:1201–1211. doi: 10.1084/jem.20031706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horwitz MA. Formation of a novel phagosome by the Legionnaires’ disease bacterium (Legionella pneumophila) in human monocytes. J Exp Med. 1983;158:1319–1331. doi: 10.1084/jem.158.4.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tilney LG, Harb OS, Connelly PS, Robinson CG, Roy CR. How the parasitic bacterium Legionella pneumophila modifies its phagosome and transforms it into rough ER: implications for conversion of plasma membrane to the ER membrane. J Cell Sci. 2001;114:4637–4650. doi: 10.1242/jcs.114.24.4637. [DOI] [PubMed] [Google Scholar]
- 8.Robinson CG, Roy CR. Attachment and fusion of endoplasmic reticulum with vacuoles containing Legionella pneumophila. Cell Microbiol. 2006;8:793–805. doi: 10.1111/j.1462-5822.2005.00666.x. [DOI] [PubMed] [Google Scholar]
- 9.Ensminger AW, Isberg RR. Legionella pneumophila Dot/Icm translocated substrates: a sum of parts. Curr Opin Microbiol. 2009;12:67–73. doi: 10.1016/j.mib.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roy CR, Berger KH, Isberg RR. Legionella pneumophila DotA protein is required for early phagosome trafficking decisions that occur within minutes of bacterial uptake. Mol Microbiol. 1998;28:663–674. doi: 10.1046/j.1365-2958.1998.00841.x. [DOI] [PubMed] [Google Scholar]
- 11.Nagai H, Kagan JC, Zhu X, Kahn RA, Roy CR. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science. 2002;295:679–682. doi: 10.1126/science.1067025. [DOI] [PubMed] [Google Scholar]
- 12.Machner MP, Isberg RR. Targeting of host Rab GTPase function by the intravacuolar pathogen Legionella pneumophila. Dev Cell. 2006;11:47–56. doi: 10.1016/j.devcel.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 13.Murata T, Delprato A, Ingmundson A, Toomre DK, Lambright DG, Roy CR. The Legionella pneumophila effector protein DrrA is a Rab1 guanine nucleotide-exchange factor. Nat Cell Biol. 2006;8:971–977. doi: 10.1038/ncb1463. [DOI] [PubMed] [Google Scholar]
- 14.Ingmundson A, Delprato A, Lambright DG, Roy CR. Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature. 2007;450:365–369. doi: 10.1038/nature06336. [DOI] [PubMed] [Google Scholar]
- 15.Hong W. SNAREs and traffic. Biochim Biophys Acta. 2005;1744:120–144. doi: 10.1016/j.bbamcr.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 16.Fukuda R, McNew JA, Weber T, Parlati F, Engel T, Nickel W, Rothman JE, Sollner TH. Functional architecture of an intracellular membrane t-SNARE. Nature. 2000;407:198–202. doi: 10.1038/35025084. [DOI] [PubMed] [Google Scholar]
- 17.Hu C, Ahmed M, Melia TJ, Sollner TH, Mayer T, Rothman JE. Fusion of cells by flipped SNAREs. Science. 2003;300:1745–1749. doi: 10.1126/science.1084909. [DOI] [PubMed] [Google Scholar]
- 18.McNew JA, Parlati F, Fukuda R, Johnston RJ, Paz K, Paumet F, Sollner TH, Rothman JE. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature. 2000;407:153–159. doi: 10.1038/35025000. [DOI] [PubMed] [Google Scholar]
- 19.Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 20.Chen YA, Scheller RH. SNARE-mediated membrane fusion. Nat Rev Mol Cell Biol. 2001;2:98–106. doi: 10.1038/35052017. [DOI] [PubMed] [Google Scholar]
- 21.Jahn R, Grubmuller H. Membrane fusion. Curr Opin Cell Biol. 2002;14:488–495. doi: 10.1016/s0955-0674(02)00356-3. [DOI] [PubMed] [Google Scholar]
- 22.Hohl TM, Parlati F, Wimmer C, Rothman JE, Sollner TH, Engelhardt H. Arrangement of subunits in 20 S particles consisting of NSF, SNAPs, and SNARE complexes. Mol Cell. 1998;2:539–548. doi: 10.1016/s1097-2765(00)80153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wimmer C, Hohl TM, Hughes CA, Muller SA, Sollner TH, Engel A, Rothman JE. Molecular mass, stoichiometry, and assembly of 20 S particles. J Biol Chem. 2001;276:29091–29097. doi: 10.1074/jbc.M011292200. [DOI] [PubMed] [Google Scholar]
- 24.Xu D, Joglekar AP, Williams AL, Hay JC. Subunit structure of a mammalian ER/Golgi SNARE complex. J Biol Chem. 2000;275:39631–39639. doi: 10.1074/jbc.M007684200. [DOI] [PubMed] [Google Scholar]
- 25.Zhang T, Wong SH, Tang BL, Xu Y, Hong W. Morphological and functional association of Sec22b/ERS-24 with the pre-Golgi intermediate compartment. MolBiol Cell. 1999;10:435–453. doi: 10.1091/mbc.10.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gagnon E, Duclos S, Rondeau C, Chevet E, Cameron PH, Steele-Mortimer O, Paiement J, Bergeron JJ, Desjardins M. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell. 2002;110:119–131. doi: 10.1016/s0092-8674(02)00797-3. [DOI] [PubMed] [Google Scholar]
- 27.Hackam DJ, Rotstein OD, Bennett MK, Klip A, Grinstein S, Manolson MF. Characterization and subcellular localization of target membrane soluble NSF attachment protein receptors (t-SNAREs) in macrophages. Syntaxins 2, 3, and 4 are present on phagosomal membranes. J Immunol. 1996;156:4377–4383. [PubMed] [Google Scholar]
- 28.Flaumenhaft R, Croce K, Chen E, Furie B, Furie BC. Proteins of the exocytotic core complex mediate platelet alpha-granule secretion. Roles of vesicle-associated membrane protein, SNAP-23, and syntaxin 4. J Biol Chem. 1999;274:2492–2501. doi: 10.1074/jbc.274.4.2492. [DOI] [PubMed] [Google Scholar]
- 29.Wang CC, Ng CP, Lu L, Atlashkin V, Zhang W, Seet LF, Hong W. A role of VAMP8/endobrevin in regulated exocytosis of pancreatic acinar cells. Dev Cell. 2004;7:359–371. doi: 10.1016/j.devcel.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 30.Braun V, Fraisier V, Raposo G, Hurbain I, Sibarita JB, Chavrier P, Galli T, Niedergang F. TI-VAMP/VAMP7 is required for optimal phagocytosis of opsonised particles in macrophages. EMBO J. 2004;23:4166–4176. doi: 10.1038/sj.emboj.7600427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rao SK, Huynh C, Proux-Gillardeaux V, Galli T, Andrews NW. Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. J Biol Chem. 2004;279:20471–20479. doi: 10.1074/jbc.M400798200. [DOI] [PubMed] [Google Scholar]
- 32.Hirose H, Arasaki K, Dohmae N, Takio K, Hatsuzawa K, Nagahama M, Tani K, Yamamoto A, Tohyama M, Tagaya M. Implication of ZW10 in membrane trafficking between the endoplasmic reticulum and Golgi. EMBO J. 2004;23:1267–1278. doi: 10.1038/sj.emboj.7600135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whiteheart SW, Rossnagel K, Buhrow SA, Brunner M, Jaenicke R, Rothman JE. N-ethylmaleimide-sensitive fusion protein: a trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J Cell Biol. 1994;126:945–954. doi: 10.1083/jcb.126.4.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Desjardins M. ER-mediated phagocytosis: a new membrane for new functions. Nat Rev Immunol. 2003;3:280–291. doi: 10.1038/nri1053. [DOI] [PubMed] [Google Scholar]
- 35.Rogers LD, Foster LJ. The dynamic phagosomal proteome and the contribution of the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2007;104:18520–18525. doi: 10.1073/pnas.0705801104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Touret N, Paroutis P, Terebiznik M, Harrison RE, Trombetta S, Pypaert M, Chow A, Jiang A, Shaw J, Yip C, Moore HP, Van Der Wel N, Houben D, Peters PJ, De Chastellier C, et al. Quantitative and dynamic assessment of the contribution of the ER to phagosome formation. Cell. 2005;123:157–170. doi: 10.1016/j.cell.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 37.Becker T, Volchuk A, Rothman JE. Differential use of endoplasmic reticulum membrane for phagocytosis in J774 macrophages. Proc Natl Acad Sci U S A. 2005;102:4022–4026. doi: 10.1073/pnas.0409219102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paumet F, Wesolowski J, Garcia-Diaz A, Delevoye C, Aulner N, Shuman HA, Subtil A, Rothman JE. Intracellular bacteria encode inhibitory SNARE-like proteins. PLoS One. 2009;4:e7375. doi: 10.1371/journal.pone.0007375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coers J, Kagan JC, Matthews M, Nagai H, Zuckman DM, Roy CR. Identification of Icm protein complexes that play distinct roles in the biogenesis of an organelle permissive for Legionella pneumophila intracellular growth. Mol Microbiol. 2000;38:719–736. doi: 10.1046/j.1365-2958.2000.02176.x. [DOI] [PubMed] [Google Scholar]
- 40.Zuckman DM, Hung JB, Roy CR. Pore-forming activity is not sufficient for Legionella pneumophila phagosome trafficking and intracellular growth. Mol Microbiol. 1999;32:990–1001. doi: 10.1046/j.1365-2958.1999.01410.x. [DOI] [PubMed] [Google Scholar]
- 41.Joiner KA, Fuhrman SA, Miettinen HM, Kasper LH, Mellman I. Toxoplasma gondii: fusion competence of parasitophorous vacuoles in Fc receptor-transfected fibroblasts. Science. 1990;249:641–646. doi: 10.1126/science.2200126. [DOI] [PubMed] [Google Scholar]
- 42.Whiteheart SW, Griff IC, Brunner M, Clary DO, Mayer T, Buhrow SA, Rothman JE. SNAP family of NSF attachment proteins includes a brain-specific isoform. Nature. 1993;362:353–355. doi: 10.1038/362353a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Establishment of the HEK293-FcγRII cell line. A) HEK293 cells transfected without (top panel) or with (middle panel) plasmid for pcDNA-FcγRII and drug-screened HEK293-FcγRII cells (bottom panel) were fixed and stained with anti-FcγRII antibody and 4, 6-diamidino-2-phenylindole (DAPI). All images were obtained with same excitation intensity. B)HEK293-FcγRII cells were infected with opsonized (anti-rabbit Legionella antibody) wild-type L. pneumophila (left column) or the dotA mutant (right column). At 1 (top and second rows) or 8 h (third and bottom rows) after infection, cells were fixed and stained with fluorescein-labeled anti-rabbit IgG secondary antibody to detect the bacteria.
Sec22b is important for maturation of the L.pneumophila-containing vacuole. A) To assess the efficiency of Sec22b silencing, cell lysates were obtained from HEK293-FcγRII cells transfected for 72 h with the siRNA indicated (top), and immunoblot analysis was used to measure the levels of the proteins indicated (right). Calnexin, Stx3 and SNAP 23 levels were used to assess equal protein loading. B) HEK293-FcγRII cells were treated with the Sec22b siRNA or mock treated at 72 h. After treatment, cells were fixed and stained with antibodies specific for Sec22b. Images show decreased Sec22b staining after silencing. Scale bar = 5 μm. C and D) HEK293-FcγRII cells were transfected with siRNA targeting Sec22b or treated with control siRNA (mock) as indicated at the bottom of the graph. Cells were fixed either 1 (C) or 4 h (D) after infection with wild-type L. pneumophila, and stained with antibodies to either calnexinor Rab1b as indicated in each panel. Colocalization of calnexin and Rab1b was assessed by fluorescence microscopy, and the data are presented as the percent of vacuoles that were positive for each marker. Data are the mean ± SEM of three independent experiments in which 100 vacuoles were scored [*p < 0.005 (C), *p < 0.004 and **p < 0.007 (D), as compared with mock-treated].
Quantitative analysis of Sec22b complexes containing plasma membrane-localized syntaxins. A) HEK293-FcγRII 3x-FLAG-Sec22b cells were infected without or with wild-type L. pneumophila or the dotA mutant. Sec22b was immunoprecipitated with anti-FLAG M2 beads from lysates prepared 1 h after infection. A portion of the cell lysate (3% of total; top rows) and the immunoprecipitated proteins (IP with anti-FLAG M2 beads; bottom rows) were separated by SDS PAGE and the amounts of the indicated proteins (Stx2, Stx3, Stx4, SNAP23, FLAG-Sec22b) were determined by immunoblot analysis. After detection, ImageJ was used to measure the band intensity of the input and that of the immunoprecipated proteins. The calculation for the amount of total protein was determined by multiplying the intensity of the band in the cell lysate by 33.33. The calculation for the percentage of total protein in the immunoprecipitated fraction was determined by dividing the band intensity of the immunoprecipated protein by calculated value for the total amount of protein in the lysate (percent of total values shown below each lane). The results from two independent experiments are shown. B) To measure the amount of Stx3 in the total cell lysate and the amount of Stx3 associated with Sec22b, cell infection and immunoprecipitations were done as described in Figure S3A. To generate a standard curve to calculate Stx3 amounts, 25, 50 or 100 ng of purified recombinant MBP-Stx3 ΔTM was loaded in duplicate and detected by immunoblot analysis with the anti-Stx3 antibody. In parallel, 0.75, 1.5 and 3% of input lysate from uninfected, wild-type or dotA-infected cells was loaded and 10 and 80% of the FLAG-tagged immunoprecipitated Sec22b was included on the blots. In addition, 1.5% of input of uninfected, wild-type or dotA-infected cell lysate were loaded with 10% of FLAG-tagged immunoprecipitated Sec22b and blotted with anti-FLAG antibody to control for equal loading. ImageJ was used to determine the band intensity of each lane. A standard curve was generated for the MBP-Stx3 ΔTM amounts and the calculated band intensities. The band intensities of Stx3 in each cell lysate and in the corresponding immunoprecipitates were used to determine protein amounts by plotting these values on the standard curve. Images shown were used to determine protein amounts and were generated from the same exposure on a single sheet of film.
The amount of plasma membrane t-SNAREs that coprecipitate with Sec22b increases with an increase in the multiplicity of infection by wild-type L. pneumophila. HEK293-FcγRII 3x-FLAG-Sec22b cells were infected without or with wild-type L. pneumophila at an MOI of 20 and 80. Sec22b was immunoprecipitated with anti-FLAG M2 beads from lysates prepared 1 h after infection. A portion of the cell lysate (5% of total; left lanes) and the immunoprecipitated proteins (IP with anti-FLAG M2 beads; right lanes) were separated by SDS PAGE and the amounts of the indicated proteins (Stx2, Stx3, Stx4, SNAP23, FLAG-Sec22b) were determined by immunoblot analysis.