

Abstract
Agouti protein and Agouti-related protein (Agrp) are paracrine-signaling molecules that normally regulate pigmentation and body weight, respectively. These proteins antagonize the effects of α-melanocyte-stimulating hormone (α-MSH) and other melanocortins, and several alternatives have been proposed to explain their biochemical mechanisms of action. We have used a sensitive bioassay based on Xenopus melanophores to characterize pharmacologic properties of recombinant Agouti protein, and have directly measured its cell-surface binding to mammalian cells by use of an epitope-tagged form (HA–Agouti) that retains biologic activity. In melanophores, Agouti protein has no effect in the absence of α-MSH, but its action cannot be explained solely by inhibition of α-MSH binding. In 293T cells, expression of the Mc1r confers a specific, high-affinity binding site for HA-Agouti. Binding is inhibited by α-MSH, or by Agrp, which indicates that α-MSH and Agouti protein bind in a mutually exclusive way to the Mc1r, and that the similarity between Agouti protein and Agrp includes their binding sites. The effects of Agouti and the Mc1r in vivo have been examined in a sensitized background provided by the chinchilla (Tyrc-ch) mutation, which uncovers a phenotypic difference between overexpression of Agouti in lethal yellow (Ay/a) mice and loss of Mc1r function in recessive yellow (Mc1re/Mc1re) mice. Double and triple mutant studies indicate that a functional Mc1r is required for the pigmentary effects of Agouti, and suggest that Agouti protein can act as an agonist of the Mc1r in a way that differs from α-MSH stimulation. These results resolve questions regarding the biochemical mechanism of Agouti protein action, and provide evidence of a novel signaling mechanism whereby α-MSH and Agouti protein or Agrp function as independent ligands that inhibit each other’s binding and transduce opposite signals through a single receptor.
Keywords: Agouti, melanocortin receptor, pigmentation, obesity, mouse coat color
A large collection of mouse coat color genes discovered over the past fifty years has proven to be a valuable resource for identifying and studying general aspects of cell biology. Two such genes, Agouti and Extension, encode a paracrine-signaling molecule and a G protein-coupled receptor (the Mc1r or melanocortin 1 receptor), respectively, and are opposing regulators of pigment production in hair follicle melanocytes (Bultman et al. 1992; Miller et al. 1993; Robbins et al. 1993). Mammalian melanocytes can produce either eumelanin (black/brown pigment) or pheomelanin (yellow/red pigment) (for review, see Prota 1992). Local production of Agouti protein by specialized cells in the dermis causes follicular melanocytes to produce pheomelanin, whereas activation of the Mc1r in melanocytes leads to the production of eumelanin (Geschwind 1966; Chhajlani and Wikberg 1992; Mountjoy et al. 1992; Millar et al. 1995). The Mc1r can be activated by melanocortin peptides such as α-melanocyte stimulating hormone (α-MSH) and adrenocorticotrophic hormone (ACTH) (for review, see Eberle 1988; Cone et al. 1996), but the extent to which these peptides normally control mammalian pigmentation is not clear.
The interaction between Agouti protein and the Mc1r is likely to be representative of similar interactions that occur between additional melanocortin receptor subtypes and agouti-related molecules in the regulation of other biological processes (Adan et al. 1996; Li et al. 1996; Fan et al. 1997; Huang et al. 1997; Ollmann et al. 1997; Shutter et al. 1997). This possibility was first suggested by the phenotype of mice carrying the dominant Agouti allele lethal yellow (Ay) in which transcripts encoding normal Agouti protein are ubiquitously expressed (Miller et al. 1993; Michaud et al. 1994). In addition to a yellow coat, mice that carry Ay display obesity, insulin resistance, premature infertility, increased body length, and increased tumor susceptibility (for review, see Silvers 1979a). Many of these phenotypes appear to be caused by inhibition of the CNS-specific melanocortin 4 receptor (Mc4r) (Gantz et al. 1993), because mice that carry a targeted mutation of the Mc4r recapitulate the metabolic and growth abnormalities observed in Ay/− mice (Huszar et al. 1997), and recombinant Agouti protein can antagonize the effects of Mc4r stimulation in vitro (Lu et al. 1994; Yang et al. 1997). Ectopic expression of Agouti in mice that carry Ay or similar alleles (for review, see Siracusa 1994) probably mimics the function of a recently discovered homologous protein, Agouti-related protein (Agrp), which is normally expressed in the hypothalamus, causes obesity when ubiquitously expressed, and can antagonize the effects of Mc4r stimulation in vitro (Ollmann et al. 1997; Shutter et al. 1997).
The exact mechanism by which Agouti protein and Agrp inhibit melanocortin-receptor signaling is not completely clear. In vitro, recombinant Agouti protein will inhibit binding of radiolabeled melanocortins to cells that express melanocortin receptors (Lu et al. 1994; Yang et al. 1997), suggesting that Agouti protein binds melanocortin receptors and acts as a competitive antagonist of ligand binding, even though Agouti protein and melanocortin peptides exhibit no sequence similarity. This model is supported by studies of Agouti protein in B16 melanoma cells, in which dose-response curves of (α-MSH)-stimulated cAMP accumulation in the presence of Agouti protein were consistent with simple competitive antagonism (Blanchard et al. 1995; Willard et al. 1995). Other studies, however, suggest alternative mechanisms of Agouti protein action. Agouti protein will inhibit the effects of forskolin or cholera toxin in cultured pigment cells (Siegrist et al. 1997; Suzuki et al. 1997), and several groups have observed that Agouti protein has physiologic effects in the absence of added melanocortin peptides (Hunt and Thody 1995; Siegrist et al. 1996; Sakai et al. 1997). In contrast to laboratory mice, where a constitutively active Mc1r mutation is epistatic to a dominant Agouti allele (for review, see Silvers 1979a), Arctic foxes that carry a constitutively active Mc1r allele still display a coat color response to Agouti (Vage et al. 1997), which suggests the existence of an as yet unidentified Agouti receptor whose downstream effectors intersect with those of the Mc1r. On the basis of the similarity of cysteine spacing between Agouti protein and invertebrate toxins that inactivate calcium channels (Olivera et al. 1991; Quistad and Skinner 1994), Zemel and colleagues have suggested that Agouti protein can regulate intracellular calcium levels (Zemel et al. 1995; Jones et al. 1996; Kim et al. 1996; Mynatt et al. 1997), possibly by binding to a specific Agouti receptor (Manne et al. 1995). Additional reports hypothesize that Agouti protein promotes melanocortin-receptor internalization, or that it functions as an inverse agonist of melanocortin receptors (Siegrist et al. 1996, 1997).
Direct measurements of Agouti protein binding could help to resolve some of the uncertainties regarding its mechanism of action. To this end, we have made use of a rapid and sensitive bioassay based on amphibian pigment cells, Xenopus melanophores (Potenza and Lerner 1992), to purify and characterize mouse Agouti protein and an epitope-tagged form of Agouti protein that retains biologic activity. We find that expression of the Mc1r confers a specific cell-surface binding site for Agouti protein in vitro. Binding is inhibited by α-MSH, or by Agrp, which shows that α-MSH and Agouti protein bind in a mutually exclusive way to the Mc1r, and that Agouti protein and Agrp bind to a common site. To investigate, in vivo, whether all the effects of Agouti protein can be explained by its ability to inhibit α-MSH binding, we have carried out genetic crosses on a background sensitized to the effects of Ay. We find that the coat color phenotype of mice that ubiquitously express Agouti differs from those that lack a functional Mc1r, and that the phenotype of double-mutants is identical to that caused by Mc1r deficiency. Thus, the effects of Agouti require a functional Mc1r, but the receptor appears to respond independently to each factor in vivo. These findings have general implications for understanding melanocortin-receptor signaling.
Results
Agouti protein specifically inhibits α-MSH-induced pigment dispersion in Xenopus melanophores
Measurements of Agouti protein activity described by us and others have been based on inhibition of α-MSH action in cultured melanocytes or in heterologous cells transfected with a mammalian melanocortin receptor (Lu et al. 1994; Blanchard et al. 1995; Sakai et al. 1997; Yang et al. 1997). As described below, we found that a bioassay based on cultured Xenopus melanophores is a more rapid and accurate method for detecting Agouti protein activity. In the permanent melanophore cell line derived by Lerner and colleagues (Potenza and Lerner 1992), agents that alter intracellular cAMP levels cause a rapid change in pigment granule dispersion that is visible to the eye and can be quantitated in a 96-well format. The melanophores are very sensitive to α-MSH (EC50 of 0.9 nm), and have been used previously to characterize small molecule melanocortin agonists and antagonists (Jayawickreme et al. 1994).
By use of inhibition of α-MSH-induced pigment dispersion in melanophores as an assay, we purified recombinant Agouti protein from conditioned media of insect cells infected with a baculovirus that contained the mouse Agouti cDNA. Two 18-kD isoforms were resolved at >99% purity after cation and anion exchange chromatography (Fig. 1B); the two isoforms differed in their charge but had identical specific activity (not shown). Amino-terminal sequencing revealed both forms to be mature Agouti protein cleaved at the predicted signal-sequence cleavage site. Mass spectrometry indicated the difference in charge and electrophoretic mobility of the two isoforms is caused by differential glycosylation (Fig. 1B), most likely at the amino-linked glycosylation site described by Wilkison and colleagues (Willard et al. 1995).
Figure 1.

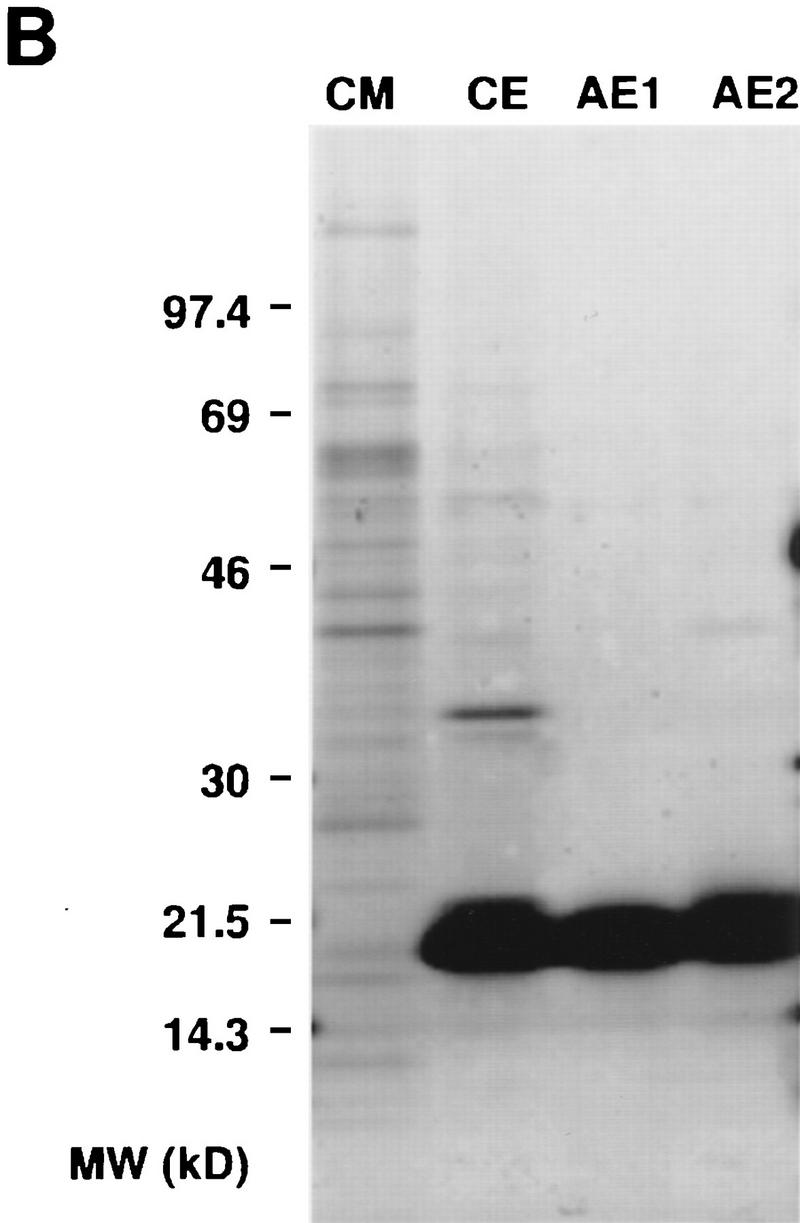

Production and characterization of recombinant Agouti protein with Xenopus melanophores. (A) Photograph of confluent melanophores in 96-well plate 45 min after treatment with the indicated concentrations of Agouti protein and α-MSH. Agouti protein has no effect by itself but inhibits pigment granule dispersion induced by α-MSH. (B) Recombinant Agouti protein analyzed by silver stained 4%–20% SDS-PAGE. Each lane contains 1 μg of protein from serial steps in the purification as described in Materials and Methods. CM, conditioned media produced by baculovirus-infected cells; CE, product of cation exchange; AE1 and AE2, products of anion exchange that differ in their extent of glycosylation but not in their primary sequence as determined by mass spectrometry and amino-terminal sequencing. (C) Effect of forskolin on melanophore pigment dispersion in the presence (○) and absence (control, ▪) of 100 nm Agouti protein. (D) Dose-response analysis of Agouti protein antagonism measured by pigment dispersion in melanophores 220 min after simultaneous addition of Agouti protein and α-MSH. The experiment was carried out as described in Materials and Methods, except that different concentrations of Agouti protein and α-MSH were mixed in a 96-well plate before addition to melanophores. The α-MSH dose-response curves at different Agouti concentrations, 0.6 nm to 150 nm, exhibit slopes that are significantly different from each other (F = 21.8, P < 0.001) as determined by a goodness-of-fit test that compares curve-fitting individual slopes for each concentration of Agouti protein with a single slope for all eight α-MSH dose-response curves. Similar results were obtained 35 min after simultaneous addition of Agouti protein and α-MSH (F = 7.9, P < 0.001).
Purified Agouti protein had no effect on melanophores by itself (Fig. 1A; data not shown), but inhibited pigment dispersion induced by α-MSH (Fig. 1A,D) or related peptides including des-Acetyl-α-MSH or [Nle4, D-Phe7]-α-MSH (NDP–MSH; not shown). Differences in Agouti protein concentration of <1 nm can be detected in the melanophore assay; the lower limit of detection is ∼0.5 nm (Fig. 1A,D). Agouti protein did not inhibit pigment dispersion caused by agents which, like α-MSH, increase adenylate-cyclase activity. For example, forskolin, a direct activator of adenylate cyclase, produces a dose-dependent dispersion of pigment granules in Xenopus melanophores that is unaffected by 100 nm Agouti protein (Fig. 1C). Similar results were observed with the agonists norepinephrine and isoproterenol (not shown), which stimulate the endogenous adrenergic receptor expressed on melanophores.
Agouti protein does not bind or inactivate α-MSH
Interaction of Agouti protein with cell-surface binding sites has been inferred by its ability to inhibit melanocortin binding or activity. These findings, however, could also be explained by ligand sequestration or inactivation as is thought to occur for several paracrine inhibitors of bone morphogenetic protein signaling such as follistatin, noggin, or chordin (Sasai et al. 1995). To investigate this possibility, we mixed Agouti protein (130 nm) and α-MSH (4 nm) under conditions identical to those used in the melanophore assay, and subjected the mixture to filter centrifugation such that free α-MSH would pass through the filter but α-MSH bound by Agouti protein would not. The amount of active α-MSH present in the filtrate was measured by the melanophore assay under conditions able to detect picomolar differences in α-MSH concentration. Compared with an equimolar amount of bovine serum albumin (BSA), Agouti protein had no effect on the ability of active α-MSH to pass through a 3-kD filter (Fig. 2). Control experiments verified that active Agouti protein was retained by the filter (not shown). Thus, inhibition of melanocortin-receptor signaling by Agouti protein cannot be explained by sequestration or inactivation of α-MSH.
Figure 2.

Lack of interaction between Agouti protein and α-MSH assayed by filtration binding. α-MSH (4 nm) was mixed with either 130 nm Agouti protein (○) or 130 nm BSA (▪) in 2 ml of 70% L-15 media, then subjected to 2 hr of filter centrifugation with a 3-kD filter (Centriprep 3, Amicon, Beverly, MA). Serial dilutions of the two filtrates were assayed for pigment dispersion activity, and no differences were apparent between the Agouti protein and the BSA samples. Testing the retentates for pigment dispersion activity (not shown) confirmed that the Agouti protein remained active after the centrifugation.
The effects of purified Agouti protein on Xenopus melanophores
Because the effects of Agouti protein on Xenopus melanophores only become apparent after pigment dispersion is stimulated with α-MSH, Agouti protein seems likely to act via the endogenous melanocortin receptor on melanophores. To investigate further if these effects could be explained entirely by inhibition of α-MSH binding, we analyzed dose-response curves of α-MSH-induced pigment dispersion in the presence of various concentrations of Agouti protein. For an antagonist that acts solely by preventing agonist binding, increasing concentrations of antagonist cause a proportionate rightward shift of the agonist dose-response curve but do not affect its minimum, maximum, or slope at equilibrium, and the antagonist is said to be competitive (Kenakin 1982). In most mammalian cell systems, depression of maximal signaling or lack thereof are the primary criteria used to assess competitive antagonism; the precision of the melanophore assay, however, offers the additional opportunity to evaluate whether or not the shape of the dose-reponse curve is independent of antagonist.
We found that concentrations of Agouti protein up to 150 nm had no effect on maximal levels of α-MSH-induced pigment dispersion, but significantly altered the slope of the dose-response curve (Fig. 1D; F = 21.5; P < 0.001). The results depicted in Figure 1D represent pigment dispersion 220 min after simultaneous addition of Agouti protein and α-MSH. Experiments in which pigment dispersion was measured for shorter periods of time (not shown) yielded similar results; at no time did the different α-MSH dose-response curves exhibit identical slopes, as expected for a competitive antagonist.
The kinetics of Agouti protein action in melanophores also suggest it has effects beyond simply inhibiting binding of α-MSH. We preincubated melanophores with 15 nm Agouti protein for various times from 25 to 360 min, added α-MSH, and then measured pigment dispersion after an additional 30 min (Fig. 3). Compared with control buffer, 15 nm Agouti caused a rightward displacement of the α-MSH dose-response curve by ∼1.2 log units after a 25-min preincubation; after a 315-min incubation, the α-MSH dose-response curve was displaced further to the right by an additional 0.8 log units (Fig. 3A). Thus, the ability of Agouti protein to inhibit α-MSH-induced pigment dispersion increases gradually over nearly 6 hr, even though Agouti protein by itself has no effect on pigment dispersion (Fig. 3; data not shown).
Figure 3.
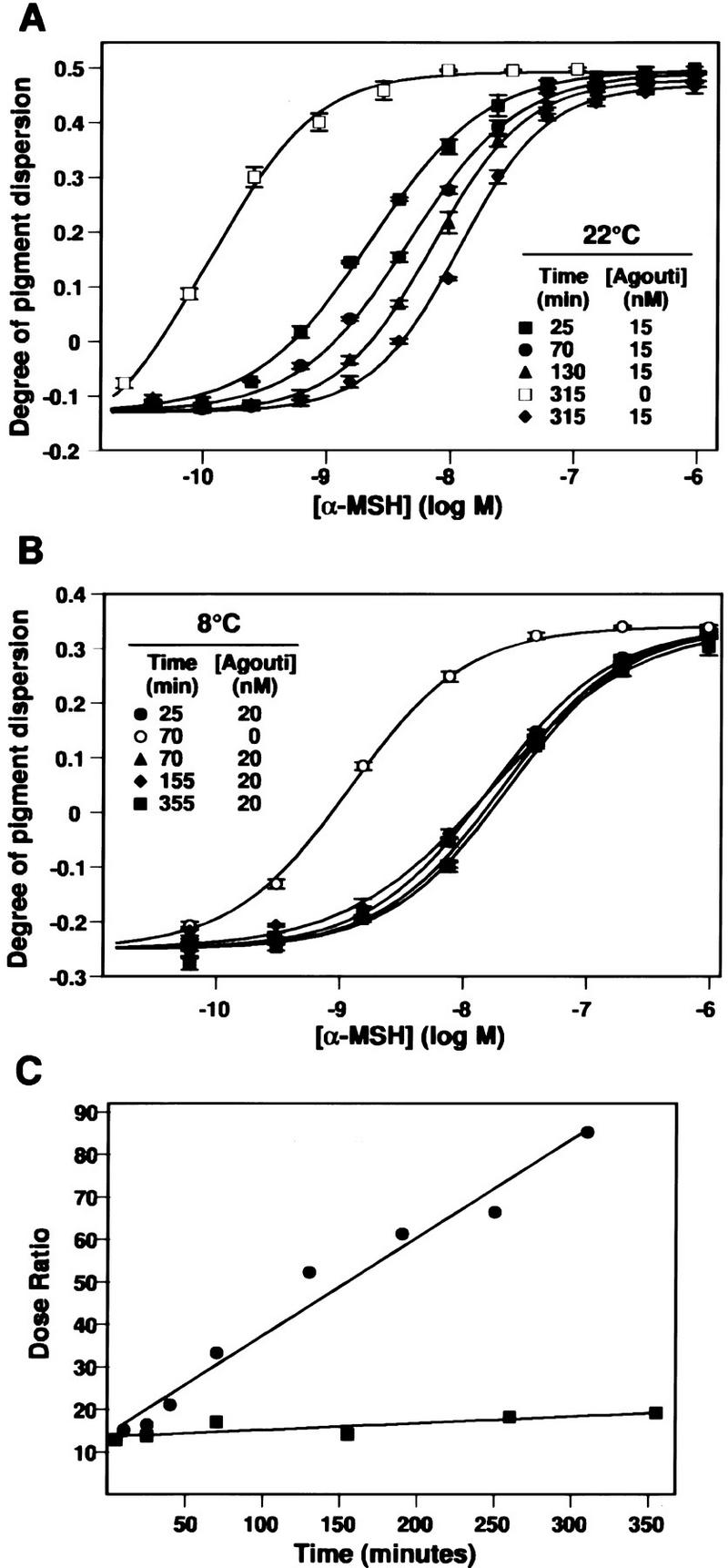
Time and temperature dependence of Agouti protein action. (A) Melanophores were preincubated with 15 or 0 nm (buffer only) Agouti protein for the indicated length of time at 22°C, various concentrations of α-MSH were added for an additional 30 min, and the degree of pigment dispersion was determined as described in Materials and Methods. Preincubation in buffer does not alter the response to α-MSH, therefore, only one time point (315 min) is shown. Only four of the eight preincubation times in Agouti protein (25, 70, 130, and 315 min) are displayed. (B) Preincubation at 8°C. Same as in A except melanophores were kept at 8°C during preincubation with 20 nm Agouti protein, then incubated at 22°C following addition of α-MSH. Qualitatively similar results (not shown) are obtained if the entire experiment is carried out at 8°C. (C) Kinetics of Agouti protein activity at 8°C (▪) and 22°C (•) as measured by the dose ratio. For each time point, the dose ratio is calculated as ([α-MSH] that yields half-maximal pigment dispersion in the presence of Agouti protein/[α-MSH] that yields half-maximal pigment dispersion in the presence of 20 nm Agouti buffer). The abscissa denotes time of preincubation with Agouti protein.
A potential explanation for the time-dependent potentiation in inhibition of α-MSH-induced pigment dispersion by Agouti protein is a slow on-rate, such that equilibrium is not reached for at least 6 hr. Alternatively, Agouti protein might reach equilibrium between bound and soluble pools quickly, but cause a gradual desensitization in the ability of the cells to respond to α-MSH by inducing, for example, Mc1r internalization and/or a post-translational modification of the Mc1r that alters coupling to downstream effectors. To help distinguish among these alternatives, we carried out the preincubation experiment at 8°C, which should reduce or eliminate desensitization, but would have no effect on, or possibly exaggerate, the time to reach equilibrium between bound and soluble pools. During a 25 min preincubation at 8°C, 20 nm Agouti protein caused a rightward displacement of the α-MSH dose-response curve by ∼1.2 log units, and continued incubation up to 355 min produced no further effect (Fig. 3B). The effects of time and temperature on Agouti protein action are most easily compared by measurements of the Dose Ratio, defined as the amount of α-MSH required to produce half-maximal pigment dispersion in the presence of Agouti protein, divided by the amount of α-MSH required to produce half-maximal pigment dispersion in the absence of Agouti protein. The dose ratio increases with continued preincubation at 22°C but not at 8°C (Fig. 3C), which is not easily explained by failure to reach equilibrium between bound and soluble pools. Instead, the effect of Agouti protein on the slope of α-MSH dose-response curves, along with the increase in Agouti protein activity following preincubation, suggests that Agouti protein can cause receptor desensitization by a time- and temperature-dependent mechanism. Taken together, these results suggest that Agouti protein affects α-MSH signaling in two ways: direct inhibition of α-MSH binding and receptor desensitization.
Specific binding of epitope-tagged Agouti protein to cells that express the Mc1r
To help resolve some of the uncertainties regarding the mechanism of Agouti protein action, we developed a system for directly measuring its cell-surface binding. Initial attempts at radioiodination rendered Agouti protein biologically inactive. Therefore, we employed an alternative approach based on immunofluorescence. We constructed a modified form of Agouti protein that contained a hemagglutinin epitope 6 residues downstream of the signal-sequence cleavage site. Recombinant epitope-containing protein (HA–Agouti) was produced in baculovirus, purified by cation exchange, and found to exhibit a specific activity in the melanophore assay equal to that of the native protein (data not shown).
To measure cell-surface binding, we incubated a 25 nm solution of HA–Agouti at 4°C for 3 hr with 293T cells transiently transfected with the Mc1r, fixed the cells with 2% formaldehyde, and detected bound HA–Agouti by immunofluorescence. To help ensure that adequate levels of Mc1r were produced after transfection, most experiments were carried out with a form of the Mc1r modified by addition of a Flag epitope at its amino terminus (Flag–Mc1r). Staining replicate samples either with an Anti-HA antibody or an Anti-Flag antibody then indicated binding of HA–Agouti or expression of the Flag–Mc1r, respectively. In addition, to help identify the 10% to 20% of cells that were transfected and express receptor at high levels, a green fluorescence protein (GFP) expression plasmid was cotransfected with each receptor plasmid in a 1:10 molar ratio.
We found that transfection of either the Mc1r or the Flag–Mc1r (Fig. 4C) conferred on 293T cells the ability to bind HA–Agouti; whereas untransfected cells or those transfected with a control Flag–β2 adrenergic receptor (Flag–B2AR, Fig. 4G) did not bind HA–Agouti (Table 1). HA–Agouti binding is specific, as it is not detected in the presence of an 8-fold (Table 1) or a 20-fold (Fig. 4E) molar excess of >99% pure native Agouti protein. Addition of HA-tagged or native Agouti protein did not visibly alter levels of Flag–Mc1r present at the cell surface (Fig. 4D,F). Nearly all the cells that bound HA–Agouti also expressed GFP (Fig. 4C; Table 1). Fixation in 2% formaldehyde was required for detection of binding, which suggests that binding of HA–Agouti is reversible (Table 1). Dissociation of HA–Agouti must occur slowly, however, because our procedure includes a 50 min wash in phosphate-buffered saline prior to fixation. We were unable to detect binding of HA–Agouti to melanophores (Table 1), possibly because of differences in receptor number and/or affinity compared with 293T cells transiently transfected with the Mc1r.
Figure 4.
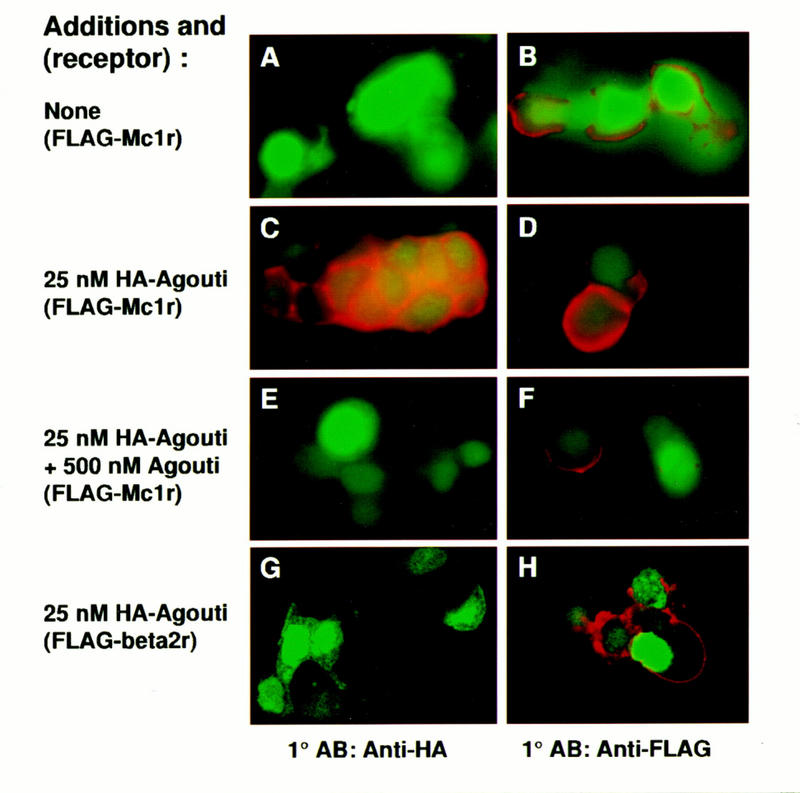
Binding of epitope-tagged Agouti protein to Mc1r-expressing cells. 293T cells were cotransfected with expression plasmids for GFP and the Flag–Mc1r (A–F) or the Flag–B2AR (G,H) as described in Materials and Methods. Transfected cells were incubated in 0 nm Agouti (A,B), 25 nm HA–Agouti (C,D,G,H), or 25 nm HA–Agouti plus 500 nm (untagged) Agouti protein (E,F). Binding of HA–Agouti was detected by immunofluorescence with an anti-HA antibody (A,C,E,G); expression of the Flag–Mc1r or the Flag–B2AR was detected by immunofluorescence with an anti-Flag antibody (B,D,F,H) as described in Materials and Methods. Green staining represents GFP expression and red staining represents bound HA–Agouti (A,C,E,G) or receptor expression (B,D,F,H). All panels represent experiments summarized in Table 1.
Table 1.
Summary of binding experiments with epitope-tagged Agouti protein
| Exp.a
|
Receptorb
|
Ligandb
|
Competitorb
|
Special Cond.c
|
AntiFlagd
|
AntiHAd
|
|---|---|---|---|---|---|---|
| I | GFP only | 25 nm HA–Agouti | none | 16° | N.A. | no |
| Mclr | 25 nm HA–Agouti | none | 16° | N.A. | yes | |
| Mclr | 25 nm HA–Agouti | excess Agoutie | 16° | N.A. | no | |
| Mclr | 25 nm HA–Agouti | 2 μm α-MSH | 16° | N.A. | no | |
| Flag–Mclr | none | none | 16° | yes | N.D. | |
| Flag–Mclr | 25 nm HA–Agouti | none | 16° | yes | yes | |
| Flag–Mclr | 25 nm HA–Agouti | excess Agouti | 16° | N.D. | no | |
| Flag–Mclr | 25 nm HA–Agouti | 2 μm α-MSH | 16° | yes | no | |
| II | Mclr | none | none | none | NA | no |
| Mclr | 25 nm HA–Agouti | none | none | N.A. | yes | |
| Mclr | 25 nm HA–Agouti | 2 μm α-MSH | none | N.A. | no | |
| Mclr | 25 nm HA–Agouti | 2 μm bombesin | none | N.A. | yes | |
| Mclr | 25 nm HA–Agouti | none | unfix.f | N.A. | no | |
| Mclr | 25 nm HA–Agouti | none | 37° | N.A. | yes | |
| Flag–B2AR | none | none | none | yes | no | |
| Flag–B2AR | 25 nm HA-Agouti | none | none | N.D. | no | |
| III | Flag–Mclr | 25 nm HA–Agouti | none | 5 hr | N.D. | yes |
| Flag–Mclr | 25 nm HA–Agouti | 2 μm α-MSH | 5 hr | N.D. | no | |
| Flag–Mclr | 25 nm HA–Agouti | 2 μm NDP–MSH | 5 hr | yes | no | |
| Melanophoresg | none | none | none | N.D. | no | |
| Melanophores | 25 nm HA–Agouti | none | none | N.D. | no | |
| IV | Flag–Mclr | none | none | none | N.D. | no |
| Flag–Mclr | 5 nm HA–Agouti | none | none | N.D. | yesh | |
| Flag–Mclr | 25 nm HA–Agouti | none | none | yes | yes | |
| V | Flag–Mclr | none | none | none | N.D. | 0/110 |
| Flag–Mclr | 33 nm HA–Agouti | none | none | yes | 92/106 | |
| Flag–Mclr | 33 nm HA–Agouti | 250 nm Agouti | none | N.D. | 0/90 | |
| Flag–Mclr | 33 nm HA–Agouti | 10 μm Agouti | none | N.D. | 0/101 | |
| Flag–Mclr | 33 nm HA–Agouti | Agrp control | none | N.D. | 87/104 | |
| Flag–Mclr | 33 nm HA–Agouti | 6 μm Agrp A+B | none | N.D. | 4/100i | |
| Flag–Mclr | 33 nm HA–Agouti | 2 μm Agrp C | none | N.D. | 2/106i | |
| VI | Flag–Mclr | none | none | none | 87/102 | 0/100 |
| Flag–Mclr | 25 nm HA–Agouti | none | none | 89/114 | 82/100 | |
| Flag–Mclr | 25 nm HA–Agouti | 500 nm Agouti | none | 87/104 | 0/100 | |
| Flag–Mclr | 25 nm HA–Agouti | 250 nm α-MSH | none | 93/109 | 0/106 | |
| Flag–Mclr | 25 nm HA–Agouti | 400 nm Agrp C | none | 91/103 | 4/103i | |
| Flag–Mclr | 25 nm HA–Agouti | 1 μm Agrp A+B | none | 84/100 | 8/103i | |
| Flag–Mclr | 25 nm HA–Agouti | Agrp control | none | N.D. | 91/113 | |
| Flag–Mclr | 25 nm HA–Agouti | 2 μm bombesin | none | N.D. | 73/102 |
Six representative experiments are depicted, from a total of 151 slides, in which each condition was tested at least twice, but usually four to five times.
Expression plasmids for the Mclr, the Flag–Mclr, or the Flag–B2AR were cotransfected with a 10-fold reduced molar ratio of an expression plasmid for GFP and binding of HA–Agouti to transfected cells in the presence or absence of various competitors was determined, as described in Materials and Methods.
All binding studies were carried out for 3 hr at 4° unless otherwise indicated.
Results of most binding experiments are described qualitatively (yes or no), based on whether or not strong immunofluorescent signal was present in multiple cells after examining at least five microscopic fields. For experiments V and VI, attempts were made to provide a semiquantitative estimate by describing the number of strongly positive immunofluorescent cells/number of GFP-expressing cells examined. (N.A.) Not applicable; (N.D.) not determined.
Partially purified Agouti protein was used as competitor for initial experiments; its concentration was not determined precisely but represented >100-fold molar excess. Experiments with a defined Agouti protein concentration used a preparation >99% pure.
(Unfixed) As described in Materials and Methods, Agouti Protein-Binding Assay, except cells were not treated with formaldehyde prior to incubation with the primary antibody.
Xenopus melanophores were used instead of 293T cells.
The number of cells staining with the anti-HA antibody was similar in experiments that compared 5 to 25 nm HA–Agouti, but the strength of the immunofluorescent signal was reduced at the lower concentration.
The few cells designated as anti-HA positive exhibited very weak immunofluorescent signals.
The effect of α-MSH and Agrp on Agouti protein binding
The findings described above strongly support a direct interaction between Agouti protein and the Mc1r, but do not exclude the possibility that Agouti protein binds to an unknown factor recruited to the cell surface by expression of the Mc1r. To address this possibility, we asked whether the melanocortin receptor ligand α-MSH could inhibit the binding of HA–Agouti. Two hundred and fifty nanomolar α-MSH had no effect on cell-surface expression of the Flag–Mc1r (Fig. 5B), but completely blocked the binding of 25 nm HA–Agouti (Fig. 5A). NDP–MSH also inhibited binding of HA–Agouti (Table 1). In contrast, addition of 2 μm bombesin, a small peptide lacking specificity for melanocortin receptors, had no effect on binding of HA–Agouti (not shown). Because all of the binding studies were carried out at 4°C, it is unlikely that the ability of α-MSH to block HA–Agouti binding is caused by internalization or degradation of an unknown protein specific for Agouti binding. Instead, we conclude that Agouti and α-MSH bind in a mutually exclusive way to the Mc1r.
Figure 5.
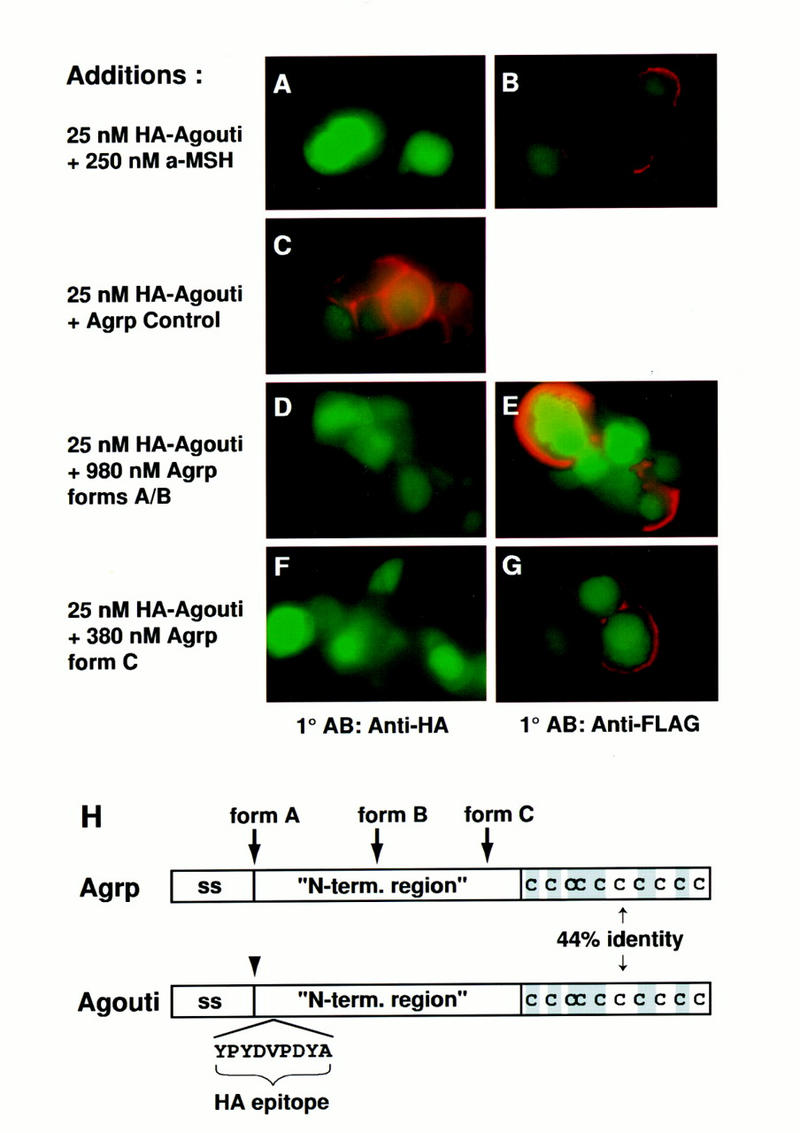
Effects of Agrp and α-MSH on Agouti protein binding. 293T cells expressing GFP and the Flag–Mc1r were incubated at 4°C in 25 nm HA–Agouti with the following additions: 250 nm α-MSH (A,B), 980 nm Agrp forms A+B (D,E), 380 nm Agrp form C (F,G), or control protein for Agrp purification (C). Binding of HA–Agouti or expression of the Flag–Mc1r was detected by immunofluorescence with anti-HA (A,C,D,F) or anti-Flag (B,E,G) antibodies as described in Materials and Methods. Green staining represents GFP expression; red staining represents bound HA–Agouti (A,C,E,G) or receptor expression (B,E,G). All panels represent experiments summarized in Table 1. (H) Diagram of Agrp and Agouti protein indicating the signal sequence (ss), placement of the HA epitope in HA–Agouti, the different forms of Agrp, and amino acid similarity between Agrp and Agouti, which is confined entirely to the cysteine-rich carboxyl terminus.
We also asked if Agrp, a newly described neuropeptide that may regulate body weight in response to leptin signaling (Ollmann et al. 1997; Shutter et al. 1997), could affect binding of HA–Agouti. Agouti and Agrp exhibit 44% amino acid identity in the carboxy-terminal cysteine-rich region required for antagonism by Agouti protein (Willard et al. 1995), and we have shown recently that recombinant Agrp can act as a potent Mc3r and Mc4r antagonist in vitro, which suggests that Agouti protein and Agrp act via a similar biochemical mechanism.
Recombinant Agrp produced in the baculovirus system can be purified in two fractions which exhibit biologic activity (Ollmann et al. 1997). One fraction contains a mixture of mature full-length protein and proteolytically cleaved forms shortened at their amino termini by 26–30 residues (Fig. 5H, forms A + B). A second fraction contains forms that are further shortened and comprised mainly of the carboxy-terminal cysteine-rich region (Fig. 5H, form C).
We found that high concentrations of either Agrp fraction would inhibit binding of HA–Agouti to cells transfected with the Flag–Mc1r (Fig. 5D,F; Table 1) without affecting cell-surface expression of the Flag–Mc1r (Fig. 5E,G; Table 1). As a control, medium conditioned by insect cells infected with an irrelevant baculovirus was subjected to procedures similar to those used to purify Agrp; we found that the control preparation had no effect on HA–Agouti binding (Fig. 5C; Table 1). These findings show that similarity between Agouti protein and Agrp includes their binding sites, and highlight the 20 residues shared between mature Agouti protein and Agrp, 10 of which are cysteine, as critical for melanocortin receptor interaction. It should be noted that in previous studies of α-MSH-induced cAMP accumulation, we found that Agrp was a potent antagonist of the MC3R and MC4R, but had little effect on the MC1R (Ollmann et al. 1997). The apparent disparity between those results and the binding studies described here may relate to structural differences between the mouse Mc1r and human MC1R, or to the phenomenon of spare receptors, whereby maximal effects of an agonist on function occur when only a fraction of the receptors are occupied. In addition, Agrp may not bind the Mc1r as avidly as Agouti protein, because a few cells weakly positive for HA–Agouti could be detected after incubation with micromolar concentrations of Agrp, whereas binding of HA–Agouti was completely abolished by 250 nm native Agouti protein (Table 1, experiment V).
The effects of Agouti on pigmentation in vivo require a functional Mc1r
The studies described above indicate that Agouti protein interacts directly with the Mc1r, but it is possible that Agouti protein may bind to and signal through additional cell-surface molecules present on pigment cells. To address this question, we asked whether the coat color phenotype of animals carrying a gain-of-function Agouti allele, lethal yellow (Ay), was modified by the presence of a loss-of-function Mc1r allele, recessive yellow (Mc1re). On most genetic backgrounds, this analysis is inconclusive because the phenotypes of each single mutant are very similar. Several observations, however, (for review, see Silvers 1979b) suggest that a hypomorphic mutation at the albino (c) locus, chinchilla (cch), produces a sensitized background in which the coat color phenotypes caused by Ay and Mc1re can be distinguished. By use of a consistent strain background, C57BL6/J, we confirmed this observation: Ay/a; cch/cch mice are cream colored, in contrast to Mc1re/Mc1re; cch/cch mice that are pale yellow with black ticking (Fig. 6B; Table 2).
Figure 6.
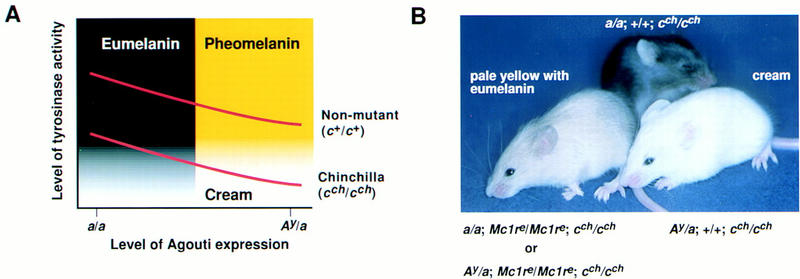
Interaction between Ay and the Mc1re mutations on a chinchilla (cch/cch) background. (A) Diagram of the relationship between Agouti expression and tyrosinase activity based on Geschwind (1966), Kobayashi et al. (1995), Movaghar (1989), and Prota et al. (1995), and as modified from Barsh (1996). During the switch from eumelanin to pheomelanin production caused by increased Agouti expression, tyrosinase activity is gradually downregulated. The exact biochemical mechanism of the switch is not clear, but may be related to abrupt cessation of other melanogenic proteins. Because tyrosinase catalyzes the rate-limiting step for production of both pigment types, animals carrying a point mutation that impairs tyrosinase activity, such as chinchilla, make very little or no pigment in the presence of high levels of Agouti, and appear cream-colored. (B) Photograph of animals that carry various combinations of coat color mutations (see text and Table 2 for further explanation).
Table 2.
Effects of the Ayand Mclremutations on a chinchilla (cch/cch) background
| Cross
|
Phenotypea
|
No.b
|
Genotypeb
|
||
|---|---|---|---|---|---|
|
Agouti
|
Mclr
|
Tyr (c)c
|
|||
| Ay/a; +/+; +/cch | yellow | Ay/a | +/+ | +/cch | |
| × | cream | Ay/a | +/+ | cch/cch | |
| a/a; +/+; cch/cch | black | a/a | +/+ | +/cch | |
| black | a/a | +/+ | cch/cch | ||
| a/a; +/Mclre, +/cch | yellow | a/a | Mclre/Mclre | +/+ or +/cch | |
| × | pale yellowd | a/a | Mclre/Mclre | cch/cch | |
| ala; +/Mclre; +/cch | black | a/a | +/+ or +/Mclre | +/+ or +/cch | |
| black | a/a | +/+ or +/Mclre | cch/cch | ||
| Ay/a; Mclre/Mclre; cch/cch | cream | 9 | Ay/a | +/Mclre | cch/cch |
| × | pale yellowd | 9 | Ay/a | Mclre/Mclre | cch/cch |
| Ay/a; +/Mclre; cch/cch | pale yellowd | 9 | a/a | Mclre/Mclre | cch/cch |
| black | 7 | a/a | +/Mclre | cch/cch | |
Subtle coat color differences exist between a/a; +/+; cch/cch and a/a; +/+; +/+ animals and between Ay/a; +/+; +/+ and a/a; Mclre/Mclre, +/+ animals.
Phenotypic classes were tallied, and genotypes were determined using molecular techniques as described in Materials and Methods only for the last cross, which represents two breeding pairs. Results similar to the first two crosses have been described previously by Feldman (1935) and by Searle and Beechey (1970), respectively.
The symbol for the albino locus (c) was recently changed to Tyr; here we refer to the chinchilla mutation with the symbol cchrather than Tyrc-ch.
Mclre/Mclre; cch/cch mice are pale yellow with eumelanin ticking and can easily be distinguished from cream-colored Ay/a; cch/cch animals by the presence of eumelanin or from yellow Mclre/Mclre; +/cch animals by reduced intensity of pheomelanin.
We constructed breeding pairs that segregated lethal yellow and recessive yellow, and found that all animals with the cream colored phenotype were Ay/a; +/Mc1re; cch/cch or Ay/a; +/+; cch/cch whereas animals with the pale yellow plus eumelanin phenotype were Ay/a; Mc1re/Mc1re; cch/cch or a/a; Mc1re/Mc1re; cch/cch (Fig. 6B, Table 2). There are several possibilities to explain the difference in coat color phenotypes between Ay and Mc1re (see below). Regardless of the exact mechanism, our results show that the effects of Ay on pigmentation in vivo require a functional Mc1r, and, therefore, argue against the presence of an additional melanocyte receptor for Agouti protein.
Discussion
Genetic and transplantation studies carried out several decades ago first suggested the possibility that gene products encoded by Agouti and Extension (now renamed as the Mc1r) might interact as ligand and receptor, respectively, because both genes affect the balance between black and yellow pigment synthesis, and because Extension was melanocyte autonomous whereas Agouti was not (for review, see Silvers 1979a). Recessive alleles of Extension, however, produce pigmentation phenotypes similar to dominant alleles of Agouti, and vice versa, which indicated that Agouti protein must inhibit, rather than activate, the Extension gene product.
More recently, molecular genetic and pharmacologic studies have shown that Agouti protein antagonizes the Mc1r (Lu et al. 1994; Blanchard et al. 1995; Willard et al. 1995; Yang et al. 1997), but the biochemical mechanism by which this takes place has been controversial (Conklin and Bourne 1993; Jackson 1993). In particular, the ability of Agouti protein to elicit cellular responses apparently independent of adenylate cyclase (Hunt and Thody 1995; Siegrist et al. 1996, 1997; Sakai et al. 1997; Suzuki et al. 1997), combined with similarity in cysteine spacing between Agouti protein and invertebrate toxins that affect calcium channels (Olivera et al. 1991; Quistad and Skinner 1994; Zemel et al. 1995), has led to uncertainty regarding the identity of the Agouti protein receptor or receptors. Our results show that the Mc1r encodes a receptor for Agouti protein, and that a functional Mc1r is required for Agouti signaling in vivo. Furthermore, our results suggest that the effects of Agouti protein cannot be explained solely by inhibition of α-MSH binding. These results are likely to apply to homologs of Agouti and the Mc1r that normally regulate other biologic processes, and have general implications for understanding melanocortin-receptor signaling.
The Mc1r is an Agouti receptor
Previous studies carried out by us and others have shown that binding of the radiolabeled melanocortins NDP–MSH or ACTH to whole cells can be inhibited by recombinant Agouti protein (Lu et al. 1994; Blanchard et al. 1995; Siegrist et al. 1997; Yang et al. 1997). As shown here, expression of the Mc1r provides a binding site for Agouti protein that can be blocked by α-MSH, which strongly suggests that Agouti protein is a ligand for the Mc1r. Although the immunofluorescence assay precludes a quantitative measurement of binding affinity, Agouti protein binding is detectable after incubation in 5 nm HA–Agouti and can be inhibited by excess native Agouti protein, showing that the binding site we describe is specific and represents a high affinity site.
Even though Agouti protein and α-MSH inhibit each other’s binding, the two molecules may not interact with identical sites on the Mc1r. The active portion of Agouti protein contains 40 amino acids and 5 disulfide bridges which, by analogy to other proteins of similar size and cysteine spacing, stabilize a hydrophilic tertiary structure that interacts with one or more extracellular domains of the receptor. In contrast, α-MSH and other small melanocortin peptides that contain the core sequence His–Phe–Arg–Trp are thought to interact with membrane-spanning portions of the receptor (Miwa et al. 1995; Prusis et al. 1995; Haskell-Luevano et al. 1996a,b). A model for the binding of omega-conotoxins, which contain three disulfide bridges with primary sequence spacing identical to Agouti protein, posits interaction with a target receptor macrosite that directly blocks access of one or more small molecule agonists to bind their microsites (Olivera et al. 1991). This same model might apply to Agouti protein, Agrp, and melanocortin receptors. Alternatively, Agouti protein and α-MSH could bind to nonoverlapping sites on the Mc1r, yet inhibit each other’s binding by inducing an allosteric change in the receptor. Additional mutagenesis studies that focus on melanocortin receptors exhibiting a differential response to Agouti protein or to Agrp (Lu et al. 1994; Ollmann et al. 1997; Yang et al. 1997) may help to clarify certain aspects of the interaction between Agouti protein and the Mc1r, although a complete understanding will require more detailed information about receptor structure.
Agouti signaling is not equivalent to inhibition of α-MSH binding in vitro
Mutually exclusive binding of Agouti protein and α-MSH to the Mc1r is consistent with a model of competitive antagonism, whereby Agouti protein would act solely to reversibly displace ligand binding. In Xenopus melanophores, however, which share some, but not all, features of melanocortin signaling with mammalian cells, our observations indicate Agouti protein has effects beyond simply inhibiting the binding of α-MSH.
Specifically, Agouti protein alters the slope of the dose-response curve for α-MSH-induced pigment dispersion, and preincubation in Agouti protein increases its ability to inhibit melanocortin activity. These findings suggest that Agouti protein not only inhibits the binding of agonists, but also alters the interaction of melanocortin receptors with intracellular effectors. The time- and temperature- dependence of Agouti protein activity revealed in the preincubation experiments might be explained by certain types of post-translational modifications that decrease receptor coupling (for review, see Lefkowitz et al. 1993). Alternatively, Agouti protein could cause internalization of the melanophore α-MSH receptor, as Eberle and colleagues have reported for the Mc1r in B16 melanoma cells (Siegrist et al. 1996).
The conclusion that Agouti protein is not a simple competitive antagonist of melanocortin receptors may help to explain the discrepancy between estimates of Agouti protein affinity based on functional compared with binding assays (Lu et al. 1994; Blanchard et al. 1995; Willard et al. 1995; Kiefer et al. 1997; Yang et al. 1997). Furthermore, the observation that Agouti protein does not act solely by inhibiting the binding of α-MSH raises the possibility that control of melanocortin receptor signaling does not require the presence of melanocortins in vivo. Injection of α-MSH near growing hairs induces eumelanin production (Geschwind 1966), but it is not known whether physiologic levels of melanocortin peptides are present in the hair follicles when transient expression of Agouti during normal hair growth gives rise to a banded pigmentation pattern. In Xenopus melanophores, the effects of Agouti protein can be detected only in the presence of melanocortin peptides. In mammalian skin, however, binding of Agouti protein to the Mc1r may be sufficient to alter receptor signaling and produce the phenotypic effects of Agouti in the absence of exogenous melanocortins.
The Mc1r is required for Agouti signaling in vivo
The lethal yellow (Ay) and recessive yellow (Mc1re) mutations usually produce a similar coat color, but we found that reduced activity of tyrosinase caused by the chinchilla (cch) mutation enhanced the difference in pigmentation phenotypes between lethal yellow and recessive yellow mice. Our comparisons were carried out on a uniform genetic background provided by the C57BL/6J strain; similar results in a mixed genetic background were reported in Mouse News Letters by Searle and Beechey (1970). Differences in pigmentation phenotypes caused by lethal yellow and recessive yellow have also been uncovered in animals heterozygous for the albino (Tyrc) mutation, or in animals homozygous for dilute (Myo5ad) or leaden (ln) (for review, see Silvers 1979b).
The different coat color phenotypes of Ay/a; cch/cch and Mc1re/Mc1re; cch/cch mice, cream-colored and pale yellow with black ticking, respectively, indicate that overexpression of Agouti protein is not equivalent to loss of Mc1r signaling. A priori, incomplete inhibition of Mc1r signaling by Agouti protein might account for a phenotypic difference between lethal yellow and recessive yellow, but this is unlikely to explain our observations because, in general, Mc1r signaling is proportionate to tyrosinase activity (Geschwind 1966; Movaghar 1989; Kobayashi et al. 1995; Prota et al. 1995), and the cream-colored phenotype is caused by a reduction rather than an increase in tyrosinase activity (see Fig. 6A).
Furthermore, the different coat color phenotypes of lethal yellow and recessive yellow are probably not caused by residual Mc1r activity in Mc1re/Mc1re mice, because the Mc1re mutation is caused by a frameshift that gives rise to a truncated protein with no detectable activity in vitro (Robbins et al. 1993; also see below). It is possible that the cream-colored phenotype of Ay/a; cch/cch mice comes about because Agouti protein causes the Mc1r to couple to an effector other than Gs and adenylate cyclase, in which case Agouti protein should be considered a potential agonist as well as an antagonist of the Mc1r.
Regardless, the ability of Agouti protein to cause a cream-colored phenotype in Ay/a; cch/cch mice requires a functional Mc1r, because Ay/a; Mc1re/Mc1re; cch/cch mice exhibit a different coat color phenotype that is indistinguishable from that observed in Mc1re/Mc1re; cch/cch mice. These findings reinforce the conclusion based on in vitro studies that the Mc1re mutation causes a complete loss of function (Robbins et al. 1993), and, more importantly, show that the ability of Agouti protein to alter pigmentation in vivo is mediated by the Mc1r and not by a receptor specific for Agouti protein.
Concluding remarks
Control of G protein-coupled receptor signaling in most biological systems is determined primarily by agonist availability. As endogenous antagonists, Agouti protein and Agouti-related protein offer some unique advantages for regulation of melanocortin receptor signaling. The limited tissue distribution and biochemical specificity of Agouti protein allows individual regulation of melanocortin receptor subtypes that could not be achieved by regulating agonist transcription, because melanocortin peptides and β-endorphin are circulating molecules derived from a single precursor, pro-opiomelanocortin (Pomc). In addition, the studies described here suggest a viewpoint whereby Agouti protein and α-MSH function as independent ligands that transduce opposite signals through the Mc1r, yet also inhibit each other’s binding to the Mc1r. Both viewpoints, that of endogenous antagonist and that of independent ligand, help to explain why genetic control of the balance between eumelanin and pheomelanin is attributable mostly to alleles of Agouti or the Mc1r rather than Pomc, because transcriptional control of α-MSH would likely affect all melanocortins as well as β-endorphin, and because Agouti-mediated signaling through the Mc1r may not require the presence of α-MSH.
To date, homologs of Agouti or Agrp have been found only in mammals, although melanocortins are widely distributed among vertebrates including avians and cartilaginous fish (for review, see Dores et al. 1990). Previously, we have suggested that an, as yet undiscovered, Agouti homolog may be responsible for dorsal–ventral differences in amphibian pigmentation (Vrieling et al. 1994), a speculation that is supported by the ability of Agouti protein and Agouti-related protein derived from mice or humans to inhibit α-MSH-induced pigment dispersion in Xenopus melanophores (Ollmann et al. 1997). Given the similarity in cysteine spacing shared between Agouti and insect neurotoxins, it is possible that a common precursor of these proteins was adapted to regulate melanocortin receptor signaling in primitive vertebrates. From this perspective, it will be interesting to determine the point at which Agouti and Agouti-related protein evolved from a common ancestor, because this may reflect adaptation of melanocortin receptor signaling for peripheral functions such as pigmentation, and central functions, such as feeding behavior.
Materials and methods
Xenopus melanophore culture and pigment dispersion assay
Xenopus melanophores were grown at 27°C in 50% L-15 media (Specialty Media, Lavallete, NJ), supplemented with 20% heat-inactivated fetal calf serum, 1 mm l-glutamine, penicillin, and streptomycin; the media had been conditioned previously by use of Xenopus fibroblasts as described by Potenza and Lerner (1992). The pigment dispersion assay developed by Lerner and colleagues (Potenza and Lerner 1992) is based on the ability of agents that cause a decrease or increase in intracellular cAMP levels to produce a dose-dependent aggregation or dispersion, respectively, of intracellular pigment granules. Because pigment granules are neither fully aggregated nor dispersed in the absence of any drug, pretreatment of the cells with melatonin to aggregate pigment granules increases the range and sensitivity of the assay for detecting agents such as α-MSH that disperse pigment granules. For a typical assay, cells were plated in 96-well plates at 25,000 cells/well 24–48 hr beforehand, washed briefly with 250 μl/well assay buffer (70% L-15 media; 0.1% BSA), 40 μl/well assay buffer was then added, followed by 40 μl/well assay buffer that contained 2 nm of melatonin (Sigma, St. Louis, MO) to provide a final melatonin concentration of 1 nm. After a 45 min incubation to aggregate pigment granules, the optical density of each well was measured at 650 nm (ODinitial) to provide a baseline optical density reading. Agouti protein samples to be assayed were added at 40 μl/well followed by the addition of various concentrations of α-MSH or forskolin at 40 μl/well. All additions were made in assay buffer supplemented with 1 nm of melatonin to maintain a constant concentration of melatonin during the assay. Optical density at 650 nm was then determined at multiple time points from 30 to 355 min (ODfinal). All steps were carried out at room temperature unless noted otherwise. A unitless parameter defined as degree of pigment dispersion was calculated as described by Potenza and Lerner (1992), (ODfinal − ODinitial)/ODfinal, which creates an internal standard for each well (ODinitial) and scales the maximal degree of pigment dispersion to 1. The effects of 1 mm melatonin occasionally increase during the course of the assay, which gives rise to negative values for the degree of pigment dispersion. Optical density at 650 nm of melanophores was measured with a Vmax kinetic microplate reader (Molecular Devices, Menlo Park, CA) in endpoint mode, and data was transferred electronically to a Microsoft Excel spreadsheet for analysis. Graphing and curve fitting of dose-response curves was carried out with DeltaGraph (DeltaPoint, Monterey, CA), with a four parameter logistic equation, y = a + [(b − a)/(1 + (10c/10x)d)]; where a = minimum, b = maximum, c = half-maximal × value, and d = slope.
Generation and purification of recombinant Agouti proteins
A 527-bp EcoRI fragment that contains the mouse Agouti ORF but lacks a polyadenylation signal [residues 122–648 in Miller et al. (1993)] was modified by insertion of a 519-bp mouse genomic fragment that contained the polyadenylation signal, with a BssHII site 15 bp 3′ of the termination codon. The entire fragment was inserted into the baculovirus transfer vector pVL1393 (Pharmingen, San Diego). For HA-tagged mouse Agouti protein, the 9-amino-acid HA epitope YPYDVPDYA was placed 5 amino acids downstream of the signal sequence cleavage site (Fig. 5H) by inserting a double-stranded DNA fragment, created by annealing together the oligonucleotides 5′-TCGAATACCCGTACGACGTCCCCGATTACGCAC-3′ and 5′-TCGAGTGCGTAATCGGGGACGTCGTACGGGTAT-3′, into a XhoI site that spans codons 26–27. The cDNAs for native Agouti or HA-tagged Agouti were cloned into the EcoRI site of baculovirus transfer vector pVL1393 (Pharmingen).
For native Agouti protein, high-titer recombinant baculovirus was produced by standard methods and added at a 60-fold dilution to 6.7 liters of Trichoplusia ni cells grown in suspension to 4 × 106 cells/ml in Ex-Cell 401 media (JR Scientific, Woodland, CA). Conditioned media harvested 50 hr after infection was centrifuged at 10,000g for 60 min, then applied at 10 ml/min to a 1.5 × 7.3-cm SP Sepharose Fast Flow cation exchange column. The column was washed with 64 ml of 50 mm Bicine, at pH 9.0; 100 mm NaCl, then eluted with a 250 ml of NaCl gradient (100 mm–600 mm NaCl in 50 mm bicine, at pH 9.0). Flowthrough, wash, and eluate fractions (6.4 ml/fraction) were diluted in melanophore assay buffer at 1:10, 1:10, and 1:100, respectively, and tested in the Xenopus melanophore assay for inhibition of α-MSH-induced pigment dispersion. A single peak of α-MSH inhibitory activity eluted from the cation exchange column at ∼470 mm NaCl, and is described below as the cation exchange product. Active fractions were buffer exchanged into 10 mm CAPS, at pH 10.5; 10 mm NaCl with a 210 ml Sephadex G-25 column, then applied to a 1 ml of HiTrap Q anion exchange column. The anion exchange column was washed with 5 ml of 10 mm CAPS at pH 10.5 and 10 mm NaCl, then eluted with a 20 ml of NaCl gradient (10 mm–450 mm NaCl in 10 mm CAPS, at pH 10.5), followed by 5 ml of 10 mm CAPS, at pH 10.5; 450 mm NaCl, and 5 ml of 10 mm CAPS, at pH 10.5; 1 m NaCl. Wash and eluate fractions (0.5 ml/fraction) were diluted in melanophore assay buffer at 1:200; α-MSH inhibitory activity was detected in the flowthrough, described below as anion exchange product 1, and in a peak eluting in ∼150 mm NaCl, described below as anion exchange product 2. Active fractions were dialyzed into 20 mm PIPES, at pH 6.8; 50 mm NaCl (storage buffer), flash frozen, and stored at −70°C. The final yield of Agouti protein from 6.7 liters of conditioned media was 8.1 mg of anion exchange product 1 and 1.2 mg of anion exchange product 2. Chromatographic media were obtained from Pharmacia (Piscataway, NJ), and all purification steps were carried out at 4°C.
One-microgram samples of conditioned media, cation exchange product, anion exchange product 1, and anion exchange product 2 were separated by SDS-PAGE on a 4%–20% gradient gel and detected by silver staining, as shown in Figure 1. On the basis of a sensitivity of 2–5 ng/band, we estimate that anion exchange products 1 and 2 are ⩾99% pure. Amino-terminal sequencing identified both products as mouse Agouti protein cleaved at the predicted signal sequence cleavage site between amino acids 22 and 23.
HA-tagged Agouti protein was produced in baculovirus by use of methods identical to those described above, but was not subjected to the final anion exchange step.
Expression of the Mc1r and Flag–Mc1r
The mouse Mc1r expression vector was constructed by inserting a 947-bp fragment that contained the mouse Mc1r protein-coding region, generously provided by Dr. Linda Rehfuss (Kline-Beecham, Philadelphia, PA), into BamHI–XbaI sites of the vector pcDNA3 (Invitrogen, San Diego, CA). For construction of the Flag–Mc1r expression vector, a 132-bp fragment that encodes a cleavable signal sequence followed by the Flag epitope was fused to the Mc1r protein-coding sequence with a Tth111I site that lies immediately 5′ of the Mc1r initiation codon. The 132-bp fragment was derived by PCR of the plasmid Sfbeta2 (Guan et al. 1992) with the oligonucleotide primers 5′-ATACTCAAGCTTGAATTCGAGCTCG-3′ and 5′-GCTCTAGAGCCGGCGTCATCATCGTCCTTG-3′, and has been shown to enhance surface expression of adrenergic receptors, which, like melanocortin receptors, lack an amino-terminal-cleavable signal sequence (Guan et al. 1992).
Receptor plasmids were transiently expressed in 293T cells by use of calcium phosphate transfection according to standard methods. Cells were plated at 9 × 105 cells/60-mm dish 16–20 hr beforehand, incubated with fresh media for 2–4 hr, then exposed to a mixture of calcium phosphate and 20 μg of plasmid DNA encoding the Mc1r, the Flag–Mc1r, or a control Flag–β 2 adrenergic receptor, generously provided by Dr. Brian Kobilka (Stanford University School of Medicine, CA). Each transfection also included 2 μg of an expression plasmid encoding GFP. After 12–16 hr, the cells were rinsed with CaCl2- and MgCl2-containing phosphate-buffered saline (PBS), then incubated in fresh media for an additional 48 hr prior to the binding assay.
Agouti protein-binding assay
HA–Agouti binds avidly to the extracellular matrix and to plastic, and, therefore, we carried out the binding assay on cells in suspension. Cells that had been transfected previously with a receptor expression plasmid were removed from the dish by rinsing with binding buffer (90% DMEM; 25 mm HEPES, at pH 7.4; 0.2% ovalbumin), washed and resuspended in binding buffer at ∼2 × 106 cells/ml, and 100-μl aliquots were placed in polystyrene tubes. To each tube, competitor protein (Agouti protein, Agrp, α-MSH, bombesin, or buffer control; see Table 1 for concentrations) was added in 50 μl of binding buffer, followed by an additional 50 μl of binding buffer that contained HA–Agouti to achieve a final concentration of 5 nm–33 nm HA–Agouti. After 3 hr at 4°C, 3 ml of cold PBS was added, cells were washed three times with 3.5 ml of PBS, then resuspended in 400 μl of PBS to which 400 μl of 4% formaldehyde in PBS was then added. After a 15 min fixation at room temperature, cells were washed twice with 3.5 ml of PBS, then resuspended in 3.5 ml of 5% goat serum in PBS. After a 60 min incubation to block nonspecific binding, cells were centrifuged and resuspended in 300 μl of 5% goat serum in PBS that contained a 1:500 dilution of primary antibody (16B12 anti-HA, BabCO, Richmond, CA; or M1 Anti-Flag, IBI/Kodak, Rochester, NY), and incubated overnight. Primary antibody was removed by centrifugation and washing three times with 3.5 ml of 5% goat serum in PBS, then cells were resuspended in 250 μl of 5% goat serum in PBS that contained a 1:300 dilution of secondary antibody, goat anti-mouse IgG conjugated to Texas Red. After a 60 min incubation, cells were washed four times with PBS, resuspended in residual PBS and mounted on a glass slide with Vectashield mounting media (Vector, Burlingame, CA). Immunofluorescent images were acquired with a Zeiss Axiophot microscope (100× objective) and CCD camera with exposure times of 1 sec for Texas Red images and 0.5 sec for GFP images. Texas Red and GFP images were merged in Adobe Photoshop with identical adjustment of brightness and contrast for all images.
Mouse strains, mutations, and breeding experiments
Mice carrying the lethal yellow (Ay), recessive yellow (Mc1re), or chinchilla (Tyrc-ch) mutations were obtained originally from The Jackson Laboratory (Bar Harbor, ME) and are maintained by one of us (M.L. Lamoreux) on the C57BL/6J background. The symbol for chinchilla was recently changed from cch to Tyrc-ch, but the older nomenclature has been used here to avoid confusion.
As summarized in Table 2, the Ay or Mc1re mutations were first placed on a chinchilla background by a cross with Ay/a; +/+; +/cch or a/a; +/Mc1re; +/cch animals, respectively. Ay/a; cch/cch animals were easily identified by their cream-colored phenotype. Presumptive Mc1re/Mc1re; cch/cch animals were selected by a coat color phenotype of pale yellow with eumelanin ticking, and their genotype confirmed by progeny testing. The results described here were obtained on a C57BL/6J background; similar results have been described for Ay and Mc1re by Feldman (1935) and Searle and Beechey (1970), respectively, for a mixed genetic background. In this earlier work, and as summarized by Silvers (1979b), the coat color phenotypes of a/a; +/+; cch/cch and Ay/a; +/+; cch/cch animals are described as sepia and ivory, respectively; here we use the designations black and cream-colored. In addition, we describe lethal yellow and recessive yellow animals as yellow; in fact, these classes exhibit subtle coat color differences and can often be distinguished. recessive yellow (a/a; Mc1re/Mc1re; +/+) animals are darker (sooty) than lethal yellow (Ay/a; +/+; +/+) animals, especially prior to weaning.
The phenotype displayed by Mc1re/Mc1re; cch/cch animals, pale yellow with eumelanin ticking, is easily distinguished from Ay/a; cch/cch animals by the presence of eumelanin, and from Mc1re/Mc1re; +/cch animals by the intensity of the yellow color.
To establish a cross in which both Ay and Mc1re were segregating, cream-colored Ay/a; +/+; cch/cch animals were bred to pale yellow a/a; Mc1re/Mc1re; cch/cch animals, a cross of (Ay/a; +/Mc1re; cch/cch × a/a; +/Mc1re; cch/cch) was set up from the F1 progeny, and breeding pairs of F2 animals were identified that produced both pale yellow and cream-colored progeny. For these F2 kindreds, genotypes at the Agouti and Mc1r loci were determined by Southern blotting and direct sequencing of PCR-amplified genomic DNA, respectively, as described previously (Miller et al. 1997).
Acknowledgments
We are grateful to Dr. Michael Lerner and members of the Lerner laboratory for providing Xenopus melanophores as well as ancillary reagents and protocols for melanophore growth. We thank Dr. Teresa Gunn for help with genotyping, Dr. Karl Reich and Cindy Harryman-Samos for their helpful advice with chromatography and binding assays, respectively, and Drs. Linda Rehfuss and Brian Kobilka for the Mc1r and SFB2 plasmids, respectively. M.M.O. and B.D.W. are supported by graduate (EY07106) and medical scientist trainee (GM07365) training grants, respectively. This work was supported in part by grants from the National Institutes of Health to M.L.L. (EY 10223) and to G.S.B. (DK28506) who is an Associate Investigator of the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL gbarsh@cmgm.stanford.edu; FAX (650) 723-1399.
References
- Adan RAH, Vanderkraan M, Doornbos RP, Bar PR, Burbach JPH. Melanocortin receptors mediate alpha-MSH-induced stimulation of neurite outgrowth in neuro 2A cells. Mol Brain Res. 1996;36:37–44. doi: 10.1016/0169-328x(95)00236-l. [DOI] [PubMed] [Google Scholar]
- Barsh GS. The genetics of pigmentation: From fancy genes to complex traits. Trends Genet. 1996;12:299–305. doi: 10.1016/0168-9525(96)10031-7. [DOI] [PubMed] [Google Scholar]
- Blanchard SG, Harris CO, Ittoop ORR, Nichols JS, Parks DJ, Truesdale AT, Wilkison WO. Agouti antagonism of melanocortin binding and action in the B16F10 murine melanoma cell line. Biochemistry. 1995;34:10406–10411. doi: 10.1021/bi00033a012. [DOI] [PubMed] [Google Scholar]
- Bultman SJ, Michaud EJ, Woychik RP. Molecular characterization of the mouse agouti locus. Cell. 1992;71:1195–1204. doi: 10.1016/s0092-8674(05)80067-4. [DOI] [PubMed] [Google Scholar]
- Chhajlani V, Wikberg JES. Molecular cloning and expression of the human melanocyte stimulating hormone receptor cDNA. FEBS Lett. 1992;309:417–420. doi: 10.1016/0014-5793(92)80820-7. [DOI] [PubMed] [Google Scholar]
- Cone RD, Lu D, Koppula S, Vage DI, Klungland H, Boston B, Chen W, Orth DN, Pouton C, Kesterson RA. The melanocortin receptors: Agonists, antagonists, and the hormonal control of pigmentation. Recent Prog Horm Res. 1996;51:287–318. [PubMed] [Google Scholar]
- Conklin BR, Bourne HR. Mouse coat colour reconsidered. Nature. 1993;364:110. doi: 10.1038/364110b0. [DOI] [PubMed] [Google Scholar]
- Dores RM, McDonald LK, Steveson TC, Sei CA. The molecular evolution of neuropeptides: Prospects for the ’90s. Brain Behav Evol. 1990;36:80–99. doi: 10.1159/000115300. [DOI] [PubMed] [Google Scholar]
- Eberle AN. The melanotropins. Chemistry, physiology and mechanism of action. Basel: Karger; 1988. [Google Scholar]
- Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- Feldman HW. A fifth allelomorph in the albino series of the house mouse. J Mammal. 1935;16:207–210. [Google Scholar]
- Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, Delvalle J, Yamada T. Molecular cloning, expression, and gene localization of a 4th melanocortin receptor. J Biol Chem. 1993;268:15174–15179. [PubMed] [Google Scholar]
- Geschwind II. Change in hair color in mice induced by injection of α MSH. Endocrinology. 1966;79:1165–1167. doi: 10.1210/endo-79-6-1165. [DOI] [PubMed] [Google Scholar]
- Guan XM, Kobilka TS, Kobilka BK. Enhancement of membrane insertion and function in a type IIIb membrane protein following introduction of a cleavable signal peptide. J Biol Chem. 1992;267:21995–21998. [PubMed] [Google Scholar]
- Haskell-Luevano C, Sawyer TK, Hendrata S, North C, Panahinia L, Stum M, Staples DJ, Castrucci AMD, Hadley ME, Hruby VJ. Truncation studies of alpha-melanotropin peptides identify tripeptide analogues exhibiting prolonged agonist bioactivity. Peptides. 1996a;17:995–1002. doi: 10.1016/0196-9781(96)00141-6. [DOI] [PubMed] [Google Scholar]
- Haskell-Luevano C, Sawyer TK, Trumpp-Kallmeyer S, Bikker JA, Humblet C, Gantz I, Hruby VJ. Three-dimensional molecular models of the hMC1R melanocortin receptor: Complexes with melanotropin peptide agonists. Drug Design Discov. 1996b;14:197–211. [PubMed] [Google Scholar]
- Huang QH, Entwistle ML, Alvaro JD, Duman RS, Hruby VJ, Tatro JB. Antipyretic role of endogenous melanocortins mediated by central melanocortin receptors during endotoxin-induced fever. J Neurosci. 1997;17:3343–3351. doi: 10.1523/JNEUROSCI.17-09-03343.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt G, Thody AJ. Agouti protein can act independently of melanocyte-stimulating hormone to inhibit melanogenesis. J Endocrinol. 1995;147:R1–R4. doi: 10.1677/joe.0.147r001. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, FairchildHuntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Jackson IJ. Molecular genetics. Colour-coded switches. Nature. 1993;362:587–588. doi: 10.1038/362587a0. [DOI] [PubMed] [Google Scholar]
- Jayawickreme CK, Quillan JM, Graminski GF, Lerner MR. Discovery and structure-function analysis of alpha-melanocyte-stimulating hormone antagonists. J Biol Chem. 1994;269:29846–29854. [PubMed] [Google Scholar]
- Jones BH, Kim JH, Zemel MB, Woychik RP, Michaud EJ, Wilkison WO, Moustaid N. Upregulation of adipocyte metabolism by agouti protein: Possible paracrine actions in yellow mouse obesity. Am J Physiol-Endocrinol Metab. 1996;33:E192–E196. doi: 10.1152/ajpendo.1996.270.1.E192. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. The Schild regression in the process of receptor classification. Can J Physiol Pharmacol. 1982;60:249–265. doi: 10.1139/y82-036. [DOI] [PubMed] [Google Scholar]
- Kiefer LL, Ittoop ORR, Bunce K, Truesdale AT, Willard DH, Nichols JS, Blanchard SG, Mountjoy K, Chen WJ, Wilkison WO. Mutations in the carboxyl terminus of the agouti protein decrease agouti inhibition of ligand binding to the melanocortin receptors. Biochemistry. 1997;36:2084–2090. doi: 10.1021/bi962647v. [DOI] [PubMed] [Google Scholar]
- Kim JH, Mynatt RL, Moore JW, Woychik RP, Moustaid N, Zemel MB. The effects of calcium channel blockade on agouti-induced obesity. FASEB J. 1996;10:1646–1652. [PubMed] [Google Scholar]
- Kobayashi T, Vieira WD, Potterf B, Sakai C, Imokawa G, Hearing VJ. Modulation of melanogenic protein expression during the switch from eu- to pheomelanogenesis. J Cell Sci. 1995;108:2301–2309. doi: 10.1242/jcs.108.6.2301. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Cotecchia S, Kjelsberg MA, Pitcher J, Koch WJ, Inglese J, Caron MG. Adrenergic receptors: Recent insights into their mechanism of activation and desensitization. Adv Second Messenger Phosphoprotein Res. 1993;28:1–9. [PubMed] [Google Scholar]
- Li SJ, Varga K, Archer P, Hruby VJ, Sharma SD, Kesterson RA, Cone RD, Kunos G. Melanocortin antagonists define two distinct pathways of cardiovascular control by alpha- and gamma-melanocyte-stimulating hormones. J Neurosci. 1996;16:5182–5188. doi: 10.1523/JNEUROSCI.16-16-05182.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu DS, Willard D, Patel IR, Kadwell S, Overton L, Kost T, Luther M, Chen WB, Woychik RP, Wilkison WO, Cone RD. Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor. Nature. 1994;371:799–802. doi: 10.1038/371799a0. [DOI] [PubMed] [Google Scholar]
- Manne J, Argeson AC, Siracusa LD. Mechanisms for the pleiotropic effects of the agouti gene. Proc Natl Acad Sci. 1995;92:4721–4724. doi: 10.1073/pnas.92.11.4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud EJ, Bultman SJ, Klebig ML, van Vugt MJ, Stubbs LJ, Russell LB, Woychik RP. A molecular model for the genetic and phenotypic characteristics of the mouse lethal yellow (Ay) mutation. Proc Natl Acad Sci. 1994;91:2562–2566. doi: 10.1073/pnas.91.7.2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar SE, Miller MW, Stevens ME, Barsh GS. Expression and transgenic studies of the mouse agouti gene provide insight into the mechanisms by which mammalian coat color patterns are generated. Development. 1995;121:3223–3232. doi: 10.1242/dev.121.10.3223. [DOI] [PubMed] [Google Scholar]
- Miller KA, Gunn TM, Carrasquillo MM, Lamoreux ML, Galbraith DB, Barsh GS. Genetic studies of the mouse mutations mahogany and mahoganoid. Genetics. 1997;146:1407–1415. doi: 10.1093/genetics/146.4.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MW, Duhl DMJ, Vrieling H, Cordes SP, Ollmann MM, Winkes BM, Barsh GS. Cloning of the mouse agouti gene predicts a secreted protein ubiquitously expressed in mice carrying the Lethal-Yellow mutation. Genes & Dev. 1993;7:454–467. doi: 10.1101/gad.7.3.454. [DOI] [PubMed] [Google Scholar]
- Miwa H, Gantz I, Konda Y, Shimoto Y, Yamada T. Structural determinants of the melanocortin peptides required for activation of melanocortin-3 and melanocortin-4 receptors. J Pharmacol Exp Ther. 1995;273:367–372. [PubMed] [Google Scholar]
- Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257:1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- Movaghar M. Tyrosinase activity in the first coat of agouti and black mice. Pigment Cell Res. 1989;2:401–407. doi: 10.1111/j.1600-0749.1989.tb00228.x. [DOI] [PubMed] [Google Scholar]
- Mynatt RL, Miltenberger RJ, Klebig ML, Zemel MB, Wilkinson JE, Wilkinson WO, Woychik RP. Combined effects of insulin treatment and adipose tissue-specific agouti expression on the development of obesity. Proc Natl Acad Sci. 1997;94:919–922. doi: 10.1073/pnas.94.3.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera BM, Rivier J, Scott JK, Hillyard DR, Cruz LJ. Conotoxins. J Biol Chem. 1991;266:22067–22070. [PubMed] [Google Scholar]
- Ollmann MM, Wilson BD, Yang Y-K, Kerns JA, Chen Y, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by Agouti-related protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- Potenza MN, Lerner MR. A rapid quantitative bioassay for evaluating the effects of ligands upon receptors that modulate cAMP levels in a melanophore cell line. Pigment Cell Res. 1992;5:372–378. doi: 10.1111/j.1600-0749.1992.tb00565.x. [DOI] [PubMed] [Google Scholar]
- Prota G. Melanins and melanogenesis. San Diego, CA: Academic Press; 1992. [Google Scholar]
- Prota G, Lamoreux ML, Muller J, Kobayashi T, Napolitano A, Vincensi MR, Sakai C, Hearing VJ. Comparative analysis of melanins and melanosomes produced by various coat color mutants. Pigm Cell Res. 1995;8:153–163. doi: 10.1111/j.1600-0749.1995.tb00657.x. [DOI] [PubMed] [Google Scholar]
- Prusis P, Frandberg PA, Muceniece R, Kalvinsh I, Wikberg JE. A three dimensional model for the interaction of MSH with the melanocortin-1 receptor. Biochem Biophys Res Comm. 1995;210:205–210. doi: 10.1006/bbrc.1995.1647. [DOI] [PubMed] [Google Scholar]
- Quistad GB, Skinner WS. Isolation and sequencing of insecticidal peptides from the primitive hunting spider, Plectreurys tristis (Simon) J Biol Chem. 1994;269:11098–11101. [PubMed] [Google Scholar]
- Robbins LS, Nadeau JH, Johnson KR, Kelly MA, Rosellirehfuss L, Baack E, Mountjoy KG, Cone RD. Pigmentation phenotypes of variant extension locus alleles result from point mutations that alter MSH receptor function. Cell. 1993;72:827–834. doi: 10.1016/0092-8674(93)90572-8. [DOI] [PubMed] [Google Scholar]
- Sakai C, Ollmann M, Kobayashi T, Abdel-Malek Z, Muller J, Vieira WD, Imokawa G, Barsh GS, Hearing VJ. Modulation of murine melanocyte function in vitro by agouti signal protein. EMBO J. 1997;16:3544–3552. doi: 10.1093/emboj/16.12.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai Y, Lu B, Steinbeisser H, De Robertis EM. Regulation of neural induction by the Chd and Bmp-4 antagonistic patterning signals in Xenopus. Nature. 1995;376:333–336. doi: 10.1038/376333a0. [DOI] [PubMed] [Google Scholar]
- Searle AG, Beechey CV. The phenotypic effects of d, ln and cch on Ay, e and wild type. Mouse News Lett. 1970;42:28. [Google Scholar]
- Shutter JR, Graham M, Kinsey AC, Scully S, Luthy R, Stark KL. Hypothalamic expression of ART, a novel gene related to agouti, is up-regulated in obese and diabetic mutant mice. Genes & Dev. 1997;11:593–602. doi: 10.1101/gad.11.5.593. [DOI] [PubMed] [Google Scholar]
- Siegrist W, Willard DH, Wilkison WO, Eberle AN. Agouti protein inhibits growth of b16 melanoma cells in vitro by acting through melanocortin receptors. Biochem Biophys Res Comm. 1996;218:171–175. doi: 10.1006/bbrc.1996.0030. [DOI] [PubMed] [Google Scholar]
- Siegrist W, Drozdz R, Cotti R, Willard DH, Wilkison WO, Eberle AN. Interactions of alpha-melanotropin and agouti on B16 melanoma cells: Evidence for inverse agonism of agouti. J Recep Signal Transductor Res. 1997;17:75–98. doi: 10.3109/10799899709036595. [DOI] [PubMed] [Google Scholar]
- Silvers WK. The coat colors of mice. New York, NY: Springer-Verlag; 1979a. The agouti and extension series of alleles, umbrous and sable; pp. 6–44. [Google Scholar]
- ————— . The coat colors of mice. New York, NY: Springer-Verlag; 1979b. Recessive yellow and lethal yellow: Similarities and differences; pp. 30–31. [Google Scholar]
- Siracusa LD. The agouti gene: Turned on to yellow. Trends Genet. 1994;10:423–428. doi: 10.1016/0168-9525(94)90112-0. [DOI] [PubMed] [Google Scholar]
- Suzuki I, Tada A, Ollmann MM, Barsh GS, Im S, Lamoreux ML, Hearing VJ, Nordlund JJ, Abdel-Malek ZA. Agouti signaling protein inhibits melanogenesis and the response of human melanocytes to alpha-melanotropin. J Invest Dermatol. 1997;108:838–842. doi: 10.1111/1523-1747.ep12292572. [DOI] [PubMed] [Google Scholar]
- Vage DI, Lu DS, Klungland H, Lien S, Adalsteinsson S, Cone RD. A non-epistatic interaction of agouti and extension in the fox, Vulpes vulpes. Nature Genet. 1997;15:311–315. doi: 10.1038/ng0397-311. [DOI] [PubMed] [Google Scholar]
- Vrieling H, Duhl DM, Millar SE, Miller KA, Barsh GS. Differences in dorsal and ventral pigmentation result from regional expression of the mouse agouti gene. Proc Natl Acad Sci. 1994;91:5667–5671. doi: 10.1073/pnas.91.12.5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willard DH, Bodnar W, Harris C, Kiefer L, Nichols JS, Blanchard S, Hoffman C, Moyer M, Burkhart W, Weiel J, et al. Agouti structure and function: Characterization of a potent alpha-melanocyte stimulating hormone receptor antagonist. Biochemistry. 1995;34:12341–12346. doi: 10.1021/bi00038a030. [DOI] [PubMed] [Google Scholar]
- Yang YK, Ollmann MM, Wilson BD, Dickinson C, Yamada T, Barsh GS, Gantz I. Effects of recombinant agouti-signaling protein on melanocortin action. Mol Endocrinol. 1997;11:274–280. doi: 10.1210/mend.11.3.9898. [DOI] [PubMed] [Google Scholar]
- Zemel MB, Kim JH, Woychik RP, Michaud EJ, Kadwell SH, Patel IR, Wilkison WO. Agouti regulation of intracellular calcium: Role in the insulin resistance of viable yellow mice. Proc Natl Acad Sci. 1995;92:4733–4737. doi: 10.1073/pnas.92.11.4733. [DOI] [PMC free article] [PubMed] [Google Scholar]