

Abstract
The Nudix hydrolase superfamily, characterized by the presence of the signature sequence GX5EX7REUXEEXGU (where U is I, L, or V), is a well studied family in which relations have been established between primary sequence and substrate specificity for many members. For example, enzymes that hydrolyze the diphosphate linkage of ADP-ribose are characterized by having a proline 15 amino acids C-terminal of the Nudix signature sequence. GDPMK is a Nudix enzyme that conserves this characteristic proline but uses GDP-mannose as the preferred substrate. By investigating the structure of the GDPMK alone, bound to magnesium, and bound to substrate, the structural basis for this divergent substrate specificity and a new rule was identified by which ADP-ribose pyrophosphatases can be distinguished from Purine-DP-mannose pyrophosphatases from primary sequence alone. Kinetic and mutagenesis studies showed that GDPMK hydrolysis does not rely on a single glutamate as the catalytic base. Instead, catalysis is dependent on residues that coordinate the magnesium ions and residues that position the substrate properly for catalysis. GDPMK was thought to play a role in biofilm formation due to its upregulation in response to RcsC signalling; however, GDPMK knockout strains show no defect in their capacity of forming biofilms.
Keywords: GDP-mannose hydrolase, Nudix, biofilm, yffH, ADP-ribose hydrolase, markless knockout
Introduction
Nudix hydrolases are a superfamily of enzymes that contain the signature sequence (GX5EX7REUXEEXGU where U is I, L, or V); Nudix enzymes utilize divalent metals to hydrolyze substrates that contain at least one nucleoside derivative and a diphosphate bond1. According to the Sanger Institute’s pfam data base, there are currently over 16,806 Nudix enzymes from over 1,186 species from all three kingdoms (http://pfam.sanger.ac.uk/) 2. This number continues to rise as the database of known sequences expands and search algorithms strictly looking for the Nudix signature sequence are refined and relaxed to encompass Nudix enzymes with less typical signature sequences.
The product of the gene yffH from E. coli was characterized as a GDP-mannose hydrolase 3. This 191 amino acids long enzyme contains the modified signature sequence GX4DX7KEUXEEXGU (residues in the Nudix motif are referred to with either their sequence numbers or as they appear in the Nudix motif: , Fig. 1A.) This sequence shows three differences from the classical signature sequence: there are four residues instead of five between and is an aspartate, and the typical arginine is replaced by a lysine (Fig. 1A). Nevertheless, this enzyme hydrolyzes GDP-mannose as its preferred substrate yielding GMP and mannose-1-phosphate 3. Since the enzyme described in this paper hydrolyzes GDP-mannose but has a lysine instead of the conserved arginine in the signature sequence, the name GDPMK is used to distinguish it from another GDP-mannose hydrolase4, GDPMH, that contains another non-canonical Nudix sequence.
Fig. 1. Structure of GDPMK dimer.

A. Structural alignment of Nudix signature sequence of GDPMK (top) and EcADPRase (bottom). Signature sequence residues are colored red. B. Dimer of GDPMK with GDP mannose bound in the active site. C.The first loop of the Nudix motif is shorter in GDPMK than in EcADPRase. GDPMK is colored yellow, EcADPRase orange. D. Structural superposition of a monomer of apo form (D152A GDPMK) and the holo form (E100A GDPMK)of GDPMK.
Although GDPMH and GDPMK prefer the same substrate, the products yielded by the two reactions are different: guanosine diphosphate plus mannose by GDPMH and guanosine monophosphate plus mannose-1-phosphate by GDPMK. Another key difference between the two enzymes is their genomic localization. The GDPMH/wcaH gene can be found within the cluster of genes in E. coli responsible for the production and diversification of O-antigen, a component of lipopolysaccharides 5. The GDPMK/yffH gene is located in an operon whose role has not yet been identified. Nevertheless, these two enzymes are most likely tied to a common biosynthetic process because they are both members of the RcsC regulon in E. coli, which has been linked to biofilm formation 6. Both GDPMK and GDPMH have similar Km values at high pH (GDPMK has 665 μM at pH 8.5 and GDPMH has 670 μM at pH 9.3) 7. Lastly, the trancripts of the genes corresponding to both of these enzymes are upregulated when E. coli binds to a solid surface in a way that is dependent on RcsC signalling (GDPMK by a factor of 3.7 and GDPMH by a factor of 3.5) 6. These observations have led us to propose that GDPMK may play a similar role in facilitating the growth of E. coli on a solid surface.
Although GDPMK 3 and GDPMH hydrolyze the same substrate, in terms of sequence homology, mechanism, and products formed, GDPMK is more similar to the family that includes the ADP-ribose pyrophosphatase from E. coli 8, a family characterized by the presence of a proline 16 amino acids C-terminal from the Nudix signature sequence. This residue is conserved in GDPMK 8–13. The homodimers of the ADPRase family exhibit domain swapping in their quaternary arrangement and residues of both monomers participate in substrate recognition and catalysis. The mechanism of this family of enzymes involves the deprotonation of an activated water and subsequent attack of the hydroxyl on a phosphorous of the diphosphate bond, leaving monophosphate products (NMP and P-X) 8. In this paper we present the results of a combination of genetic, biochemical, and structural studies aimed at characterizing GDPMK and at defining sequence and structural motifs that will allow Nudix ADP-ribose hydrolases to be distinguished from Nudix GDP-mannose hydrolases. To further characterize GDPMK, we carried out experiments to determine wheather or not GDPMK has a significant role in biofilm formation.
Matherial and Methods
Cloning
The E. coli yffH gene was cloned into the pET 24a vector as described by Xu et al 3. All mutants were produced with the QuickChange® site directed mutagenesis kit from Stratagene.
Expression and Purification of GDPMK and its mutants
Expression and purification was the same for the wild type and all mutants. Briefly, BL21 (DE3) cells were transformed with the plasmid, a single colony was inoculated in 10 ml of LB broth containing 30 μg/ml kanamycin, and grown at 37 °C. The overnight culture was used to inoculate 1L of the same medium and grown at 37 °C to an absorbance of 0.6 at which point 0.1 mM IPTG was added. The cells were grown for three hours, centrifuged, and the pellet was frozen at −80 °C. After thawing, cells were resuspended in 50 mM Tris pH 7.5, 1 mM EDTA, 1 mM DTT (TED) and lysed by microfluidization. The lysate was centrifuged and the pellet discarded. The lysate was fractionated by bringing the solution to 60% and 90% saturation of ammonium sulfate. The 90% pellet was redissolved in TED buffer and loaded onto a HiPrep 16/10 Phenyl FF (high sub) column (™ GE), The eluted protein was dialyzed against TED buffer and loaded onto a Source Q. The eluate from the Source Q column was loaded onto a gel filtration column equilibrated with TED + 0.15 M NaCl. The protein eluted at approximately as a mono-disperse dimer. The eluate was concentrated to aproximatedly 4 mg/mL and stored at −80 °C. Purity was assessed at each step by SDS-PAGE electrophoresis. Visual inspection revealed a purity of at least 95%.
Enzymatic Assays
Determination of substrate specificity
GDPMK activity was assayed by measuring the conversion of a phosphatase resistant substrate (e.g. GDP-mannose) into two phosphate sensitive products (GMP + mannose -1-phosphate). The ammount of phosphate released was determined by the method of Ames and Dubin 14–16 with a few minor variations. Briefly, a reaction mixture containing 40 μL of 62.5 mM Tris pH 8.5, 6.25 mM MgCl2, 1.25 mM DTT, 0.03 U/μL CIP (Calf Intestinal Phosphatase), and 1.4 nM GDPMK enzyme was incubated with 10 μL of 5 mM substrate (GDP-mannose, GDP-glucose, UDP-glucose, ADP-ribose, ADP-glucose, FAD, CDP-ethanolamine, CDP-choline, NAD, diadenosine pentaphosphate.) For the nucleoside triphosphates reactions (GTP, UTP, ATP or CTP) the CIP was replaced by inorganic pyrophosphatase. Each mixture was incubated at 37 °C for 150 seconds. 10 μL of 100 mM EDTA was added to quench the reaction and inorganic phosphate was assayed. Briefly, 240 μL of water was added to each sample followed by 700 μL of 1:7 v/v 10% abscorbic acid, 0.42% ammonium Molybdate tetrahydrate in 1 N H2SO4. The samples were incubated at 42 °C for 20 minutes and the amount of phosphomolybdate was estimated from the absorbance at 820 nm.
Determination of Km
The protocol described above was used to assay the initial rates of hydrolysis of GDP-mannose at concentrations varying from 50 μM to 1.5 mM. 40 μL of the reaction mixture was added to 10 μL of 8 different substrate concentrations to initiate the reaction and then the phosphate released was measured as detailed above. Measurements were done in triplicate and a non linear least-squares fit to the Michaelis Menten equation with the program Gnuplot was used to determine Km and Vmax17.
Determination of the pH-rate profile
A mixture containing 6.25 mM MgCl2, 1.25 mM DTT, 0.03 U/μL CIP, and 2.5 mM GDP-mannose was added to 5 μL of either 1 M sodium MES pH 6, 1 M Bis Tris pH 6.5, 1 M Hepes pH 7, 1 M Tris pH 7.5, 1 M Tris pH 8, 1 M Tris pH 8.5, or 1 M Bicine pH 9. To these mixtures 5 μL of enzyme (either wild type or mutants) was added in different amounts to ensure that the rates measured was the initial rate. In all reactions, final enzyme concentrations were close to 1.2 nM with the exception of E100A, R44S, and K176A, which were 25.9 μM, 56 nM, and 1.46 μM respectively. Reactions were conducted for 6 or 7 minutes and the phosphate released was measured as described above. The kcat values were computed at each pH.
Crystallization
Crystals of wild type protein (wt) were grown at 18 °C by vapor diffusion using the hanging drop method. The solution in the drop contained 2 μL of the enzyme and 1 μL of the reservoir solution, composed of 1.5 M K+/Na+ Tartrate and 0.1 M Bis Tris propane pH 7.5. These crystals had five monomers in the asymmetric unit (A to E). Three of the chains had a tartrate molecule bound.
For preparing crystals of enzyme/substrate complex, wt protein and all mutants expressed and purified as described above, were incubated with 4 mM GDP-mannose and 4 mM MgCl2 on ice for 5 minutes. Crystals were grown at 18 °C by vapor diffusion using the hanging drop method. The final solution in the drop for all mutants contained 4 μL of the substrate-protein mixture and 1 μL of the reservoir, composed of 0.1 M Tris pH 8.5, 0.2 M MgCl2, and 20%–26% PEG 3350. WT and D152A mutant crystals grew to maximum size in 5 days. Some E100A mutant crystals were time sensitive. Large time insensitive E100A mutant crystals appeared after two weeks and did not contain substrate. Large, time sensitive E100A mutant plates grew to their maximal size in 24 hours and contained GDP-mannose in the active site. Data on the time sensitive crystals had to be collected (or the crystals frozen) within the first 24 hours because the crystals degraded after that time. All crystals were flash frozen with the addition of 5% PEG 3350 as a cryoprotectant.
Structure Determination
Data for E100A and D152A were collected at the home source (FRE+ with Saturn CCD detector; Rigaku USA, Inc) and, for the complex of wt+tartrate and the complex of E100A+substrate, remotely at BNL beamline X6A using the ALS type sample automounter. Data were processed using the HKL suite18.The structures were determined by molecular replacement (MR) using the program AMoRe 19 with PDB entry 1GOS and 1VIU as the search model 8. Refinement was done with Refmac5 20 and iterative rounds of rebuilding were done with Coot 21. Data statistics are shown in Table 1. The quality of the structures was evaluated by Procheck, Whatcheck, and Molprobity 22–24. Figures were drawn with Molscript, Povscript, and Raster 3D 25–27. Coordinates and structure factors were deposited in the Protein Data Bank under the following accession codes: wild type enzyme (PDB ID 3O52), GDPMK D152A (PDB ID 3O6Z), GDPMK E100A (PDB ID 3O69), GDPMK E100A-Mg2+-GDP-mannose complex (PDB ID 3O61).
Table 1.
Data collection and refinement statistics for GDPMK wild type and its mutants
| Crystal | wt + tartrate | D152A plus GDP-mannose | E100A (grown for 3 weeks) | E100A plus GDP-mannose (grown for 24 hrs) |
|---|---|---|---|---|
| Space group | C2221 | P212121 | P212121 | P21 |
| Cell dimensions | a = 75.3 Å | a = 58.9 Å | a=60.3 | a=54.2 |
| b = 103.2 Å | b = 69.3 Å | b=69.2 | b=55.8 | |
| c = 254.5 Å | c = 99.6 Å | c=98.5 | c=158.1 | |
| α=b=γ = 90° | α=b=γ = 90° | α=b=γ = 90° | α=γ = 90° | |
| b=91.7 | ||||
| Data Collection Statistics | ||||
| Wavelength (Å) | 0.9809 | 1.54 | 0.9787 | |
| Resolution(Å) | 30.0-2.5 | 50.0-2.03 | 50.0-2.1 | 50.0-2.45 |
| (HighRes shell)a | (2.54-2.50) | (2.10-2.03) | (2.18-2.10) | (2.49-2.45) |
| Measured Reflect. | 240,244 | 161,406 | 134,487 | 104,724 |
| Unique Reflections | 33,433 | 26,521 | 23,871 | 32,636 |
| I/σ | 28.6 (2.9) | 40.7 (6.4) | 30.1 (5.2) | 17.8 (4.4) |
| Completeness (%) | 96.4 (80.7) | 98 (98.1) | 96 (75.1) | 93 (59.0) |
| Rsym/Rmerge (%)b | 6.9 (34.4) | 7.7 (37.7) | 8.9 (33.1) | 9.8(16.5) |
| Refinement | ||||
| Rcryst (%) | 0.21 | 0.23 | 0.20 | 0.22 |
| Rfree | 0.28 | 0.30 | 0.28 | 0.29 |
| R.m.s deviations | ||||
| Bond length (Å) | 0.015 | 0.014 | 0.011 | 0.014 |
| Angle (°) | 1.5 | 1.4 | 1.3 | 1.6 |
| Monomer in ASU | 5 | 2 | 2 | 4 |
| Total Atoms | 7420 | 3343 | 3342 | 6246 |
| Protein atoms | 7153 | 2987 | 2943 | 5971 |
| Water molecules | 197 | 329 | 375 | 112 |
| Ligand | Na+, Cl−, Tartrate | Mg2+, Cl−, PEG, Tris | Mg2+, PEG, Na+, Cl− | Mg2+, GDP-mannose, Na+, Cl− |
| Bfactor (protein)Å2 | 44.4 | 33.2 | 24.0 | 46.2 |
| Bfactor (ligand) Å2 | 65.1 | 49.5 | 42.7 | 52.7 |
| Bfactor (H20) Å2 | 41.6 | 39.6 | 31.0 | 45.4 |
| Ramachandran(MF, allowed) % c | 87.1, 100 | 88.4, 100 | 89.8, 99.4 | 88.6, 99.7 |
| PDB Accession Code | 3O52 | 3O6Z | 3O69 | 3O61 |
Data in parentheses correspond to the outer resolution shell.
Rsym=ΣhklΣJ|Ij-<I>|/ΣhklΣJ Ij where <I> is the mean intensity of j observations from a reflection hkl and its symmetry equivalents.
MF=Percentage of residues in most favorable region allowed= Percentage of residues in allowed region
Biofilm Assays
Quantitative Biofilm Assay
Biofilm formation was assayed by crystal violet staining of adherent cells in microtiter wells, as described previously with modifications 28,29. Overnight cultures (37° C, 250 rpm) were diluted 1:100 into medium without antibiotics and incubated at 26° C for 24 h without shaking in a 96 well microtiter plate. Bacterial growth was determined by measuring the absorbance at 600 nm using the Synergy-HT plate reader (BioTek, Winooski, VT). To measure biofilm, planktonic cells were removed by discarding the medium and the wells rinsed with water three times. Bound cells were stained with 1.25 volumes (250 μl) of crystal violet solution (Fisher Scientific; Cat # 255–960B) for 2 min and the wells were rinsed again three times with water to remove unbound and excess dye. Bound dye was solubilised with 1.25 volumes of 33% acetic acid and absorbance at 630 nm determined using a microtiter plate reader. Background staining was corrected by subtracting the crystal violet bound to uninoculated controls. Each experiment was conducted at least twice in replicates of three.
Bacterial Strains and Growth conditions
All the E. coli strains used in this study are listed in Table 2. The strains were grown either in LB medium (g/L: Tryptone: 10; Yeast Extract: 5; Sodium Chloride: 10; pH: 7.4) or CFA medium (g/L: Casamino acids: 10; Yeast Extract: 1.5; MgSO4: 0.05; MnCl2: 0.005; pH: 7.4) or KB medium (g/L: K2HPO4: 11; KH2PO4: 8.5; Yeast Extract: 6.0; Glucose: 5. 0) or M9 minimal medium + thiamine (g/L: Na2HPO4.7H2O: 12.8; KH2PO4: 3; NaCl: 0.5; NH4Cl: 1.0; MgSO4: 0.24; CaCl2: 0.011; Glucose: 4; Thiamine: 0.001 g). Cultures were grown overnight at 37° C, 250 rpm in medium with or without antibiotics (100 μg/ml of kanamycin for overnights of csrA::kan strains). To test for biofilm formation in different media, overnights of cultures grown in one medium were used for inoculating into fresh medium of the same type for biofilm assay (i.e. overnight cultures in LB into fresh LB) except for assay in M9 medium where overnights grown in LB were used to inoculate into fresh M9 minimal medium.
Table 2.
E. coli strains used in this study
| Strain | Genotype |
|---|---|
| MG1655 | Wild type E. coli K-12 |
| MG1655 ΔyffH::cam | ΔyffH:: cam |
| MG1655 ΔyffH-1 | ΔyffH, marker-less, colony 1 |
| MG1655 ΔyffH-2 | ΔyffH, marker-less, colony 2 |
| TRMG-1 | MG1655 csrA::kan, colony 1 |
| TRMG-2 | MG1655 csrA::kan, colony 2 |
| TRMG ΔyffH-1 | MG1655 csrA::kan, ΔyffH, colony 1 |
| TRMG ΔyffH-2 | MG1655 csrA::kan, ΔyffH, colony 2 |
Construction of the yffH Knockout Strain
The gene coding for GDPMK, yffH, was deleted by homologous recombination from E. coli strain MG1655 using upstream and downstream primers encompassing the entire gene30. Complete deletion was verified by PCR analysis
Construction of markerless mutants of yffH
To avoid effects of polarity from yffH gene replacement, the chloramphenicol marker in MG1655 ΔyffH::cam was removed with the help of FLP recombinase following standard procedures to generate markerless mutants of yffH. P1 transduction was used to move the csrA::kan into MG1655 and MG1655 ΔyffH markerless mutant.
Results
GDPMK Overall Structure
GDPMK is a dimer in solution. In the wt crystals, monomers A and B, and monomers C and D form non-crystallographic dimers, while monomer E forms a crystallographic dimer with a monomer in another asymmetric unit. The N-terminal domain of each monomer (residues 1–44) is formed by a three-strand antiparallel βsheet (β-strands 1,2, and 3) and a Nudix domain (amino acids 45–191). The monomers are related by an approximate (or crystallographic) two-fold axis of symmetry. The N terminal domains are swapped between the two monomers (Fig. 1B), such that residues of both chains contribute to the active site. The 5-strand mixed β sheet of the Nudix domain is flanked between the helix of the Nudix motif and the C-terminal helices. The catalytic L9 loop is disorded in one of the monomers. The structure of free GDPMK observed in the apo E100A structure shows a shift of 2.5 Å of the loop L2 between β2 and β3 with respect to the E100A GDPMK mutant in complex with GDP-mannose, (Fig. 1D). The L9 loop moves 2.73 Å to accommodate the substrate in the pocket and Y17 moves 2.11 Å towards the substrate likely bringing Y17 close enough to form a hydrogen bond. The loop containing D30 moves 2.4 Å possibly aiding in positioning the beta strand that contains the guanosine-recognizing residues, R39 and E40, (Fig. 1D).
Structure of the Nudix Motif
In GDPMK, the Nudix motif starts with G86 at the end of the β-strand containing residues S83 through G86 ( in Fig. 1A). The sequence of this β-strand (SCAG) is closest to that of the Nudt14 UGPPase (UDP-glucose pyrophosphatase) consensus sequence identified recently (LCAG) 31. This strand contains G86 ( ) as well as the non-signature residue A85 whose carbonyl is involved in metal binding. The loop of the Nudix motif is composed of residues L87-E92, which contains the glutamate that interacts with K99 and only one signature sequence residue, D91 ( ).
The canonical Nudix signature sequence is longer than the one observed in GDPMK by one amino acid inserted between the first glycine ( ) and the first glutamate ( ), which in GDPMK is an aspartate (GX5EX7REU-EE-GU vs GX4DX7KEU-EE-GU) (Fig. 1A). The difference in sequence length is reflected in the length of the loop of the motif but it does not alter the presence of the salt bridge D91–K99 ( to ), nor the relative position of the phosphorous being attacked in the reaction (Fig. 1C).
Metal Binding Site
The structure of the D152A mutant of the GDPMK shows two octahedrally coordinated magnesium ions (Fig. 2A). OE1 of E100, OE1 of E151, and two water molecules, W1 and W2, coordinate Mg1 in a plane while OE2 of E104 and water molecule, W3, occupying the axial positions. Mg2 has OE2 of E104, the main chain carbonyl of A85, water molecule W4, and water molecule W5 as plane ligands with water molecules W6 and W1 occupying the axial positions. A85 directly precedes the first glycine G86 ( ) of the Nudix signature sequence and it is characteristic for the main chain carbonyl of the residue at this position to be a ligand of one of the metals required for catalysis.
Fig. 2. Mg2+ binding sites.

A. Magnesium binding sites in the structure of the GDPMK D152A mutant. The two magnesium ions are bound to residues of the Nudix signature sequence. Mg1 coordination is completed by E151 from loop L9. B. Metal binding site in the structure of the GDPMK E100A mutant in the absence of substrate. C. Metal binding site in the structure of the GDPMK E100A mutant in the presence of substrate.
Mutation of E100 to alanine ( ) of the GDPMK Nudix signature sequence abolishes Mg1 binding (Fig. 2B). The presence of the substrate in the structure replaces two of the ligand water molecules by oxygens from the α and β phosphorous atoms while removing a coordinating water molecule, leading to incomplete coordination of Mg2 (Fig. 2C).
Recognition of GDP-mannose
The structure of the complex of the GDPMK E100A mutant with GDP-mannose and magnesium displays GDP-mannose bound in a horseshoe conformation in both active sites (Figs. 3A, B). The mannose is recognized by residues of the same monomer that contain the Nudix motif (R67, K176, E127) but the guanine base is recognized by residues of the other monomer (K’38, E’40) (In this discussion, residues 45–191 correspond to the monomer that contributes the Nudix motif while resudes 1–44 and P122, also identified by a “prime” belong to the other monomer). A conserved proline, P’122, forms the back of the sugar binding site.
Fig. 3. Recognition of GDP-mannose.

A. Electron density omit σA map of the GDP-mannose in the active site of the GDPMK E100A mutant (contour level =1σ). Red and yellow ribbons show the two monomers. The magnesium ion is shown as a white sphere. B. GDP-mannose is shown as turquoise sticks. The residues of both chains that form the binding pocket are shown as sticks. Hydrogen bonds to both the guanosine base and the mannose are shown as black dashes. C. Guanine bound to GDPMK by π- π interactions to R’39 and Y’17 of the opposite monomer. The guanine base is further stabilized by hydrogen bonds to K’38 and E’40. D. GDPMK recognition of mannose mediated by hydrogen bonds to conserved E127 and to K176, a residue that is not found in the ADP-ribose pyrophosphatase family.
The carbonyl of K’38 is at hydrogen bonding distance of the N-H of guanosine and the peptide amide of E’40 is at hydrogen bonding distance of the oxygen of guanosine (Fig. 3C). E’40 is a conserved residue of the fold whose role is to hold the hairpin of a 3-stranded β-sheet of the opposite monomer, forming the domain swapped guanosine binding site. A strong π-π interaction with Y’17 and a stacking interaction with the guanidinium of R’39 secure the base of the sugar nucleotide in place. D150 is at H-bonding distance of the oxygens of the ribose of the base (Fig. 4A). R’29 stabilizes D150 in place by making a hydrogen bond to its backbone oxygen when substrate is bound; however, in the two structures without substrate where loop L9 can be seen, H’37 forms this hydrogen bond instead. Additionally, in the abscence of substrate, hydrogen bonds are made between R’29 and E149 and E151. A conserved arginine, R67, uses a bifurcated hydrogen bond to oxygens 3B and 2B of the β phosphate of GDP-mannose (Fig. 3D, only one is seen). The mannose forms a H-bond to conserved residue E127. K176 makes hydrogen bond to an oxygen atom of mannose, and may further stabilized the sugar in the binding pocket by electrostatic interactions (Fig. 3D).
Fig. 4. Mechanistic insight.

A. GDPMK active site showing all residues and water molecules that are present near the diphosphate bond. E151 is in an ideal position to act as the catalytic base, but its mutation shows no large reduction of activity. B. Tartrate recognition in wild type structure. The bound tartrate is coordinated by hydrogen bonds to K176, R44, and E127 of one monomer and the amide NH of G’123 in the other monomer. All of these residues are involved in the mannose binding pocket.
Recognition of Tartrate
Significant electron density not corresponding to the protein was found in chains A, B and E of the wt enzyme crystals. Upon comparison of the shape of the density with the shapes of the various chemicals in the crystallization mixture, a tartrate molecule was successfully refined into the unidentified density. This tartrate is found with identical relative position in chains A, B, and E. In chain C, no density corresponding to the tartrate was found and in chain D the tartrate binds in the pocket but not at the same position as in chains A, B, and E. In chain A, on one side of the pocket, nitrogen Nz of K176 is at hydrogen bonding distance of the O1 of a tartrate. On the opposite side of the pocket, the O4 of the tartrate is at hydrogen bonding distance of the nitrogen Nh1 of R’44. E127 and the amide of G123 are at hydrogen bonding distance from O2 and O3 of the tartrate, respectively (Fig. 4B). Since tartrate is coordinated by R’44, E127, and K176, which are all residues that interact with the mannose substrate, it is clear that tartrate binds in the mannose binding cleft.
Kinetic Studies
GDPMK showed a preference for GDP-mannose as a substrate 3, and no activity towards other Nudix substrates including FAD, CDP-ethanolamine, GDP-glucose, CDP choline, NAD+, diadenosine pentaphosphate, GTP, UTP, ATP, or CTP (data not shown). There was low but significant activity for sugar nucleotides such as GDP-glucose, ADP-glucose, UDP-glucose, and GDP-mannose (Table 3) and for ADP-ribose.
Table 3.
Relative rates of hydrolysis by GDPMK for GDP mannose compared to other sugar nucleotides
| GDPMK | |
|---|---|
| GDPM-mannose | 1.0 |
| ADP-glucose | 0.15 |
| ADP-ribose | 0.26 |
| UDP-glucose | 0.13 |
| GDP-glucose | 0.45 |
Based on the structure of the wt GDPMK, the following residues were mutated to probe the role of individual residues in substrate binding and in the catalytic mechanism: Nudix conserved residue E100 ( ), a metal ligand, (Fig. 2); R44 and K176 at H-bonding distance of the mannose (Fig. 3c and d); E149, D150, E151, and D152 present in loop L9 which contains the catalytic base in the ADPRase family 32 (Figs. 2, 6).
Fig. 6. Structural Alignment of GDPMK (CAQ32837.1), HsADPRase from H. sapiens (CAG33476.1), and EcADPRase from E. coli (NP_417506.1).

A. GDPMK is colored red and yellow. Two hydroxyl oxygens of the GDP-mannose make hydrogen bonds to E’127 and S’121 of the other monomer. These residues are conserved in the other two enzymes. B. HsADPRase is gray and darkblue; C. EcADPRase is colored dark cyan and brown. The main difference between the three enzymes is the presence of K176 in GDPMK, which is partly conserved as R196 in HsADPRase but not conserved in EcADPRase where it is an alanine (seen in brown). D. Sequences of the three enzymes. Nudix identities are colored blue, substrate recognition identities are in green, and residues in a brown background represent the putative mannose recognition site.
The E100A mutation abolishes GDP-mannose hydrolase activity (Table 4). Mutations R44S and K176A decreased kcat by a factor of 33.5 and 631.5 respectively, but neither affected it as much as E100A for which kcat was reduced by a factor of 4.4 × 104. The Km for the R44S and K176A mutants was at least twice of the wt; however, the E100A activity was too low for measuring its Km. Mutations E149A, D150A, and D152A did not have a significant effect on either Km or kcat (Fig. 4A). The GDPMK E151A mutant had a 3 fold reduced kcat but its Km was unchanged suggesting that E151A could help position a water molecule to be the catalytic base but its presence is not absolutely required for catalysis.
Table 4.
Effects of mutations on the kinetic parameters of GDPMK with Mg2+ activation and GDP-mannose as substrate at pH 8.5 and 37° C
| kcat (s−1) | Km (μM) | |
|---|---|---|
| WT | 90.2±6.7 | 659±65 |
| E151A | 21.3±4.6 | 594±140 |
| K176A | 0.11±0.04 | >1200 |
| R44S | 1.69±0.12 | >1200 |
| E100A | <0.01 | NDa |
| E149A | 80.6±6.3 | 555±85 |
| D150A | 126.4±14.2 | NDa |
| D152A | 42.6±6.1 | NDa |
| Q65A | 24.4±17.4 | NDa |
: The Kms labeled “ND” were not determined. In the case of E100A the activity was too low to be measured
The pH profile of the wt enzyme showed that activity increased with pH reaching a maximum at pH 8.5 and decreasing for higher pH values (Fig. 7). All mutants had a pH profile whose shape approximated that of the wt except the K176A mutant, which showed, in addition to the peak at pH 8.5 a shoulder at pH 7.0 (data not shown).
Fig. 7. Initial rate vs. pH for GDPMK.
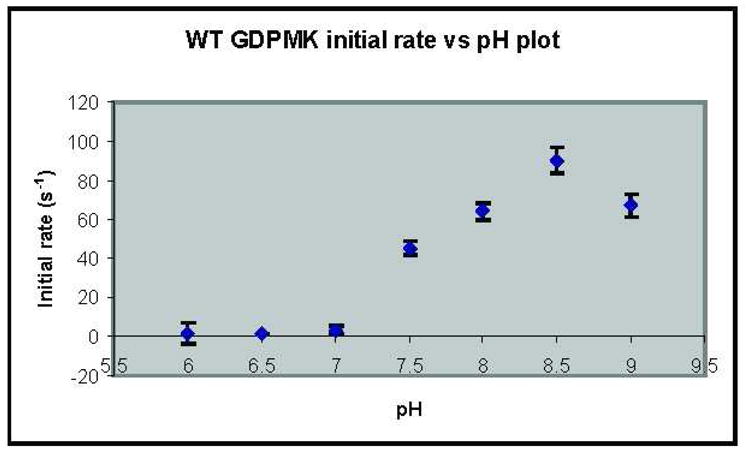
Initial rates were measured at pH 6, 6.5, 7, 7.5, 8, 8.5, and 9 at 37 °C and using 5 mM GDP mannose as the substrate.
Activity of GDPMK in Biofilm Formation
The presence of the GDPMK gene in the RcsC regulon and the observation that GDPMK transcripts are upregulated when E. coli grows on a solid surface suggested that the enzyme may be involved in biofilm formation. To test this hypothesis we constructed markerless deletion mutants of the gene yffH in order to assess the role of this gene product on biofilm formation in wild type E. coli K-12 MG1655 and its csrA mutant (prefixed with a TR in the strain name), which is derepressed for the synthesis of biofilm adhesin, PGA. Biofilm formation was tested in four different media (LB, CFA, KB and M9 + Glucose + Thiamine) to test for possible role of this gene product in biofilm formation in any of these conditions.
Deletion of yffH did not have a substantial effect on biofilm formation (Fig. 5A) or growth (Fig. 5B) of MG1655 ΔyffH or TRMG1655 ΔyffH strains. Biofilm formation of the wild type E. coli, MG1655 and its yffH mutants were low in all the media compared to their respective csrA mutant derivatives. The csrA mutant derivatives exhibited ~10 to 25 fold higher biofilm formation in LB, CFA and KB medium compared to their wild type isogenic strains and ~2 to 4 fold higher biofilm formation in M9 minimal medium supplemented with thiamine. Kornberg medium supported higher biofilm formation followed by LB and CFA.
Fig. 5. Effect of yffH deletion on biofilms formation.

A. Growth of yffH mutants of MG1655 or TRMG1655 in LB/CFA/KB/M9 minimal medium. B. Biofilm formation of yffH mutants of MG1655 or TRMG1655 in LB/CFA/KB/M9 minimal medium. Error bars depict SD.
Discussion
Until now, it as been difficult to distinguish GDP-mannose pyrophosphatases from ADP-ribose pyrophosphatases based on their sequences. This difficulty leads to wrong annotations of enzymes belonging to these families. In this paper we present the results of a combination of genetic, biochemical, and structural studies aimed at defining sequences and structural motifs that will allow Nudix GDP-mannose pyrophosphatases to be distinguished from Nudix ADP-ribose hydrolases.
Members of the ADP-ribose pyrophosphatase family of Nudix enzymes are dimeric proteins with two domains per monomer: an N-terminal domain and a Nudix domain. In the dimer these two domains are swapped. Their sequences are characterized by having the canonical Nudix sequence and a proline 16 residues downstream from the sequence. Curiously, GDPMKs, with the same three characteristics, have maximal activity with GDP-mannose instead of ADP-ribose. Our studies show that both families of enzymes use an equivalent glutamate residue to make hydrogen bonds to the nucleotide base (E52 in EcADPRase and E40 in GDPMK(2.7 Å)) with two hydrogen bonds formed to E52 in EcADPRase and one hydrogen bond to E40 (2.7 Å) and one to K38 in GDPMK (3.03 Å). In both cases, the base is stacked between an arginine of β-strand 3 (R51 in EcADPRase and R39 in GDPMK) and a hydrophobic residue at the top of loop L2 (F29 in EcADPRase and Y17 in GDPMK). A glutamate from loop L9 completes the coordination of the magnesium (E164 in EcADPRase and E151 in GDPMK) but mutation of this residue in GDPMK lowers the activity only 5-fold. E164 has not been mutated in EcADPRase. The oxygens of the diphosphate are hydrogen bonded to equivalent residues in both structures (R79 in EcADPRase and R67 in GDPMK(2.90 Å)). In both families of enzymes, the pocket for the sugar is formed by residues of the opposite monomer, but the recognition of the sugar is different (Fig. 6). In GDPMK, K176 is involved in sugar recognition and the equivalent residue in EcADPRase is an alanine (Fig. 6A). The presence of K176 allows the sugar binding pocket to interact with 3 oxygens (O6A with K176 (3.17 Å), O41 (2.90 Å) and O31 with E127 (2.77 Å)) of the mannose ring instead of the two oxygens used by EcADPRase.
If K176 were the key to mannose recognition over ribose recognition, then other enzymes that prefer mannose would also contain this lysine. There is only one other documented ADP-ribose pyrophosphatase family member that prefers mannose over ribose and that is human HsADPRase with a kcat towards ADP mannose 1.5 times greater than the kcat towards ADP-ribose 33. Structural alignment of HsADPRase to GDPMK bound to GDP-mannose shows an arginine in the place of K176, providing similar three oxygen interactions to stabilize a mannose preferentially over a ribose. It should be noted that GDPMK has the sequence D174G175K176 with a salt bridge between the aspartate and lysine (2.79 Å). Similarly, HsADPRase has the sequence D194A195R196 also with a salt bridge between the aspartate and arginine (Fig. 6B). In contrast, EcADPRase has the sequence N187A188A189 for these residues, neither conserving the aspartate nor the basic residue (Fig. 6C). In all cases, the DGK sequence is located at the first turn of α4 of the Nudix fold. Thus, this study suggests the rules by which to predict the preferred substrate of a particular unknown Nudix sugar nucleotide pyrophosphatase knowing only its primary sequence. If the enzyme sequence has the characteristics of those of an ADPRase but, in addition, it contains an aspartate followed by a small amino acid a lysine or an arginine at around 67 and 78 residues C-terminal to the final glycine of the Nudix signature sequence, the enzyme most likely prefers a substrate that contains mannose and should be initially classified as a purine-diphospho-mannose hydrolase; otherwise, it can be classified as an ADP-ribose pyrophosphatase. Of course actual kinetic studies must be performed in each case to ensure the correct specificity. In summary, the distinguishing characteristic between these two groups is the presence of an aspartate-small amino acid-basic amino acid combination at the beginning of the final α helix. This new motif is important because it will allow more accurate annotation of the many ADP-ribose pyrophosphatase enzymes for which the sequence is known but whose substrate specificity and physiological function are not known.
Implications for Catalysis by GDPMK
GDPMK and ADP-ribose pyrophosphatase seem to use similar catalytic mechanism. In the case of GDPMK, however, mutagenesis studies do not provide a clear candidate for the catalytic base. To this date, two different catalytic mechanisms have been observed for ADP-ribose pyrophosphatases. In the first, found in the enzymes that use three magnesium ions such as HsADPRase (human Nudt5 gene), MtADPRase, and EcADPRase (E. coli ORF209), a glutamate residue from the loop L9 is used to abstract a proton from an activated water molecule 11,32,34. This hydroxyl, stabilized by the presence of two magnesium ions, attacks the α phosphate, cleaving the diphosphate bond. This mechanism was strongly supported by mutational data that showed the requirement of an L9 loop aspartate or glutamate for catalytic activity. The enzymes Ndx2 and Ndx4, are thought to perform this same hydrolysis by a different mechanism 9,13,30. They bind magnesium ions in the same position as the two activating magnesium ions described above; however, they lack a third magnesium ion. It is proposed that these two magnesium ions are activating enough to promote the spontaneous conversion of a water molecule into a hydroxyl that subsequently cleaves the diphosphate bond independently of a general base in the L9 loop. The data supporting this mechanism was the mutation of all candidate acidic residues in L9 loop and the observation that none of the mutations had a large effect on catalytic activity for Ndx2 or Ndx4. In the case of GDPMK, the structural evidence points to a catalytic base from the L9 loop, E151, deprotonating a water molecule, which would then attack a phosphate (Fig. 4A). However, kinetic studies show that mutation of each of the L9 loop residues failed to show a substantial change in kcat. Suggesting that GDPMK is in the same family as the Ndx2 and Ndx4 enzymes. Consequently, inactivating mutations are found at positions E100, K176, and R44. The E100A mutation predictably abolished activity since this residue, conserved in other nudix structures, plays a similar essential role of chelating a magnesium. The lack of activity in the absence of magnesium chelation in the mutant enzyme allowed the crystallization with GDP-mannose in the active site. Mutations of K176 and R44 leading to a loss of catalytic activity, is unexpected since these residues are far away from the active site. In the structure, K176 provides a hydrogen bond to one of the oxygens of the mannose. Evidence for the impaired binding and positioning of the substrate in the K176A and R44A mutants comes from kinetic studies that showed that Km of these mutants is approximately twice that of WT. The activity of the E100A mutant was too low to assess its effect on substrate binding. Additionally, R44 could help promote binding of the mannose in the catalytic pocket through favorable interactions with the oxygens of the sugar. We hypothesize that the enzyme requires that GDP-mannose adopts the precise conformation necessary for hydrolysis to occur. If K176 or R44 are substituted, the substrate cannot be positioned adequately for hydrolysis, leading to the large change in kcat for these mutants.
Finally, despite being in the RCSC regulon GDPMK deletion has no effect on biofilm formation. These results suggest that either GDPMK is involved in another function associated with growth in solid substrates or the regulon contains redundant enzymes capable of carrying out equivalent functions.
Table 5.
kcat of different substrates relatives to kcat of lowest -Km substrate for each enzyme. ADP-ribose pyrophosphatase enzyme with the characteristic basic residue near the mannose prefer mannose to ADP-ribose. EcADPRase data from (Moreno-Bruna et al., 2001). HsADPRase data from Yang et al., 2000).
| GDP-mannose | ADP-mannose | ADP-ribose | |
|---|---|---|---|
| GDPMK | 100 | ND | 25.6 |
| HsNudt5 | 15 | 149 | 100 |
| EcAdprase | 1 | 58.5 | 100 |
Acknowledgments
Data collection was carried out in part at X6A beam line, funded by the National Institute of General Medical Sciences, National Institute of Health under agreement GM-0080. The NSLS, Brookhaven National Laboratory is supported by the US Department of energy under contract No. DE AC02-98CH10886. We kindly acknowledge the support of Gustavus and Louis Pfeiffer Research Foundation (A.N.B.), NIH grant NS061827 (L.M.A.) and NIH GM066794 and FLA-MCS-004949 from University of Florida CRIS project (T.R.).
References
- 1.Bessman MJ, Frick DN, O’Handley SF. The MutT proteins or “Nudix” hydrolases, a family of versatile, widely distributed, “housecleaning” enzymes. J Biol Chem. 1996;271(41):25059–25062. doi: 10.1074/jbc.271.41.25059. [DOI] [PubMed] [Google Scholar]
- 2.Eddy S, editor. Wellcome Trust SI. Pfam 24.0. 24.0. Cambridge; UK: 2009. http://pfam.sanger.ac.uk/clan/NUDIX#tabview=tab2. [Google Scholar]
- 3.Xu W, Dunn CA, O’Handley SF, Smith DL, Bessman MJ. Three new Nudix hydrolases from Escherichia coli. J Biol Chem. 2006;281(32):22794–22798. doi: 10.1074/jbc.M603407200. [DOI] [PubMed] [Google Scholar]
- 4.Legler PM, Massiah MA, Bessman MJ, Mildvan AS. GDP-mannose mannosyl hydrolase catalyzes nucleophilic substitution at carbon, unlike all other Nudix hydrolases. Biochemistry. 2000;39(29):8603–8608. doi: 10.1021/bi000537p. [DOI] [PubMed] [Google Scholar]
- 5.Wang L, Reeves PR. The Escherichia coli O111 and Salmonella enterica O35 gene clusters: gene clusters encoding the same colitose-containing O antigen are highly conserved. J Bacteriol. 2000;182(18):5256–5261. doi: 10.1128/jb.182.18.5256-5261.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrieres L, Clarke DJ. The RcsC sensor kinase is required for normal biofilm formation in Escherichia coli K-12 and controls the expression of a regulon in response to growth on a solid surface. Mol Microbiol. 2003;50(5):1665–1682. doi: 10.1046/j.1365-2958.2003.03815.x. [DOI] [PubMed] [Google Scholar]
- 7.Xia Z, Azurmendi HF, Lairson LL, Withers SG, Gabelli SB, Bianchet MA, Amzel LM, Mildvan AS. Mutational, structural, and kinetic evidence for a dissociative mechanism in the GDP-mannose mannosyl hydrolase reaction. Biochemistry. 2005;44(25):8989–8997. doi: 10.1021/bi050583v. [DOI] [PubMed] [Google Scholar]
- 8.Gabelli SB, Bianchet MA, Bessman MJ, Amzel LM. The structure of ADP-ribose pyrophosphatase reveals the structural basis for the versatility of the Nudix family. Nat Struct Biol. 2001;8(5):467–472. doi: 10.1038/87647. [DOI] [PubMed] [Google Scholar]
- 9.Ooga T, Yoshiba S, Nakagawa N, Kuramitsu S, Masui R. Molecular mechanism of the Thermus thermophilus ADP-ribose pyrophosphatase from mutational and kinetic studies. Biochemistry. 2005;44(26):9320–9329. doi: 10.1021/bi050078y. [DOI] [PubMed] [Google Scholar]
- 10.Dunn CA, O’Handley SF, Frick DN, Bessman MJ. Studies on the ADP-ribose pyrophosphatase subfamily of the nudix hydrolases and tentative identification of trgB, a gene associated with tellurite resistance. J Biol Chem. 1999;274(45):32318–32324. doi: 10.1074/jbc.274.45.32318. [DOI] [PubMed] [Google Scholar]
- 11.Kang LW, Gabelli SB, Cunningham JE, O’Handley SF, Amzel LM. Structure and mechanism of MT-ADPRase, a nudix hydrolase from Mycobacterium tuberculosis. Structure (Camb) 2003;11(8):1015–1023. doi: 10.1016/s0969-2126(03)00154-0. [DOI] [PubMed] [Google Scholar]
- 12.Okuda K, Nishiyama Y, Morita EH, Hayashi H. Identification and characterization of NuhA, a novel Nudix hydrolase specific for ADP-ribose in the cyanobacterium Synechococcus sp. PCC 7002. Biochim Biophys Acta. 2004;1699(1–2):245–252. doi: 10.1016/j.bbapap.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 13.Yoshiba S, Ooga T, Nakagawa N, Shibata T, Inoue Y, Yokoyama S, Kuramitsu S, Masui R. Structural insights into the Thermus thermophilus ADP-ribose pyrophosphatase mechanism via crystal structures with the bound substrate and metal. J Biol Chem. 2004;279(35):37163–37174. doi: 10.1074/jbc.M403817200. [DOI] [PubMed] [Google Scholar]
- 14.Ames BN, Dubin DT. The Role of Polyamines in the Neutralization of Bacteriophage Deoxyribonucleic acid. Journal Biological Chemistry. 1960;235(3):769–775. [PubMed] [Google Scholar]
- 15.O’Handley SF, Frick DN, Bullions LC, Mildvan AS, Bessman MJ. Escherichia coli orf17 codes for a nucleoside triphosphate pyrophosphohydrolase member of the MutT family of proteins. Cloning, purification, and characterization of the enzyme. J Biol Chem. 1996;271(40):24649–24654. doi: 10.1074/jbc.271.40.24649. [DOI] [PubMed] [Google Scholar]
- 16.Frick DN, Townsend BD, Bessman MJ. A novel GDP-mannose mannosyl hydrolase shares homology with the MutT family of enzymes. J Biol Chem. 1995;270(41):24086–24091. doi: 10.1074/jbc.270.41.24086. [DOI] [PubMed] [Google Scholar]
- 17.Williams T, Kelly C. Gnuplot 4th Berkeley Distribution- January 3rd, 2001. 2001. Gnuplot: command-driven interactive function plotting program. [Google Scholar]
- 18.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;277:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 19.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallographica. 1994;A50:157–163. [Google Scholar]
- 20.CCP4. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallographica D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 21.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 22.Laskowski R, MacArthur M, Moss D, Thornton J. PROCHECK: a program to check the sterochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 23.Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381(6580):272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 24.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35(Web Server issue):W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kraulis J. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structure. Journal Applied Crystallography. 1991;24:946–950. [Google Scholar]
- 26.Merrit E, Bacon D. Raster3D: Photorealistic molecular graphics. Methods in Enzymology. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 27.Fenn TD, Ringe D, Petsko GA. POVScript+: a program for model and data visualization using persistence of vision ray-tracing. J Appl Cryst. 2003;36:944–947. [Google Scholar]
- 28.Jackson DW, Simecka JW, Romeo T. Catabolite repression of Escherichia coli biofilm formation. J Bacteriol. 2002;184(12):3406–3410. doi: 10.1128/JB.184.12.3406-3410.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jackson DW, Suzuki K, Oakford L, Simecka JW, Hart ME, Romeo T. Biofilm formation and dispersal under the influence of the global regulator CsrA of Escherichia coli. J Bacteriol. 2002;184(1):290–301. doi: 10.1128/JB.184.1.290-301.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wakamatsu T, Nakagawa N, Kuramitsu S, Masui R. Structural basis for different substrate specificities of two ADP-ribose pyrophosphatases from Thermus thermophilus HB8. J Bacteriol. 2008;190(3):1108–1117. doi: 10.1128/JB.01522-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heyen CA, Tagliabracci VS, Zhai L, Roach PJ. Characterization of mouse UDP-glucose pyrophosphatase, a Nudix hydrolase encoded by the Nudt14 gene. Biochem Biophys Res Commun. 2009;390(4):1414–1418. doi: 10.1016/j.bbrc.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gabelli SB, Bianchet MA, Ohnishi Y, Ichikawa Y, Bessman MJ, Amzel LM. Mechanism of the Escherichia coli ADP-ribose pyrophosphatase, a Nudix hydrolase. Biochemistry. 2002;41(30):9279–9285. doi: 10.1021/bi0259296. [DOI] [PubMed] [Google Scholar]
- 33.Yang H, Slupska MM, Wei YF, Tai JH, Luther WM, Xia YR, Shih DM, Chiang JH, Baikalov C, Fitz-Gibbon S, Phan IT, Conrad A, Miller JH. Cloning and characterization of a new member of the Nudix hydrolases from human and mouse. J Biol Chem. 2000;275(12):8844–8853. doi: 10.1074/jbc.275.12.8844. [DOI] [PubMed] [Google Scholar]
- 34.Zha M, Guo Q, Zhang Y, Yu B, Ou Y, Zhong C, Ding J. Molecular mechanism of ADP-ribose hydrolysis by human NUDT5 from structural and kinetic studies. J Mol Biol. 2008;379(3):568–578. doi: 10.1016/j.jmb.2008.04.006. [DOI] [PubMed] [Google Scholar]