

Abstract
Humans are exposed to N-nitroso compounds (NOCs) both endogenously and exogenously from a number of environmental sources, and NOCs are both mutagenic and carcinogenic. After metabolic activation, some NOCs can induce carboxymethylation of nucleobases through a diazoacetate intermediate, which could give rise to similar p53 mutations as those seen in human gastrointestinal cancers. It was previously found that the growth of polymerase η-deficient human cells was inhibited by treatment with azaserine, a DNA carboxymethylation agent, suggesting the importance of this polymerase in bypassing the azaserine-induced carboxymethylated DNA lesions. In the present study, we examined how carboxymethylated DNA lesions, which included N6-carboxymethyl-2′-deoxyadenosine (N6-CMdA), N4-carboxymethyl-2′-deoxycytidine (N 4-CMdC), N3-carboxymethylthymidine (N3-CMdT) and O4-carboxymethylthymidine (O4-CMdT), perturbed the efficiency and fidelity of DNA replication mediated by Saccharomyces cerevisiae polymerase η (pol η). Our results from steady-state kinetic assay showed that pol η could readily bypass and extend past N6-CMdA, and incorporated the correct nucleotides opposite the lesion and its neighboring 5′ nucleoside with high efficiency. By contrast, the polymerase could bypass N4-CMdC inefficiently, with substantial misincorporation of dCMP followed by dAMP, though pol η could extend past the lesion with high fidelity and efficiency when dGMP was incorporated opposite the lesion. On the other hand, yeast pol η experienced great difficulty in bypassing O4-CMdT and N3-CMdT and the polymerase inserted preferentially the incorrect dGMP opposite these two DNA lesions; the extension step, nevertheless, occurred with high fidelity and efficiency when the correct dAMP was opposite the lesion, as opposed to the preferentially incorporated incorrect dGMP. The above results suggest that these lesions may contribute significantly to diazoacetate-induced mutations and those in p53 gene observed in human gastrointestinal tumors.
Normal metabolic activities and environmental factors can cause DNA damage, which can result in up to one million molecular lesions per cell per day (1). These lesions, such as nucleobase modifications, strand breaks, and cross-link lesions, can induce single-base substitutions, frameshifts, and deletions into the genome, affecting the cell's ability to replicate its DNA, transcribe genes, stimulate apoptosis, and regulate cell division (2, 3). To counteract the deleterious effects of DNA lesions, cells are equipped with various DNA damage response pathways, including a battery of DNA repair pathways and translesion synthesis (TLS) (4, 5). TLS is a type of damage tolerance that provides a way to replicate past potentially harmful DNA lesions as opposed to repairing them (2, 5, 6). This prevents long-term blockage of replication fork and ensures continuing DNA replication (2, 5, 6). This pathway is critical for maintaining replication fork progression and increasing cell survival.
TLS DNA polymerases, many of which belong to the Y-family, are a group of specialized DNA polymerases which can bypass DNA damage by facilitating nucleotide insertion opposite damaged bases and enabling extension past the damage site (5, 7). The insertion and extension steps often involve different polymerases, though the same polymerase may be responsible for both steps (8). These polymerases typically have larger active sites than traditional replicative polymerases which allow them to accommodate erroneous base pairs and bulky DNA lesions (9). Consequently, TLS polymerases often lack the efficiency and fidelity of replicative polymerases (5), though in some instances the nucleotide incorporation mediated by these polymerases opposite DNA lesions can be both accurate and efficient (10-12). Among the Y-family polymerases, polymerase η (pol η) is capable of bypassing a variety of DNA lesions (9). The predominant established cellular role of pol η involves bypassing, with high efficiency and accuracy, thymine-thymine cyclobutane pyrimidine dimers (CPDs) that accumulate following exposure to UV light (13, 14). The importance in bypassing these dimers is pronounced in those with XPV syndrome, as these patients lack a functional pol η and exhibit a dramatic increase in susceptibility to developing skin tumors (13-15). In addition to replicating across CPDs with high efficiency and accuracy, pol η is also capable of bypassing a broad spectrum of other DNA lesions with varying degrees of efficiency and fidelity (9).
N-nitroso compounds (NOCs) cause damage to DNA and are both mutagenic and carcinogenic (16, 17). Humans are exposed to NOCs both endogenously and exogenously through diet, tobacco smoke, and other environmental sources (18). Endogenous sources account for 45-75% of total NOC exposure, and an European Prospective Investigation into Cancer and Nutrition (EPIC) study revealed that NOC exposure is associated with elevated risk of developing non-cardia gastric cancer (19). Some endogenously formed NOCs could be converted metabolically to diazoacetate (20). A study by Gottschalg and coworkers (21) demonstrated that the replication of potassium diazoacetate (KDA)-treated, human p53 gene-carrying plasmid in yeast cells could lead to substantial single-base substitutions at both GC and AT base pairs. More importantly, the patterns of mutations induced by KDA at non-CpG sites resemble closely those found in p53 gene of human stomach and colorectal cancers (21), indicating that diazoacetate may be an important etiological factor for the development of human gastrointestinal cancers (21). Diazoacetate could induce carboxymethylation and, to a lesser extent, methylation of DNA (20); nevertheless, the mutation spectrum induced by KDA differs markedly from that induced by a methylating agent, suggesting that carboxymethylation is the major source for KDA-induced mutations (21). The known carboxymethylated DNA lesions include N7-carboxymethylguanine (N7-CMGua), O6-carboxymethyl-2′-deoxyguanosine (O6-CMdG), N6-carboxymethyl-2′-deoxyadenosine (N6-CMdA), N4-carboxymethyl-2′-deoxycytidine (N4-CMdC), N3-carboxymethylthymidine (N3-CMdT), and O4-carboxymethylthymidine (O4-CMdT) (20, 22-26).
In the present study, we examined how four site-specifically inserted, carboxymethylated DNA lesions previously synthesized by our laboratory, which include N-CMdA, N4-CMdC, N3-CMdT, and O4-CMdT (Structures depicted in Figure 1) (23, 24), perturb DNA replication by hindering yeast pol η and inducing mutations. We chose pol η for the present study based on a previous observation that the growth of human lymphoblastoid cells deficient in XPV gene, which encodes human pol η (14), was inhibited substantially upon treatment with 10-20 μM azaserine, a DNA carboxymethylating agent (25). We assessed, by using steady-state kinetic assay, the efficiency and fidelity of yeast pol η to incorporate nucleotides opposite the four carboxymethylated DNA lesions and to extend past these lesions.
Figure 1.
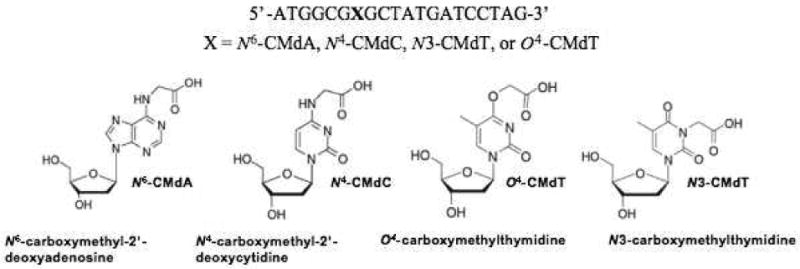
Structures of the four carboxymethylated nucleosides, N6-CMdA, N4-CMdC, N3-CMdT, and O4-CMdT, as well as the 20mer sequences of the lesion-containing substrates used for the in vitro replication studies.
Experimental Procedures
Materials
All unmodified oligodeoxyribonucleotides (ODNs) were purchased from Integrated DNA Technologies (Coralville, IA), and [γ-32P]ATP was acquired from Perkin-Elmer (Boston, MA). Saccharomyces cerevisiae DNA polymerase η (pol η) was expressed and purified following previously published procedures (27, 28). The Bio-Rad Protein Assay kit (Bio-Rad, Hercules, CA) was used to quantify the concentration of pol η, and enzyme purity was confirmed to be ∼95% based on SDS-PAGE analysis. All other enzymes used in this study were purchased from New England BioLabs (Ipswich, MA).
Preparation of Lesion-bearing ODN Substrates
ODNs carrying carboxymethylated 2′-deoxyribonucleoside derivatives were previously synthesized (23, 24). The 12mer lesion-bearing substrates, d(ATGGCGXGCTAT) [X represents an N6-CMdA, N4-CMdC, or O4-CMdT] were ligated with the 5′-phosphorylated d(GATCCTAG) in the presence of a template ODN, d(CCGCTCCCTAGGATCATAGCYCGCCAT) (Y is a dT, dG, or dA), using previously described procedures (29). The resultant lesion-bearing 20mer substrates were purified using 20% denaturing polyacrylamide gel electrophoresis (PAGE). Purity and identities of the substrates were further confirmed by PAGE and LC-MS/MS analysis. The corresponding 20mer N3-CMdT-containing substrate was synthesized in our previous study (23).
Primer Extension Assay
The 20mer lesion-containing ODNs or the corresponding unmodified ODNs (0.05 μM) were annealed with a 5′-32P-labeled 13mer primer (0.05 μM), by heating to 95°C and cooling slowly to room temperature. A mixture of all four dNTPs (250 μM each) and yeast pol η (concentrations indicated in Figure 2 and Supplementary Figure 1) were added to the duplex mixture. The reaction was carried out under standing-start conditions in the presence of a buffer containing 10 mM Tris-HCl (pH 7.5), 5 mM MgCl2, and 7.5 mM dithiothreitol (DTT) at 37°C for 1 hr. The reaction was terminated with an equal volume of formamide gel-loading buffer (80% formamide, 10 mM EDTA, pH 8.0, 1 mg/mL xylene cyanol, and 1 mg/mL bromophenol blue). The products were resolved on 20% denaturing polyacrylamide gels with 8 M urea, and gel-band intensities were quantified using a Typhoon 9410 Variable Mode Imager (Amersham Biosciences Co.).
Figure 2.
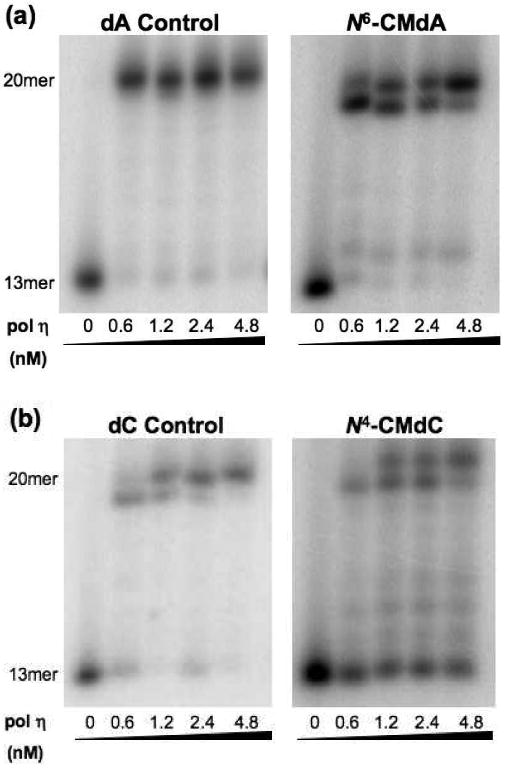
Primer extension assays opposite the (a) N6-CMdA and (b) N4-CMdC lesions and corresponding controls with yeast polymerase η in the presence of all four dNTPs [250 μM each]. The products were resolved with 20% denaturing polyacrylamide gels.
Steady-state Kinetic Measurements
Steady-state kinetic assays were performed following previously published procedures (30). The 32P-labeled primer-template complex (0.05 μM, produced using procedures described in primer extension assay) was incubated with yeast pol η (1.2 nM) at 37°C for 10 min in the presence of individual dNTPs at various concentrations indicated in Figures 3-4 and Figures S2-S4 in the Supporting Information. The reaction was terminated with an equal volume of formamide gel-loading buffer used in the primer extension assay. Extension products were separated on 20% denaturing polyacrylamide gels containing 8 M urea. Gel-band intensities of the primer and its extension products were quantified using a Typhoon 9410 Variable Mode Imager and ImageQuant 5.2 Software (Amersham Biosciences Co.). The concentration of dNTP was optimized to allow for less than 20% nucleotide incorporation. The kinetic parameters, Vmax and Km, for the incorporation of incorrect and correct nucleotides were determined by plotting the observed rate of nucleotide incorporation (Vobs) as a function of dNTP concentration using non-linear curve regression with Origin 6.0 (OriginLab, Northampton, MA). The data was then fit with the Michaelis-Menten equation:
Figure 3.
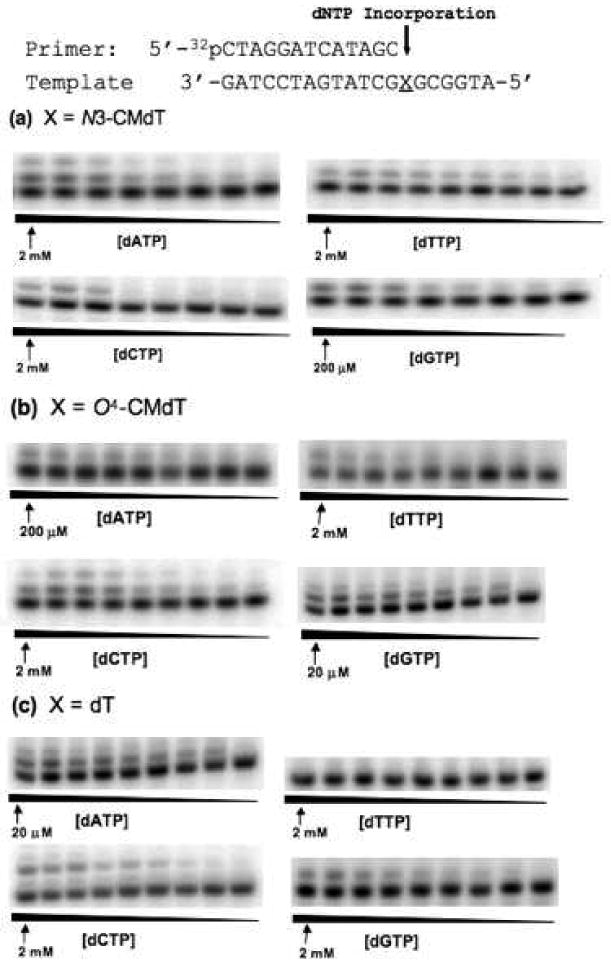
Representative gel images for steady-state kinetic assays measuring nucleotide incorporation opposite the N3-CMdT and O4-CMdT lesions and unmodified dT using 1.2 nM yeast polymerase η. Reactions were carried out in the presence of individual dNTPs with the highest concentrations indicated in the figures. The concentration ratios between neighboring lanes were 0.50.
Figure 4.
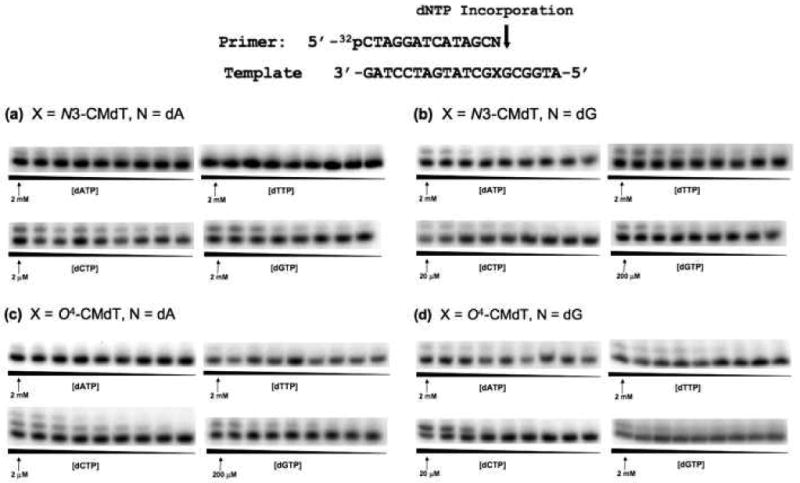
Representative gel images for steady-state kinetic assays measuring extension past N3-CMdT and O4-CMdT lesions with base opposite the lesions indicated in the figure as ‘N’. Reactions were carried out using 1.2 nM yeast polymerase η and in the presence of individual dNTPs with the highest concentrations indicated in the figures. The concentration ratios between neighboring lanes were 0.50.
The kcat values were calculated by dividing the Vmax with the concentration of pol η used. The efficiency of nucleotide incorporation was calculated by the ratio of kcat/Km and frequency of nucleotide misincorporation (finc) and frequency of extension (fext) were determined based on the following equations:
Results
To understand how the carboxymethylated lesions N6-CMdA, N4-CMdC, N3-CMdT, and O4-CMdT are recognized by yeast pol η, we prepared 20mer ODN substrates containing these lesions at defined sites as previously described (23, 24), and examined the effect of these lesions on the fidelity and efficiency of DNA replication by using primer extension and steady-state kinetic assays.
Primer Extension across N6-CMdA, N4-CMdC, N3-CMdT, and O4-CMdT with Yeast Pol η
We first performed primer extension assays to assess the ability of yeast pol η to extend a 13mer primer in the presence of 20mer DNA templates containing a site-specifically inserted N6-CMdA, N4-CMdC, N3-CMdT, O4-CMdT and the corresponding unmodified nucleosides. The primer extension results indicate that, in the presence of all four dNTPs, yeast pol η can bypass successfully N6-CMdA and N4-CMdC, as well as generate full-length products (Figure 2a,b). However, N4-CMdC was much more difficult for yeast pol η to bypass than N6-CMdA, which is manifested by the presence of significant amount of unextended primer (Figure 2b). On the other hand, yeast pol η is extremely hindered by N3-CMdT and O4-CMdT, only generating extension products up to three nucleotides past the lesion (Figure S1); no full-length extension product(s) could be detected.
Steady-state Kinetic Analyses of Yeast Pol η-mediated Nucleotide Insertion opposite N6-CMdA, N4-CMdC, N3-CMdT, and O4-CMdT
We next employed steady-state kinetic assays to measure the kinetic parameters for nucleotide incorporation by yeast pol η opposite the four carboxymethylated DNA lesions, and unmodified dA, dC, and dT (see Experimental Procedures, and representative gel images are shown in Figure 3 and Figure S2).
Significant differences were found in the accuracy and efficiency of nucleotide insertion opposite the four carboxymethylated DNA lesions. Yeast pol η preferentially incorporated the correct nucleotide, dTMP, opposite N6-CMdA, at an efficiency that is ∼87% of that for dTMP incorporation opposing unmodified dA (Table 1). Misinsertion of dAMP, dCMP, and dGMP opposite N6-CMdA occurred at very low frequencies (0.075%, 0.44%, and 0.84%, respectively). By contrast, yeast pol η substantially misincorporated dCMP (132%, relative to the correct dGMP insertion) and dAMP (31%) opposite N4-CMdC (Table 1). Efficiency for the correct dGMP incorporation opposite N4-CMdC is also ∼7 fold lower than that for the corresponding insertion opposite undamaged dC. Additionally, nucleotide insertion across N3-CMdT and O4-CMdT occurred with compromised efficiency, which was accompanied with substantial misincorporation of dGMP (Table 1). Yeast pol η incorporates dGMP ∼80 times more efficiently than dAMP opposite O4-CMdT, and a 1.3-fold higher efficiency in dGMP than dAMP insertion opposite N3-CMdT was observed. Yeast pol η also incorporates dTMP and dCMP opposite O4-CMdT and N3-CMdT, though at efficiencies lower than that for the corresponding insertion of the correct dAMP (Table 1). Relative to nucleotide incorporation opposite unmodified dT, efficiencies for correct nucleotide insertion opposite N3-CMdT and O4-CMdT dropped substantially to ∼0.08% and ∼0.015%, respectively. The reduced efficiency and fidelity underscore the mutagenic potential of N3-CMdT and O4-CMdT, and significant difficulty experienced by yeast pol η in bypassing the two lesions.
Table 1.
Steady-state kinetic parameters for S. cerevisiae pol η-mediated nucleotide incorporation opposite N4-CMdC, N6-CMdA, N3-CMdT, and O4-CMdT, as well as opposite unmodified dA, dC, and dTa
| dNTP | kcat (min-1) | Km (μM) | kcat/Km (μM-1min-1) | finc |
|---|---|---|---|---|
| N4-CMdC-containing substrate | ||||
| dTTP | 12 ± 1 | 93 ± 7 | 0.13 ± 0.05 | 6.4 × 10-2 |
| dGTP | 14 ± 1 | 7 ± 1 | 2.0 ± 0.2 | 1 |
| dCTP | 14 ± 1 | 5.1 ± 0.5 | 2.6 ± 0.2 | 1.3 |
| dATP | 15 ± 1 | 25 ± 2 | 0.62 ± 0.08 | 0.31 |
| dC-containing substrate | ||||
| dTTP | 20 ± 1 | 470 ± 40 | 0.045 ± 0.005 | 1.9 × 10-3 |
| dGTP | 24 ± 0.2 | 1.0 ± 0.06 | 24 ± 0.9 | 1 |
| dCTP | 10 ± 1 | 120 ± 10 | 0.082 ± 0.001 | 3.5 × 10-3 |
| dATP | N/A | N/A | N/A | N/A |
| N6-CMdA-containing substrate | ||||
| dTTP | 16 ± 1 | 0.50 ± 0.07 | 32 ± 4 | 1 |
| dGTP | 8.7 ± 0.7 | 33 ± 2 | 0.27 ± 0.04 | 8.4 × 10-3 |
| dCTP | 19 ± 2 | 130 ± 20 | 0.14 ± 0.01 | 4.4 × 10-3 |
| dATP | 13 ± 1 | 540 ± 40 | 0.024 ± 0.002 | 7.5 × 10-4 |
| dA-containing substrate | ||||
| dTTP | 15 ± 1 | 0.41 ± 0.01 | 37 ± 4 | 1 |
| dGTP | 8.2 ± 0.5 | 210 ± 20 | 0.039 ± 0.001 | 1.1 × 10-3 |
| dCTP | 40 ± 3 | 400 ± 30 | 0.089 ± 0.002 | 2.5 × 10-3 |
| dATP | 10 ± 1 | 310 ± 13 | 0.033 ± 0.001 | 8.7 × 10-4 |
| N3-CMdT-containing substrate | ||||
| dTTP | 13 ± 1 | 87 ± 6 | 0.15 ± 0.01 | 0.4 |
| dGTP | 11 ± 1 | 23 ± 2 | 0.48 ± 0.03 | 1.3 |
| dCTP | 5.2 ± 1 | 63 ± 5 | 0.083 ± 0.008 | 0.2 |
| dATP | 13 ± 1 | 35 ± 3 | 0.38 ± 0.02 | 1 |
| O4-CMdT-containing substrate | ||||
| dTTP | 11 ± 1 | 260 ± 30 | 0.043 ± 0.003 | 0.57 |
| dGTP | 11 ± 1 | 1.8 ± 0.2 | 6.1 ± 0.8 | 84 |
| dCTP | 7.1 ± 1 | 280 ± 20 | 0.025 ± 0.002 | 0.35 |
| dATP | 14 ± 1 | 200 ± 15 | 0.072 ± 0.003 | 1 |
| dT-containing substrate | ||||
| dTTP | N/A | N/A | N/A | N/A |
| dGTP | 28 ± 2 | 20 ± 2 | 1.4 ± 0.01 | 2.5 × 10-3 |
| dCTP | 43 ± 2 | 35 ± 3 | 1.3 ± 0.02 | 2.3 × 10-3 |
| dATP | 78 ± 4 | 0.16 ± 0.01 | 490 ± 20 | 1 |
kcat and Km values based on three independent measurements
‘N/A’ indicates data not available, where no nucleotide incorporation was detected even when high concentrations of dNTP was used (2 mM).
Steady-state Kinetic Analyses of Yeast Pol η-mediated Extension past N6-CMdA, N4-CMdC, N3- CMdT, and O4-CMdT
We next employed steady-state kinetic assays to determine the efficiency and fidelity of yeast pol η-mediated extension past the carboxymethylated lesions and their respective controls (representative gel images shown in Figures 4, S3 & S4). Based on the kinetic parameters for the insertion step opposite N4-CMdC, N3-CMdT, and O4-CMdT, we utilized multiple primers for the extension past these lesions. In addition to measuring the kinetics for extension when the correct dNTP is inserted opposite these lesions, we also included a mismatched base pair based on the nucleotide preferentially incorporated during the insertion step (Figure 4 and Figure S3) to more accurately account for the polymerase's role in insertion and extension.
Significant differences were found in the extension past the four carboxymethylated lesions depending on the nature of the unmodified nucleoside placed opposite the lesion. For the extension past the N6-CMdA:dT pair, yeast pol η incorporated preferentially the correct dCMP opposite dG, the next nucleoside in the template sequence after the lesion site (Figure S3 and Table 2). Misincorporation of dAMP, dTMP, and dGMP occurred at low frequencies (0.48%, 0.60%, and 0.88%, respectively, Table 2). The kcat/Km for dCMP insertion was calculated to be 25 μM-1 min-1, slightly lower than the 50 μM-1 min-1 observed in the insertion of dCMP opposite dG while extending the lesion-free dA:dT pair (Table 2), indicating that pol η can both bypass and extend past N6-CMdA with high efficiency and fidelity. Extension past N4-CMdC occurred with varying degrees of fidelity, which is dependant on the nucleotide inserted opposite the lesion. In the presence of the N4-CMdC:dC mispair (dCMP was the most preferentially inserted opposite N4-CMdC, vide supra), extension occurred at low efficiency and fidelity; dGMP was incorrectly inserted opposite the template dG ∼52% of the time, which was accompanied with a ∼2% misincorporation of dTMP (Table 2). In contrast, with the correct N4-CMdC:dG base pair, the correct dCMP was predominantly incorporated opposite the dG in the template, with misinsertion of dAMP, dTMP, and dGMP occurring at frequencies of 0.22%, 0.67%, and 1.4% respectively (Table 2).
Table 2.
Steady-state kinetic parameters for S. cerevisiae pol η-mediated extension past N4-CMdC, N6-CMdA, N3-CMdT, and O4-CMdT, as well as unmodified dA, dC, and dT (designated by ‘X’). ‘N’ represents the nucleoside placed opposite the ‘X’ (See Figure 4). a
| dNTP | kcat (min-1) | Km (μM) | kcat/Km (μM-1min-1) | fext | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| X = N6-CMdA, N = dT | |||||||||||||||
| dTTP | 3 ± 0.2 | 20 ± 1 | 0.15 ± 0.01 | 6.0 × 10-3 | |||||||||||
| dGTP | 10 ± 1 | 45 ± 4 | 0.22 ± 0.02 | 8.8 × 10-3 | |||||||||||
| dCTP | 28 ± 2 | 1.1 ± 0.01 | 25 ± 2 | 1 | |||||||||||
| dATP | 14 ± 1 | 110 ± 10 | 0.12 ± 0.01 | 4.8 × 10-3 | |||||||||||
| X = N4-CMdC, N = dC | |||||||||||||||
| dTTP | 14 ± 1 | 92 ± 6 | 0.15 ± 0.01 | 0.016 | |||||||||||
| dGTP | 12 ± 1 | 2.5 ± 0.21 | 4.9 ± 0.3 | 0.52 | |||||||||||
| dCTP | 16 ± 1 | 1.7 ± 0.1 | 9.4 ± 0.9 | 1 | |||||||||||
| dATP | 12 ± 1 | 500 ± 50 | 0.024 ± 0.001 | 2.6 × 10-3 | |||||||||||
| X = N4-CMdC, N = dG | |||||||||||||||
| dTTP | 13 ± 1 | 110 ± 9 | 0.12 ± 0.01 | 6.7 × 10-3 | |||||||||||
| dGTP | 21 ± 1 | 82 ± 7 | 0.26 ± 0.02 | 0.014 | |||||||||||
| dCTP | 25 ± 2 | 1.4 ± 0.13 | 18 ± 2 | 1 | |||||||||||
| dATP | 15 ± 1 | 380 ± 20 | 0.039 ± 0.003 | 2.2 × 10-3 | |||||||||||
| X = N3-CMdT, N = dG | |||||||||||||||
| dTTP | 17 ± 1 | 82 ± 8 | 0.21 ± 0.01 | 7.5 × 10-3 | |||||||||||
| dGTP | 15 ± 1 | 3 ± 0.3 | 5 ± 0.4 | 0.18 | |||||||||||
| dCTP | 12 ± 1 | 0.44 ± 0.03 | 28 ± 2 | 1 | |||||||||||
| dATP | 15 ± 1 | 64 ± 5 | 0.24 ± 0.02 | 8.6 × 10-3 | |||||||||||
| X = O4-CMdT, N = dG | |||||||||||||||
| dTTP | 20 ± 2 | 140 ± 12 | 0.14 ± 0.01 | 7.4 × 10-3 | |||||||||||
| dGTP | 13 ± 1 | 10 ± 1 | 1.3 ± 0.13 | 0.068 | |||||||||||
| dCTP | 15 ± 1 | 0.79 ± 0.06 | 19 ± 3 | 1 | |||||||||||
| dATP | 12 ± 1 | 140 ± 10 | 0.085 ± 0.007 | 4.5 × 10-3 | |||||||||||
| X = N3-CMdT, N = dA | |||||||||||||||
| dTTP | N/A | N/A | N/A | N/A | |||||||||||
| dGTP | 22 ± 2 | 95 ± 8 | 0.23 ± 0.1 | 9.6 × 10-3 | |||||||||||
| dCTP | 27 ± 2 | 1.1 ± 0.1 | 24 ± 2 | 1 | |||||||||||
| dATP | 21 ± 2 | 700 ± 60 | 0.031 ± 0.002 | 1.3 × 10-3 | |||||||||||
| X = O4-CMdT, N = dA | |||||||||||||||
| dTTP | 20 ± 2 | 330 ± 20 | 0.061 ± 0.003 | 3.1 × 10-3 | |||||||||||
| dGTP | 21 ± 2 | 94 ± 8 | 0.22 ± 0.02 | 0.011 | |||||||||||
| dCTP | 28 ± 2 | 1.4 ± 0.1 | 20 ± 1 | 1 | |||||||||||
| dATP | 23 ± 2 | 550 ± 50 | 0.042 ± 0.002 | 2.0 × 10-3 | |||||||||||
| X = dC, N = dG | |||||||||||||||
| dTTP | N/A | N/A | N/A | N/A | |||||||||||
| dGTP | 16 ± 1 | 260 ± 20 | 0.062 ± 0.005 | 1.9 × 10-3 | |||||||||||
| dCTP | 28 ± 2 | 0.87 ± 0.08 | 32 ± 2 | 1 | |||||||||||
| dATP | 10 ± 1 | 450 ± 30 | 0.022 ± 0.002 | 6.9 × 10-4 | |||||||||||
| X = dA, N = dT | |||||||||||||||
| dTTP | 19 ± 1 | 770 ± 10 | 0.024 ± 0.001 | 4.9 × 10-4 | |||||||||||
| dGTP | 13 ± 1 | 220 ± 10 | 0.059 ± 0.005 | 1.2 × 10-3 | |||||||||||
| dCTP | 30 ± 2 | 0.61 ± 0.03 | 50 ± 3 | 1 | |||||||||||
| dATP | N/A | N/A | N/A | N/A | |||||||||||
| X = dT, N = dA | |||||||||||||||
| dTTP | 25 ± 2 | 300 ± 20 | 0.083 ± 0.006 | 1.8 × 10-3 | |||||||||||
| dGTP | 13 ± 1 | 110 ± 10 | 0.12 ± 0.01 | 2.7 × 10-3 | |||||||||||
| dCTP | 17 ± 1 | 0.38 ± 0.03 | 45 ± 3 | 1 | |||||||||||
| dATP | N/A | N/A | N/A | N/A | |||||||||||
kcat and Km values based on three independent measurements
‘N/A’ indicates data not available, where no nucleotide incorporation was detected even when high concentrations of dNTP was used (2 mM).
A similar trend was observed for extension past the N3- and O4-CMdT lesions. Along this line, when the preferentially inserted nucleotide (i.e., dGMP) is placed opposite the two dT lesions, extension occurred at low fidelity, with dGMP being incorporated opposite the dG on the 5′ side of N3-CMdT and O4-CMdT at frequencies of 18% and 6.8%, respectively (Table 2). However, when the correct dA was placed opposite N3-CMdT and O4-CMdT, there is only 0.96% and 1.1% misinsertion of dGMP (Table 2). Taken together, the extension past all four carboxymethylated lesions has better efficiency and accuracy when the correct nucleotide was incorporated opposite the lesions.
Discussion
We previously reported the formation of the four carboxymethylated DNA lesions in calf thymus DNA following treatment with diazoacetate (23, 24). In this paper, we performed in vitro replication studies on these lesion-containing DNA substrates using purified yeast pol η. Results from primer extension and steady-state kinetic assays for nucleotide insertion demonstrate that, depending on the structures of the carboxymethylated DNA lesions, bypass by yeast pol η can be error-prone or error-free. Replication across N6-CMdA was both accurate and efficient, whereas yeast pol η exhibited reduced ability to extend the primer across N4-CMdC in the template strand and the polymerase preferentially inserted the incorrect dCMP opposite the lesion. In contrast, N3-CMdT and O4-CMdT hindered yeast pol η from generating full-length replication products and the polymerase displayed high frequencies of nucleotide misincorporation opposite the two lesions.
We also demonstrated that pol η is capable of extending past the carboxymethylated lesions with high efficiency and fidelity when the correct nucleotide was inserted opposite the lesions; however, the pol η-mediated extension beyond the damage site is neither efficient nor accurate when a wrong nucleotide was incorporated opposite the lesion. In this context, the placement of the preferentially inserted, incorrect nucleotide (dCMP) opposite the N4-CMdC results in a misincorporation frequency of over 50% at the extension step. Likewise, the placement of the preferentially inserted dGMP opposite the N3-CMdT and O4-CMdT led to lower fidelity in extension than when the correct dAMP is incorporated opposite the lesions. Efficiency of extension is also considerably higher than that of insertion for N4-CMdC, N3-CMdT, and O4-CMdT, regardless of whether the correct or the preferentially inserted incorrect nucleotide was placed opposite the lesion.
It is important to discuss the results obtained from the steady-state kinetic measurements in the context of previous studies about this type of DNA lesions. Along this line, a recent study on the mutagenic potential of these carboxymethylated lesions in E. coli cells revealed that N6-CMdA did not block DNA replication or induce mutations (31), which is in keeping with what we observed from the in vitro replication experiments using purified S. cerevisiae pol η. N4-CMdC only slightly blocked DNA replication in E. coli cells and it was not mutagenic (31), a finding distinct from steady-state kinetic measurement results showing that this lesion directs significant frequency of dCMP misincorporation. In line with what we found from primer extension and steady-state kinetic measurements, N3-CMdT and O4-CMdT are strong blockades to DNA replication in E. coli cells (31). From the replication study in E. coli cells, the major mutation observed for N3-CMdT was T→A transversion at ∼65% frequency, along with T→C transition at a frequency of ∼5%, whereas O4-CMdT induced predominantly T→C transition at ∼85% frequency (31). Consistent with these findings, results from our steady-state kinetic measurements revealed substantial misincorporation of dGMP opposite O4-CMdT. On the other hand, steady-state kinetic analysis revealed the preferential insertion of dGMP opposite N3-CMdT, whereas this lesion was found to direct considerable misincorporation of dTMP in E. coli cells (vide supra).
Together, aside from the similarities in results obtained from replication studies in E. coli cells and those from the in vitro replication studies using yeast pol η, there are some notable differences revealed by these two assays, particularly for nucleotide misincorporation opposite N3-CMdT and N4-CMdC. This is not unexpected viewing that the steady-state kinetic measurements were carried out with purified S. cerevisiae pol η in the presence of one type of nucleotide at a time, whereas the in vivo replication experiment involved the participation of the entire replication machinery of E. coli cells. Along this line, it is worth noting that E. coli has two Y-family polymerases, Pol IV (encoded by the dinB gene) and Pol V (UmuD′2C, encoded by the umuDC genes) (32). In addition, E. coli pol V and eukaryotic pol η exhibit a very similar pattern of dNTP insertion opposite a variety of lesions (33).
It is also of interest to compare our results with previous steady-state kinetic measurements on other structurely related DNA lesions, particularly the corresponding methylated DNA lesions. In this context, while there were no previous steady-state kinetic measurements on nucleotide incorporation opposite N6-methyladenine or N4-methylcytosine, such assays on O4-methylthymine showed that this lesion could direct significant misincorporation of dGMP by Klenow fragment of E. coli DNA polymerase I or Drosophila melanogaster polymerase α-primase complex (34). In addition, N3-methylthymine was found to be a very strong block to Klenow fragment, and the lesion could direct the misinsertion of dAMP or dTMP (35). We found that the addition of a carboxymethyl group onto the O4 and N3 positions of thymine hinders severely yeast pol η, in addition to directing the polymerase to misincorporate dGMP.
The mutation spectra induced by diazoacetate in the yeast functional p53 mutation assay displayed high frequencies of mutations observed at both AT and GC base pairs, including AT→TA, AT→GC, AT→CG as well as GC→AT, GC→TA, and GC→CG mutations (21). Additionally, the types and frequencies of these mutations at non-CpG sites are remarkably similar to those observed for p53 gene in human gastrointestinal tract tumors, and are distinct from the mutation spectra induced by the methylating agent N-methyl-N-nitrosourea (MNU) (21). Our steady-state kinetic measurement results suggest that N4-CMdC, N3-CMdT and O4-CMdT may contribute to the diazoacetate-induced mutations in human p53 gene and those mutations observed in human gastrointestinal tract tumors.
A previous study demonstrated that deficiency in pol η confers an elevated sensitivity of human lymphoblastoid cells toward azaserine exposure, suggesting the importance of this polymerase in bypassing the carboxymethylated DNA lesions induced by azaserine (25). Our results revealed the efficient bypass of N6-CMdA and, to a lower degree, N4-CMdC by S. cerevisiae pol η. It can be envisaged that the failure in bypassing these and other carboxymethylated DNA lesions induced by azaserine may result in replication fork stalling, which may ultimately lead to increased cell death in pol η-deficient cells. Along this line, future studies about the formation and replication of these DNA lesions in mammalian cells will reveal the role of pol η and other TLS polymerases in bypassing the carboxymethylated DNA lesions in mammalian cells (36), and further illuminate the biological consequences of these carboxymethylated DNA lesions.
Supplementary Material
Acknowledgments
Funding Support: This work was supported by the National Institutes of Health (R01 DK082779 to Y. W.) and Ashley L. Swanson was supported by an NRSA Institutional Training Grant (T32 ES018827).
Footnotes
Supporting Information Available: Primer extension and steady-state kinetic assay results. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 2.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2nd. ASM Press; Washington, D.C.: 2006. [Google Scholar]
- 3.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 4.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 5.Lehmann AR, Niimi A, Brown S, Sabbioneda S, Wing JF, Kannouche PL, Green CM. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair. 2007;6:891–899. doi: 10.1016/j.dnarep.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 6.Friedberg EC. DNA damage and repair. Nature. 2003;421:436–440. doi: 10.1038/nature01408. [DOI] [PubMed] [Google Scholar]
- 7.Ling H, Boudsocq F, Plosky BS, Woodgate R, Yang W. Replication of a cis-syn thymine dimer at atomic resolution. Nature. 2003;424:1083–1087. doi: 10.1038/nature01919. [DOI] [PubMed] [Google Scholar]
- 8.Prakash S, Prakash L. Translesion DNA Synthesis in eukaryotes: A one- or two-polymerase affair. Genes Dev. 2002;16:1872–1883. doi: 10.1101/gad.1009802. [DOI] [PubMed] [Google Scholar]
- 9.Yang WW, R What a difference a decade makes: insights into translesion DNA synthesis. Proc Natl Acad Sci USA. 2007;104:15591–15598. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson RE, Prakash S, Prakash L. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Pol η. Science. 1999;283:1001–1004. doi: 10.1126/science.283.5404.1001. [DOI] [PubMed] [Google Scholar]
- 11.Jarosz DF, Godoy VG, Delaney JC, Essigmann JM, Walker GC. A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature. 2006;439:225–228. doi: 10.1038/nature04318. [DOI] [PubMed] [Google Scholar]
- 12.Yuan B, Cao H, Jiang Y, Hong H, Wang Y. Efficient and accurate bypass of N2-(1-carboxyethyl)-2′-deoxyguanosine by DinB DNA polymerase in vitro and in vivo. Proc Natl Acad Sci USA. 2008;105:8679–8684. doi: 10.1073/pnas.0711546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson RE, Kondratick CM, Prakash S, Prakash L. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 1999;285:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- 14.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 15.Cleaver JE. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat Rev Cancer. 2005;5:564–573. doi: 10.1038/nrc1652. [DOI] [PubMed] [Google Scholar]
- 16.Lijinsky W. Carcinogenicity and mutagenicity of N-nitroso compounds. Mol Toxicol. 1987;1:107–119. [PubMed] [Google Scholar]
- 17.Mirvish SS. Role of N-nitroso compounds (NOC) and N-nitrosation in etiology of gastric, esophageal, nasopharyngeal and bladder cancer and contribution to cancer of known exposures to NOC. Cancer Lett. 1995;93:17–48. doi: 10.1016/0304-3835(95)03786-V. [DOI] [PubMed] [Google Scholar]
- 18.Tricker AR. N-nitroso compounds and man: sources of exposure, endogenous formation and occurrence in body fluids. Eur J Cancer Prev. 1997;6:226–268. [PubMed] [Google Scholar]
- 19.Jakszyn P, Bingham S, Pera G, Agudo A, Luben R, Welch A, Boeing H, Del Giudice G, Palli D, Saieva C, Krogh V, Sacerdote C, Tumino R, Panico S, Berglund G, Siman H, Hallmans G, Sanchez MJ, Larranaga N, Barricarte A, Chirlaque MD, Quiros JR, Key TJ, Allen N, Lund E, Carneiro F, Linseisen J, Nagel G, Overvad K, Tjonneland A, Olsen A, Bueno-de-Mesquita HB, Ocke MO, Peeters PH, Numans ME, Clavel-Chapelon F, Trichopoulou A, Fenger C, Stenling R, Ferrari P, Jenab M, Norat T, Riboli E, Gonzalez CA. Endogenous versus exogenous exposure to N-nitroso compounds and gastric cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST) study. Carcinogenesis. 2006;27:1497–1501. doi: 10.1093/carcin/bgl019. [DOI] [PubMed] [Google Scholar]
- 20.Shuker DE, Margison GP. Nitrosated glycine derivatives as a potential source of O6-methylguanine in DNA. Cancer Res. 1997;57:366–369. [PubMed] [Google Scholar]
- 21.Gottschalg E, Scott GB, Burns PA, Shuker DE. Potassium diazoacetate-induced p53 mutations in vitro in relation to formation of O6-carboxymethyl- and O6-methyl-2′-deoxyguanosine DNA adducts: relevance for gastrointestinal cancer. Carcinogenesis. 2007;28:356–362. doi: 10.1093/carcin/bgl150. [DOI] [PubMed] [Google Scholar]
- 22.Zurlo J, Curphey TJ, Hiley R, Longnecker DS. Identification of 7-carboxymethylguanine in DNA from pancreatic acinar cells exposed to azaserine. Cancer Res. 1982;42:1286–1288. [PubMed] [Google Scholar]
- 23.Wang J, Wang Y. Chemical synthesis of oligodeoxyribonucleotides containing N3- and O4-carboxymethylthymidine and their formation in DNA. Nucleic Acids Res. 2009;37:336–345. doi: 10.1093/nar/gkn946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Wang Y. Synthesis and characterization of oligodeoxyribonucleotides containing a site-specifically incorporated N6-carboxymethyl-2′-deoxyadenosine or N4-carboxymethyl-2′-deoxycytidine. Nucleic Acids Res. 2010;38:6774–6784. doi: 10.1093/nar/gkq458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Driscoll M, MacPherson P, Xu Y, Karran P. The cytotoxicity of DNA carboxymethylation and methylation by the model carboxymethylating agent azaserine in human cells. Carcinogenesis. 1999;20:1855–1862. doi: 10.1093/carcin/20.9.1855. [DOI] [PubMed] [Google Scholar]
- 26.Harrison KL, Fairhurst N, Challis BC, Shuker DE. Synthesis, characterization, and immunochemical detection of O6-(carboxymethyl)-2′-deoxyguanosine: a DNA adduct formed by nitrosated glycine derivatives. Chem Res Toxicol. 1997;10:652–659. doi: 10.1021/tx960203u. [DOI] [PubMed] [Google Scholar]
- 27.Cannistraro VJ, Taylor JS. DNA-thumb interactions and processivity of T7 DNA polymerase in comparison to yeast polymerase η. J Biol Chem. 2004;279:18288–18295. doi: 10.1074/jbc.M400282200. [DOI] [PubMed] [Google Scholar]
- 28.Jiang Y, Wang Y. In vitro replication and repair studies of tandem lesions containing neighboring thymidine glycol and 8-oxo-7,8-dihydro-2′-deoxyguanosine. Chem Res Toxicol. 2009;22:574–583. doi: 10.1021/tx8003449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu C, Wang Y. LC-MS/MS identification and yeast polymerase eta bypass of a novel gamma-irradiation-induced intrastrand cross-link lesion G[8-5]C. Biochemistry. 2004;43:6745–6750. doi: 10.1021/bi0497749. [DOI] [PubMed] [Google Scholar]
- 30.Goodman MF, Creighton S, Bloom LB, Petruska J. Biochemical basis of DNA replication fidelity. Crit Rev Biochem Mol Biol. 1993;28:83–126. doi: 10.3109/10409239309086792. [DOI] [PubMed] [Google Scholar]
- 31.Yuan B, Wang J, Cao H, Sun R, Wang Y. High-throughput analysis of the mutagenic and cytotoxic properties of DNA lesions by next-generation sequencing. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fuchs RP, Fujii S, Wagner J. Properties and functions of Escherichia coli: Pol IV and Pol V. Adv Protein Chem. 2004;69:229–264. doi: 10.1016/S0065-3233(04)69008-5. [DOI] [PubMed] [Google Scholar]
- 33.Lee CH, Chandani S, Loechler EL. Homology modeling of four Y-family, lesion-bypass DNA polymerases: the case that E. coli Pol IV and human Pol κ are orthologs, and E. coli Pol V and human Pol η are orthologs. J Mol Graph Model. 2006;25:87–102. doi: 10.1016/j.jmgm.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 34.Dosanjh MK, Essigmann JM, Goodman MF, Singer B. Comparative efficiency of forming m4T.G versus m4T.A base pairs at a unique site by use of Escherichia coli DNA polymerase I (Klenow fragment) and Drosophila melanogaster polymerase α-primase complex. Biochemistry. 1990;29:4698–4703. doi: 10.1021/bi00471a026. [DOI] [PubMed] [Google Scholar]
- 35.Shrivastav N, Li D, Essigmann JM. Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis. 2010;31:59–70. doi: 10.1093/carcin/bgp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan B, You C, Andersen N, Jiang Y, Moriya M, O'Connor TR, Wang Y. The roles of DNA polymerases κ and ι in the error-free bypass of N2-carboxyalkyl-dG lesions in mammalian cells. J Biol Chem. 2011;286:17503–17511. doi: 10.1074/jbc.M111.232835. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.