

Summary
Diagonal discriminant rules have been successfully used for high-dimensional classification problems, but suffer from the serious drawback of biased discriminant scores. In this paper, we propose improved diagonal discriminant rules with bias-corrected discriminant scores for high-dimensional classification. We show that the proposed discriminant scores dominate the standard ones under the quadratic loss function. Analytical results on why the bias-corrected rules can potentially improve the predication accuracy are also provided. Finally, we demonstrate the improvement of the proposed rules over the original ones through extensive simulation studies and real case studies.
Keywords: Bias correction, Diagonal discriminant analysis, Discriminant score, Large p small n, Tumor classification
1. Introduction
Class prediction using high-dimensional data such as microarrays has been recognized as an important problem since the seminal work of Golub et al. (1999). A variety of methods have been developed and compared, including Discriminant Analysis and its extensions (Dudoit et al., 2002; Ghosh, 2003; Zhu and Hastie, 2004; Huang and Zheng, 2006; Wu, 2006; Shen et al., 2006; Guo et al., 2007; Pang et al., 2009), Random Forests (Breiman, 2001; Statnikov et al., 2008), Support Vector Machines (Furey et al., 2000; Lee et al., 2004; Vapnik and Kotz, 2006), Dimension Reduction Methods (Antoniadis et al., 2003; Dai et al., 2006) and Nearest Shrunken Centroids Methods (Tibshirani et al., 2002, 2003; Wang and Zhu, 2007; Dabney and Storey, 2007). Also see review papers with extensive comparison studies by Dudoit et al. (2002), Lee et al. (2005), and Statnikov et al. (2008).
In high-dimensional microarray data classification, it is common that the number of training samples, n, is much smaller than the number of features examined, p. This “large p small n” paradigm has posed numerous statistical challenges to most classical classification methods, such as the well-known linear discriminant analysis (LDA) and the quadratic discriminant analysis (QDA), because the sample covariance matrices are singular. This greatly limits the usage of both methods in high-dimensional data classification. To overcome the singularity problem, various approaches that rely on a diagonal approximation to the covariance matrices have been proposed. This leads to the so-called diagonal discriminant rules, which have been widely used for high-dimensional data (Dudoit et al., 2002; Speed, 2003; Tibshirani et al., 2003; Dettling, 2004; Ye et al., 2004; Dabney, 2005; Lee et al., 2005; Pique-Regi et al., 2005; Asyali et al., 2006; Noushath et al., 2006; Shieh et al., 2006; Wang and Zhu, 2007; Natowicz et al., 2008; Pang et al., 2009). In practice, the most commonly used diagonal discriminant rules for high-dimensional data are the diagonal LDA (DLDA) and the diagonal QDA (DQDA) rules introduced by Dudoit et al. (2002). Due to the relatively small n, the diagonal discriminant rules, which ignore the correlation among features, performed remarkably well compared with the more sophisticated methods in terms of both accuracy and stability (Dudoit et al., 2002; Dettling, 2004; Lee et al., 2005; Pang et al., 2009). In addition, DLDA and DQDA are easy to implement and not very sensitive to the number of predictor variables (Dudoit et al., 2002). Bickel and Levina (2004) conducted a theoretical study of this phenomenon and proved that diagonal discriminant rules can indeed outperform Fisher’s LDA when p > n.
The diagonal discriminant rules have been shown to perform well for high-dimensional data with small sample sizes, but suffer from the serious drawback of biased discriminant scores. In this paper we propose to correct the biases in the discriminant scores of diagonal discriminant analysis. Before we proceed, it is worth pointing out that the idea of bias correction in discriminant analysis is not entirely new (Ghurye and Own, 1969; Moran and Murphy, 1979; McLachlan, 1992). For instance, Moran and Murphy (1979) proposed several bias correction methods for the plug-in discriminant scores under the condition that the sample size for each class, nk, is larger than p. However, the improvement of their bias-corrected rules is not significant (James, 1985; McLachlan, 1992), mainly because the dominant term of the bias, p/nk, is not large. This has, at least partially, discouraged the popularity of the previously proposed bias-corrected discriminant rules. For microarray data, however, the ratio p/nk can be very large. As a consequence, the commonly used discriminant rules, e.g., DQDA and DLDA, may result in low prediction accuracy, especially when the design is fairly unbalanced.
The remainder of the paper is organized as follows. In Section 2, we introduce the notation and briefly review the diagonal discriminant rules. In Section 3, we derive the bias-corrected estimators of the discriminant scores and show that they dominate the original ones. In Section 4, we present some analytical results on why the bias-corrected rules can potentially increase the overall prediction accuracy. We then conduct extensive simulation studies to investigate the performance of the proposed methods in Section 5, and apply them to three real microarray data sets in Section 6. Finally, we conclude the paper in Section 7 with discussions and future directions.
2. Diagonal Discriminant Analysis
Suppose we have K distinct classes and samples from each class follow a p-dimensional multivariate normal distribution with mean vector μk and covariance matrix Σk, where k = 1, ···, K. Assume we observe nk i.i.d. random samples from the kth class, that is,
The total sample size is then . The principal goal of discriminant analysis is to predict the class label for a new observation, y. Let πk denote the prior probability of observing a sample from the kth class with . The QDA decision rule is to assign y to class , where is the discriminant score defined as in Friedman (1989), that is,
Minimizing over k is equivalent to maximizing the corresponding posterior probabilities.
In practice, the population parameters of the multivariate normal distributions are unknown and usually are estimated from the training data set, with μk by the sample means, , and Σk by the sample covariance matrices, . In addition, the prior probability πk is commonly estimated by nk/n and treated as a constant in classification problems (Friedman, 1989; Guo et al., 2007). The above estimates of parameters lead to the following sample version of ,
| (1) |
One important special case of QDA is to assume that the covariance matrices are all the same, i.e., Σk = Σ for all k. This leads to LDA, with the simplified discriminant score given by
The corresponding sample version of is then
| (2) |
with the pooled sample covariance matrix estimate .
QDA and LDA are expected to perform well if the multivariate normal assumption is satisfied and good “plug-in” estimates of the population parameters are available (Friedman, 1989). In general, LDA is more popular than QDA, largely due to its simplicity and robustness to the violations of the underlying distribution assumption and the common covariance matrices assumption (James, 1985). To make LDA work, we require that n ≥ p to ensure the non-singularity of Σ̂. Similarly for QDA, we require that nk ≥ p for each class.
When p is greater than n, we may regularize the covariance matrix estimates with generalized matrix inverse or shrinkage to address the singularity problem. However, these estimators are usually unstable due to the limited number of observations (Guo et al., 2007). In 2002, Dudoit et al. proposed to use DQDA and DLDA for classifying tumors using microarray data. Specifically, they assumed the covariance matrices to be diagonal by replacing the off-diagonal elements of Σ̂k or Σ̂ with zeros. For DQDA, we have , which simplifies Equation (1) to
| (3) |
For DLDA, we have , which simplifies Equation (2) to
| (4) |
3. Bias-Corrected Diagonal Discriminant Analysis
In this section, we first show that and are biased. We then propose several bias-corrected estimators for the discriminant scores and demonstrate their superiority over the original ones. Denote Equation (3) as
where and . Denote the true discriminant score as
where and . In Web Appendix A, we show that the following two estimators are unbiased for Lk1 and Lk2, respectively,
where Ψ(·) is the digamma function (Abramowitz and Stegun, 1972). Based on the above two unbiased estimators, we define
which is a bias-corrected discriminant score of DQDA. We refer to the corresponding rule as the bias-corrected DQDA (BQDA).
For DLDA, we denote Equation (4) as and the corresponding true discriminant score as , where and . Also in Web Appendix A, we show that the following estimator is unbiased for Lk,
which leads to the bias-corrected DLDA (BLDA) with
Further, in Appendix A we have that
Theorem 1
Under the quadratic loss function, we have
the discriminant score of BQDA, , dominates the discriminant score of DQDA, , when nk > 5; and
the discriminant score of BLDA, , dominates the discriminant score of DLDA, , when n > K + 4.
The maximum likelihood estimators (MLE), , are also common for estimating (Guo et al., 2007). By plugging into Equation (3), we obtain the discriminant score of MLE-based DQDA (MQDA). In practice, there is usually no clear indication between and , as to which estimator performs better when n is small. It is worth pointing out that, when the bias correction technique is applied, DQDA and MQDA lead to the same discriminant score so that we do not need to distinguish the two methods any more. A similar result can be established for the MLE-based DLDA (MLDA).
4. Prediction Accuracy
In this section we compare the performance of the bias-corrected discriminant rules with that of the original ones. The prediction accuracy is a common measure for evaluating the performance of a discriminant rule. It is defined as the proportion of samples classified correctly in the test set and is usually used for a balanced experimental design (Dudoit et al., 2002). However, when the design is unbalanced, a classification method favoring the majority class may have a high prediction accuracy (Qiao and Liu, 2009). There are many evaluation criteria for unbalanced designs, e.g., G-mean, F-measure, recall, class-weighted accuracy, among others (Chen et al., 2004; Cohen et al., 2006; Qiao and Liu, 2009). All of the above performance metrics can be viewed as functions of the classification matrix formed by the probabilities Pr(True Class = i, Predicted Class=j). Each metrix has its own advantages and limitations (Chen et al., 2004; Cohen et al., 2006). In this study, we apply the class-weighted accuracy (CWA) criterion (Cohen et al., 2006), which is defined as
where ak are the per-class predication accuracies and wk are non-negative weights with . For simplicity, we assume equal weights, i.e., wk = 1/K, and set the prior probability πk = 1/K as well. Note that CWA is equivalent to one of the criteria proposed by Qiao and Liu (2009), which they referred to as the “mean within group error with one-step fixed weights” criterion.
In what follows we establish some analytical results for the bias-corrected rules. For simplicity of exposition, we consider the binary classification (K = 2) with the following three assumptions:
the variances are known and equal (without loss of generality, we assume that );
n1 < n2, i.e., the class 1 is the minority class and the class 2 is the majority class; and
the covariance matrix of the test data is diagonal.
Under the above assumptions, we have for DLDA, and for BLDA. Denote . For DLDA, we assign y to the minority class if D̂ < 0; otherwise, we assign it to the majority class. For BLDA, the decision boundary is instead of a usual zero. It is easy to see that the expected change of prediction accuracy caused by the bias correction, Pr D̂,k, is given as
Note that for an unbalanced design, the prediction accuracy of the minority class always increases and that for the majority class always decreases because of the bias correction. The overall CWA change, PrΔ, is given as
| (5) |
where a positive PrΔ indicates an overall improvement on the classification performance.
By the Lindeberg condition of the central limit theorem (Lehmann, 1998), it can be shown that when p → ∞, D̂ converges in distribution to N (−δ + U, 4b1δ + c) if y is from class 1, and D̂ converges in distribution to N (δ + U, 4b2δ + c) if y is from class 2, where , b1 = 1 + 1/n2, b2 = 1 + 1/n1 and . Note that δ is the squared Euclidean distance between two samples. Further, we have
Theorem 2
Under Assumptions (i) – (iii), the overall CWA change, PrΔ, is positive when 0 < δ/p ≤ 2 and p → ∞.
The proof of Theorem 2 is shown in Appendix B. Theorem 2 suggests that the bias correction improves the overall predication accuracy as p goes large and δ is bounded by 2p. It is also worth mentioning that the proposed decision boundary U is asymptotically optimal under certain situations. By the definition of PrΔ, it is easy to see that the optimal decision boundary, Uopt, can be achieved at the intersection of the two limiting normal distributions, N (−δ + U, 4b1δ + c) and N (δ + U, 4b2δ + c). When δ is not large and/or the sample sizes, n1 and n2, are at least moderately large, we have 4b1δ + c ≈ 4b2δ + c and thus Uopt ≈ U. In general, as 4b1δ + c < 4b2δ + c, Uopt is slightly larger than U when δ is close to zero, and vice versa when δ is large. Note that Uopt depends on the quantity of δ so it is unknown in practice. Simulation study (not shown) indicates that the discriminant rules based on U and an estimated Ûopt perform similarly when an accurate estimate of the unknown δ can be obtained. While if a less accurate estimate of δ is employed, the performance of Ûopt can be unsatisfactory. In addition, Uopt is obtained only in the asymptotic sense so it may not work well when p is small. For the above reasons, in what follows we will only focus on the decision boundary U but not Ûopt. Note that the cross-validation (CV) method can be used as an alternative to select the decision boundary. Simulation study (not shown) indicates that it performs similarly as U when the sample size of each class is large. While for a small n1 or n2, CV is unstable and consequently the performance of BLDA is not satisfactory, as indicated in Braga-Neto and Dougherty (2004), Fu et al. (2005) and Isaksson et al. (2008).
When p is small, the overall change of CWA is given as
| (6) |
where is the joint density function, and PrD̂|y is the expected change of prediction accuracy given an observation y. One way to obtain PrΔ in Equation (6) is to use the numerical integration approach as shown in Appendix C.
5. Simulation Studies
In this section, we conduct extensive simulation studies to assess the performance of different discriminant rules under various settings. We explain in detail both simulation designs and results of the simple binary classification, as well as some more complicated scenarios such as the multiple classification.
5.1 Simulation Design
We draw nk training samples, xk,i, and mk test samples, yk,j, from a G-dimensional multivariate normal distribution, , where i = 1, …, nk and j = 1, …, mk. Usually G is large for microarray studies. For binary classification problem, we have K = 2. Note that we only choose p genes from all G genes for classification based on certain feature selection criteria.
We first evaluate CWA directly under different simulation settings with the assumptions stated in Section 4. We assume that all p genes are informative and the differences of the two group means are the same across all p genes. Note that if we increase p, the overall strength of the signal, , becomes stronger and eventually both the bias-corrected methods and the biased methods will classify samples with 100% accuracy. To visualize the comparison results for different p values, we fix δ as a constant. For large p, we compute the change of CWA directly from Equation (5); otherwise, CWA is approximated by integrating Equation (6) numerically. In both cases, we assume genes are independent from each other with variances equal to one. When the genes are dependent with unknown variances, we go through the regular classification procedure to estimate the prediction accuracy as outlined below.
Next we consider simulation settings that are closer to real data structures where genes are correlated to each other. We set the first g genes are informative, e.g., μ1i = 0.5 and μ2i = 0, i = 1, …, g, and the rest of (G − g) genes have μ1i = μ2i = 0, i = g + 1, …, G. Note that no feature selection procedure is involved here yet. We select the first p genes for classification. If p ≤ g, all p genes are informative. If p > g, all of the g informative genes and (p − g) non-informative genes are selected. Usually we let g ≪ G due to the fact that most of the genes are not differentially expressed in microarray experiments, e.g., G = 10, 000 and g = 50. Similarly as in Guo et al. (2007), we use block diagonal correlation structures to model the dependence among genes. Specifically, we partition the G genes into H equal sized blocks with H = G/g. We have
where the hth block on the diagonal line is defined as
with and the pre-defined correlation coefficient ρ. We simulate the diagonal elements from the uniform distribution, U(0.5, 1.5). To model the situation with equal covariance matrices between two classes, we set Σ = Σ1 = Σ2; otherwise, we use Σ1 ≠ Σ2.
The simulation design of multiple classification is similar to that of binary classification. For simplicity, we consider the following three-class case where the first g genes are informative with μ1i = 0.5, μ2i = 0, and μ3i = −0.5, i = 1, …, g. We choose the two-fold cross-validation scheme to estimate CWA. Specifically for each simulation, we randomly take two-thirds of the samples from each class as the training set and the rest as the test set, i.e., nk/(mk +nk)=2/3. The average CWA is computed by repeating this random division and testing procedure 100 times for each simulation and then averaging for 1,000 simulations.
5.2 Simulation Results
Results that assess the CWA change assuming constant variances are shown in Figure 1. The sum of squared mean vector difference is set as δ = 10 (except for the lower right panel of Figure 1). The positive PrΔ values, i.e., overall changes of CWA, indicate that the bias-corrected discriminant rules outperform the original ones (the top panels). The PrΔ values computed from Equation (5) are very close to those from Equation (6) even when p is as small as 10. In the upper left panel, we fix the degree of unbalance, n2/n1 = 5, and vary p. We observe that as p increases, PrΔ increases sharply first and then decreases slowly after reaching its maximum value. The tail becomes heavier when n1 increases, that is, the improvement always keeps for large sample sizes. For example, with n1 = 20 and n2 = 100, we still have about 5% gain of CWA at p = 100 and the maximum 20.6% is reached at p = 874. In the upper right panel, n2 is fixed at 40. When n1 changes from 4 to 40, PrΔ decreases as n1 increases for small p. For large p, PrΔ increases first then decreases as n1 increases. For example, when p = 500, the maximum improvement of 19.4% can be obtained at n1 = 10. The bottom panels show that we may have PrΔ < 0 under certain conditions. The lower left panel shows that PrΔ is negative when p < 5 and increases with p (n1 = 4 and n2 = 20). The lower right panel shows that PrΔ becomes negative when δ > 133.9 and reaches a minimum at δ = 142.9 (n1 = 4, n2 = 20, and p = 50).
Figure 1.

PrΔ as functions of different factors (p, n1 and δ). The solid lines represent results using Equation (5). The symbols on the lines represent results using Equation (6). The dashed lines represent PrΔ = 0. We set n2/n1 = 5 except for the upper right panel and δ = 10 except for the lower right panel. Upper left: the results for different values of n1 are shown. Upper right: the results for different values of p are shown (n2 = 40). Lower left: use Equation (6) for small p (n1 = 4). Lower right: use Equation (5) for large δ (n1 = 4 and p = 50).
The bias-corrected discriminant scores do not always outperform the original ones (Section 4). Under certain conditions, we may have PrΔ < 0 (Figure 1, the bottom panels). This implies that either (i) a very small number of features is selected, or (ii) a strong signal exists in differentiating the two classes. In practice, a classifier with more than 50 features is often used for classification in microarray analysis (Dudoit et al., 2002; Lee et al., 2005; Golub et al., 1999). Simulation studies suggest PrΔ increases rapidly as p increases (Figure 1, the left panels). When the signal is strong, e.g., 2p = 100 < δ, both bias-corrected methods and the original methods work quite well. Simulation studies suggest that PrΔ ≈ 0 (Figure 1, the lower right panel).
For the more general simulation settings, we set ρ = 0.3, H = 200, and G = 10, 000. We use the equal diagonal covariance matrix to generate samples for both classes. The left column of Figure 2 shows the simulation results for p = 100, n2 = 40, and n1 varying from 4 to 40. We examine the accuracy of the proposed bias-corrected discriminant scores in terms of the squared biases (Bias2) and the mean squared errors (MSE) in logarithmic scales for the top and middle panels. We observe that both BQDA and BLDA have smaller Bias2 and MSE compared with their biased counterparts. When n1 increases, the difference between unbiased and biased discriminant rules decreases. The bottom panel shows the corresponding results of CWA. Similar as those in Figure 1, the improved prediction accuracy of bias-corrected discriminant scores is consistent, with larger improvement happens at smaller sample sizes with higher degrees of unbalance, and becomes indistinguishable for the balanced data.
Figure 2.

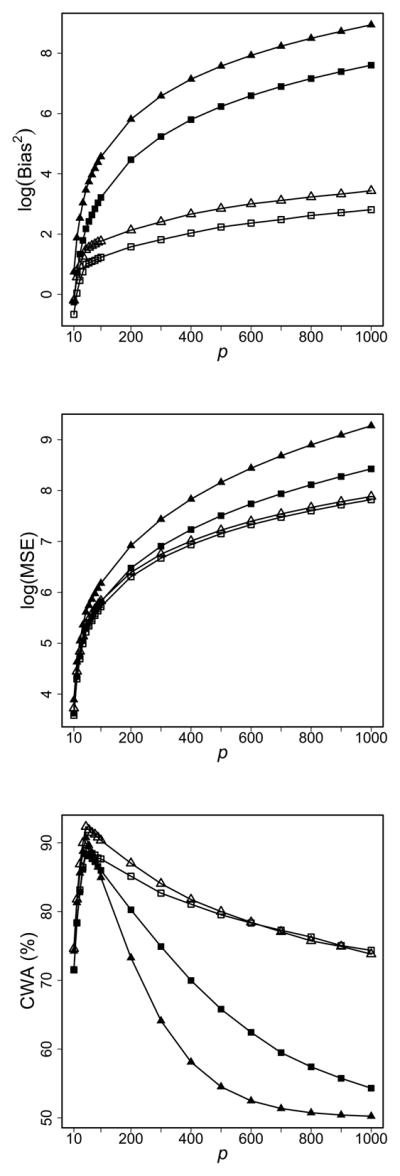
Comparison between bias-corrected discriminant rules and the original ones. Left column: n2 = 40 and p = 100. Right column: n1 = 20 and n2 = 100. Top row: Bias2 in logarithmic scale. Middle row: MSE in logarithmic scale. Bottom row: CWA.
The right column of Figure 2 displays the effect of p on the estimation and prediction accuracy (n1 = 20 and n2 = 100). It is clear that the bias-corrected discriminant scores provide more accurate and more stable estimates than the original ones consistently (the top and middle panels). The bottom panel shows that the bias-corrected scores have slight improvement when p is small, e.g., CWA increases about 1% for BQDA versus DQDA when p = 10. The improvement becomes more evident when p increases, e.g., CWA increases 6.9% for BQDA versus DQDA at p = 100. CWA of all methods peak around p = g. As more non-informative genes are included in the classifier, the class predication will tend to be random with a final CWA at 50%. However, we observe that even for such situations the bias-corrected discriminant rules still outperform the biased ones.
For multiple classification, we consider three sets of designs: 1) keep n1 = n3; 2) keep n2 = n3; 3) keep n2/n3 = 1/2. For all settings, we vary n1 and n2 the same way as in the binary classification settings. In the left columns of Web Figures 1–19, n1 varies from 4 to 40 with n2 = 40 and p = 100. In the right columns, p varies from 10 to 1000 with sample sizes fixed. We observe similar patterns as in binary classification simulation studies. For the results of using unequal covariance matrices and MLE-based discriminant rules (MLDA and MQDA), see Web Figures. As in Guo et al. (2007), we also conduct simulations with a feature selection procedure (Section 6) and incorporate different degrees of correlations, e.g., ρ = 0.5 or 0.7. The comparisons also have similar patterns as shown for binary classifications (see Web Figures). As the simulation results suggest, the improved performance of the bias-corrected discriminant rules over the original ones is evident for unbalanced classification analyses, especially when the degree of unbalance, e.g., the ratio n2/n1, is far from 1.
6. Case Studies
In this section, we apply the proposed bias-corrected methods to three real microarray data sets and compare them with several other popular classification methods, including the original diagonal discriminant analysis (DQDA, DLDA, MQDA and MLDA), support vector machines (SVM), and k-nearest neighbors (kNN). SVM is a supervised machine learning method that aims to find a separating hyperplane into the input space which maximizes the margin between classes (Boser et al., 1992). It is one commonly used classification method for high-dimensional data with small sample sizes. See for example in Ye et al. (2004), Lee et al. (2005) and Shieh et al. (2006). kNN is a simple algorithm that classifies a sample by the majority voting of its neighbors. This non-parametric classification method is widely used in discriminant analysis and works well in many studies (Dudoit et al., 2002; Lee et al., 2005). In this study we use the radial basis kernel for SVM and take the 3 nearest neighbors in Euclidean distance for kNN.
For the binary classification, we first analyze the B-cell lymphoma (BCL) data set in Shipp et al. (2002). The authors applied the weighted voting classification algorithm to differentiate diffuse large B-cell lymphoma (DLBCL) from follicular lymphoma (FL), a related germinal centers B-cell lymphoma. The gene expression data based on oligonucleotide microarray are available for 58 DLBCL and 19 FL pre-treatment biopsy samples with 6,817 genes. Although DLBCL and FL have different responses to cancer therapy, they share similar morphologic and clinical features over time. The authors showed that the two types of tumors may be distinguished by using their molecular markers. The second data set studied embryonal tumor of central nervous system (CNS), about which little is known biologically, but is believed to have heterogeneous pathogenesis (Pomeroy et al., 2002). The authors investigated the molecular heterogeneity of the most common brain tumor type, medulloblastomas, including primarily the desmoplastic subclass and the classic subclass. The desmoplastic subclass is often seen with a high frequency with Gorlin’s syndrome. They analyzed 9 desmoplastic samples and 25 classic samples with oligonucleotide microarrays of 6,817 genes. The results suggested that the Sonic Hedgehog (SHH) signaling pathway is involved in the pathogenesis of desmoplastic medulloblastoma. In the same study, the authors also investigated the problem of distinguishing multiple types of embryonal CNS tumors at gene expression level. In the original data set, there are 60 medulloblastomas, 10 malignant gliomas, 10 AT/RT (5 CNS, 5 renal-extrarenal), 6 supratentorial PNETs and 4 normal cerebellums. We exclude the class of normal samples in the study as BQDA requires a minimum of four training samples and one test sample for each class.
All data sets with raw intensity values can be downloaded from the Broad institute website (http://www.broad.mit.edu) and are pre-processed with the standard microarray data preprocessing R package from Bioconductor (http://www.bioconductor.org). We normalize all of the data sets with Robust Multichip Average (RMA) as described in Irizarry et al. (2003). The array control probe sets are removed from analysis after normalization. As in Dudoit et al. (2002), we perform a simple gene selection procedure using the ratios of the between-groups sum of squares (BSS) to the within-groups sum of squares (WSS) for the training set. Specifically, for the jth gene, the ratio is
where x̄..j is the averaged expression values across all samples and x̄k.j is that across samples belonging to the kth class. We select the top p genes with the largest BSS/WSS ratios for classification. Similar to the simulation studies in Section 5, we randomly divide the samples of each class into the training set and the test set. The training sample size for the smallest class varies from 4 to n1 + m1 − 1 (we always set the first class be the smallest one), where n1 + m1 is the total sample size for the smallest class. For other classes, we hold the same number of samples, m1, for testing, and use the rest for training. We repeat this procedure 1,000 times and report the average CWA for each method.
The results for the binary classification are summarized in Figure 3, where CWA is treated as a function of n1 with p = 100.
Figure 3.


CWA (%) as a function of n1 for the DLBCL (left) and CNS (right) data sets. The top two panels show the comparison of QDA-based methods; the bottom two panels show the comparison of LDA-based methods. All panels include SVM and kNN.
It is clear that the performance of the bias-corrected rules is consistenly better than that of the original ones. It is also interesting to see that the large improvement by the bias correction may result in the change of order of CWA. For example, in the top right panel, when n1 = 4, BQDA performs the best, even when SVM and kNN have higher CWA than DQDA and MQDA. Similar patterns of CWA keep for different p values (results not shown). Although there is little difference between the MLE and the sample variance estimator when nk is large, for data sets with small sample sizes, MQDA usually has lower prediction accuracy than DQDA (the top panels) while MLDA performs slightly better than DLDA (the bottom panels).
For the multi-class classification, we show the results with only one test sample but at a series number of selected features p in Table 1. We observe that the bias-corrected discriminant rules outperform the other methods for all p values, among which BQDA performs the best when p ≥ 100 and BLDA performs the best when p < 100 (Table 1 with the top ranked CWA highlighted).
Table 1.
CWA (%) for the multi-class CNS data set
| p | 10 | 50 | 100 | 150 | 200 |
|---|---|---|---|---|---|
| BQDA | 68.480 | 73.618 | 78.438 | 76.160 | 75.180 |
| DQDA | 63.517 | 68.630 | 69.735 | 69.253 | 69.303 |
| MQDA | 62.923 | 68.152 | 69.165 | 68.955 | 69.185 |
| BLDA | 68.643 | 77.082 | 76.040 | 73.807 | 72.655 |
| DLDA | 68.135 | 74.703 | 75.415 | 73.715 | 72.137 |
| MLDA | 68.230 | 74.807 | 75.422 | 73.723 | 72.155 |
| SVM | 63.415 | 71.405 | 74.838 | 74.038 | 72.473 |
| kNN | 61.520 | 61.830 | 62.817 | 63.770 | 63.570 |
7. Discussion
For high-dimensional data such as microarrays, we face the challenge of building a reliable classifier with a limited number of samples. For instance, a typical microarry study has expression levels for thousands of genes but less than one hundred samples. Much smaller sample sizes (< 10) are also common in practice. Diagonal discriminant analysis has been recommended for high-dimensional data classification problem with remarkably good performance (Dudoit et al., 2002; Lee et al., 2005). However, the conventional estimators of diagonal discriminant scores may not be reliable as they are all biased. In this paper, we proposed several bias-corrected discriminant rules that improve the overall prediction accuracy in both simulation studies and real case studies. The bias-corrected methods improve the prediction of the minority class, but sacrifice some performance for the majority class in terms of the per-class prediction accuracy. In reality, the minority class, e.g., representing some rare disease samples, is often of interest and may deserve more weight. Here we show that generally the bias-corrected methods offer higher CWA than the corresponding biased ones, even with the equal weights. The improvement may be affected by many factors, among which the sample size of minority class, n1, the degree of unbalance, n1/n2, and the number of features selected, p, are the most important ones.
When the design is balanced, the bias-corrected rules perform similarly as the original ones, even though the bias-corrected rules provide better estimator of discriminant scores. Specifically, BLDA performs exactly the same as DLDA, and BQDA performs similarly as DQDA. For unbalanced designs, the change of CWA is non-trivial. As shown in Sections 5 and 6, the bias-corrected methods outperform their biased counterparts under all simulation settings and real case studies when n2/n1 > 1 and p is large. The improvement is evident when the sample size of the minority class in the training class (n1) is small. When n1 is large, the improvement is still not trivial as long as the ratio of n2/n1 keeps. For the DLBCL data set with 18 training samples in the minority class, the overall performance improvement is still observable with only 100 genes selected for classification. To make the bias-corrected rules work, BQDA requires nk ≥ 4, and BLDA requires n ≥ K + 2 which is less restrictive than BQDA. Most of the public available microarray data sets satisfy such requirements.
One possible future research is to propose a regularization between BLDA and BQDA as those in Friedman (1989), Guo et al. (2007) and Pang et al. (2009). To stabilize the variances of and , or equivalently to correct their second-order biases is also of interest. Specifically, we will incorporate the bias correction together with the shrinkage technique in Tong and Wang (2007) and Pang et al. (2009). The rationale behind the shrinkage estimation is to trade off the increased bias for a possible “significant decrease” in the variance (James and Stein, 1961; Radchenko and James, 2008). As a consequence, the good performance of the shrinkage-based discriminant rules is mainly because of the largely reduced variances in the corresponding discriminant scores (Pang et al., 2009). Nevertheless, the bias terms still remain, and more likely, the biases will be larger than that in the original diagonal discriminant scores owing to the impact of shrinkage. Motivated by this, we expect that to correct the biases for the shrinkage-based discriminant rules can be of great interest.
Finally, we reiterate that the diagonal matrix assumption used is somewhat restrictive, so it might be necessary to drop such condition and obtain similar results for more general covariance matrices. Storey and Tibshirani (2001) suggested that the clumpy dependence (i.e., the block diagonal matrix) is a likely form of dependence in the setting of microarray data analysis. This is also mentioned in Langaas et al. (2005). Inspired by this, one natural extension is to propose the bias-corrected rules for the covariance matrix Σk = diag(Σk,1, …, Σk,H), where H is the total number of blocks. To calculate the expectation of L̂k1 and L̂k2 under the block diagonal covariance matrix, the following well-known results can be used (Das Gupta, 1968),
where Σ̂k is the sample covariance matrix of class k. In addition, to avoid the singularity problem, it needs to be assumed that each block size is not bigger than the sample size.
8. Supplementary Materials
Web Appendix and Figures referenced in Sections 3–5 are available under the Paper Information link at the Biometrics website http://www.biometrics.tibs.org.
Supplementary Material
Acknowledgments
The research was supported in part by NIH grant GM59507 and NSF grant DMS0714817. The authors thank Dr. Xin Qi for helpful suggestions, and Dr. Joshua Sampson for a critical reading and extensive discussions of the paper. Part of the simulations were run on the Yale High Performance Computing Cluster, supported by NIH grant RR19895-02. The authors also thank the editor, the associate editor and two referees for their constructive comments and suggestions that have led to a substantial improvement in the article.
Appendix A: the proof of theorem 1
-
Recall that is an unbiased estimator of while is biased. To verify , it suffices to show
(A1) Denote , when nk > 5, we haveFor Cov(L̂k1, L̂k2), note that (Chan, 2006). We haveThusThen Equation A1 can be simplified towhich holds for any nk > 5.
The proof of (ii) is skipped since it is essentially the same as that of (i).
Appendix B: the proof of theorem 2
For ease of notation, denote
Note that for any integers 0< n1 < n2, we have . We establish Theorem 2 via the following two steps.
-
When 0 < δ ≤ U < 2p (i.e., δ − U ≤ 0 < δ). As τ1 < τ2, we have
The second inequality is obtained as the standard normal density is a unimodal function and the interval [ ] contains the mode. The last equality is obtained by the symmetry of the standard normal density function.
When U < δ ≤ 2p (i.e., 0 < δ − U < δ ≤ 2p). Denote the length of interval [ ] as I1 = U/τ1, and the length of interval [ ] as I2 = U/τ2. We have I1 > I2 as τ1 < τ2. Thus by the monotone decreasing property of the N(0, 1) density on (0, ∞), as long as the lower bound of I1 is not larger than that of I2, i.e., if , we can claim that Theorem 2 holds.
In what follows we verify the condition , or equivalently to verify that
| (A2) |
By the condition that δ ≤ 2p, the left hand side of the Equation (A2) is
Meanwhile, by Lemmas 1 and 2 shown below, the right hand side of Equation (A2) is
Hence, Equation (A2) is established and Theorem 2 holds.
Lemma 1
For any 0 < a < b, the function f(x) = (a + x)/(b + x) is a monotone increasing function of x on (0, ∞).
Lemma 2
For any integers 0 < n1 < n2, we have
Appendix C: PrΔ in equation (6)
Under the assumptions in Section 4, note that if y is given, we can write as a linear combination of two independent non-central chi-square random variables, both with p degrees of freedom, i.e.,
where . The expected change of CWA for any fixed observation is defined as Pr D̂|y = Pr(0 < D̂ < U|y). One way to obtain Pr D̂|y is to use the inversion formula of probability characteristic function (Durrett, 1996), i.e.,
where
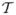 (η) is the characteristic function for D̂, and
(η) is the characteristic function for D̂, and
To compute PrΔ in Equation (6), we can sample y from both classes and integrate Pr D̂|y numerically.
References
- Abramowitz M, Stegun IA. Handbook of Mathematical Functions. New York: Dover; 1972. [Google Scholar]
- Antoniadis A, Lambert-Lacroix S, Leblanc F. Effective dimension reduction methods for tumor classification using gene expression data. Bioinformatics. 2003;19:563–570. doi: 10.1093/bioinformatics/btg062. [DOI] [PubMed] [Google Scholar]
- Asyali MH, Colak D, Demirkaya O, Inan MS. Gene expression profile classification: a review. Current Bioinformatics. 2006;1:55–73. [Google Scholar]
- Bickel PJ, Levina E. Some theory of Fisher’s linear discriminant function, ‘naive Bayes’, and some alternatives when there are many more variables than observations. Bernoulli. 2004;10:989–1010. [Google Scholar]
- Boser BE, Guyon IM, Vapnik VN. A training algorithm for optimal margin classifiers. COLT ’92: Proceedings of the Fifth Annual Workshop on Computational Learning Theory.1992. [Google Scholar]
- Braga-Neto UM, Dougherty ER. Is cross-validation valid for small-sample microarray classification? Bioinformatics. 2004;20:374–380. doi: 10.1093/bioinformatics/btg419. [DOI] [PubMed] [Google Scholar]
- Breiman L. Random forests. Machine Learning. 2001;45:5–32. [Google Scholar]
- Chen C, Liaw A, Breiman L. Technical Report. Department of Statistics, University of California; Berkeley: 2004. Using random forest to learn imbalanced data; p. 666. [Google Scholar]
- Cohen G, Hilario M, Sax H, Hugonnet S, Geissbuhler A. Learning from imbalanced data in surveillance of nosocomial infection. Artificial Intelligence in Medicine. 2006;37:7–18. doi: 10.1016/j.artmed.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Dabney AR. Classification of microarrays to nearest centroids. Bioinformatics. 2005;21:4148–4154. doi: 10.1093/bioinformatics/bti681. [DOI] [PubMed] [Google Scholar]
- Dabney AR, Storey JD. Optimality driven nearest centroid classification from genomic data. PLoS ONE. 2007;2:e1002. doi: 10.1371/journal.pone.0001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Lieu L, Rocke D. Dimension reduction for classification with gene expression microarray data. Statistical Applications in Genetics and Molecular Biology. 2006;5:6. doi: 10.2202/1544-6115.1147. [DOI] [PubMed] [Google Scholar]
- Das Gupta S. Some aspects of discrimination function coefficients. Sankhya. 1968;30:387–400. [Google Scholar]
- Dettling M. Bagboosting for tumor classification with gene expression data. Bioinformatics. 2004;20:3583–3593. doi: 10.1093/bioinformatics/bth447. [DOI] [PubMed] [Google Scholar]
- Dudoit S, Fridlyand J, Speed TP. Comparison of discrimination methods for the classification of tumors using gene expression data. Journal of the American Statistical Association. 2002;97:77–87. [Google Scholar]
- Durrett R. Probability: Theory and Examples. 2 California: Duxbury Press; 1996. [Google Scholar]
- Friedman JH. Regularized discriminant analysis. Journal of the American Statistical Association. 1989;84:165–175. [Google Scholar]
- Fu W, Dougherty ER, Mallick B, Carroll RJ. How many samples are needed to build a classifier: a general sequential approach. Bioinformatics. 2005;21:63–70. doi: 10.1093/bioinformatics/bth461. [DOI] [PubMed] [Google Scholar]
- Furey TS, Cristianini N, Duffy N, Bednarski DW, Schummer M, Haussler D. Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics. 2000;16:906–914. doi: 10.1093/bioinformatics/16.10.906. [DOI] [PubMed] [Google Scholar]
- Ghosh D. Penalized discriminant methods for the classification of tumors from gene expression data. Biometrics. 2003;59:992–1000. doi: 10.1111/j.0006-341x.2003.00114.x. [DOI] [PubMed] [Google Scholar]
- Ghurye SG, Own I. Unbiased estimation of some multivariate probability densities and related functions. Annals of Mathematical Statistics. 1969;40:1261–1271. [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Guo Y, Hastie T, Tibshirani R. Regularized linear discriminant analysis and its application in microarrays. Biostatistics. 2007;8:86–100. doi: 10.1093/biostatistics/kxj035. [DOI] [PubMed] [Google Scholar]
- Huang D, Zheng C. Independent component analysis-based penalized discriminant method for tumor classification using gene expression data. Bioinformatics. 2006;22:1855–1862. doi: 10.1093/bioinformatics/btl190. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of affymetrix genechip probe level data. Nucleic Acids Research. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaksson A, Wallman M, Göransson H, Gustafsson MG. Cross-validation and bootstrapping are unreliable in small sample classification. Pattern Recognition Letters. 2008;29:1960–1965. [Google Scholar]
- James M. Classification Algorithms. New York: Wiley; 1985. [Google Scholar]
- James W, Stein C. Estimation with quadratic loss. Proceedings of the Third Berkeley Symposium on Mathematical Statistics and Probability. 1961;1:361–379. [Google Scholar]
- Langaas M, Lindqvist BH, Ferkingstad E. Estimating the proportion of true null hypotheses, with application to DNA microarray data. Journal of the Royal Statistical Society B. 2005;67:555–572. [Google Scholar]
- Lee JW, Lee JB, Park M, Song SH. An extensive comparison of recent classification tools applied to microarray data. Computational Statistics & Data Analysis. 2005;48:869–885. [Google Scholar]
- Lee YK, Lin Y, Wahba G. Multicategory support vector machines: theory and application to the classification of microarray data and satellite radiance data. Journal of the American Statistical Association. 2004;99:67–81. [Google Scholar]
- Lehmann EL. Elements of Large Sample Theory. New York: Springer; 1998. [Google Scholar]
- McLachlan GJ. Discriminant Analysis and Statistical Pattern Recognition. New York: Wiley; 1992. [Google Scholar]
- Moran MA, Murphy BJ. A closer look at two alternative methods of statistical discrimination. Applied Statistics. 1979;28:223–232. [Google Scholar]
- Natowicz R, Incitti R, Horta EG, Charles B, Guinot P, Yan K, Coutant C, Andre F, Pusztai L, Rouzier R. Prediction of the outcome of preoperative chemotherapy in breast cancer using DNA probes that provide information on both complete and incomplete responses. BMC Bioinformatics. 2008;9:149. doi: 10.1186/1471-2105-9-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushath S, Kumar GH, Shivakumara P. Diagonal Fisher linear discriminant analysis for efficient face recognition. Neurocomputing. 2006;69:1711–1716. [Google Scholar]
- Pang H, Tong T, Zhao H. Shrinkage-based diagonal discriminant analysis and its applications in high-dimensional data. Biometrics. 2009;65:1021–1029. doi: 10.1111/j.1541-0420.2009.01200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pique-Regi R, Ortega R, Asgharzadeh S. Sequential diagonal linear discriminant analysis (SeqDLDA) for microarray classification and gene identification. Proceedings of the 2005 IEEE Computational Systems Bioinformatics Conference.2005. [Google Scholar]
- Pomeroy S, Tamayo P, Gaasenbeek M, Sturla L, Angelo M, McLaughlin M, Kim J, Goumnerova L, Black P, Lau C, Allen J, Zagzag D, Olson J, Curran T, Wetmore C, Biegel J, Poggio T, Califano A, Stolovitzky G, Louis D, Mesirov J, Lander E, Golub T. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–442. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- Qiao X, Liu Y. Adaptive weighted learning for unbalanced multicategory classification. Biometrics. 2009;65:159–168. doi: 10.1111/j.1541-0420.2008.01017.x. [DOI] [PubMed] [Google Scholar]
- Radchenko R, James GM. Variable inclusion and shrinkage algorithms. Journal of the American Statistical Association. 2008;103:1304–1315. [Google Scholar]
- Shen R, Ghosh D, Chinnaiyan A, Meng Z. Eigengene-based linear discriminant model for tumor classification using gene expression microarray data. Bioinformatics. 2006;22:2635–2642. doi: 10.1093/bioinformatics/btl442. [DOI] [PubMed] [Google Scholar]
- Shieh G, Jiang Y, Shih YS. Comparison of support vector machines to other classifiers using gene expression data. Communications in Statistics: Simulation and Computation. 2006;35:241–256. [Google Scholar]
- Shipp MA, Ross KN, Tamayo P, Weng AP, Kutok JL, Aguiar RC, Gaasenbeek M, Angelo M, Reich M, Pinkus GS, Ray TS, Koval MA, Last KW, Norton A, Lister TA, Mesirov J, Neuberg DS, Lander ES, Aster JC, Golub TR. Diffuse large b-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nature Medicine. 2002;8:68–74. doi: 10.1038/nm0102-68. [DOI] [PubMed] [Google Scholar]
- Speed TP. Statistical Analysis of Gene Expression Microarray Data. London: Chapman and Hall; 2003. [Google Scholar]
- Statnikov A, Wang L, Aliferis CF. A comprehensive comparison of random forests and support vector machines for microarray-based cancer classification. BMC Bioinformatics. 2008;9:319. doi: 10.1186/1471-2105-9-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Technical Report 2001-28. Department of Statistics, Stanford University; 2001. Estimating the positive false discovery rate under dependence, with applications to DNA microarrays. [Google Scholar]
- Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proceedings National Academic Science. 2002;99:6567–6572. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R, Hastie T, Narasimhan B, Chu G. Class prediction by nearest shrunken centroids, with applications to DNA microarrays. Statistical Science. 2003;18:104–117. [Google Scholar]
- Tong T, Wang Y. Optimal shrinkage estimation of variances with applications to microarray data analysis. Journal of the American Statistical Association. 2007;102:113–122. [Google Scholar]
- Vapnik V, Kotz S. Estimation of Dependences Based on Empirical Data. New York: Springer; 2006. [Google Scholar]
- Wang S, Zhu J. Improved centroids estimation for the nearest shrunken centroid classifier. Bioinformatics. 2007;23:972–979. doi: 10.1093/bioinformatics/btm046. [DOI] [PubMed] [Google Scholar]
- Wu B. Differential gene expression detection and sample classification using penalized linear regression models. Bioinformatics. 2006;22:472–476. doi: 10.1093/bioinformatics/bti827. [DOI] [PubMed] [Google Scholar]
- Ye J, Li T, Xiong T, Janardan R. Using uncorrelated discriminant analysis for tissue classification with gene expression data. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2004;1:181–190. doi: 10.1109/TCBB.2004.45. [DOI] [PubMed] [Google Scholar]
- Zhu J, Hastie T. Classification of gene microarrays by penalized logistic regression. Biostatistics. 2004;5:427–443. doi: 10.1093/biostatistics/5.3.427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.