

Abstract
Checkpoints that respond to DNA structure changes were originally defined by the inability of yeast mutants to prevent mitosis following DNA damage or S-phase arrest. Genetic analysis has subsequently identified subpathways of the DNA structure checkpoints, including the reversible arrest of DNA synthesis. Here, we show that the Cds1 kinase is required to slow S phase in the presence of DNA-damaging agents. Cds1 is phosphorylated and activated by S-phase arrest and activated by DNA damage during S phase, but not during G1 or G2. Activation of Cds1 during S phase is dependent on all six checkpoint Rad proteins, and Cds1 interacts both genetically and physically with Rad26. Unlike its Saccharomyces cerevisiae counterpart Rad53, Cds1 is not required for the mitotic arrest checkpoints and, thus, defines an S-phase specific subpathway of the checkpoint response. We propose a model for the DNA structure checkpoints that offers a new perspective on the function of the DNA structure checkpoint proteins. This model suggests that an intrinsic mechanism linking S phase and mitosis may function independently of the known checkpoint proteins.
Keywords: Cds1 kinase, S-phase arrest, DNA structure checkpoints, S. pombe
Checkpoint pathways that respond to changes in DNA structure ensure the integrity of the DNA. After detection of specific DNA or DNA–protein structures, a signal is transduced to effector molecules that implement checkpoint-dependent responses such as cell-cycle arrest (Elledge 1996). Many components of the DNA-structure checkpoint pathways have been identified (Carr and Hoekstra 1995). In the fission yeast Schizosaccharomyces pombe, a group of six checkpoint Rad proteins (Rad1, Rad3, Rad9, Rad17, Rad26, and Hus1) are thought to participate in the monitoring and signaling processes that detect both DNA damage and incomplete DNA replication (Al-Khodairy and Carr 1992; Enoch et al. 1992; Rowley et al. 1992; Al-Khodairy et al. 1994). Central to this group is the Rad3 protein, which shares homology with both budding yeast and human checkpoint proteins (Savitsky et al. 1995; Bentley et al. 1996; Cimprich et al. 1996). Rad3 is a member of a larger subfamily of protein kinases that share structural similarities. This subfamily consists of large proteins with a lipid kinase-related domain at the carboxyl terminus. One member, DNA–PKcs, is well characterized as a protein kinase that is activated by association with DNA-binding subunits (Jeggo et al. 1995). By analogy with DNA–PK, we have proposed that Rad3 is activated by the other checkpoint Rad proteins, which may interact with the specific DNA or DNA–protein structures generated by DNA damage and DNA synthesis (Carr 1997).
DNA structure checkpoints respond to several distinct signals. The best characterized are DNA damage caused by UV or γ-irradiation and S-phase arrest resulting from hydroxyurea (HU) exposure. In response to DNA damage, but not S-phase arrest, Chk1 kinase becomes phosphorylated in a manner dependent on Rad3 and the other checkpoint Rad proteins (Walworth and Bernards 1996). Thus, in this case, an effector is activated only in response to specific inputs. Thus, the damage and replication checkpoints can be formally considered as distinct pathways.
The DNA structure checkpoints were first identified by their effects on mitosis (Weinert and Hartwell 1988; Enoch and Nurse 1990). The checkpoint pathways, however, control other responses. For example, following treatment with HU, the checkpoint Rad proteins are essential to enable cells to survive S-phase arrest. In the absence of the checkpoint Rad proteins, HU causes rapid cell death during S phase (Enoch et al. 1992; Carr 1994), even though DNA synthesis can continue after the HU is removed. Thus, checkpoint-dependent reversible S-phase arrest is vital to survival when replication is perturbed. A related phenotype is also seen when checkpoint mutants are exposed to DNA damage during S phase: Survival of S-phase cells is dependent on a functional checkpoint pathway (Al-Khodairy et al. 1994).
In Saccharomyces cerevisiae, two proteins (Mec1 and Rad53) have been identified that are involved in both the DNA damage and the DNA replication checkpoints. Mec1 (Kato and Ogawa 1994; Weinert et al. 1994) is a homolog of Schizosaccharomyces pombe Rad3. Rad53 is a protein kinase that is phosphorylated and activated in response to DNA damage and HU exposure (Allen et al. 1994; Sanchez et al. 1996; Sun et al. 1996). Rad53 activation is dependent on Mec1, but null mutants of RAD53 are only partially defective in G2 arrest after DNA damage (Pati et al. 1997). In S. pombe, a structural homolog of Rad53, called Cds1, has been identified as a multicopy suppresser of a temperature-sensitive DNA polymerase α mutation (Murakami and Okayama 1995). Cds1 was proposed to link DNA replication to the mitotic machinery. Cds1 is not required to prevent mitosis following DNA damage.
In this report we show that Cds1 is a protein kinase that is phosphorylated and activated by DNA-replication arrest and activated by DNA damage. The activation by DNA damage is only seen in S phase, suggesting that the Cds1 response is specific to replication structures. The major effect of Cds1 deletion is not the inappropriate onset of mitosis, but an inability of cells to properly regulate S phase when challenged during replication. We propose a model for Cds1 function in which Cds1 defines a subpathway of the DNA structure checkpoints that prevents irreversible lethal DNA damage from occurring when DNA synthesis is perturbed.
Results
S-phase arrest causes mitotic delay in the absence of Cds1
Cds1 has been reported to link S phase to mitosis (Murakami and Okayama 1995). In cds1 null mutants arrested in S phase by HU, however, mitotic arrest is essentially normal during the first 6 hr (Fig. 1A and Fig. 3 of Murakami and Okayama 1995), approximately the length of time that HU delays bulk DNA synthesis (Sazer and Sherwood 1990). Despite this mitotic delay, there is rapid and irreversible loss of viability associated with entry into S phase in the presence of HU (Murakami and Okayama 1995). Rapid loss of viability during S phase is also seen with the checkpoint rad class of mutants (Enoch et al. 1992; Carr 1994).
Figure 1.
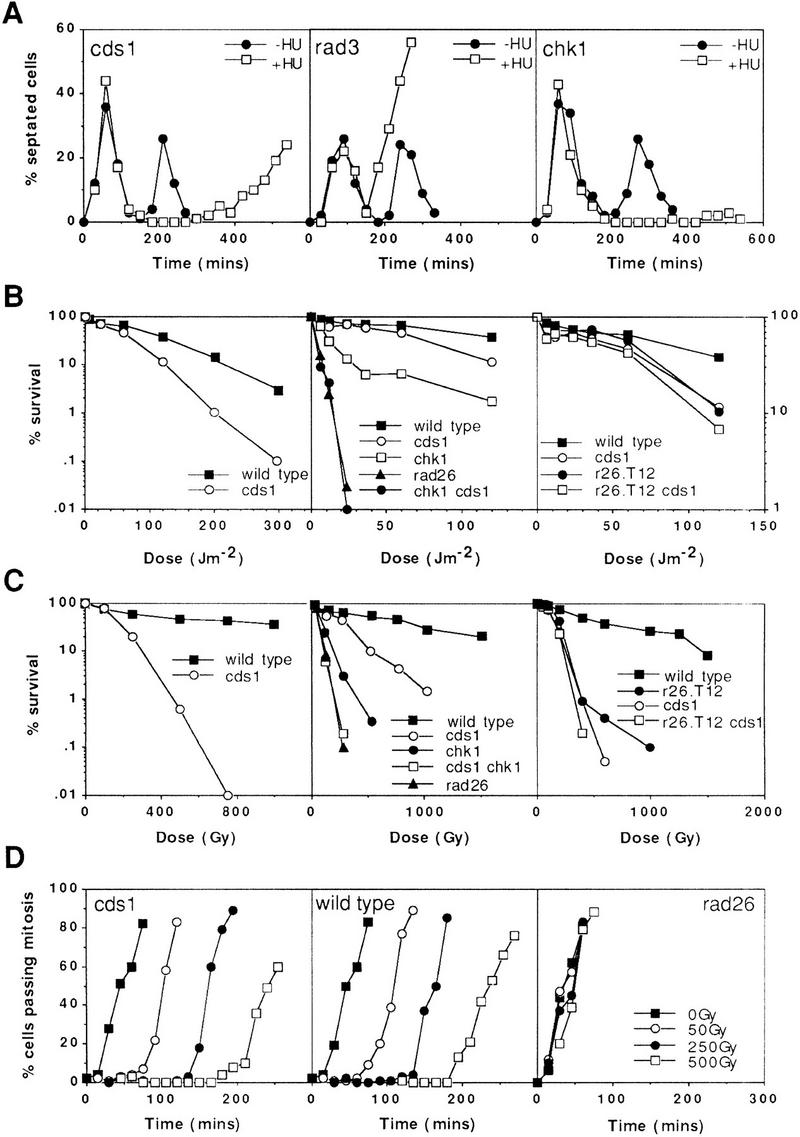
Checkpoint and survival analysis of cds1 mutants. All designations indicate null alleles, with the exception of r26.T12, which designates the rad26.T12 allele. (A) cds1 null mutant cells display mitotic arrest following inhibition of S phase with HU. Synchronous cultures of G2 cells were generated on lactose gradients (Barbet and Carr 1993) and divided into two samples, one of which was incubated in 10 mm HU. Septation index and DAPI staining were used to follow progress through the cell cycle. cds1 mutants arrested mitosis for over 6 hr following S-phase arrest. In contrast, a representative of the checkpoint rad group of mutants, rad3, entered mitosis without completing S phase. (B,C) Radiation survival of logarithmically growing cells show that cds1 null mutant cells are sensitive to damaging agents and show increased sensitivity with chk1, but not rad26.T12. (D) cds1 cells, in contrast to checkpoint-defective (rad26) cells have a normal DNA-damage checkpoint at mitosis. Synchronous cultures of G2 cells were generated on lactose gradients and dividend into four samples. One sample was not irradiated, whereas one each was irradiated with 50, 250, or 500 Gy of ionizing radiation.
Figure 3.


Cds1 is phosphorylated during S-phase arrest. (A) (Left) Immunoprecipitation (top) and kinase activity (bottom) from wild-type and Cds1 null cells by use of affinity-purified Cds1 sera or preimmune sera identifies Cds1 as a single band migrating at 52 kD. In extracts from cells treated with HU, Cds1 undergoes an apparent increase in molecular weight (second lane). Kinase activity against MBP in the IPs correlates with the presence of Cds1 and increases in cells treated with HU. To show the specificity of the kinase activity to Cds1, wild-type or kinase-dead Cds1 protein were expressed in cds1 null cells, immunoprecipitated (top, right) and assayed for kinase activity (bottom, right). (B) The Cds1 mobility shift can be reversed by phosphatase treatment. Ten and twenty units of CIAP restored the mobility of Cds1 from HU-treated cells to that of Cds1 from untreated cells. Twenty units of heat-treated CIAP does not affect Cds1 mobility. (C) Cds1 kinase can be activated by DNA damage. Asynchronous cultures of wild-type and chk1 null cells were subjected to 500 Gy of ionizing radiation. Cell extracts were made at time points following irradiation and assayed for Cds1 kinase activity. Wild-type cells showed a small but reproducible increase in Cds1 kinase activity. chk1 cells showed a greater increase in Cds1 kinase activity. (D,E) Cds1 phosphorylation (D) and activation (E) following HU treatment is dependent on the function of the checkpoint Rad proteins. Cds1 phosphorylation is not seen in immunoprecipitates from rad1, rad3, rad9, rad17, rad26, or hus1 null mutants but is evident in wild-type (WT) and chk1 mutants. IP-kinase activity is much reduced or absent in the checkpoint rad null strains when compared with wild-type and chk1 null cells.
Cds1 is required for the response to DNA damage
It has been reported that cds1 mutant cells are not sensitive to DNA damage (Murakami and Okayama 1995). These studies used UV as the DNA-damaging agent. We find that Cds1 null cells are significantly sensitive to UV (Fig. 1B) and ionizing radiation (Fig. 1C), despite the fact that they maintain a normal G2 checkpoint when irradiated (Fig. 1D). To characterize this sensitivity further, we have created double mutants with a number of well characterized checkpoint mutants (Fig. 1B,C).
The Chk1 mutant is unable to arrest mitosis after DNA damage, but is more resistant to radiation than the checkpoint rad group of mutants (Al-Khodairy et al. 1994). Interestingly, a specific allele of one of the checkpoint rad genes, rad26.T12, has been characterized that retains a normal mitotic arrest after DNA damage, but is still sensitive to DNA-damaging agents (Al-Khodairy et al. 1994). Genetic studies of chk1.d rad26.T12 double mutants show that these cells have a similar phenotype to the rad26.d null mutant. This led us to propose that rad26.T12 is defective in a subpathway of the checkpoint response that is independent of mitotic arrest (Al-Khodairy et al. 1994).
We have created cds1.d chk1.d double mutants and compared the sensitivities with the respective single mutants and with the rad26.d null mutant. cds1.d chk1.d double-mutant cells are more sensitive to DNA damage than the two single mutants, as would be expected from their distinct phenotypes. The double mutant shows an equivalent sensitivity to rad26.d (Fig. 1B,C). We have also created cds1.d rad26.T12 double mutants and compared the sensitivity of these with the respective single mutants. The double mutants are no more sensitive to DNA damage than the most sensitive single mutant (Fig. 1B,C). This is also true for the cds1.d rad26.d double mutant (data not shown).
The classical interpretation of epistasis analysis in repair studies is as follows: Two mutants that do not give additional sensitivity when combined (compared with the most sensitive single mutant) act in the same pathway. Conversely, two null mutations that give increased sensitivity when combined act in distinct pathways. The simplest interpretation of our data is that the cds1-dependent DNA-damage response and the chk1-dependent DNA-damage response both require the correct function of the checkpoint rad pathway. Therefore, we can separate the checkpoint rad pathway into two subpathways, one mediated by Chk1, which arrests mitosis, and one mediated by Cds1, which is independent of mitotic arrest. rad26.T12 mutant cells would appear to be defective in the Cds1, but not the Chk1, dependent pathway. We will refer to the Cds1-mediated response as the checkpoint rad-dependent S-phase recovery pathway.
cds1 is a multicopy suppressor of rad26.T12
Sequence analysis of the rad26.T12 allele identified a single arginine to tryptophan mutation at position 541 of the Rad26 protein (see Materials and Methods). This site is 74 amino acids from the carboxyl terminus of the protein. An allele of rad26 with a defect in sensing slowed DNA replication has been described recently (Uchiyama et al. 1997). This phenotype results from a duplication of 6 amino acids within Rad26, at position 511, 30 amino acids distant from the rad26.T12 mutation. To investigate the potential involvement of a carboxy-terminal Rad26 domain in the checkpoint rad-dependent recovery pathway, we deleted incremental carboxy-terminal regions. Removal of the carboxy-terminal 11 amino acids of rad26 (604–614) did not affect function. Removal of the 31 amino acids (584–614) resulted in a phenotype similar to the null mutant (Fig. 2B; see Materials and Methods), despite the fact that a correct size protein is produced (data not shown). Therefore, the carboxyl terminus does not encode a defined functional domain involved in the recovery pathway.
Figure 2.
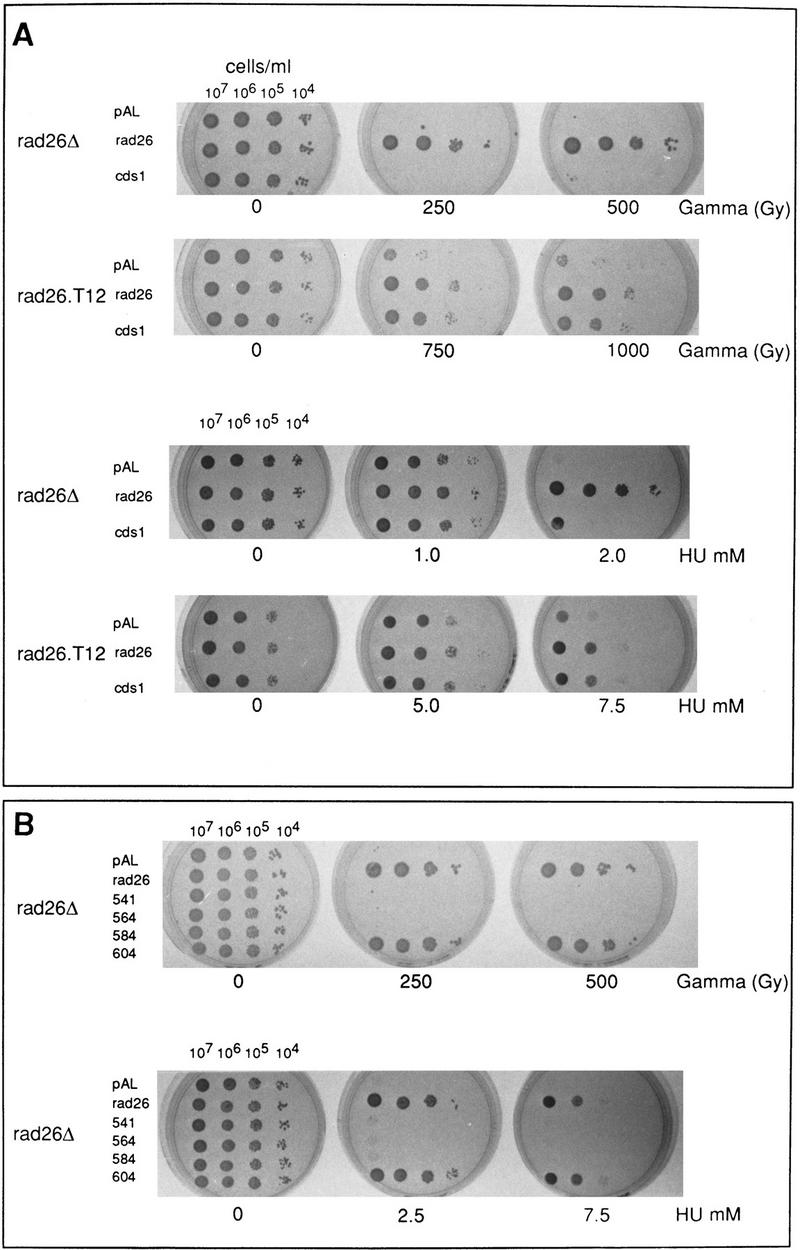

cds1 is a multicopy suppresser of rad26.T12 mutant cells. (A) rad26 null and rad26.T12 mutant cells were transformed with rad26 genomic DNA, cds1 genomic DNA, or an empty vector control (pAL). Cells were serially diluted (from 107 cells/ml to 104 cells/ml), spotted onto plates and irradiated at the doses indicated. Cells were also spotted onto plates containing a range of HU concentrations. (B) rad26 null cells were transformed with the rad26 genomic clone, the empty vector (pAL) or rad26 constructs containing carboxy-terminal deletions. Numbers correspond to the length of the constructs (amino acids). Full-length Rad26 is 614 amino acids. Cells were diluted to the concentrations shown and either spotted onto plates and irradiated at the doses indicated or spotted onto plates containing different concentrations of HU. (C) Cds1 immunoprecipitates with Rad26 in S. pombe extracts. Wild-type S. pombe was transformed with REP41 (leucine selection) and REP42 (uracil selection) plasmids containing cds1 and rad26 cDNA tagged with either HA or Myc epitopes. Immunoprecipitation was carried out on extracts from logarithmically growing cells, after 18 hr growth without thiamine. Immunocomplexes were processed for Western blotting with anti-Myc antibody. Rad26 associates with itself (lane 3) and with Cds1 (lanes 7,8). The empty vector (lanes 1,2,4,5) was used as control. We estimate overexpression at ∼50× of wild-type protein levels. Comparing the amount of Cds1 precipitated with Rad26 to total protein, we estimate ∼3%–5% recovery. (D) Immunoprecipitations were repeated by use of proteins expressed in rabbit reticulocyte lysate. In this case, Rad26 was seen to associate with itself, but not with Cds1, suggesting bridging proteins or posttranslational modification are required for the Rad26–Cds1 interaction. This would be consistent with levels of recovery.
To further investigate the relationship between Cds1 and the checkpoint rad-dependent recovery pathway, we transformed rad26.d and rad26.T12 cells with a plasmid containing a cds1 genomic clone. As controls, we used an empty vector and a wild-type rad26 gene. cds1 overexpression rescues much of the radiation sensitivity and HU sensitivity of the rad26.T12 allele (Fig. 2A), consistent with Cds1 functioning either in parallel or downstream of Rad26. This effect is unlikely to be the result of any cell-cycle delay imposed by the overexpression, as cell-cycle delay following irradiation is proficient in the rad26.T12 strain (Al-Khodairy et al. 1994). Furthermore, Cds1 does not unduly affect the cell cycle as judged by cell length at mitosis in these experiments.
The common involvement of Rad26 and Cds1 in the checkpoint rad-dependent recovery pathway, and the allele-specific suppression of rad26.T12 phenotypes, suggest that Rad26 and Cds1 may physically interact. To test this hypothesis, we have expressed HA- and Myc-tagged constructs of Rad26 and Cds1 at moderate levels in S. pombe and assayed for coimmunoprecipitation (Fig. 2C). HA–Rad26 is able to precipitate Myc–Cds1, and HA–Cds1 is able to precipitate Myc–Rad26. In addition, Rad26, but not Cds1, appears to form homomultimers, as HA–Rad26 precipitates Myc–Rad26 when the two constructs are expressed in the same cell. To determine if these interactions were direct, or require either bridging proteins or specific posttranslational modification, we translated Rad26 and Cds1 in rabbit reticulocyte lysates for coimmunoprecipitation (Fig. 2D). Rad26 did not precipitate with Cds1, but still interacted with itself. Thus, we conclude that bridging proteins or modifications are required for Cds1–Rad26, but not Rad26–Rad26 interactions.
Cds1 is a protein kinase and phosphoprotein activated by S-phase arrest and DNA damage
To verify that Cds1 is a protein kinase, we purified overexpressed GST–Cds1 fusion protein from S. pombe and established conditions under which it will phosphorylate myelin basic protein (MBP) in vitro (data not shown). Specificity for Cds1 was shown by an equivalent purification of a GST fusion protein carrying an Asp to Glu mutation in residue 312 of the kinase domain. This kinase dead protein did not have kinase activity for MBP (data not shown). A rabbit polyclonal antibody to Cds1 protein was used to detect Cds1 protein in asynchronous wild-type cells. By immunoprecipitation (IP) and Western blotting after SDS-PAGE, a single band is seen that migrates at ∼50 kD, near the predicted molecular weight of 51.9 kD for Cds1 (Fig. 3A). This band is absent in IP’s from cds1 null mutant cells. In addition, the antibody recognizes overexpressed Cds1 (data not shown), verifying its specificity.
Following incubation of cells in HU, a mobility shift for Cds1 is evident by SDS-PAGE; Cds1 migrates as a more diffuse band, with an apparent increase in molecular mass of several kilodaltons (Fig. 3A,B). The mobility shift can be reversed by phosphatase treatment (Fig. 3B), indicating that it is a result of phosphorylation. To ascertain if Cds1 phosphorylation correlates with increased kinase activity, IP kinase assays against an MBP substrate was used to measure the Cds1-dependent kinase activity from treated and untreated cells. Cds1-dependent activity increases markedly in IP kinase assays performed on extracts prepared from cells treated with HU (Fig. 3A). By overexpressing Cds1 and kinase-dead Cds1 ∼25-fold in cds1 null cells, we have shown that the activity observed is dependent on the integrity of the Cds1 kinase domain (Fig. 3A). These data indicate that the phosphorylation event signifies the activation of the Cds1 kinase.
Because cds1.d mutants are sensitive to DNA damage, we have investigated Cds1 activation following treatment with ionizing radiation. By use of IP kinase assays, we can detect activation of the Cds1 kinase in wild-type cells after irradiation (Fig. 3C). Because wild-type cells accumulate in G2 following irradiation, we have also tested chk1 mutant cells, which do not arrest in G2 after DNA damage (Al-Khodairy et al. 1994). Increased levels of activation were seen. We have been unable to detect a mobility shift of Cds1 following DNA damage (data not shown). The kinase activity of Cds1 following DNA damage does not reach the levels seen following treatment with HU. Thus, we cannot determine if the phosphorylation event observed following HU treatment is distinct from that which occurs after DNA damage, or if Cds1 is partially phosphorylated in response to DNA damage.
The increases in Cds1 phosphorylation and kinase activity are dependent on the checkpoint Rad proteins
The Cds1 homolog in S. cerevisiae, Rad53, is a phosphoprotein that is activated by HU treatment and DNA damage. Rad53 activation is dependent on Mec1, the homolog of S. pombe Rad3 (Sanchez et al. 1996; Sun et al. 1996). By use of null mutants of the six checkpoint rad genes, we have shown that the phosphorylation (Fig. 3D) and activation (Fig. 3E) of Cds1 induced by HU is largely dependent on the function of the six checkpoint Rad proteins, Rad1, Rad3, Rad9, Rad17, Rad26, and Hus1. Both rad3.d and rad26.d mutants are completely unable to phosphorylate or activate Cds1. Low, but reproducible, levels of activation are seen in rad9.d, rad17.d, and hus1.d mutants. A moderate level of activity is seen in rad1.d cells. In no case was a Cds1 mobility shift seen.
Our genetic analysis places Cds1 function in the checkpoint rad-dependent recovery pathway, a subpathway of the checkpoint response. This subpathway has also been identified as defective in the rad26.T12 mutant, and we find evidence of an association between Cds1 and Rad26. It is therefore possible that the major function of Rad26 is in the recovery pathway. To address this, we followed the kinetics of Cds1 phosphorylation and activation in response to HU in rad26.T12 cells (Fig. 4A). Compared with wild type, there is a reduction in Cds1 activity, and Cds1 does not become phosphorylated. Quantification (Fig. 4B) of Cds1 kinase activity shows less than half the activity seen in wild-type cells. These data are consistent with a role for Rad26 in mediating the Cds1 response.
Figure 4.
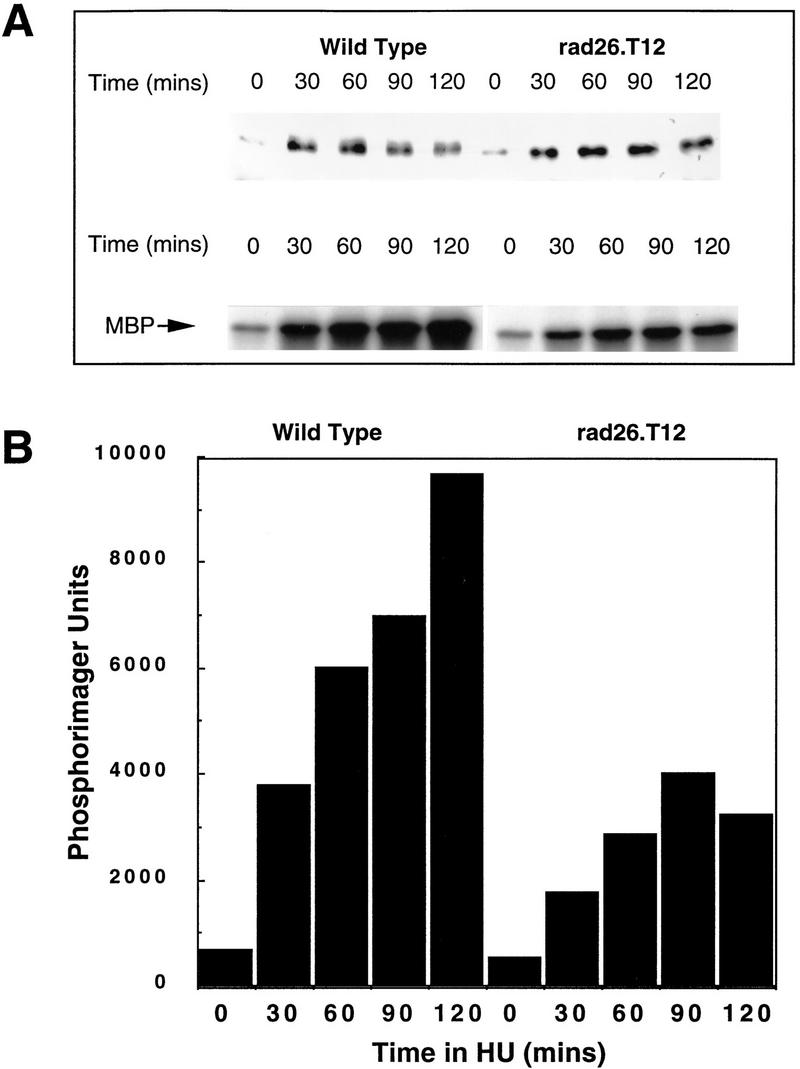
Activation of Cds1 kinase is decreased in rad26.T12 mutant cells. (A) Logarithmically growing wild-type and rad26.T12 cells were treated with 20 mm HU and Cds1 mobility (top) and kinase activity (bottom) were assayed from cell extracts prepared at 30 min intervals. (B) The activation of Cds1 kinase is reduced in rad26.T12 cells in response to HU when quantified by PhosphoImager analysis of the autoradiograph shown in A.
Cds1 is not active during normal S phase
In S. cerevisiae, it is not clear if Rad53 is activated during S phase of the normal cell cycle (Sanchez et al. 1996; Sun et al. 1996). If Cds1 is activated and phosphorylated during unperturbed S phase, the increased activity following HU treatment or DNA damage may be a consequence of cells accumulating in S phase. We have studied Cds1 phosphorylation and activity in synchronized cells. Cells were synchronized in two ways; either by a temperature shift and release strategy with a temperature-sensitive cdc10.V50 mutant [cdc10 mutants arrest the cell cycle at start, during G1 (Aves et al. 1985)] or by centrifugal elutriation. No evidence of either a mobility shift or reproducible activation of Cds1 kinase was seen as cells proceeded through unperturbed S phase (Fig. 5B,D; data not shown). On occasion, low levels of Cds1 activity, but not phosphorylation, are detected in late S phase [i.e., Fig. 5B, 90(−)]. This is not consistent, however, and may represent experimental artefacts or low levels of spontaneous replication stalling in synchronous cultures.
Figure 5.

Activation of Cds1 kinase is S-phase dependent. (A) Septation of wild-type cells synchronized in G2 by elutriation. The culture was divided into three and incubated at 30°. HU was added (20 mm) at either t = 0 (□) or t = 60 (•) min; control (▪). The cell-cycle stage of the culture was followed by septation index by Calcofluor and DAPI staining. Untreated cells show two peaks in septation. The second peak is missing in the HU-treated culture indicating the activation of the replication checkpoint. (B) Samples were taken from all three cultures and processed for Cds1 kinase assay. Cds1 kinase does not activate during G2 despite the presence of 20 mm HU (30+, 60+). Cds1 kinase activity accumulates after 90 min when the majority of the cells have entered or are entering S phase. (C) Septation index of wild-type cells synchronized in G2 by elutriation (30°C). (D) Cds1 kinase activity of irradiated or unirradiated cells removed at the times indicated and treated with 250 Gy of radiation. Cell extracts were prepared after 40 min of recovery incubation at 30°C. Cells irradiated in G2 at t = 0 do not activate Cds1 kinase after irradiation, whereas cells entering S phase after irradiation develop significant activity. Consistent with the S-phase dependency of the kinase activation cells irradiated late in S phase/G2 (t = 90,110) fail to generate significant kinase activity compared with the unirradiated control. (E) cdc10.V50 cells irradiated at the permissive temperature (26°C) show activation of Cds1 kinase whereas cells irradiated and held at the restrictive temperature (37°C) do not, thus showing that Cds1 kinase is not activated by DNA damage during G1 phase. (F) Activation of Cds1 by temperature shift in various cdc mutants defective in DNA synthesis. Logarithmic cultures of wild-type, cdc6, cdc17, cdc20, cdc21, cdc22, and polα cells grown at 27°C were split into two samples, one was incubated for a further 2 hr, 30 min at 27°C, whereas the other was incubated at 37°C for 2 hr, 30 min before extracts were prepared and tested for IP kinase activity.
The most likely explanation for Cds1 activation during HU treatment is that Cds1 is activated in response to stalled DNA replication. To rule out the alternative possibility that HU treatment itself will activate Cds1 irrespective of cell-cycle position, we synchronized wild-type cells by elutriation and treated aliquots with HU during either G2 or G1/S phase. We observed that the activation of Cds1 and its associated mobility shift (data not shown) occurs at a time commensurate with the effect of HU during S phase (Fig. 5A,B), and is not an effect of the HU itself. Thus, we conclude that HU induced Cds1 activation is specific to perturbed S phase.
Cds1 is activated by DNA damage during S phase
In S. cerevisiae, Rad53 is activated by DNA damage during G1, S phase, and post S phase. Because there are significant phenotypic differences between rad53 and cds1 mutants in the two yeasts, we investigated the cell-cycle specificity of Cds1 activation by DNA damage. In synchronous wild-type cells generated by elutriation, Cds1 activation was only seen when cells were irradiated during the G1/S period of the cell cycle, and did not occur in cells irradiated during G2 phase (Fig. 5C,D at 0 min). Thus, Cds1 activation occurs only in G1 and/or S phase. To distinguish between G1 and S phase, we arrested cells at start with the cdc10.V50 mutant before treating them with radiation (Fig. 5E). G1 arrested cells did not activate Cds1 after irradiation when held in G1. These two experiments allow us to conclude that Cds1 is activated by DNA damage only during S phase.
Cds1 activation in a range of temperature-sensitive cdc mutants
To define more precisely at which point during S phase Cds1 is able to be activated, we have studied the activation of Cds1 in a range of temperature-sensitive cdc mutants defective in various replication proteins (Fig. 5F). We find that Cds1 is strongly activated only in temperature-sensitive polymerase α (D’Urso et al. 1995; Francesconi et al. 1997) and temperature-sensitive polymerase ε (cdc20) mutants following a temperature shift. Cds1 is not activated in temperature-sensitive polymerase δ (cdc6) (Iino and Yamamoto 1997) or in cdc21 (Mcm protein) (Mairorano et al. 1996) and is only moderately activated in late S phase cdc17 (DNA ligase) mutant cells (Barker et al. 1987). Cds1 is activated in cdc22 (ribonucleotide reductase) mutants (Fernandez-Sarabia et al. 1993), consistent with the data by use of HU.
Cds1 null mutants are defective in an S phase DNA-damage checkpoint
S. cerevisiae mec1 and rad53 mutants are defective in slowing down S phase when treated with the DNA-damaging agent methylmethane sulfonate (MMS). We have tested the ability of wild-type, rad3, chk1, cds1, rad26, and rad26.T12 mutant cells to slow S phase in response to MMS. By use of two different methods to synchronize cells in G1, we have shown that wild-type cells have this checkpoint response, and that it is lost in rad3 mutants (Fig. 6A,B). By use of one of the synchronization methods, we have further shown that cds1 mutant cells, but not chk1 mutant cells, have also lost this checkpoint. As expected, rad26 null mutant cells behave in a similar manner to rad3. Consistent with our genetic data, rad26.T12 mutant cells are also defective in slowing S phase when DNA is damaged (Fig. 6B). These data are consistent with Cds1 and Chk1 defining specific subpathways of the checkpoint Rad response. They also show that rad26.T12 mutants are specifically defective in the Cds1-dependent, but not the Chk1-dependent response.
Figure 6.

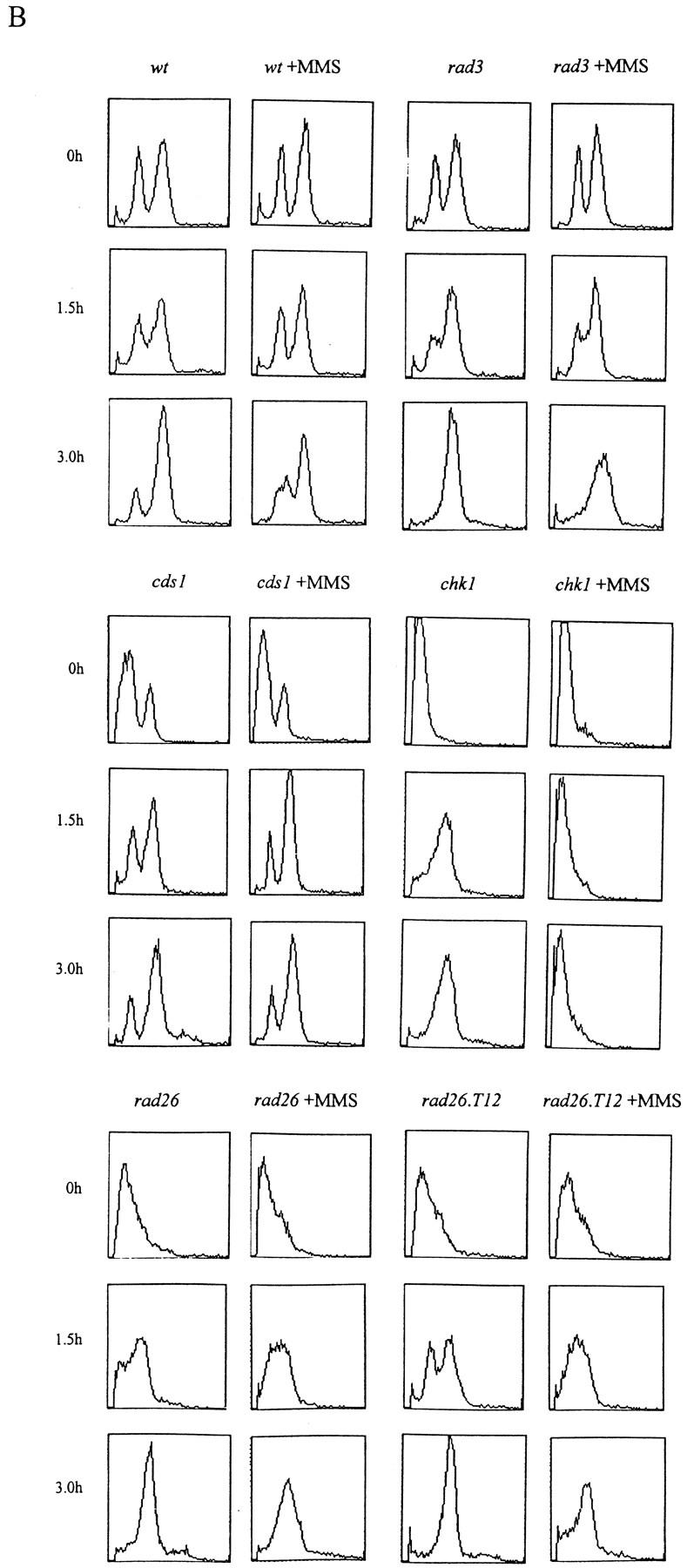
The S-phase progression checkpoint requires rad3, rad26, and cds1. (A) rad+ cells (h− cyr1 sxa2) and rad3 mutant cells (h−cyr1 sxa2 rad3) were synchronized in G1 by centrifugal elutriation (collecting early G2 cells) followed by exposure to P factor. Cells were released from P factor-induced G1 arrest in either the presence or absence of 0.033% MMS and S-phase progression followed by FACS. Attempts to synchronize other checkpoint mutants this way were complicated by the morphological changes associated with the cyr1 sxa2 background. (B) Wild-type, rad3, cds1, chk1, rad26, and rad26-T12 cells were synchronized in G1 by nitrogen starvation, released into the cell cycle in the presence or absence of 0.033% MMS and followed by FACS. As before, rad3 cells are unable to slow S phase in response to DNA damage. Similarly, cds1 and rad26 are required for this checkpoint, whereas chk1 is not. Cells carrying the rad26.T12 mutation also have a defect in this checkpoint.
Loss of Cds1 results in the abnormal activation of Chk1 by HU
A clue to the biological significance of Cds1 activation can be seen by examining the phenotype of the cds1.d chk1.d double mutant, and the behavior of the DNA damage-specific Chk1 kinase in cds1 null cells. Unexpectedly, after HU treatment, cds1.d chk1.d double mutants enter mitotic catastrophe and form cuts in a manner identical to the checkpoint rad null mutants such as rad3.d and rad26.d (Fig. 7A). From this analysis, it is apparent that the mitotic delay seen in cds1 null mutants is dependent on Chk1, which is primarily involved in the DNA-damage checkpoint at mitosis (Walworth et al. 1993; Al-Khodairy et al. 1994). There are two possible explanations for this. Either Cds1 and Chk1 play redundant overlapping roles to arrest mitosis in the presence of the S-phase inhibitor HU, or the Chk1 kinase is activated when cds1 null mutant cells are treated with HU.
Figure 7.

Mitotic arrest in cds1 mutants is dependent on Chk1 activation. (A) Synchronous cultures of appropriate strains were tested for the mitotic checkpoint in response to HU as in Fig. 1. (•) −Hu; (□) + HU. (B) The phosphorylation-dependent mobility shift of Chk1 kinase was assayed in cds1+ and cds1 null mutant cells containing an integrated triple HA-tagged chk1 gene (Walworth and Bernards 1996). Samples from asynchronous cultures treated with 12 mm HU were taken at the times indicated. Chk1 was detected, as described previously (Walworth and Bernards 1996), with anti-HA monoclonal. In cds1+ cells, Chk1 remains unphosphorylated, whereas in cds1 null mutant cells Chk1 is rapidly phosphorylated.
Chk1 kinase has been shown to be phosphorylated in response to DNA damage, and this phosphorylation event correlates to Chk1 function. Furthermore, Chk1 has been shown not to be phosphorylated in response to HU treatment, which is consistent with its lack of a HU-sensitive phenotype (Walworth and Bernards 1996). These observations allow us to distinguish between the two possibilities discussed above. We have created a cds1 null mutant strain, in which the chk1 gene is tagged in the genome by three HA epitopes (Walworth and Bernards 1996) and compared the phosphorylation of Chk1 in the presence and absence of Cds1 function following HU treatment (Fig. 7B). Chk1 becomes rapidly phosphorylated in response to HU only when Cds1 is absent, and not when it is present. Thus, the function of Cds1 during HU treatment prevents a situation in which Chk1 becomes phosphorylated. Because Chk1 normally responds specifically to DNA damage, we speculate that the activity of Cds1 prevents the occurrence of DNA-damage structures during S-phase arrest.
Discussion
We have shown that HU and DNA damage both activate the Cds1 kinase during S phase, but not during G1 or G2. At least in the case of HU arrest, this is reflected by a phosphorylation event that results in a mobility shift of Cds1 on SDS-PAGE. We have also shown that Cds1 is activated at the restrictive temperature in temperature-sensitive mutants of DNA polymerase α and ε, but not DNA polymerase δ. Cds1 activation is dependent on the correct functioning of the checkpoint rad pathway, and Cds1 is able to interact with Rad26. Whereas cds1 mutant cells have a normal mitotic checkpoint after DNA damage, they are defective in regulating S phase in the presence of DNA-damaging agents. Together, these data show that the cds1 defines a subpathway of the checkpoint rad response that is specific to S phase. The evolutionary conservation of the DNA structure-dependent checkpoint pathways may have significant consequences for our understanding of carcinogenesis. During evolution there have been significant alterations in checkpoint protein function between different species (Carr and Hoekstra 1995). This is most clearly documented for the large lipid kinase motif proteins, such as Rad3 (S. pombe), Mec1/Tel1 (S. cerevisiae), and Atm/Atr (mammalian species) (Zakian 1995). Our data suggest that the same is true for the Cds1–Rad53 kinases.
Activation of Cds1 is specific to S phase
In S. cerevisiae, Rad53 is involved in all the known DNA structure checkpoint responses including G1 and G2 arrest after DNA damage, and G2 arrest during HU treatment (Allen et al. 1994). The Rad53 protein can be phosphorylated and activated in response to DNA damage at different points in the cell cycle, and is similarly phosphorylated and activated during S phase and G2 arrest (Sanchez et al. 1996; Sun et al. 1996). In contrast, Cds1 is not involved in the DNA damage-dependent arrest of mitosis and is only activated by DNA damage during S phase. Furthermore, whereas S-phase arrest by HU or temperature-sensitive polymerase α and ε mutants causes activation of Cds1, the cds1 null mutant does not have a defect in mitotic arrest when S phase is blocked by HU. Interestingly, a temperature-sensitive DNA polymerase δ mutant did not result in Cds1 activation at the restrictive temperature, and activation was low in mutants arrested in late S phase.
Cds1 defines a subpathway of the DNA-structure checkpoint response
The phosphorylation and activation of Rad53 in response to S-phase arrest is dependent on Mec1, and in response to DNA damage is dependent on Mec1 and Mec3 (Sanchez et al. 1996; Sun et al. 1996). Furthermore, Tel1 overexpression can restore HU and damage dependent Rad53 phosphorylation (Sanchez et al. 1996). In S. pombe, we find that Cds1 activation and phosphorylation in response to HU arrest is dependent on all six of the checkpoint Rad proteins. In S. pombe, Rad1 and Rad17 are required for all the checkpoint-dependent responses to DNA damage and S-phase arrest (Al-Khodairy and Carr 1992), whereas in S. cerevisiae the homologous proteins are only required for the DNA-damage responses and not the HU response (Weinert et al. 1994). Interestingly, a slight activation of Cds1 is evident in rad1, rad9, rad17, and hus1 null mutants.
Genetic analysis shows that Cds1 acts in a separate branch of the checkpoint response from Chk1. Combining the phenotypic observations of chk1 and cds1 mutants with the biochemistry of the kinases suggests that the checkpoint Rad proteins are involved in sensing and signaling the presence of specific DNA or DNA protein structures, and activate Chk1 or Cds1 as appropriate. Chk1 appears to be activated by DNA damage-specific structures, whereas Cds1 is activated by DNA replication-specific structure. This could reflect cell cycle and DNA metabolism-specific localization of the checkpoint Rad proteins, and suggests that the putative complex(es) formed between these proteins are capable of monitoring a range of distinct structures. By comparison with DNA–PK, in which the catalytic subunit (also a large lipid kinase motif protein) is activated by the Ku heterodimer when it associates with DNA ends, it is interesting to speculate that the Rad3 kinase is activated by association with distinct subunits (perhaps Rad1, Rad9, Rad17, Rad26, and Hus1), which may interact with different DNA or DNA–protein structures. In this context, it would also be possible to envisage an explanation for the different requirements in S. pombe and S. cerevisiae for these proteins. Perhaps in S. pombe, Rad3 function is compromised by loss of any associated subunits, whereas in S. cerevisiae, Mec1 function could be more tolerant.
The genetics and cell-cycle specific activation of Cds1 offers an opportunity to dissect out the specific function of Cds1 within the network of closely related checkpoint responses. Cds1 acts primarily to ensure the reversible arrest of DNA synthesis. This subpathway of the checkpoint rad response was first observed during studies on the hus1 mutant (Enoch et al. 1992). hus1 null mutants, in common with other checkpoint rad mutants, lose viability very rapidly on exposure to HU during S phase. Furthermore, by use of the rad26.T12 mutant, which retains a normal G2 arrest after DNA damage (Al-Khodairy et al. 1994), it has been possible to associate an element of the radiation sensitivity seen in checkpoint rad mutants with radiation exposure during the G1/S phase of the cell cycle. Because epistasis and biochemical analysis shows that the radiation sensitivity associated with loss of Cds1 function is caused by a defect in the same pathway as that defined by the rad26.T12 mutation, and because cds1 and rad26.T12 mutants share a defect in slowing S phase when exposed to DNA-damaging agents, it is now possible to formally propose two distinct checkpoint rad dependent DNA metabolism responses: Chk1 mediated G2 arrest and Cds1 mediated S-phase recovery.
Cds1 function may prevent DNA damage accumulating when S phase is arrested
Contrary to the published report, we show that cds1 null mutant cells do not have a significant defect in mitotic arrest following HU exposure. cds1 cells, however, die during S-phase arrest, in a manner independent of mitosis (Murakami and Okayama 1995). A further effect of HU treatment in cds1 null mutant cells is the activation of the DNA-damage checkpoint kinase Chk1. Because the double mutant cds1.d chk1.d does not arrest mitosis after exposure to HU, we can conclude that this unscheduled activation of Chk1 in response to HU is the reason cds1 single-mutant cells arrest mitosis in response to HU. The fact that Chk1 is not normally phosphorylated when cells are treated with HU, indicates that this is not because Cds1 and Chk1 kinase play overlapping redundant roles during S-phase arrest. Instead, we propose that the inappropriate activation of the Chk1 kinase in cds1.d cells in response to HU is a consequence of the accumulation of unrepairable DNA-damage structures during S-phase arrest, which, in turn, activate the DNA-damage checkpoint.
Cds1 may interact with replication proteins to inhibit the formation of, or stabilize, replication structures during S-phase perturbations
Cds1 acts downstream of the checkpoint Rad group of proteins and enables cells to arrest DNA synthesis reversibly in response to HU or DNA damage, thereby preventing the accumulation of unrepairable DNA lesions. It is not yet clear what the substrates of Cds1 kinase are. Possible substrates would be the components of the replication apparatus such as polymerase α-primase (Marini et al. 1997) or even upstream activators of replicon initiation such as the Hsk1 kinase (the S. pombe homolog of S. cerevisiae Cdc7) (Masai et al. 1995). In response to DNA-replication inhibition or DNA damage, the Cds1-dependent checkpoint response may prevent the initiation of new replicons, and/or stabilize existing replicons to prevent replication fork collapse and irreparable damage. It has been pointed out recently that if two replication forks that are converging collapse, this will create a region of unreplicated, and potentially unrepairable, DNA (Elledge 1996). The prevention of such a scenario during perturbations to S phase is certainly an important consideration.
Two models for the DNA-structure checkpoint pathway(s) during S-phase perturbations
In these studies, we have defined a role for Cds1 in an S phase-specific pathway that allows cells to arrest S phase reversibly, and, thus, tolerate the consequence of HU or DNA-damage treatments during S phase. Whereas cds1 null mutants do not show a significant mitotic arrest defect in the first 6 hr when DNA replication is inhibited, the delay seen in these cds1 null cells is unexpectedly dependent on the Chk1 kinase. On the basis of the current data relating to S. pombe DNA structure checkpoints, we propose two models for the action of the checkpoints in response to perturbations in S phase. (Fig. 8A,B).
Figure 8.
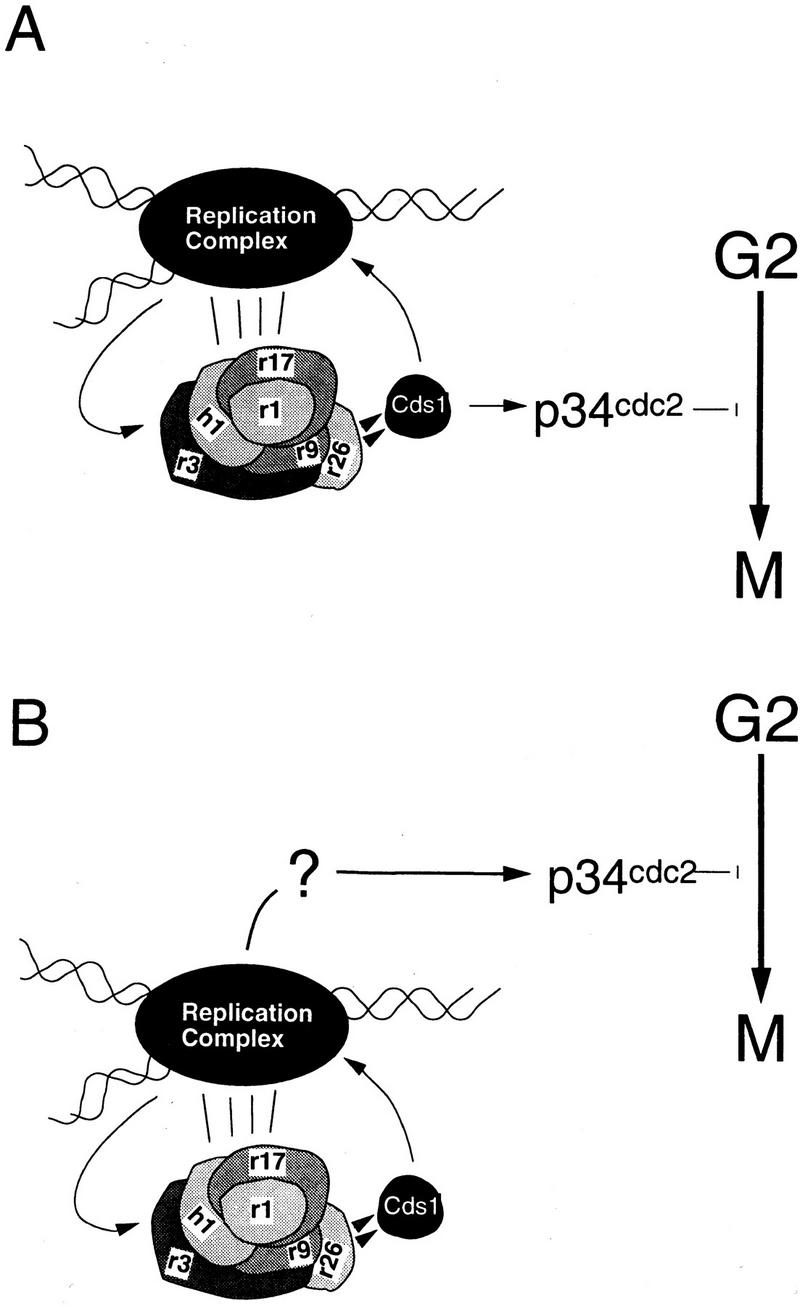
Model for the function of Cds1 in the S-phase arrest response. (A) Checkpoint Rad-dependent activation of Cds1 by perturbations to S-phase acts to ensure both S-phase arrest, by phosphorylating components of the replication complex, and mitotic arrest by phosphorylating a substrate that leads to inactivation of the mitotic apparatus. (B) Cds1 only acts to phosphorylate and stabilize the replication machinery. This prevents inappropriate DNA damage and maintains the structure(s) that are activating a Cds1 independent mitotic arrest signal. This signal would also be independent of the checkpoint Rad proteins, until such time as replication was perturbed, when maintenance of the appropriate replication structures becomes a prerequisite.
Model A
Cds1 has two separate targets. The first is the replication machinery. Here, Cds1 acts to stabilize S-phase events in response to DNA-replication arrest or DNA damage. The second is the mitotic apparatus. Here, Cds1 targets proteins that regulate p34cdc2 activity, as has been proposed for Chk1 (Furnari et al. 1997; O’Connell et al. 1997; Peng et al. 1997; Sanchez et al. 1997). If Cds1 is directly transducing the mitotic arrest signal, in its absence the unscheduled activation of Chk1 is obscuring the phenotypic effect.
Model B
Cds1 has a single target, the replication machinery, in which Cds1 would act to stabilize S-phase events. To account for the loss of mitotic checkpoint function after HU exposure in cds1.d chk1.d double mutants, we suggest that a, as yet uncharacterized, mechanism links replication directly to p34cdc2 through a checkpoint Rad-independent pathway. This intrinsic mechanism would, however, require stable replication structures to maintain the link between S phase and mitosis. During normal S phase, this may be active replication itself. During S-phase arrest, the maintenance of S-phase structures would require the checkpoint Rad and Cds1-dependent pathway. An easily conceptualized mechanism for the link between S phase and mitosis in such a model is the sequestration of p34cdc2 complexes to replication structures, which would keep it unavailable for mitotic initiation. Interestingly, Orp2, Cdc18, and p34cdc2 may be complexed together during S-phase initiation (Leatherwood et al. 1996; Brown et al. 1997), and Orp2 overexpression is able to arrest the cell cycle independently of the checkpoint rad pathway (Leatherwood et al. 1996).
At present, either variation of our model is consistent with the reported data. One important prediction of the first model is that there is no intrinsic mechanism that ensures the alternation of S phase and M phase in unperturbed cells, because Cds1 (and probably Rad53) activity is not detected during normal S phase. In yeast, there are no published data that allow us to conclude whether such an intrinsic checkpoint is operating. In mammalian cells, however, fusing S phase and G2 cells shows that normal S phase inhibits mitosis, consistent with an intrinsic checkpoint (Rao and Johnson 1970).
At present, we favor our second model. One reason for this is that it allows for the activity of an intrinsic checkpoint mechanism. Furthermore, it explains an anomaly in the phenotype of the checkpoint rad mutants. When S phase is inhibited in synchronized S-phase cells, mitosis is apparently advanced by ∼30 min relative to untreated cells in rad1, rad3, rad9, rad17, rad26, and hus1 mutants (i.e., Figs. 1A and 7A). This suggests an intrinsic (checkpoint Rad independent) signal is being lost. This effect is not seen in cdc2.3w cells (Enoch and Nurse 1990; Sheldrick and Carr 1993), which are also defective in the mitotic checkpoint, but in a downstream event independent of the effects within S phase. An important consequence of the second model is that it determines that the activity of the checkpoint Rad proteins does not have a direct role in the intrinsic checkpoint mechanism linking S phase and mitosis. The checkpoint Rad proteins would function as ancillary replication proteins required to coordinate S phase only when S phase is perturbed.
At first sight, the checkpoint Rad proteins are directly involved in the DNA-damage checkpoint, which acts through Chk1. Recent evidence in S. cerevisiae, however, clearly shows that the downstream effectors of the checkpoint pathway can operate normally in the complete absence of Mec1 and many other checkpoint proteins when a fragment of an unrelated protein is expressed (Pati et al. 1997). This apparent anomaly of a checkpoint without the key checkpoint proteins is consistent with our model, and could be explained if the role of the checkpoint Rad proteins in the DNA-damage checkpoint were analogous to what we propose in the DNA-replication checkpoint, that is, they act to stabilize specific structures in order that these are available to an as yet uncharacterized mechanism. In S. pombe, this could be (as an example) the direct association of Chk1 with damage/repair structures.
Materials and methods
Genetics, molecular biology, and cell biology techniques
Double mutants were created by standard genetic techniques (Gutz et al. 1974). Cell-survival analysis and checkpoint measurements were performed as described previously (Al-Khodairy et al. 1994). Cells were synchronized by elutriation (Aves et al. 1985), or by a 3 hr temperature shift from 27°C to 35°C of either a cdc25.22 mutant strain or a cdc10.V50 mutant strain.
Analysis of rad26
The mutation responsible for the rad26.T12 phenotype was identified by sequencing genomic PCR products generated with PCR primers 5′-TTCAAACAACTAAGCTTACGCATAAACGAG-3′, TCTAATGAATGGATCCAACCACCAAATACG, GAGCCTGATGAAAGCTTTACTTTCTCTACA, TAATTTGCGAGGATCCGGGTGCGGGACGGG with rad26.T12 and wild-type DNA. Nested deletion analysis of rad26 was performed by site-directed mutation (primers: GGAAGTTGCGTCTCTTAGATAATGAAACAGATGA, TGAACAAAAGCGTTTTAGATAATGAAACAGATGA, TGAAAATGCGTTACAATAGATAAT-GAAACAGATGA, TTTAGTATGTCTTGTTTAGATAATGAAACAGATGA) to generate genomic clones of rad26 (based on plasmid pRad26) lacking the appropriate regions of the rad26 ORF. The following mutants were tested for function by complementation of the radiation and HU-sensitive phenotypes (C indicates carboxyl terminus): rad26.d604-C, rad26.d584-C, rad26.d564-C, rad26.d541-C. Suppression analysis was performed with a genomic clone of cds1 kindly provided by T. Wang (Stanford University, CA).
Antibodies for Cds1
GST–Cds1 protein was expressed in S. pombe, purified on glutathione agarose, and used to immunize rabbits. Sera was tested for reactivity against purified protein, S. pombe extracts containing expressed epitope-tagged protein, wild-type S. pombe cell extracts, and cds1 null mutant cells. When compared with preimmune serum, the antibody clearly recognized a Cds1-specific band, which could also be seen in wild-type cells. Several nonspecific bands of approximately the same molecular weight were also seen on Western blots of total wild-type cell extract, even when the antibody was affinity purified against the original antigen. Immunoprecipitation prior to Western analysis abolished these bands, leaving a single Cds1-specific band.
Preparation of protein extract
Logarithmically growing cells were washed in PBS, then washed in lysis buffer [50 mm Tris (pH 7.5), 80 mm β-glycerophosphate, 250 mm NaCl, 15 mm nitrophenylphosphate, 50 mm NaF, 5 mm EDTA, 1 mm DTT, and 0.1% NP-40 supplemented with protease inhibitor cocktail (AEBSF, leupeptin, aprotinin, pepstatin, bestatin, and E-64 all at 10 μg/ml final concentration)]. Cells were disrupted with glass beads (BDH), in lysis buffer in a mikrodismembranator (Braun) for 3 × 1 min at 2000 rpm. Protein extract was cleared at 14,000 rpm in a microfuge at 4°C for 5 min and either used fresh or from frozen (LN2 and storage at −80°C). Preparation of cell extracts from cells expressing tagged proteins from the nmt1 promotor: Cells were grown in the presence of thiamine (promotor repressed), washed several times in distilled water and then inoculated into minimal medium lacking thiamine with the relevant selection. Cells were grown in the absence of thiamine (promoter active) for 18 hr.
Immunoprecipitation of Cds1 and immunoblotting
One milligram of total protein was diluted to 500 μl with lysis buffer and incubated with Cds1 affinity-purified antibody at a dilution of 1:250 at 4°C for 2 hr. Immunocomplexes were collected with protein A agarose (Sigma) for 1 hr at 4°C with mixing. Pellets were washed five times with lysis buffer, boiled in SDS sample buffer, and loaded onto 10% [200:1 acrylamide/bis acrylamide (National Diagnostics)], SDS–polyacrylamide gels. Proteins were transferred to nitrocellulose (MSI). The membranes were blocked with Blotto (PBS, 1% fat free milk powder, 0.05% Tween 20) and incubated in Blotto plus Cds1 antisera (1:1000 dilution), washed in Blotto, and incubated with peroxidase-conjugated secondary antibody (1:5000 dilution. DAKO). The procedure for coimmunoprecipitations was essentially the same as above, except 1 μl of anti-HA (BaBco) was used per sample. Anti c-Myc at a dilution of 1:1000 (Pharmingen) was used to probe the subsequent Western blots. Chemiluminescent detection of HRP-conjugated secondary antibodies was carried out by mixing 10 ml of 100 mm Tris at pH 8.5, 5.4 mm H2O2 with 25 μl of 90 mm p-coumaric acid (Sigma) and 50 μl of 250 mm luminol (Fluka).
Cds1 kinase assay
Alternatively, immunoprecipitates were processed for Cds1 kinase activity. Protein A pellets were washed three times in lysis buffer and three times in kinase buffer (10 mm HEPES at pH 7.5, 75 mm KCl, 5 mm MgCl2, 0.5 mm EDTA, 1 mm DTT). Twenty microliters (50% slurry) of bead pellet was incubated with 10 μl of ×2 kinase buffer, 5 μCi of α-32P]ATP (ICN), 1 μl 2 mm ATP, 5 μl of myelin basic protein (1 mg/ml of stock) at 30°C for 15 min. The reaction was stopped by the addition of 20 μl of 2× SDS sample buffer. Samples were run on 15% polyacrylamide gels, fixed in 40% methanol, 10% acetic acid, and dried down, before exposure to film (Hyperfilm, Amersham). Kinase assays were also carried out on GST and 2myc6His–Cds1 fusion proteins. Protein expressed from the nmt1 promoter was purified on glutathione–agarose (Sigma) or nickel NTA agarose (Qiagen). DTT and EDTA were omitted from lysis, and wash buffers used for nickel NTA purification.
The kinase activity observed is Cds1 specific, because a Cds1 kinase dead mutant, when expressed in cds1 null cells, does not have appreciable kinase activity when compared with similar experiments with wild-type Cds1. During the production of the cDNA by PCR (primers 5′-GAGAGACATATGGAAGAACCA-3′ and 5′-AGAGAGGTCGACACTCGA-3′) and the site directed mutagenesis to produce the kinase dead allele cds1.KD (Primer 5′-GCCAAGCCAAATTCAGATATTTTAAGATGG-3′), we sequenced the constructs to avoid unwanted mutations. During this analysis, we identified two separate changes compared with the published sequence. These were verified as correct by sequencing the same region of the genomic clone. The modified sequence, including introns, of the cds1 gene has been submitted to the database with the accession no. AJ222869.
Phosphatase treatment
Immunoprecipitates were washed three times in lysis buffer and three times in calf alkaline phosphatase (CIAP) buffer (50 mm Tris at pH 8.0, 5 mm MgCl2), treated with the indicated units of CIAP (GIBCO BRL) for 30 min at 37°C and then processed for Western blotting.
S-phase checkpoint analysis
Method 1
Early G2 cells from log-phase cultures of cyr1 sxa2 strains were collected by centrifugal elutriation. After 30 min incubation in minimal media at 30°C, P-factor was added to a final concentration of 1μg/ml. Samples were scored for septation every 30 min. After the peak of septation, the culture was divided in two. One-half was treated with 0.033% MMS (final concentration), and cultures were incubated for 30 min at 30°C. Cells were harvested by centrifugation, washed three times with water to release them from the P-factor-induced G1 arrest, resuspended in minimal media or minimal media + 0.033% MMS, and incubated for 4 hr at 30°C. Samples were taken at the time of release and then every 30 min and fixed in 70% EtOH (Sazer and Sherwood 1990). Fixed cells were treated with RNase, stained with propidium iodide, and analyzed for DNA content with a Coulter Epics Elite flow cytometer.
Method 2
For starvation-induced synchronization in G1, log-phase cells grown in minimal media were harvested, washed three times with water, resuspended in minimal media lacking a nitrogen source and incubated 18 hr at 27°C. Afterward, cells were harvested, washed with water, and allowed to recover by incubation in complete media at 30°C for 1.5 hr. The cells were then harvested, washed twice in water, and divided into two cultures in minimal media, one containing 0.033% MMS. The cultures were incubated at 30°C and samples were taken every half hour and fixed in 70% EtOH. Fixed cells were processed as described above including one step of sonication and analyzed for DNA content with a Becton-Dickinson FACScan flow cytometer.
Acknowledgments
F.O. was supported by project grant 043822/Z/95/Z (Wellcome Trust), J.M.M. by project grant G9409713MA (MRC), N.W. by funds from the University of Medicine and Dentistry of New Jersey (UMDNJ) Foundation and an award to the Cancer Institute of New Jersey from the American Cancer Society. A.M.C. and H.D.L. acknowledge European Community contract F14PCT950010.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL a.m.carr@sussex.ac.uk; FAX 01273 678 121.
References
- Al-Khodairy F, Carr AM. DNA repair mutants defining G2 checkpoint pathways in Schizosaccharomyces pombe. EMBO J. 1992;11:1343–1350. doi: 10.1002/j.1460-2075.1992.tb05179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khodairy F, Fotou E, Sheldrick KS, Griffiths DJF, Lehmann AR, Carr AM. Identification and characterisation of new elements involved in checkpoints and feedback controls in fission yeast. Mol Biol Cell. 1994;5:147–160. doi: 10.1091/mbc.5.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JB, Zhou Z, Siede W, Friedberg EC, Elledge SJ. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes & Dev. 1994;8:2401–2415. doi: 10.1101/gad.8.20.2401. [DOI] [PubMed] [Google Scholar]
- Aves SJ, Durkacz BW, Carr A, Nurse P. Cloning, sequencing and transcriptional control of the Schizosaccharomyces pombe cdc10 “start” gene. EMBO J. 1985;4:457–463. doi: 10.1002/j.1460-2075.1985.tb03651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbet NC, Carr AM. Fission yeast wee1 protein kinase is not required for DNA damage-dependent mitotic arrest. Nature. 1993;364:824–827. doi: 10.1038/364824a0. [DOI] [PubMed] [Google Scholar]
- Barker DG, White JHM, Johnston LH. Molecular characterisation of the DNA ligase gene, CDC17 from the fission yeast Schizosaccharomyces pombe. Eur J Biochem. 1987;162:659–667. doi: 10.1111/j.1432-1033.1987.tb10688.x. [DOI] [PubMed] [Google Scholar]
- Bentley NJ, Holtzman DA, Flaggs G, Keegan KS, DeMaggio A, Ford JC, Hoekstra M, Carr AM. The S. pombe rad3 checkpoint gene. EMBO J. 1996;15:6641–6651. [PMC free article] [PubMed] [Google Scholar]
- Brown GW, Jallepalli PV, Huneycutt BJ, Kelly TJ. Interaction of the S phase regulator cdc18 with cyclin-dependent kinase in fission yeast. Proc Natl Acad Sci. 1997;94:6142–6147. doi: 10.1073/pnas.94.12.6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr AM. Radiation checkpoints in model systems. Int J Radiat Biol. 1994;66:S133–S139. [PubMed] [Google Scholar]
- ————— Control of cell cycle arrest by the Mec1sc/Rad3sp DNA structure checkpoint pathway. Curr Opin Genet Dev. 1997;7:93–98. doi: 10.1016/s0959-437x(97)80115-3. [DOI] [PubMed] [Google Scholar]
- Carr AM, Hoekstra MF. The cellular responses to DNA damage. Trends Cell Biol. 1995;5:32–40. doi: 10.1016/s0962-8924(00)88934-5. [DOI] [PubMed] [Google Scholar]
- Cimprich KA, Shin TB, Keith CT, Schreiber SL. cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc Natl Acad Sci. 1996;93:2850–2855. doi: 10.1073/pnas.93.7.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Urso G, Grallert B, Nurse P. DNA polymerase alpha, a component of the replication initiation complex, is essential for the checkpoint coupling S phase to mitosis in fission yeast. J Cell Sci. 1995;108:3109–3118. doi: 10.1242/jcs.108.9.3109. [DOI] [PubMed] [Google Scholar]
- Elledge SJ. Cell cycle checkpoints: Preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Enoch T, Nurse P. Mutation of fission yeast cell cycle control genes abolishes dependece of mitosis on DNA replication. Cell. 1990;60:665–673. doi: 10.1016/0092-8674(90)90669-6. [DOI] [PubMed] [Google Scholar]
- Enoch T, Carr AM, Nurse P. Fission yeast genes involved in coupling mitosis to completion of DNA replication. Genes & Dev. 1992;6:2035–2046. doi: 10.1101/gad.6.11.2035. [DOI] [PubMed] [Google Scholar]
- Fernandez-Sarabia MJ, McInerny C, Harris P, Gordon C, Fantes P. The cell cycle genes cdc22+ and suc22+ of fission yeast Schizosaccharomyces pombe encode the large and smal subunits of ribonucleotide reductase. Mol & Gen Genet. 1993;238:241–251. doi: 10.1007/BF00279553. [DOI] [PubMed] [Google Scholar]
- Francesconi S, Grenon M, Bouvier D, Baldacci G. p56chk1 protein kinase is required for the DNA replication checkpoint at 37°C in fission yeast. EMBO J. 1997;16:1332–1341. doi: 10.1093/emboj/16.6.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari B, Rhind N, Russell P. Cdc25 mitotic inducer targeted by Chk1 DNA damage checkpoint kinase. Science. 1997;277:1495–1497. doi: 10.1126/science.277.5331.1495. [DOI] [PubMed] [Google Scholar]
- Gutz H, Heslot H, Leupold U, Loprieno N. Schizosaccharomyces pombe. In: King RC, editor. Handbook of genetics. New York, NY: Plenum Press; 1974. [Google Scholar]
- Iino Y, Yamamoto M. The Schizosaccharomyces pombe cdc6 gene encodes the catalytic subunit of DNA polymerase delta. Mol & Gen Genet. 1997;254:93–97. doi: 10.1007/s004380050395. [DOI] [PubMed] [Google Scholar]
- Jeggo AP, Taccioli GE, Jackson SP. Menage à trois: Double strand break repair, V(D)J recombination and DNA-PK. BioEssays. 1995;17:949–957. doi: 10.1002/bies.950171108. [DOI] [PubMed] [Google Scholar]
- Kato R, Ogawa H. An essential gene, ESR1, is required for mitotic cell growth, DNA repair and meiotic recombination in Saccharomyces cerevisiae. Nucleic Acids Res. 1994;22:3104–3112. doi: 10.1093/nar/22.15.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leatherwood J, Lopez-Girona A, Russell P. Interaction of Cdc2 and Cdc18 with a fission yeast ORC2- like protein. Nature. 1996;379:360–363. doi: 10.1038/379360a0. [DOI] [PubMed] [Google Scholar]
- Mairorano D, Van Assendelft GB, Kearsey SE. Fission yeast cdc21, a member of the MCM protein family, is required for onset of S phase and is located in the nucleus throughtout the cell cycle. EMBO J. 1996;15:863–872. [PMC free article] [PubMed] [Google Scholar]
- Marini F, Pellicioli A, Paciotti V, Lucchini G, Plevani P, Stern DF, Foiani M. A role for DNA primase in coupling DNA replication to DNA damage response. EMBO J. 1997;16:639–650. doi: 10.1093/emboj/16.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masai H, Miyake T, Arai K. hsk1+, a Schizosaccharomyces pombe gene related to Saccharomyces cerevisiae CDC7, is required for chromosomal replication. EMBO J. 1995;14:3094–3104. doi: 10.1002/j.1460-2075.1995.tb07312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami H, Okayama H. A kinase from fission yeast responsible for blocking mitosis in S phase. Nature. 1995;374:817–819. doi: 10.1038/374817a0. [DOI] [PubMed] [Google Scholar]
- O’Connell MJ, Raleigh JM, Verkade HM, Nurse P. Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J. 1997;16:545–554. doi: 10.1093/emboj/16.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pati D, Keller C, Groudine M, Plon SE. Reconstitution of a MEC1-independent checkpoint in yeast by expression of a novel human fork head cDNA. Mol Cell Biol. 1997;17:3037–3046. doi: 10.1128/mcb.17.6.3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C-Y, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: Regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- Rao PN, Johnson RT. Mammalian cell fusion studies on the regulation of DNA synthesis and mitosis. Nature. 1970;225:159–164. doi: 10.1038/225159a0. [DOI] [PubMed] [Google Scholar]
- Rowley R, Subramani S, Young PG. Checkpoint controls in Schizosaccharomyces pombe: rad1. EMBO J. 1992;11:1335–1342. doi: 10.1002/j.1460-2075.1992.tb05178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science. 1996;271:357–360. doi: 10.1126/science.271.5247.357. [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ. Conservation of the Chk1 checkpoint pathway in mammals: Linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–1501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. A single ataxia telangiectasia gene with a product similar to PI 3-kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Sazer S, Sherwood SW. Mitochondrial growth and DNA synthesis occur in the absence of nuclear DNA replication in fission yeast. J Cell Sci. 1990;97:509–516. doi: 10.1242/jcs.97.3.509. [DOI] [PubMed] [Google Scholar]
- Sheldrick KS, Carr AM. Feedback controls and G2 checkpoints: Fission yeast as a model system. BioEssays. 1993;15:775–782. doi: 10.1002/bies.950151202. [DOI] [PubMed] [Google Scholar]
- Sun Z, Fay DS, Marini F, Foiani M, Stern DF. Spk1/Rad53 is regulated by Mec1-dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes & Dev. 1996;10:395–406. doi: 10.1101/gad.10.4.395. [DOI] [PubMed] [Google Scholar]
- Uchiyama M, Galli I, Griffiths DFJ, Wang TSF. A novel mutant allele of Schizosaccharomyces pombe rad26 defective in monitoring S-phase progression to prevent premature mitosis. Mol Cell Biol. 1997;17:3101–3115. doi: 10.1128/mcb.17.6.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth N, Bernards R. rad-dependent responses of the chk1-encoded protein kinase at the DNA damage checkpoint. Science. 1996;271:353–356. doi: 10.1126/science.271.5247.353. [DOI] [PubMed] [Google Scholar]
- Walworth N, Davey S, Beach D. Fission yeast chk1 protein kinase links the rad checkpoint pathway to cdc2. Nature. 1993;363:368–371. doi: 10.1038/363368a0. [DOI] [PubMed] [Google Scholar]
- Weinert TA, Hartwell LH. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science. 1988;241:317–322. doi: 10.1126/science.3291120. [DOI] [PubMed] [Google Scholar]
- Weinert TA, Kiser GL, Hartwell LH. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes & Dev. 1994;8:652–665. doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]
- Zakian VA. ATM-related genes: What do they tell us about functions of the human gene? Cell. 1995;82:685–687. doi: 10.1016/0092-8674(95)90463-8. [DOI] [PubMed] [Google Scholar]