

Abstract
Estrogen- and antiestrogen-regulated, AF-2-dependent transcriptional activation by purified full-length human estrogen receptor (ER) was carried out with chromatin templates in vitro. With this system, the ability of purified human p300 to function as a transcriptional coactivator was examined. In the absence of ligand-activated ER, p300 was found to have little effect (less than twofold increase) on transcription, whereas, in contrast, p300 was observed to act synergistically with ligand-activated ER to enhance transcription. When transcription was limited to a single round, p300 and ER were found to enhance the efficiency of transcription initiation in a cooperative manner. On the other hand, when transcription reinitiation was allowed to occur, ER, but not p300, was able to increase the number of rounds of transcription. These results suggest a two-stroke mechanism for transcriptional activation by ligand-activated ER and p300. In the first stroke, ER and p300 function cooperatively to increase the efficiency of productive transcription initiation. In the second stroke, ER promotes the reassembly of the transcription preinitiation complex. Therefore, ER exhibits distinct, dual functions in transcription initiation and reinitiation.
Keywords: RNA polymerase II, in vitro transcription, estrogen receptor, p300, CBP, coactivator, nuclear receptors
The transduction of extracellular signals into intracellular responses is a fundamental and important process. Lipophilic hormones, which include both steroids (such as sex steroids, corticosteroids, mineralocorticoids, and ecdysteroids) and nonsteroids (such as retinoic acid, thyroid hormone, and vitamin D3), are a group of molecules involved in signal transduction in both vertebrates and invertebrates (Reichel and Jacob 1993; Mangelsdorf et al. 1995). The intracellular targets for the lipophilic hormones are a group of structurally related receptors that bind specifically to the hormones (Beato et al. 1995; Mangelsdorf and Evans 1995; Mangelsdorf et al. 1995). These receptors, which collectively constitute the nuclear hormone receptor superfamily, are sequence-specific DNA-binding transcription factors that alter patterns of gene expression in response to their cognate ligands (Evans 1988; Mangelsdorf et al. 1995).
The superfamily of nuclear receptors has been subdivided into several different classes on the basis of the dimerization and DNA-binding properties of the proteins (Mangelsdorf et al. 1995). Class I receptors are steroid hormone receptors that homodimerize and bind to response elements organized as inverted repeats, whereas class II receptors include vitamin and thyroid hormone receptors that share the common heterodimerization partner retinoid X receptor (RXR) and typically bind to response elements organized as direct repeats (Truss and Beato 1993; Glass 1994). The estrogen receptor (ER) is a class I receptor that binds to a structurally related group of steroidal ligands known as estrogens, which includes the abundant natural ligand, 17β-estradiol (E2). The ER is a ligand-modulated transcription factor that is transcriptionally inactive in the absence of ligand (Gronemeyer 1991; Beato et al. 1996). Two independent activation functions, termed AF-1 and AF-2, have been identified in the ER. AF-1 is a ligand-independent activation region in the amino-terminal portion of the protein, whereas AF-2 is a ligand-dependent activation region in the carboxyl terminal ligand-binding domain (Webster et al. 1988; Tora et al. 1989; Danielian et al. 1992; Tzukerman et al. 1994). The available data suggest that an estrogen-induced conformational change in the ER promotes a synergistic interaction between AF-1 and AF-2 that leads to transcriptional activation (Beekman et al. 1993; Kraus et al. 1995).
Several transcriptional coactivators have been found to interact with nuclear hormone receptors in a ligand-dependent manner and to enhance the transcriptional activity of ER and other nuclear receptors (Horwitz et al. 1996). These factors include the steroid receptor coactivator 1 (SRC1)-like family of proteins (such as SRC1/NCoA1, GRIP1/TIF2/NCoA2, pCIP/ACTR/RAC3/AIB1) (Oñate et al. 1995; Hong et al. 1996, 1997; Kamei et al. 1996; Voegel et al. 1996; Chen et al. 1997; Anzick et al. 1997; Li et al. 1997; Torchia et al. 1997) as well as p300 and CREB-binding protein (CBP; these related proteins are collectively referred to as p300/CBP) (Chakravarti et al. 1996; Hanstein et al. 1996; Kamei et al. 1996). p300/CBP functions as a coactivator both for nuclear receptors and for transcription factors that are not in the receptor superfamily (Kwok et al. 1994; Bhattacharya et al. 1996; Dai et al. 1996; Eckner et al. 1996; Oliner et al. 1996; Akimaru et al. 1997). p300 and CBP have been found to possess histone acetyltransferase activity (Bannister and Kouzarides 1996; Ogryzko et al. 1996) as well as to interact with RNA polymerase II-containing complexes (Kee et al. 1996; Nakajima et al. 1997a,b). It will be interesting and important to understand the mechanisms by which these coactivators participate in ligand-activated transcription by nuclear receptors.
In this work, we have used a biochemical approach to study the function of the human ER and p300. We initially established a chromatin transcription system that recreates estrogen- and anti-estrogen-regulated and AF-2-dependent transcription by purified, full-length ER. Then, we investigated the mechanisms of ligand-regulated transcriptional activation by ER and p300. These studies revealed two differential strategies by which ER and p300 function cooperatively to activate transcription.
Results
Ligand-dependent transcriptional activation by ER is specific for chromatin templates
To study the mechanism of ER function, it was first necessary to establish a ligand-dependent in vitro transcription system with purified, full-length human ER. We therefore, synthesized Flag epitope-tagged versions of wild-type ER and an AF-2 single amino acid substitution mutant ER (Leu-540 to Gln; designated L540Q) in Spodoptera frugipenda (Sf9) cells by using a baculovirus expression vector (Fig. 1A). The L540Q amino acid substitution impairs the AF-2 activation domain but does not affect the ligand-binding activity of ER (Wrenn and Katzenellenbogen 1993). Immunoaffinity purification of the recombinant proteins yielded preparations of ligand-free ER and ER(L540Q) of ∼90% homogeneity (Fig. 1B) that exhibited nearly equivalent ligand-binding activity (Fig. 1C) as well as DNA-binding activity (Fig. 1D).
Figure 1.
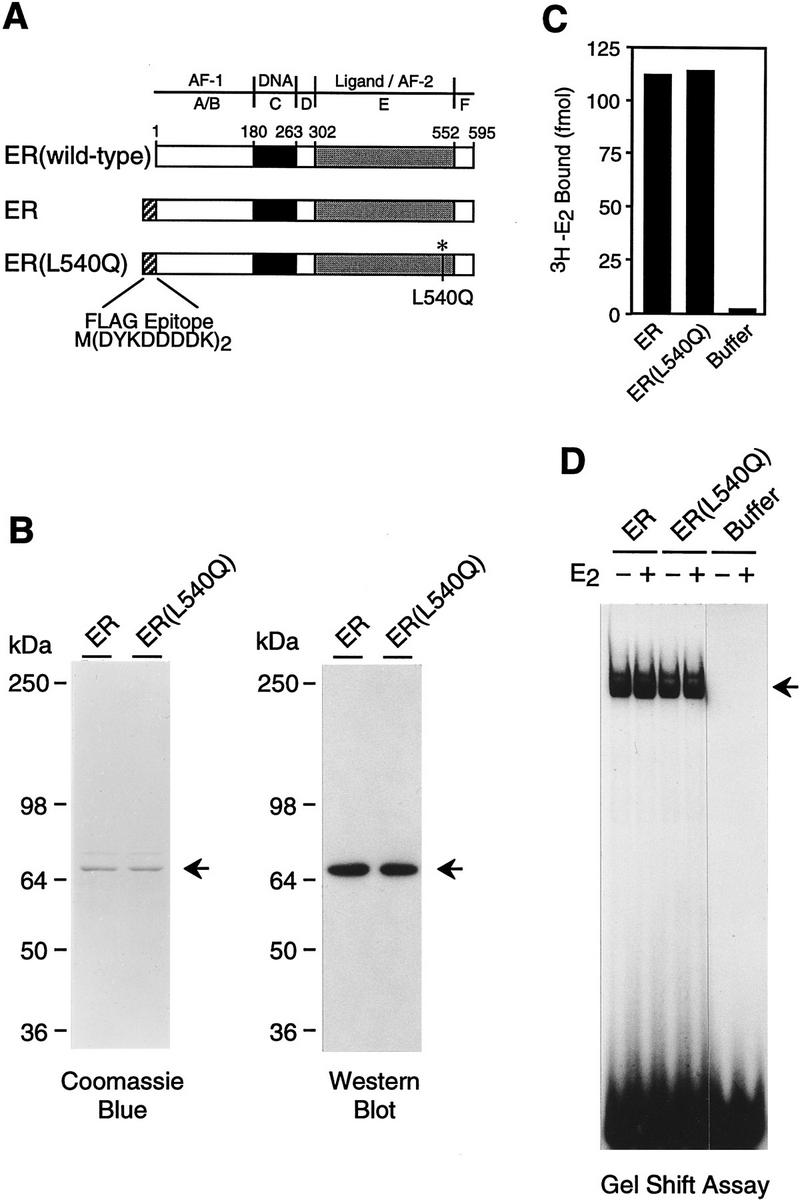
Purification of human ER and L540Q variant. (A) Schematic diagram of wild-type and epitope-tagged ERs. The locations of the amino-terminal Flag epitope tag and the Leu-540 → Gln (L540Q) point mutation are indicated. (B) Synthesis and purification of epitope-tagged ERs. Flag-tagged ER and ER(L540Q) were synthesized in Sf9 cells by using a baculovirus expression vector and affinity-purified with monoclonal antibodies that recognize the FLAG epitope. The proteins were subjected to 8% polyacrylamide–SDS gel electrophoresis. (Left) Total protein was visualized by staining with Coomassie Brilliant Blue R-250; (right) ERs were detected by Western blot analysis with anti-ER antibodies. (C) Ligand-binding activity of purified recombinant ERs. The 3H–E2 binding activity of approximately equimolar amounts of purified ERs was determined by ligand binding assays. Specific binding activity was determined by analyzing a second set of samples in the presence of a 200-fold excess of unlabeled E2. The net binding of 3H–E2 (in fmoles) is shown. (D) DNA-binding activity of purified recombinant ERs. Gel mobility shift assays were performed with approximately equimolar amounts of purified ERs. The samples were incubated with a 32P-end-labeled double-stranded oligonucleotide containing a consensus ERE sequence in the presence or the absence of E2 and then analyzed with a nondenaturing 4.8% polyacrylamide gel.
Because of the potentially important role of chromatin in the regulation of transcription by ER, we analyzed the transcriptional activity of the purified ER with chromatin templates. Periodic nucleosomal arrays were assembled by using a chromatin assembly extract termed S190 (Kamakaka et al. 1993; Bulger and Kadonaga 1994), and transcription reactions were carried out with a HeLa nuclear extract (Dignam et al. 1983). As a reporter template, we used the plasmid pERE, which contains four copies of the Xenopus vitellogenin A2 gene estrogen response element (ERE) upstream of the adenovirus E4 core promoter. As shown in Figure 2A, we observed potent activation of transcription by purified ER when the factor was added either to naked DNA prior to chromatin assembly (added during assembly) or to preassembled chromatin (added after assembly). Full transcriptional activation by ER was dependent on 17β-estradiol (E2), although a severalfold increase in transcription was seen in the absence of ligand. (In this experiment, there was ∼75-fold activation, though we typically observed ∼30- to 50-fold activation by ER ± E2.) The concentrations of ER and E2 in these transcription reactions were 4.5 and 30 nm, respectively, which are approximately the optimal concentrations of these components, as determined by titration studies (data not shown). By Western blot analysis, the ER remained intact as the full-length polypeptide throughout the course of chromatin assembly (Fig. 2B). In addition, the inclusion of ER in the assembly reactions did not affect the efficiency or quality of chromatin assembly (Fig. 2C).
Figure 2.
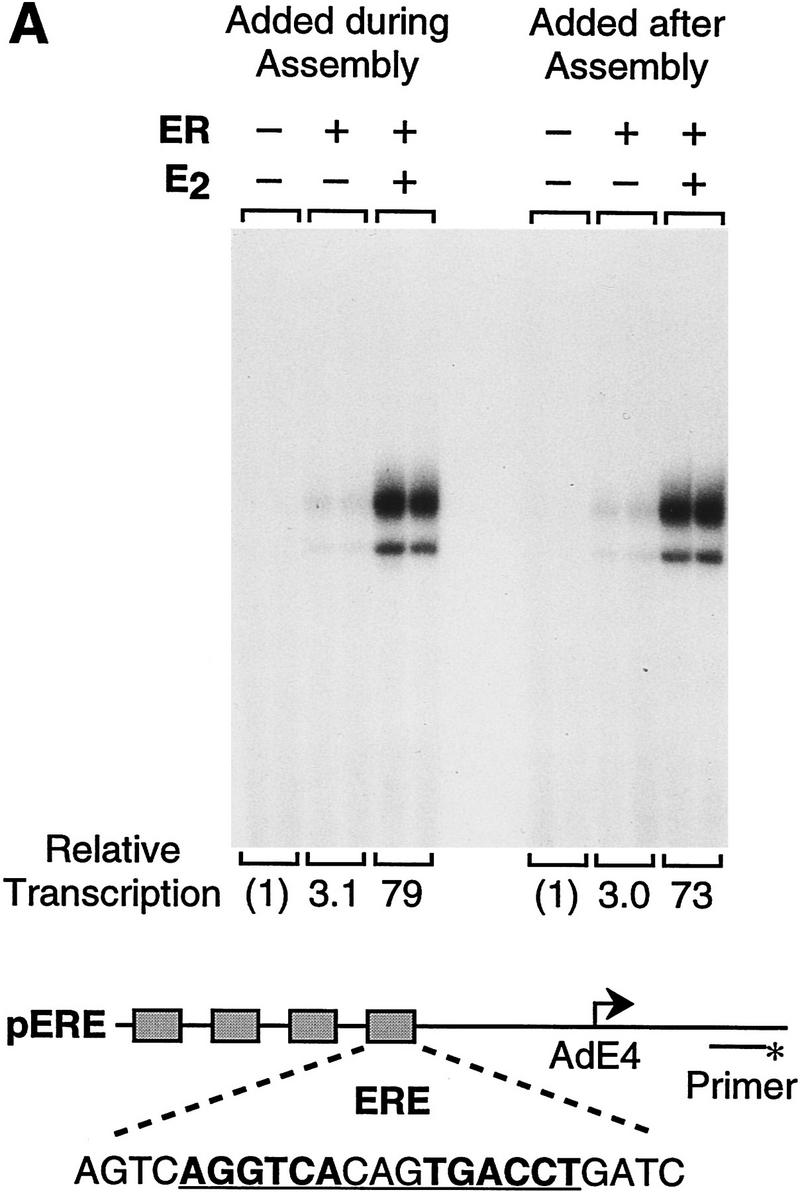


Ligand-dependent activation by ER with chromatin but not with nonchromatin templates. (A) Ligand-dependent transcriptional activation by ER with chromatin templates in vitro. The plasmid pERE was assembled into chromatin and then subjected to in vitro transcription–primer extension analysis. Where indicated, ER and E2 were added to naked DNA before chromatin assembly (added during assembly) or to preassembled chromatin (added after assembly). The final concentrations of ER and E2 in the transcription reactions were 4.5 and 30 nm, respectively. (B) ER is not degraded during chromatin assembly. Aliquots of assembly reaction mixtures containing the indicated components were taken either before (0 hr) or after (4.5 hr) chromatin assembly was complete. The samples were subjected to Western blot analysis with anti-ER antibodies. (C) The efficiency of chromatin assembly is not affected by ER and E2. Chromatin assembled in the presence or the absence of ER (10 nm) and E2 (100 nm) was subjected to micrococcal nuclease digestion and DNA supercoiling analyses. Where noted, the DNA supercoiling gel contained 2 μm chloroquine. The positions of nicked circular (N) and supercoiled (S) DNAs are indicated. (D) Ligand-stimulated transcription by ER requires packaging of the DNA template into chromatin. Chromatin assembly and in vitro transcription reactions were performed as in A. To inhibit assembly of the pERE tem-plate DNA into chromatin, a nontemplate competitor DNA (pUC118; at a pUC118:pERE mass ratio of 3) was added to the assembly reactions 30 min prior to the addition of pERE template DNA (before assembly). As a control, pUC118 was added to the assembly reactions subsequent to assembly of pERE into chromatin (after assembly). The amounts of transcription in each lane are directly comparable in terms of autoradiography exposure time and the amounts of reaction products that were applied to each gel lane. The relative transcription levels for each set of reaction conditions (i.e., the presence or absence of competitor DNA added before or after chromatin assembly) are normalized to the transcription reactions in which ER and E2 were not included, as designated by (1). The final concentrations of ER and E2 in the transcription reactions were 4.5 and 30 nm, respectively.
To test whether this transcriptional activation is dependent on packaging of the template into chromatin, we compared the transcriptional activity of ER with chromatin and nonchromatin templates. In these experiments, a nontemplate competitor DNA (pUC118) was added to the chromatin assembly reactions prior to the addition of the template DNA (pERE) at a 3:1 mass ratio of pUC118/pERE. Chromatin was initially assembled onto pUC118 DNA for 30 min to deplete the free histones prior to addition of pERE template DNA. As shown in Figure 2D, depletion of histones by the addition of pUC118 before pERE (competitor DNA added before assembly) led to an increase in the amount of basal transcription in the absence of ER and a low (about twofold) amount of transcriptional activation by ER ± E2. In contrast, as a control, when pERE was assembled into chromatin prior to the addition of a threefold mass excess of pUC118 (competitor DNA added after assembly), strong activation of transcription by ER ± E2 was observed. We have also found that ligand-dependent transcriptional activation by ER does not occur in standard in vitro transcription assays with naked DNA templates in the absence of the S190 assembly extract (data not shown). Therefore, these data suggest that E2-stimulated transcription by ER is specific for chromatin templates and that the ER functions effectively with a preassembled chromatin template, as approximately the same amount of transcription was observed when ER was added either to naked DNA prior to chromatin assembly or to preassembled chromatin templates.
Transcriptional activation by ER in vitro requires the AF-2 domain and is blocked by anti-estrogens
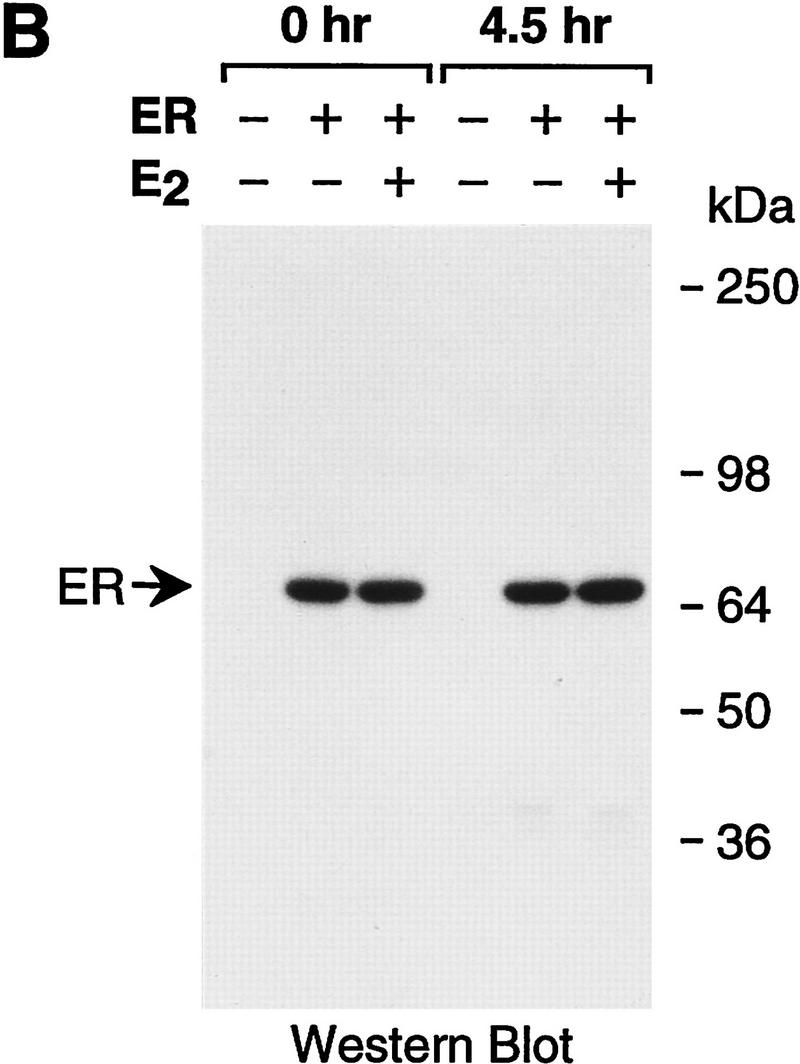
To test the specificity of ER function with the chromatin transcription system, we examined the relative ability of wild-type ER and L540Q mutant ER to activate transcription. The Leu-540 residue is in a conserved region of ER [in the putative twelfth helix of the ligand-binding domain (Wurtz et al. 1996)] that is critical for AF-2 function (Danielian et al. 1992). In transient transfection assays, L540Q ER is transcriptionally inactive in response to E2 (Wrenn and Katzenellenbogen 1993); however, L540Q ER possesses DNA- and ligand-binding activities that are approximately equivalent to those of the wild-type ER (Fig. 1C,D). As seen in Figure 3A (left), L540Q ER is transcriptionally inactive in the presence of E2. Western blot analysis revealed that both the wild-type and L540Q ERs remained intact throughout the course of chromatin assembly (Fig. 3B). Therefore, transcriptional activation (but not DNA or ligand binding) by ER is specifically affected by alteration of the Leu residue at position 540 in the AF-2 domain.
Figure 3.
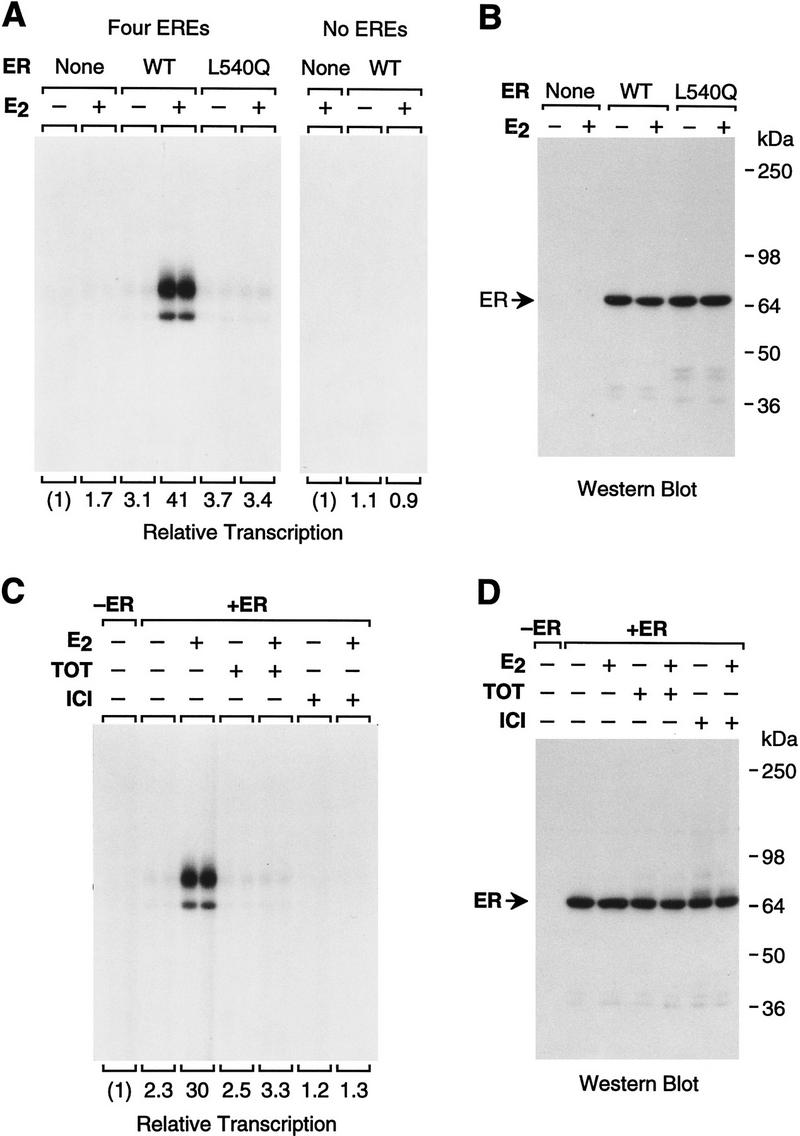
In vitro transcriptional activation by ER requires the AF-2 domain and is blocked by anti-estrogens. (A) Transcriptional activation by ER is ERE- and AF-2-dependent. pERE (four EREs) and pIE0 [which is identical to pERE, except that it does not contain any EREs (no EREs)] were assembled into chromatin with ER, ER(L540Q), and/or E2 and then subjected to in vitro transcription–primer extension analysis. The final concentrations of receptor and E2 in the transcription reactions were 4.5 and 30 nm, respectively. (B) ER and ER(L540Q) are not degraded during chromatin assembly. Aliquots of chromatin assembled with the indicated components were subjected to Western blot analysis with anti-ER antibodies. (C) Anti-estrogens inhibit E2-dependent activation by ER with chromatin templates in vitro. Chromatin assembly–in vitro transcription reactions were performed with ER, E2, TOT, and ICI, as indicated. The final concentrations of ER, E2, and anti-estrogens in the transcription reactions were 4.5 nm, 15 nm, and 3 μm, respectively. (D) ER is not degraded during chromatin assembly in the presence of anti-estrogens. Aliquots of chromatin assembled with the indicated components were subjected to Western blot analysis with anti-ER antibodies.
We also examined whether activation by ER is dependent on the presence of EREs in the reporter template. To this end, we tested the transcriptional activity of ER with pERE (containing four EREs) or pIE0 (which is equivalent to pERE, except that it lacks EREs) and found that the ER-mediated activation is dependent on the EREs in the pERE reporter plasmid (Fig. 3A). Thus, transcriptional activation by ER in the chromatin transcription assay requires binding of the factor to the chromatin template.
We then tested the specificity of the transcriptional response to estrogens and anti-estrogens. Anti-estrogens compete with estrogens for binding to the ER and are thought to configure the receptor in a transcriptionally inactive conformation (McDonnell et al. 1994). We examined the effects of two anti-estrogens, transhydroxytamoxifen (TOT) and ICI 164,384 (ICI). TOT is a nonsteroidal triphenylethylene derivative that has partial agonist activity in some assays, whereas ICI is a steroidal compound with pure antagonist activity (Santen et al. 1990; Wakeling 1990; McDonnell et al. 1995). As depicted in Figure 3C, neither TOT nor ICI induced transcriptional activation by ER. Moreover, TOT and ICI were each able to block E2-induced transcription by ER (at a 200:1 molar ratio of anti-estrogen/E2). Western blot analysis revealed that there was no apparent difference in the stability of ER in the presence or the absence of E2, TOT, or ICI (Fig. 3D). Interestingly, ICI consistently inhibited transcription to a greater extent than TOT. Hence, the differing antagonist effects of TOT and ICI, as observed in vivo, appear to be evident in vitro. Furthermore, inhibition of transcription by anti-estrogens was observed with chromatin templates but not with nonchromatin templates (data not shown). These results suggest that packaging of the DNA template into chromatin is an important component of anti-estrogen function. Therefore, on the basis of the transcriptional requirement for ER and E2 (Fig. 2), EREs in the reporter gene (Fig. 3A), and AF-2 (Leu-540) of ER (Fig. 3A), as well as the inhibition of transcriptional activation by anti-estrogens (Fig. 3C), we conclude that the mechanism of transcriptional activation by ER with chromatin templates in vitro resembles the natural mechanism of action of ER in vivo.
Binding of ER to chromatin is not sufficient for transcriptional activation
To address whether or not binding of ER to chromatin correlates with transcriptional activation, we directly subjected the chromatin templates (assembled with the same concentrations of ER and ligands used in the transcriptional analyses shown in Figure 3) to partial digestion with DNase I and resolved the resulting cleavage patterns by primer extension analysis (Gralla 1985; Pazin et al. 1994). As shown in Figure 4A, wild-type ER binds to the EREs in pERE in either the presence or the absence of E2. In addition, DNA binding by ER is not inhibited by the presence of either TOT or ICI (Fig. 4B). Moreover, L540Q mutant ER binds to chromatin in a manner that is indistinguishable from wild type ER (Fig. 4C). These data indicate that the presence of ER is sufficient for binding to chromatin and that this binding is not significantly affected by E2, TOT, ICI, or the L540Q mutation in AF-2. It is particularly important to note that the conditions for chromatin assembly in the footprinting experiments are identical to those used in the chromatin transcription studies. Therefore, binding of the ER to chromatin is not sufficient for transcriptional activation, because the factor binds to chromatin under conditions in which it is not transcriptionally active. These data suggest a model for ER function in which the critical step for transcriptional activation is not the binding of ER to DNA but is instead the generation of the estrogen-dependent active conformation of the DNA-bound factor.
Figure 4.

The sequence-specific binding of ER to chromatin is not affected by estrogens or anti-estrogens. Chromatin was assembled onto pERE with the indicated components and then directly subjected to DNase I–primer extension footprinting analysis. The concentrations of ER and ligands are identical to those used in the corresponding transcriptional analyses (Fig. 3), for which the samples were subsequently diluted by a factor of 0.3. The schematic diagrams denote the locations of the EREs, TATA box, and RNA start site. (A) The binding of ER to chromatin is not affected by E2. The positions of increased (solid arrowheads) and decreased (open arrowheads) sensitivity to DNase I are shown. (B) The binding of ER to chromatin is not affected by anti-estrogens. (C) Both ER and ER(L540Q) bind to chromatin.
p300 synergistically enhances ligand-dependent transcriptional activation by ER
To investigate the function of p300/CBP as a coactivator of ER, we tested the ability of p300 to enhance ligand-dependent transcriptional activation by ER. We therefore synthesized and purified full-length, His6-tagged human p300 (Fig. 5A). The effect of the purified p300 on ligand-dependent activation by ER was then examined in the chromatin transcription assay. As shown in Figure 5B, the inclusion of p300 enhances transcriptional activation by ER ± E2 (>7-fold enhancement at 30 nm p300) yet only modestly affects (≤2-fold increase) transcription in the absence of ER + E2 or with ER alone. Thus, p300 acts cooperatively with ER + E2 to enhance transcription (as the amount of transcriptional activation with p300 and ER + E2 together is greater than the multiplicative product of the respective amounts of activation that are seen with p300 alone and with ER + E2 alone).
Figure 5.


The transcriptional coactivator p300 functions synergistically with ER to enhance ligand-dependent transcription with chromatin templates in vitro. (A) Synthesis and purification of human p300. His6-tagged, human p300 was synthesized in Sf9 cells by using a baculovirus expression vector and purified by nickel chelate chromatography. The protein was subjected to 8% polyacrylamide–SDS gel electrophoresis and staining with Coomassie Blue R-250. (B) Purified, exogenous p300 stimulates ligand-dependent transcription by ER. pERE was assembled into chromatin with ER and E2, as noted, and the samples were subjected to in vitro transcription analysis in the presence or the absence of p300. The final concentrations of ER and E2 in the transcription reactions were 4.5 and 30 nm, whereas p300 varied from 0 to 30 nm. (C) Transcriptional enhancement by p300 requires a chromatin template. Chromatin assembly–in vitro transcription reactions were performed as in B. (Right, non-chromatin) pUC118 competitor DNA was added to the assembly reactions prior to pERE, as in Fig. 2D. The final concentrations of ER, E2, and p300 in the transcription reactions were 4.5, 30, and 15 nm. (D) Anti-estrogens inhibit transcriptional synergism between ER and p300. Chromatin assembly–in vitro transcription reactions were performed as in B, with the indicated components. The final concentrations of factors and ligands in the transcription reactions were ER (4.5 nm), p300 (15 nm), E2 (15 nm), and anti-estrogens (3 μm).
We then tested whether the p300-mediated enhancement of transcription was specific for chromatin templates by competitor DNA (pUC118) analysis, as in Figure 2D. These experiments revealed that p300 enhances ligand-dependent transcriptional activation by ER with chromatin but not with nonchromatin templates (Fig. 5C). We also examined whether anti-estrogens affect the ability of p300 to enhance ER-activated transcription (Fig. 5D). These experiments revealed that a 200-fold molar excess of either TOT or ICI (relative to E2) blocked transcriptional activation by ER + E2 in the presence of p300. Moreover, as in Figure 3C, the amount of inhibition with ICI was consistently greater than that seen with TOT. Thus, these experiments demonstrate that p300 acts cooperatively with ER + E2 to enhance transcription in a chromatin-specific and ligand-regulated manner.
A dual function of ER in transcription initiation and reinitiation
To examine the steps in the transcription process that are regulated by ER and p300, we used the detergent Sarkosyl to analyze the ability of these factors to increase the efficiency of transcription in a single round as well as to promote transcription reinitiation. Sarkosyl was used at a concentration [0.2% (wt/vol)] that inhibits the assembly of the transcription preinitiation complex but not the elongation of the transcriptionally engaged polymerase (Hawley and Roeder 1985, 1987; Kadonaga 1990). Therefore, the addition of Sarkosyl immediately after transcription initiation (upon addition of ribonucleoside 5′-triphosphates) results in a single round of transcription, because the detergent inhibits the reassembly of the preinitiation complex. In this manner, we measured the amount of transcription that occurs in a single round of transcription (+Sarkosyl) as well as in multiple rounds of transcription (−Sarkosyl) (Fig. 6A). Then, by division of the amount of transcription that is seen in the absence of Sarkosyl (multiple rounds) by the amount of transcription seen in the presence of Sarkosyl (single round), we were able to determine the average number of rounds of transcription for each experimental condition. It is also important to indicate that the single round transcription experiments reveal the efficiency of the transcription initiation process, because transcriptional elongation occurs readily in the presence of Sarkosyl.
Figure 6.


Ligand-activated ER promotes transcription reinitiation, whereas p300 enhances transcription initiation. (A) Single and multiple round transcriptional analysis with ER and p300. Chromatin assembly–in vitro transcription reactions were carried out as in Fig. 5B with the indicated components. (Right, single round) Sarkosyl was added 10 sec after initiation of transcription by the addition of rNTPs. Under these conditions, Sarkosyl inhibits transcription reinitiation but not elongation and, therefore, a single round of transcription is obtained. For each reaction condition, the number of rounds of transcription was determined by dividing the amount of transcription in the absence of Sarkosyl by the corresponding amount of transcription in the presence of Sarkosyl. In these experiments, Sarkosyl was used at 0.2% (wt/vol) final concentration, but identical results were observed with 0.05% and 0.4% Sarkosyl (data not shown). The final concentrations of ER, E2, and p300 in the transcription reactions were 4.5, 30, and 15 nm. (B) p300 enhances the efficiency of transcription but does not promote transcription reinitiation. Reactions were performed with the indicated components, as in A. The final concentrations of ER and E2 were 4.5 and 30 nm. (C) p300 does not enhance transcriptional elongation. Reactions were performed with the indicated components, as in A. Primer extension analyses were performed with four different primers that yield reverse transcription products of the indicated lengths. The final concentrations of ER, E2, and p300 in the transcription reactions were 4.5, 30, and 15 nm.
We thus analyzed the basis for transcriptional activation by ER. In a single round of transcription, only a 3.8-fold activation of transcription by ER + E2 was seen (Fig. 6A, lane 9). Thus, ER + E2 has a modest effect upon increasing the efficiency of transcription initiation. When transcription was not limited to a single round, however, a 44-fold stimulation of transcription by ER + E2 was observed (Fig. 6A, lane 3). These experiments revealed an average of three rounds of transcription in the absence of ER + E2 (Fig. 6A, lane 1), an average of six rounds of transcription with ER only (Fig. 6A, lane 2), and an average of 33 rounds of transcription in the presence of ER + E2 (Fig. 6A, lane 3). Hence, these results show that an important component of ligand-activated transcription by ER is to promote transcription reinitiation. [It is also interesting to note, parenthetically, that the major RNA start site in the first round of transcription appears to be a few nucleotides upstream of the major RNA start site that is used in subsequent rounds of transcription (cf., e.g., Fig. 6A, lanes 3, 6, and 12).]
We then examined the coactivator function of p300. In a single round of transcription, p300 synergistically enhances (3.8-fold activation by ER + E2 only, 1.3-fold activation by p300 only, and 100-fold activation by ER + E2 and p300) ligand-dependent transcriptional activation by ER (Fig. 6A, cf. lanes 9, 10, and 12). Hence, ligand-activated ER and p300 act cooperatively to increase the efficiency of productive transcription initiation. Yet, in contrast to ER, p300 does not increase the number of rounds of transcription (Fig. 6A, cf. lanes 3 and 6). In that experiment, however, it was possible that the amount of transcription (and thus, the number of rounds of transcription) was limited by a factor other than p300 (e.g., such as polymerase). We therefore analyzed the effect of p300 on transcription reinitiation under conditions where the transcriptional capacity was not limiting, and these experiments revealed that p300 increases the amount of transcription but not the number of rounds of transcription (Fig. 6B, 12 nm p300). Thus, these results show that ligand-activated ER stimulates transcription by promoting transcription reinitiation, whereas p300 functions as a coactivator that acts cooperatively with ER to enhance transcriptional initiation but not reinitiation.
Although it appears that ER and p300 function synergistically to activate the transcription initiation process, we also sought to test the ability of p300 to increase the extent of transcriptional elongation. To this end, we carried out chromatin transcription reactions and subjected the reaction products to primer extension analysis with a series of primers that detect transcripts that vary in length from 52 to 333 nucleotides. As shown in Figure 6C, the transcriptional enhancement by p300 that we observe does not occur at the latter stages of transcriptional elongation. Thus, the data collectively indicate that p300 can enhance the efficiency of productive transcription initiation, which involves preinitiation complex assembly, initiation of RNA synthesis, and promoter clearance.
These results therefore reveal a dual function of ER in both transcription initiation and reinitiation. ER functions cooperatively with p300 to increase the efficiency of productive initiation. In addition, ER, but not p300, promotes the reassembly of transcription complexes to facilitate reinitiation.
Discussion
This study describes ligand-regulated, AF-2-dependent transcriptional activation with purified, full-length human ER by using chromatin templates in vitro. With this system, purified human p300 functions cooperatively with estrogen-activated ER to enhance transcription. Single versus multiple round transcriptional analyses revealed that ER has the dual function of increasing transcription initiation in a cooperative manner with p300 as well as promoting transcription reinitiation. p300 was found to enhance transcription initiation, but not reinitiation, in conjunction with ER. In addition, p300 had little effect on transcription in the absence of ER. These results suggest a two-stroke model for transcriptional activation by ER and p300 (Fig. 7). In the first stroke, ER and p300 function cooperatively to increase the efficiency of productive transcription initiation. In the second stroke, ER promotes the reassembly of the transcription preinitiation complex. These processes involve two levels of transcriptional enhancement by ligand-activated ER. First, there is the cooperativity between ER and p300 in the activation of transcription initiation. Second, there is the multiplicative ability of ER to promote transcription reinitiation.
Figure 7.
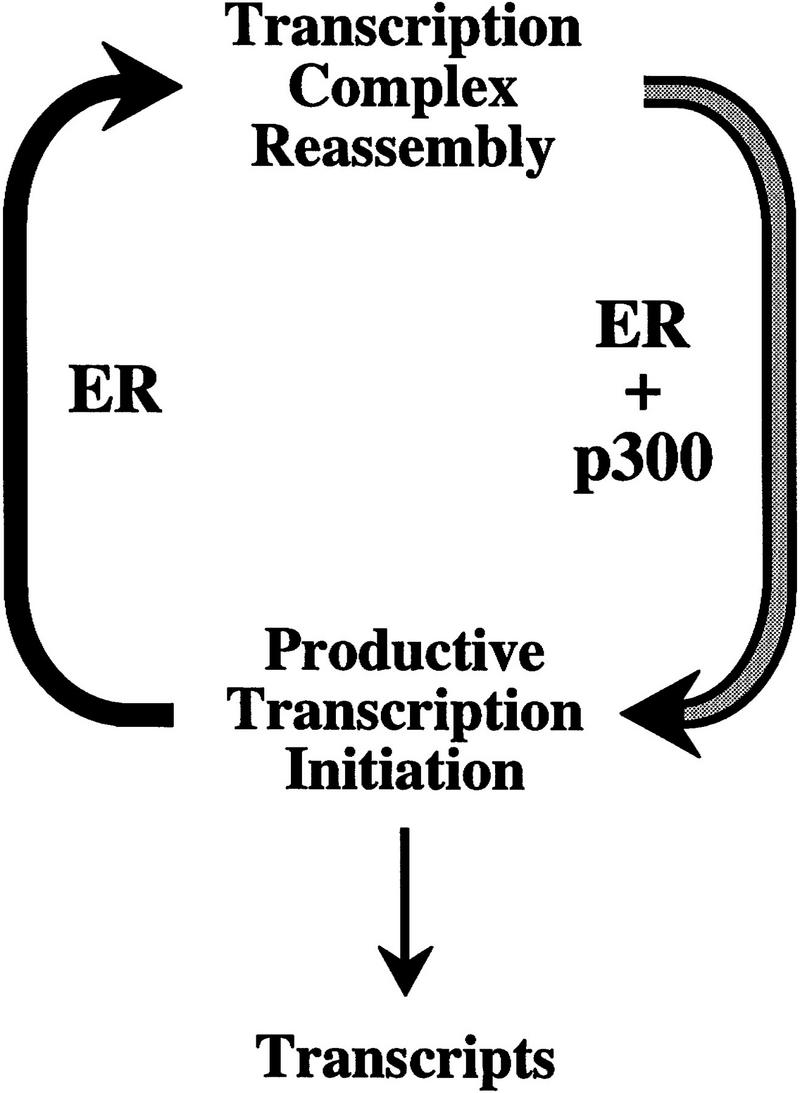
A two-stroke model for transcriptional activation by ER and p300. In this model ER and p300 function cooperatively to increase productive transcription initiation in the first stroke, whereas ER promotes the reassembly of the transcription preinitiation complex in the second stroke.
Chromatin is an integral component of transcriptional regulation by the ER
Estrogen- and anti-estrogen-regulated transcriptional activation by ER was observed with chromatin templates, but not with nonchromatin templates. These findings reveal the central importance of chromatin structure in the function of the ER. Earlier biochemical studies of the transcriptional activity of steroid receptors (e.g., estrogen, glucocorticoid, progestin, and androgen receptors) were generally carried out with nonchromatin templates. In those experiments, the amount of activation was found to correlate with the binding of the factors to the naked DNA templates. In some instances, the receptor bound constitutively to DNA and ligand-independent transcriptional activation was observed (see, e.g., Bagchi et al. 1990a; Elliston et al. 1990; Klein-Hitpass et al. 1990; Tsai et al. 1990; Shemshedini et al. 1992; Schmitt and Stunnenberg 1993; De Vos et al. 1994). In other instances, ligand-dependent transcriptional activation was seen upon ligand-dependent binding of the receptor to DNA (see, e.g., Bagchi et al. 1990b; Elliston et al. 1992; Beekman et al. 1993). In studies with naked DNA templates partial or no antagonism of progesterone receptor or glucocorticoid receptor by antihormones has also been found in situations where the antihormone-receptor complexes remained bound to DNA (Bagchi et al. 1990b; Tsai et al. 1990; Klein-Hitpass et al. 1991; Shemshidini et al. 1992). In addition, the transcriptional activity of ER + E2 was examined with chromatin templates that were reconstituted by salt–urea dialysis (Schild et al. 1993). In that work, a greater extent (sevenfold) of transcriptional activation by ER + E2 was observed with chromatin templates than with nonchromatin templates, but it was not shown whether or not transcription was regulated by estrogens and anti-estrogens.
In this study we have analyzed the properties of purified human ER with periodic nucleosome arrays. Transcriptional activity of the ER was observed to be regulated by estrogens and anti-estrogens (Figs. 2 and 3), as well as to be abolished by a single amino acid substitution (Leu-540 to Gln) in AF-2 (Fig. 3). In addition, the coactivator p300 was able to enhance transcriptional activation by ER with chromatin, but not with nonchromatin, templates (Fig. 5). These results suggest that the role of chromatin in the function of nuclear receptors is more specific than that of a general transcriptional repressor. It will be interesting to determine, especially with regard to the structure and function of the core histones, whether or not there are specific, functionally important interactions between ligand-activated receptors and components of chromatin.
Relation between binding of ER to chromatin and transcriptional activation
The binding of purified recombinant ER to chromatin was found to occur efficiently in the presence or the absence of estrogens or anti-estrogens (Fig. 4). These findings are generally consistent with the results of previous experiments on the binding of ER (in the absence of associated heat shock proteins) to naked DNA in vitro (see, e.g., Murdoch et al. 1990; Furlow et al. 1993; Metzger et al. 1995; Cheskis et al. 1997). We do not, however, extrapolate these data to the behavior of ER in vivo, as these experiments with free polypeptide ER do not involve issues such as the association of heat shock proteins with ER and nuclear localization. Nevertheless, these studies do indicate that transcriptionally inactive ER (i.e., unliganded, anti-estrogen-bound, or AF-2 mutant version ER) can bind to chromatin templates. Thus, the binding of ER to chromatin is necessary (Fig. 3) but not sufficient (Fig. 4) for transcriptional activation. These findings are in contrast to earlier results obtained in biochemical studies of steroid receptors that did not distinguish between DNA binding and transcriptional activation (as discussed above). These experiments therefore provide biochemical evidence for the importance of the proper estrogen-induced, transcriptionally active conformation of chromatin-bound ER to achieve gene activation. It is also notable that these data are consistent with the observation that DNA binding by the progesterone receptor is not sufficient to mediate transcriptional activation in cultured cells (Mymryk and Archer 1995).
ER promotes transcription reinitiation with chromatin templates
An important component of transcriptional activation by ER is its ability to promote transcription reinitiation. Upon activation by ER + E2, there was a >10-fold increase in the number of rounds of transcription (from ∼3 to ∼35 rounds) (Fig. 6). Because transcriptional activation by the ER occurs with chromatin but not with nonchromatin templates, we suggest that chromatin provides an environment that is conducive to multiple cycles of transcription, such as that mediated by the ER. It has also been found that ER interacts with TBP, hTAFII30, hTAFII28, and TFIIB (Ing et al. 1992; Jacq et al. 1994; Sadovsky et al. 1995; Beato and Sánchez-Pacheo 1996; May et al. 1996). Thus, the results collectively suggest that there might be a complex containing ER and TFIID and/or TFIIB that remains bound at the promoter for multiple cycles of transcription and facilitates reinitiation.
It is interesting to note that we consistently observed ∼33–39 rounds of transcription by E2-induced ER in a 30-min transcription reaction, which corresponds to greater than one round of transcription per minute. By comparison, only about two to five rounds of transcription are typically seen under comparable conditions with HeLa extracts and naked DNA templates (see, e.g., Hawley and Roeder 1987; White et al. 1992; Yean and Gralla 1997). In addition, the transcriptional activator Gal4–VP16 was found to increase the amount of transcription reinitiation only by a factor of ∼1.4 (7.2 rounds in the presence of Gal4–VP16 and 5.2 rounds in the absence of Gal4–VP16 in a 45-min reaction) with nonchromatin templates (White et al. 1992). Hence, the number of cycles of transcription that is achieved by ER with the chromatin templates is significantly higher than that seen previously with other transcription systems.
The coactivator function of p300 with ligand- activated ER
Estrogen-induced ER and p300 act in a cooperative fashion to increase the efficiency of productive transcription initiation (Figs. 5 and 6). p300-Mediated transcriptional enhancement is dependent on ligand-activated ER as well as a chromatin template. In the absence of ER, p300 has little or no effect on basal transcription and therefore, does not stimulate transcription nonspecifically. It should also be noted that there is endogenous p300/CBP in the HeLa transcription extract used in these experiments and, hence, the amount of transcriptional enhancement by p300 is likely to be higher than that which we have observed.
It is worthwhile to consider how ligand-activated ER and p300 increase the efficiency of transcription initiation (Fig. 5). That is, how might ER and p300 increase the number of transcripts that are generated in a single round of transcription? A few possible explanations, which are not mutually exclusive, are as follows. First, ER and p300 might increase the propensity to assemble a transcription preinitiation complex at a promoter, possibly via interactions with the RNA polymerase II complex (Kee et al. 1996; Nakajima et al. 1997a,b). Second, ER and p300 might increase the ratio of productive (i.e., leading to a transcript that has cleared the promoter) versus nonproductive transcription. It is known, for instance, that there is a significant amount of abortive initiation in transcription by RNA polymerase II (see, e.g., Luse and Jacob 1987). Third, p300 might covalently modify transcription factors and/or components of chromatin by protein acetylation to yield more active transcription factors and/or less repressive chromatin (Bannister and Kouzarides 1996; Ogryzko et al. 1996).
In the future it will be important to extend our understanding of the function of ER and p300. For instance, it will be interesting to examine the role of p300/CBP-associated factor (PCAF), SRC-1, and related factors in this process. In this manner, we hope to obtain an increasingly accurate view of the molecular events that occur during transcriptional activation by ER and other nuclear receptors.
Materials and methods
Purification of ER and p300
The ligand-binding, DNA-binding, and transactivation activities of wild-type and Flag epitope-tagged ERs were compared by Scatchard analyses, gel mobility shift assays, and transient transfection assays, and the amino-terminal Flag tag did not affect the activity of the ERs in these assays (data not shown). Flag-tagged ER and ER(L540Q) were prepared by infection of Sf9 cells with the appropriate recombinant baculovirus followed by immunoaffinity chromatography of a high salt cell extract with an anti-Flag M2 affinity resin (Kodak/IBI). Protein was eluted with 0.2 mg/ml of Flag peptide (Kodak/IBI), frozen in liquid nitrogen, and stored at −80°C. [High salt (0.5 m NaCl) extraction removes associated heat shock proteins from the ER (Bresciani et al. 1990; Pratt et al. 1996).] A 500-ml culture typically yielded ∼7–10 μg of purified ER or ER(L540Q). Ligand-binding and DNA-binding assays were performed as described previously (Pakdel and Katzenellenbogen 1992; Kraus et al. 1994). Western blot assays were carried out with a monoclonal antibody (H-151; Stressgen) directed against the carboxyl-terminus of ER or with a polyclonal antiserum raised against a bacterially expressed, affinity-purified, His6-tagged amino-terminal fragment (amino acids 1–113) of ER.
His6-tagged human p300 was prepared by infection of Sf9 cells with the appropriate recombinant baculovirus followed by affinity chromatography of a whole cell extract with Ni–NTA affinity resin (Qiagen). Protein was eluted with buffer containing 250 mm imidazole, frozen in liquid nitrogen, and stored at −80°C. p300 concentrations and purity were estimated by BCA protein assays (Pierce) and by comparison to BSA standards on SDS gels stained with Coomassie Brilliant Blue R-250. Typical yields were ∼40–50 μg of protein/15-cm-diam. plate.
Chromatin assembly and analysis
pERE contains four tandem copies of the Xenopus vitellogenin A2 gene ERE (Walker et al. 1984) located 67 bp upstream of the adenovirus E4 core promoter in pIE0 (Pazin et al. 1994). Chromatin assembly reactions were performed with the S190 extract derived from Drosophila embryos (Kamakaka et al. 1993; Bulger and Kadonaga 1994). DNA supercoiling and micrococcal nuclease digestion analyses were performed as described (Kamakaka et al. 1993; Bulger and Kadonaga 1994). The binding of ER to chromatin was analyzed by DNase I primer extension footprinting (Pazin et al. 1994).
In a standard chromatin assembly reaction (500 ng of template DNA; 100 μl final volume), S190 (30 μl) was incubated with purified Drosophila core histones (0.7 μg) in a final volume of 55 μl for 30 min at room temperature. In a separate tube, ER (or corresponding buffer, as a control), pERE or pIE0 (500 ng), and ligands (or corresponding buffer, as a control) in a final volume of 30 μl were mixed and incubated on ice for 15 min. Then, the S190–histone mix (55 μl) and the ER–DNA template mix (30 μl) for each reaction were combined with ATP mix (15 μl; 0.2 m phosphocreatine, 20 mm ATP, 650 μg/ml of creatine phosphokinase, 30 mm MgCl2), and the mixture (100 μl) was incubated at 27°C for 4.5 hr. Where specifically indicated, components such as ER, ligand, and/or p300 were not included in the initial assembly reaction, but were added to chromatin after the assembly reactions were complete (after four hours reaction time). In such cases, the reactions were incubated for an additional 30 min at 27°C after the components were added to allow interaction of the factors with the preassembled chromatin. From a single assembly reaction, aliquots of chromatin were removed for DNA supercoiling, micrococcal nuclease digestion, footprinting, and/or transcription analyses. Also, in some cases, the integrity of the ER in the reaction mixtures was monitored by Western blot analysis. In experiments with mock-assembled nonchromatin templates (Figs. 2D and 5C), competitor DNA (1.5 μg of pUC118; a threefold mass excess relative to the specific template DNA) was incubated at 27°C for 30 min with the S190–histone mix (55 μl) and ATP mix (15 μl). Then, the ER–template mix (30 μl; containing 500 ng of pERE) was added, and the reactions were then incubated for an additional 4 hr at 27°C.
In vitro transcription
in vitro transcription reactions were performed with HeLa cell nuclear extracts (Dignam et al. 1983), as modified slightly by the use of 0.42 m KCl for the extraction of nuclei instead of 0.42 m NaCl. Transcription of chromatin (or mock-assembled nonchromatin templates) with the HeLa nuclear extract was performed as described elsewhere (Sheridan et al. 1995; Pazin et al. 1996). Also, in the initial experiments (Figs. 2 and 3), chromatin was assembled in the presence of HMG-17 (10 molecules/180 bp of specific template DNA) (Paranjape et al. 1995). When added at this amount, the HMG-17 increased the E2-dependent activity of the ER with the chromatin templates by a factor of ∼1.5–2. All reaction conditions were performed in duplicate, and each experiment was performed a minimum of two separate times to ensure reproducibility. Quantitation of the data was carried out with a PhosphorImager (Molecular Dynamics).
Acknowledgments
We thank M. Pazin, A. Kutach, and J. Tyler for critical reading of the manuscript. We are most grateful to B. Katzenellenbogen for the ER cDNAs and ligands. W.L.K. is the recipient of a National Institutes of Health (NIH) postdoctoral fellowship (National Research Service Award DK09275). This work was supported by a grant from the NIH (GM46995) to J.T.K.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jkadonaga@ucsd.edu; FAX (619) 534-0555.
References
- Akimaru H, Chen Y, Dai P, Hou D-X, Nonaka M, Smolik SM, Armstrong S, Goodman RH, Ishii S. Drosophila CBP is a co-activator of cubitus interruptus in hedgehog signalling. Nature. 1997;386:735–738. doi: 10.1038/386735a0. [DOI] [PubMed] [Google Scholar]
- Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan X-Y, Sauter G, Kallioniemi O-P, Trent JM, Meltzer PS. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–968. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- Bagchi MK, Tsai SY, Weigel NL, Tsai M-J, O’Malley BW. Regulation of in vitro transcription by progesterone receptor. J Biol Chem. 1990a;265:5129–5134. [PubMed] [Google Scholar]
- Bagchi MK, Tsai SY, Tsai M-J, O’Malley BW. Identification of a functional intermediate in receptor activation in progesterone-dependent cell-free transcription. Nature. 1990b;345:547–550. doi: 10.1038/345547a0. [DOI] [PubMed] [Google Scholar]
- Bannister A, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Eckner R, Grossman S, Oldread E, Arany Z, D’Andrea A, Livingston DM. Cooperation of Stat2 and p300/CBP in signalling induced by interferon alpha. Nature. 1996;383:344–347. doi: 10.1038/383344a0. [DOI] [PubMed] [Google Scholar]
- Beato M, Sánchez-Pacheo A. Interaction of steroid hormone receptors with the transcription initiation complex. Endocr Rev. 1996;17:587–609. doi: 10.1210/edrv-17-6-587. [DOI] [PubMed] [Google Scholar]
- Beato M, Herrlich P, Schütz G. Steroid hormone receptors: Many actors in search of a plot. Cell. 1995;83:851–857. doi: 10.1016/0092-8674(95)90201-5. [DOI] [PubMed] [Google Scholar]
- Beato M, Chavez S, Truss M. Transcriptional regulation by steroid hormones. Steroids. 1996;61:240–251. doi: 10.1016/0039-128x(96)00030-x. [DOI] [PubMed] [Google Scholar]
- Beekman JM, Allan GF, Tsai SY, Tsai M-J, O’Malley BW. Transcriptional activation by the estrogen receptor requires a conformational change in the ligand-binding domain. Mol Endocrinol. 1993;7:1266–1274. doi: 10.1210/mend.7.10.8264659. [DOI] [PubMed] [Google Scholar]
- Bresciani F, Medici N, Abbondanza C, Moncharmont B, Puca GA. Receptor purification. Vol. 2. Clifton, NJ: Humana Press; 1990. Purification of the estrogen receptor; pp. 181–192. [Google Scholar]
- Bulger M, Kadonaga JT. Biochemical reconstitution of chromatin with physiological nucleosome spacing. Methods Mol Genet. 1994;5:241–262. [Google Scholar]
- Chakravarti D, LaMorte VJ, Nelson MC, Nakajima T, Schulman IG, Juguilon H, Montminy M, Evans RM. Role of CBP/p300 in nuclear receptor signalling. Nature. 1996;383:99–103. doi: 10.1038/383099a0. [DOI] [PubMed] [Google Scholar]
- Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y, Evans RM. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell. 1997;90:569–580. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- Cheskis BJ, Karathanasis S, Lyttle CR. Estrogen receptor ligands modulate its interaction with DNA. J Biol Chem. 1997;272:11384–11391. doi: 10.1074/jbc.272.17.11384. [DOI] [PubMed] [Google Scholar]
- Dai P, Akimaru H, Tanaka Y, Hou D-X, Yasukawa T, Kanei-Ishii C, Takahashi T, Ishii S. CBP as a transcriptional coactivator of c-Myb. Genes & Dev. 1996;10:528–540. doi: 10.1101/gad.10.5.528. [DOI] [PubMed] [Google Scholar]
- Danielian PS, White R, Lees JA, Parker MG. Identification of a conserved region required for hormone dependent transcriptional activation by steroid hormone receptors. EMBO J. 1992;11:1025–1033. doi: 10.1002/j.1460-2075.1992.tb05141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos P, Schmitt J, Verhoeven G, Stunnenberg HG. Human androgen receptor expressed in HeLa cells activates transcription in vitro. Nucleic Acids Res. 1994;22:1161–1166. doi: 10.1093/nar/22.7.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckner R, Yao T-P, Oldread E, Livingston DM. Interaction and functional collaboration of p300/CBP and bHLH proteins in muscle and B-cell differentiation. Genes & Dev. 1996;10:2478–2490. doi: 10.1101/gad.10.19.2478. [DOI] [PubMed] [Google Scholar]
- Elliston JF, Fawell SE, Klein-Hitpass L, Tsai SY, Tsai M-J, Parker MG, O’Malley BW. Mechanism of estrogen receptor-dependent transcription in a cell-free system. Mol Cell Biol. 1990;10:6607–6612. doi: 10.1128/mcb.10.12.6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliston JF, Beekman JM, Tsai SY, O’Malley BW, Tsai M-J. Hormone activation of baculovirus expressed progesterone receptors. J Biol Chem. 1992;267:5193–5198. [PubMed] [Google Scholar]
- Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlow JD, Murdoch FE, Gorski J. High affinity binding of the estrogen receptor to a DNA response element does not require homodimer formation or estrogen. J Biol Chem. 1993;268:12519–12525. [PubMed] [Google Scholar]
- Glass CK. Differential recognition of target genes by nuclear receptor monomers, dimers, and heterodimers. Endocrine Rev. 1994;15:391–407. doi: 10.1210/edrv-15-3-391. [DOI] [PubMed] [Google Scholar]
- Gralla JD. Rapid “footprinting” on supercoiled DNA. Proc Natl Acad Sci. 1985;82:3078–3081. doi: 10.1073/pnas.82.10.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronemeyer H. Transcription activation by estrogen and progesterone receptors. Annu Rev Genet. 1991;25:89–123. doi: 10.1146/annurev.ge.25.120191.000513. [DOI] [PubMed] [Google Scholar]
- Hanstein B, Eckner R, Direnzo J, Halachmi S, Jiu H, Searcy B, Kurokawa R, Brown M. p300 is a component of an estrogen receptor coactivator complex. Proc Natl Acad Sci. 1996;93:11540–11545. doi: 10.1073/pnas.93.21.11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley DK, Roeder RG. Separation and partial characterization of three functional steps in transcription initiation by human RNA polymerase II. J Biol Chem. 1985;260:8163–8172. [PubMed] [Google Scholar]
- ————— Functional steps in transcription initiation and reinitiation from the major late promoter in a HeLa nuclear extract. J Biol Chem. 1987;262:3452–3461. [PubMed] [Google Scholar]
- Hong H, Kohli K, Trivedi A, Johnson D, Stallcup MR. GRIP1, a novel mouse protein that serves as a transcriptional coactivator in yeast for the hormone binding domains of steroid hormone receptors. Proc Natl Acad Sci. 1996;93:4948–4952. doi: 10.1073/pnas.93.10.4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H, Kohli K, Garabedian MJ, Stallcup MR. GRIP1, a transcriptional coactivator for the AF-2 transactivation domain of steroid, thyroid, retinoid, and vitamin D receptors. Mol Cell Biol. 1997;17:2735–2744. doi: 10.1128/mcb.17.5.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz KB, Jackson TA, Bain DL, Richer JK, Takimoto GS, Tung L. Nuclear receptor coactivators and corepressors. Mol Endocrinol. 1996;10:1167–1177. doi: 10.1210/mend.10.10.9121485. [DOI] [PubMed] [Google Scholar]
- Ing NH, Beekman JM, Tsai SY, Tsai M-J, O’Malley BW. Members of the steroid hormone receptor superfamily interact with TFIIB (S300-II) J Biol Chem. 1992;267:17617–17623. [PubMed] [Google Scholar]
- Jacq X, Brou C, Lutz Y, Davidson I, Chambon P, Tora L. Human TAFII30 is present in a distinct TFIID complex and is required for transcriptional activation by the estrogen receptor. Cell. 1994;79:107–117. doi: 10.1016/0092-8674(94)90404-9. [DOI] [PubMed] [Google Scholar]
- Kadonaga JT. Assembly and disassembly of the Drosophila RNA polymerase II complex during transcription. J Biol Chem. 1990;265:2624–2631. [PubMed] [Google Scholar]
- Kamakaka RT, Bulger M, Kadonaga JT. Potentiation of RNA polymerase II transcription by Gal4–VP16 during but not after DNA replication and chromatin assembly. Genes & Dev. 1993;7:1779–1795. doi: 10.1101/gad.7.9.1779. [DOI] [PubMed] [Google Scholar]
- Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin S-C, Heyman RA, Rose DW, Glass CK, Rosenfeld MG. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- Kee BL, Arias J, Montminy MR. Adaptor-mediated recruitment of RNA polymerase II to a signal-dependent activator. J Biol Chem. 1996;271:2373–2375. doi: 10.1074/jbc.271.5.2373. [DOI] [PubMed] [Google Scholar]
- Klein-Hitpass L, Tsai SY, Weigel NL, Allan GF, Riley D, Rodriguez R, Schrader WT, Tsai M-J, O’Malley BW. The progesterone receptor stimulates cell-free transcription by enhancing the formation of a stable preinitiation complex. Cell. 1990;60:247–257. doi: 10.1016/0092-8674(90)90740-6. [DOI] [PubMed] [Google Scholar]
- Klein-Hitpass L, Cato ACB, Henderson D, Ryffel GU. Two types of antiprogestins identified by their differential action in transcriptionally active extracts from T47D cells. Nucleic Acids Res. 1991;19:1227–1234. doi: 10.1093/nar/19.6.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus WL, Montano MM, Katzenellenbogen BS. Identification of multiple, widely spaced estrogen-responsive regions in the rat progesterone receptor gene. Mol Endocrinol. 1994;8:952–969. doi: 10.1210/mend.8.8.7997237. [DOI] [PubMed] [Google Scholar]
- Kraus WL, McInerney EM, Katzenellenbogen BS. Ligand-dependent, transcriptionally productive association of the amino- and carboxyl-terminal regions of a steroid hormone nuclear receptor. Proc Natl Acad Sci. 1995;92:12314–12318. doi: 10.1073/pnas.92.26.12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok RPS, Lundblad JR, Chrivia JC, Richards JP, Bachinger HP, Brennan RG, Roberts SGE, Green MR, Goodman RH. Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature. 1994;370:223–226. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- Li H, Gomes PJ, Chen JD. RAC3, a steroid/nuclear receptor-associated coactivator that is related to SRC-1 and TIF2. Proc Natl Acad Sci. 1997;94:8479–8484. doi: 10.1073/pnas.94.16.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luse DS, Jacob GA. Abortive initiation by RNA polymerase II in vitro at the adenovirus 2 major late promoter. J Biol Chem. 1987;262:14990–14997. [PubMed] [Google Scholar]
- Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: The second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May M, Mengus G, Lavigne A-C, Chambon P, Davidson I. Human TAFII28 promotes transcriptional stimulation by activation function 2 of the retinoid X receptors. EMBO J. 1996;15:3093–3104. [PMC free article] [PubMed] [Google Scholar]
- McDonnell DP, Clemm DL, Imhof MO. Definition of the cellular mechanisms which distinguish between hormone and antihormone activated receptors. Semin Cancer Biol. 1994;5:327–336. [PubMed] [Google Scholar]
- McDonnell DP, Clemm DL, Hermann T, Goldman ME, Pike JW. Analysis of estrogen receptor function in vitro reveals three distinct classes of antiestrogens. Mol Endocrinol. 1995;9:659–669. doi: 10.1210/mend.9.6.8592512. [DOI] [PubMed] [Google Scholar]
- Metzger D, Berry M, Ali S, Chambon P. Effects of antagonists on DNA binding properties of the human estrogen receptor in vitro and in vivo. Mol Endocrinol. 1995;9:579–591. doi: 10.1210/mend.9.5.7565805. [DOI] [PubMed] [Google Scholar]
- Murdoch FE, Meier DA, Furlow JD, Grunwald KAA, Gorski J. Estrogen receptor binding to a DNA response element in vitro is not dependent upon estradiol. Biochemistry. 1990;29:8377–8385. doi: 10.1021/bi00488a026. [DOI] [PubMed] [Google Scholar]
- Mymryk JS, Archer TK. Dissection of progesterone receptor-mediated chromatin remodeling and transcriptional activation in vivo. Genes & Dev. 1995;9:1366–1376. doi: 10.1101/gad.9.11.1366. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Uchida C, Anderson SF, Parvin JD, Montminy M. Analysis of a cAMP-responsive activator reveals a two component mechanism for induction via signal-dependent factors. Genes & Dev. 1997a;11:738–747. doi: 10.1101/gad.11.6.738. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Uchida C, Anderson SF, Lee C-G, Hurwitz J, Parvin JD, Montminy M. RNA helicase A mediates association of CBP with RNA polymerase II. Cell. 1997b;90:1107–1112. doi: 10.1016/s0092-8674(00)80376-1. [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- Oliner JD, Andresen JM, Hansen SK, Zhou S, Tjian R. SREBP transcriptional activity is mediated through an interaction with the CREB-binding protein. Genes & Dev. 1996;10:2903–2911. doi: 10.1101/gad.10.22.2903. [DOI] [PubMed] [Google Scholar]
- Oñate SA, Tsai SY, Tsai M-J, O’Malley BW. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science. 1995;270:1354–1357. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- Pakdel F, Katzenellenbogen BS. Human estrogen receptor mutants with altered estrogen and antiestrogen ligand discrimination. J Biol Chem. 1992;267:3429–3437. [PubMed] [Google Scholar]
- Paranjape SM, Krumm A, Kadonaga JT. HMG-17 is a chromatin-specific transcriptional coactivator that increases the efficiency of transcription initiation. Genes & Dev. 1995;9:1978–1991. doi: 10.1101/gad.9.16.1978. [DOI] [PubMed] [Google Scholar]
- Pazin MJ, Kamakaka RT, Kadonaga JT. ATP-dependent nucleosome reconfiguration and transcriptional activation from preassembled chromatin templates. Science. 1994;266:2007–2011. doi: 10.1126/science.7801129. [DOI] [PubMed] [Google Scholar]
- Pazin MJ, Sheridan PL, Cannon K, Cao Z, Keck JG, Kadonaga JT, Jones KA. NF-κB-mediated chromatin reconfiguration and transcriptional activation of the HIV-1 enhancer in vitro. Genes & Dev. 1996;10:37–49. doi: 10.1101/gad.10.1.37. [DOI] [PubMed] [Google Scholar]
- Pratt WB, Gehring U, Toft DO. Stress-inducible cellular responses. Basel, Switzerland: Birkhäuser Verlag; 1996. Molecular chaperoning of steroid hormone receptors. [DOI] [PubMed] [Google Scholar]
- Reichel RR, Jacob ST. Control of gene expression by lipophilic hormones. FASEB J. 1993;7:427–436. doi: 10.1096/fasebj.7.5.8385039. [DOI] [PubMed] [Google Scholar]
- Sadovsky Y, Webb P, Lopez G, Baxter JD, Fitzpatrick PM, Gizang-Ginsberg E, Cavailles V, Parker MG, Kushner PJ. Transcriptional activators differ in their responses to overexpression of TATA-box-binding protein. Mol Cell Biol. 1995;15:1554–1563. doi: 10.1128/mcb.15.3.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen RJ, Manni A, Harvey H, Redmond C. Endocrine treatment of breast cancer in women. Endocrine Rev. 1990;11:221–265. doi: 10.1210/edrv-11-2-221. [DOI] [PubMed] [Google Scholar]
- Schild C, Claret F-X, Wahli W, Wolffe AP. A nucleosome-dependent static loop potentiates estrogen regulated transcription from the Xenopus vitellogenin B1 promoter in vitro. EMBO J. 1993;12:423–433. doi: 10.1002/j.1460-2075.1993.tb05674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt J, Stunnenberg HG. The glucocorticoid receptor hormone binding domain mediates transcriptional activation in the absence of ligand. Nucleic Acids Res. 1993;11:2673–2681. doi: 10.1093/nar/21.11.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemshedini L, Ji J, Brou C, Chambon P, Gronemeyer H. In vitro activity of the transcription activation functions of the progesterone receptor. J Biol Chem. 1992;267:1834–1839. [PubMed] [Google Scholar]
- Sheridan PL, Sheline CT, Cannon K, Voz ML, Pazin MJ, Kadonaga JT, Jones KA. Activation of the HIV-1 enhancer by the LEF-1 HMG protein on nucleosome-assembled DNA in vitro. Genes & Dev. 1995;9:2090–2104. doi: 10.1101/gad.9.17.2090. [DOI] [PubMed] [Google Scholar]
- Tora L, White J, Brou C, Tasset D, Webster N, Scheer E, Chambon P. The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell. 1989;59:477–487. doi: 10.1016/0092-8674(89)90031-7. [DOI] [PubMed] [Google Scholar]
- Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature. 1997;387:677–684. doi: 10.1038/42652. [DOI] [PubMed] [Google Scholar]
- Truss M, Beato M. Steroid hormone receptors: Interactions with deoxyribonucleic acid and transcription factors. Endocrine Rev. 1993;14:459–479. doi: 10.1210/edrv-14-4-459. [DOI] [PubMed] [Google Scholar]
- Tsai SY, Srinivasan G, Allan GF, Thompson EB, O’Malley BW, Tsai M-J. Recombinant human glucocorticoid receptor induces transcription of hormone response genes in vitro. J Biol Chem. 1990;265:17055–17061. [PubMed] [Google Scholar]
- Tzukerman MT, Esty A, Santiso-Mere D, Danielian P, Parker MG, Stein RB, Pike JW, McDonnell DP. Human estrogen receptor transactivational capacity is determined by both cellular and promoter context and mediated by two functionally distinct intramolecular regions. Mol Endocrinol. 1994;8:21–30. doi: 10.1210/mend.8.1.8152428. [DOI] [PubMed] [Google Scholar]
- Voegel JJ, Heine MJS, Zechel C, Chambon P, Gronemeyer H. TIF2, a 160 kDa transcriptional mediator for the ligand-dependent activation function AF-2 of nuclear receptors. EMBO J. 1996;15:3667–3675. [PMC free article] [PubMed] [Google Scholar]
- Wakeling AE. Novel pure antiestrogens: Mode of action and therapeutic prospects. Ann NY Acad Sci. 1990;595:384–356. doi: 10.1111/j.1749-6632.1990.tb34308.x. [DOI] [PubMed] [Google Scholar]
- Walker P, Germond J-E, Brown-Luedi M, Givel F, Wahli W. Sequence homologies in the region preceding the transcription initiation site of the liver estrogen-responsive vitellogenin and apo-VLDLII genes. Nucleic Acids Res. 1984;12:8611–8626. doi: 10.1093/nar/12.22.8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster NJG, Green S, Jin JR, Chambon P. The hormone-binding domains of the estrogen and glucocorticoid receptors contain an inducible transcription activation function. Cell. 1988;54:199–207. doi: 10.1016/0092-8674(88)90552-1. [DOI] [PubMed] [Google Scholar]
- White J, Brou C, Wu J, Lutz Y, Moncollin V, Chambon P. The acidic transcriptional activator GAL-VP16 acts on preformed template-committed complexes. EMBO J. 1992;11:2229–2240. doi: 10.1002/j.1460-2075.1992.tb05282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrenn CK, Katzenellenbogen BS. Structure-function analysis of the hormone binding domain of the human estrogen receptor by region-specific mutagenesis and phenotypic screening in yeast. J Biol Chem. 1993;268:24089–24098. [PubMed] [Google Scholar]
- Wurtz J-M, Bourguet W, Renaud J-P, Vivat V, Chambon P, Moras D, Gronemeyer H. A canonical structure for the ligand-binding domain of nuclear receptors. Nature Struct Biol. 1996;3:87–94. doi: 10.1038/nsb0196-87. [DOI] [PubMed] [Google Scholar]
- Yean D, Gralla JD. Transcription reinitiation rate: A special role for the TATA box. Mol Cell Biol. 1997;17:3809–3816. doi: 10.1128/mcb.17.7.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]