

Abstract
The rational design of DNA/RNA aptamers for use as molecular probes depends on a clear understanding of their structural elements in relation to target-aptamer binding interactions. We present a simple method to create aptamer probes that can occupy two different structural states. Then, based on the difference in binding affinity between these states, target-aptamer binding interactions can be elucidated. The basis of our two-state system comes from the incorporation of azobenzene within the DNA strand. Azobenzene can be used to photo-regulate the melting of DNA-duplex structures. When incorporated into aptamers, the light-regulated conformational change of azobenzene can be used to analyze how aptamer secondary structure is involved in target binding. Azobenzene modified aptamers showed no change in target selectivity, but showed differences in binding affinity as a function of the number, position, and conformation of azobenzene modifications. Aptamer probes that can change binding affinity on demand may have future uses in targeted drug delivery and photodynamic therapy.
INTRODUCTION
To use DNA/RNA aptamers as biosensors, it is necessary to engineer functional capabilities into the aptamer molecular structures. This engineering is facilitated when there is a clear understanding of the structural elements of the aptamer and the aptamer-target binding interaction. For example, structural information has been used to engineer biosensors for molecular targets including ions, small molecules, and proteins (1–8). The use of these robust biosensor designs is limited to targets for which the aptamer secondary structure is easily predictable or atomic structures are known. The most common targets being potassium ion (K+), adenosine triphosphate (ATP), cocaine, thrombin, and platelet derived growth factor (PDGF). The atomic structures that have been solved are primarily RNA-aptamers with small molecule targets (9–10). The only existing aptamer-protein structures are the thrombin, nucleolin, and NF-κB aptamer-protein complexes, of which, thrombin is the only protein that has not been shown to naturally bind nucleotides (11–16). This lack of structural information for most aptamers severely limits the application of current biosensor designs and the development of new biosensor designs. Therefore, a method that can yield information about how the secondary structure of an aptamer relates to protein binding would facilitate the implementation of aptamer biosensor designs.
The structural properties of molecules can be controlled by conjugation with azobenzene moieties. This control is based on the ability of azobenzene molecules to switch between two conformations, cis- or trans-, after absorbing ultraviolet (UV) or visible light, respectively. Azobenzene has been used to photo-regulate molecular interactions for diverse applications ranging from building molecular motors to regulation of gene translation (17–20). In particular, when covalently inserted into the phosphate backbone of DNA, azobenzene moieties have been shown to reversibly regulate the conversion from dsDNA to ssDNA as well as DNA triplex structures (21–22). The duplex regulation is based on the stabilization of the dsDNA by trans-azobenzene, which intercalates within dsDNA and makes stacking interactions that offset the change in conformation of the double helix (23). Some groups are using this structure switching ability to create DNA nanomachines and chemical switches powered by light (24–30). In this work, we incorporate azobenzene into the phosphate backbone of an aptamer and use the light-regulated structural change of azobenzene to analyze how the predicted secondary structure of the aptamer is involved in protein binding.
Previous aptamer research from our lab has shown that the minimal sequence needed for the Sgc8 aptamer to bind to CCRF-CEM T-cell Acute Lymphoblastic Leukemia (T-ALL) cells has a predicted hairpin structure with 8 bases in the stem and 24 bases in the loop (31–32). This truncated sequence was named Sgc8c. Although this hairpin has a predicted melting temperature of 46°C, it is not possible to increase the temperature beyond 37°C to test whether the melting of this stem structure affects cell binding. Experiments of this nature would lead to artifacts as cells react biologically to increasing temperature, which could lead to different protein expression. Therefore, the relevance of this hairpin structure to the protein binding interaction could not be probed directly. The protein target for this aptamer was found to be Protein Tyrosine Kinase-like 7 (PTK7) and this protein is up-regulated in various cancer cell lines (33). Some information about the structure of the Sgc8c/PTK7 binding interaction can be ascertained from the literature. First, this aptamer does not compete with the binding of PTK7 antibodies that bind to a region between amino acids 36–146 (33). Second, Sgc8c has been used in many biosensor applications, and is frequently immobilized at the 5′ or 3′ end to nanoparticles and microfluidic channels, indicating that the ends of the aptamer do not directly interact with the protein target (34–37). These previous results suggest that the predicted stem-loop structure Sgc8c is important for protein binding.
In this work, we inserted azobenzene at various positions within the predicted stem and loop of Sgc8c as depicted in the schematic. Our intent was to probe the necessity of the stem-loop structure and learn more about the aptamer-protein interaction. We saw a clear decrease in affinity between the UV and visible states of probes that had azobenzene in the stem region. Further, azobenzene modifications around the T-G mismatch near the stem-loop intersection and in the GC-rich portion of the loop resulted in significant changes in binding affinity.
MATERIALS AND METHODS
Azobenzene phosphoramidite monomer synthesis
D-Threoninol was chosen as the linker for synthesizing optically pure diols based on the different properties from L- conformation. The synthesis routes are similar to published protocols (Supporting Information Scheme S1) (38). Compound 1. In a round-bottom flask, a solution of D-threoninol (0.91 g, 9.0 mmol), 4-(phenylazo) benzoic acid (2.25 g, 10.0 mmol), Dicyclohexyl Carbodiimide (2.05 g, 10.0 mmol), and hydroxybenzotriazole (1.32 g, 10.0 mmol) in dimethylformamide (50 mL) was stirred under an argon atmosphere at room temperature for 24 h. The reaction mixture was filtered and then concentrated. The residue was purified by column chromatography (ethyl acetate/methanol 20/1) and dried to afford compound 1 (2.34 g, 7.48 mmole, 83%) as an orange solid. 1H NMR (CDCl3): δ 7.96–7.38 (m, 9H), δ 7.12 (d, 1H), δ 4.33 (m, 1H), δ 4.09 (m, 1H), δ 3.98 (d, 2H), δ 1.29 (d, 3H). Compound 2. To a solution containing compound 1 (0.8 g, 2.4 mmol) and 4-dimethylaminopyridine (0.015 g, 0.12 mmol) in dry pyridine (10 mL) at 0 °C, 3-[2-(dimethylamino)ethyl]indole-Cl (1.0 g, 3.0 mmol) in CH2Cl2 (4 mL) was added dropwise. The mixture was stirred for 1 h at 0 °C and then at room temperature for another 24 h. The solvent was evaporated, and the residue was an orange-red oil, which was purified by column chromatography (ethyl acetate/hexane/triethylamine 50:50:3) and dried to afford compound 2 (0.76 g, 1.24 mmol, 52%) as an orange-red solid. 1H NMR (CDCl3): δ 8.00–6.78 (m, 23H), δ 4.25 (m, 1H), δ 4.17 (m, 1H), δ 3.77 (s, 6H), δ 3.60 and 3.42 (dd, 2H), 1.23 (d, 3H). Compound 3. To a solution containing compound 2 (0.62 g, 1.0 mmol) in anhydrous CH3CN (20 mL) at 0 °C, N, N' Diisopropylethylamine (0.39 g, 3.0 mmol) was added in 15 minutes. Then, 2-cyanoethyl diisopropyl chlorophosphoramidite (290 μl, 1.3 mmol) was added dropwise, and the reaction mixture was stirred at 0 °C for 5 h. After removing the solvent, the residue was dissolved in ethyl acetate, and the organic phase was washed with NaHCO3 standard solution and NaCl solution and dried over anhydrous magnesium sulfate. The solvent was evaporated, and the residue was purified by column chromatography (ethyl acetate/hexane/triethylamine 40:60:3), and dried to afford the title compound (0.52 g, 0.64 mmol, 64%) as orange red solid. 1H NMR (CDCl3): δ 8.00–6.79 (m, 22H), δ 6.62 (d, 1H), δ 4.48 (m, 1H), δ 4.39 (m, 1H), δ 4.21–4.10 (m, 2H), δ 3.77 (s, 6H), δ 3.57–3.34 (m, 4H), δ 2.76–2.72 (m, 2H), δ 1.30–1.25 (m, 15H). 31P (CDCl3): δ 149.
Synthesis and Purification of DNA
DNA sequences were synthesized using a traditional DNA/RNA synthesizer ABI3400 (Applied Biosystems) at 1 μmole scale. 3′Biotin-CPG (Controlled Pore Glass) was purchased from ChemGenes Corp. (Wilmington, MA). A proper amount of azobenzene phosphoramidite was dissolved in dry acetonitrile in a vial connected to the synthesizer (20 mg of azobenzene monomer can make a single incorporation in the DNA at 1.0 μmole scale synthesis). The azobenzene-tethered monomer is regarded as a normal base for insertion in programming the synthesizer. A coupling program of longer reaction time was applied to couple the Azo-moieties. After the synthesis, the CPG substrate was transferred to a glass vial, and standard AMA (ammonium hydroxide: methylamine = 1:1) deprotection solutions were added and incubated in a water bath at 50 °C for 12 hours. After centrifuge to separate the CPG beads from DNA in the solution, the clear supernatant was carefully collected. The DNA was then concentrated by ethanol precipitation. The precipitate was dissolved in triethylammonium acetate solution and delivered to reverse phase High Pressure Liquid Chromatography (HPLC) using a C18 column with a linear elution. The collected product was vacuum dried, detritylated, and stored at −20°C for future use. HPLC was performed with a ProStar HPLC (Varian) using a C18 column (Econosil, 5U, 250 x 4.6 mm) from Alltech Associates. To measure the concentration of azobenzene modified DNA, the extinction coefficient of azobenzene modified DNA was calculated by adding 4100 M−1cm−1 for each azobenzene moiety to the extinction coefficient of the original DNA sequence. UV-Vis measurements were performed with a Cary Bio-30 UV spectrometer (Varian).
Cell Culture and Buffers
CCRF-CEM (CCL-119, T cell line, human ALL) and Ramos (CRL-1596, Burkitt’s lymphoma) cell lines were obtained from American Type Culture Collection. Both cell types were cultured in RPMI medium 1640 (American Type Culture Collection) supplemented with 10% Fetal Bovine Serum (FBS) (heat-inactivated; GIBCO) and 100 units/mL penicillin-streptomycin (Cellgro). Cultures were routinely tested for Mycoplamsa infection. Immediately before experiments, cells were harvested, rinsed via centrifugation at 450 x g with washing buffer [4.5 g/L glucose and 5 mM MgCl2 in Dulbecco’s Phosphate Buffered Saline (PBS) with calcium chloride and magnesium chloride (Sigma)], and re-suspended at 12,500,000 cell/mL in binding buffer [washing buffer supplemented with yeast tRNA (0.1 mg/mL; Sigma), Bovine Serum Albumin (BSA) (1mg/mL; Fisher), and 10% FBS]. Cell concentrations were determined using a hemacytometer (Hauser Scientific).
Flow cytometry
A two-step procedure was used to label the cells with aptamers and then Streptavidin-Phycoerythrin (Streptavidin-PE) (Invitrogen). Slight modifications to our previous protocols were needed to illuminate the probes prior to cell binding. Since aptamer secondary structure may inhibit conformational changes of azobenzene-moieties upon illumination due to steric hindrance, aptamers were stored at low salt conditions, i.e. in de-ionized water. Since cells cannot be exposed to low salt conditions, aptamer solutions were stored as 10X solutions and diluted to the final concentration only when mixed with cells in binding buffer immediately after illumination. At the start of the experiment, 20 μL aliquots of 10X aptamer stock solutions were transferred to round-bottom falcon tubes (BD Biosciences) and positioned on top of a UVGL-58 UV lamp (UVP, LLC Upland, CA) emitting 366 nm light for 20 minutes or in front of a common green laser pointer for 5 minutes. During the 20 minute UV illumination, cells were harvested and re-suspended to a final concentration of 12,500,000 cell/mL in binding buffer as described in the Cell culture and buffers section. Immediately after illumination, 80 μL of cell solution containing 1,000,000 cells was added to each tube, vortexed, and incubated for 20 minutes on ice in the dark. After rinsing twice via centrifugation with washing buffer, cells were incubated with Streptavidin-PE for 20 minutes according to manufacturer’s instructions. After a final rinse in washing buffer, cells were re-suspended in 200 uL binding buffer and analyzed using a FACScan cytometer (BD Immunocytometry Systems). For each sample, a non-binding control aptamer was used to set the voltage and gain on the photomultiplier tube for channel 2 such that the entire background histogram was clearly visible and had a mean of roughly 2–3 fluorescence units. After 10,000 counts were measured, the mean fluorescence intensity was recorded for each sample.
Determination of dissociation constants
To reduce errors from variations in ambient light and cell culturing conditions, the dissociation constants for the cis-aptamer, trans-aptamer, and non-binding control aptamer were measured simultaneously on the same batch of cells. For accurate curve fitting, it was also beneficial to perform each data point in duplicate. Therefore, we created binding curves by incubating duplicates of 6 different concentrations of cis-aptamer, trans-aptamer, and control aptamer on one batch of cells. Since modified aptamers varied significantly in their binding affinities, preliminary experiments were performed on each trans-aptamer to estimate the dissociation constant and determine a suitable range of concentrations with which to create final binding curves. For the preliminary experiments, trans-aptamers were incubated with cells at 1, 5, 20, 100, and 500 nM. Based on these results, 6 concentrations were chosen to create the final binding curve for each aptamer. Typical concentrations for high affinity aptamers were 1, 2, 5, 10, 20, and 250 nM and for low affinity aptamers were 50, 100, 200 500, 1000, and 2000 nM. Serial dilutions of the cis-, trans-, and control aptamers at each concentration were incubated with cells and measured using flow cytometry. For each concentration the mean fluorescence intensity of duplicate measurements was averaged and the background signal of the control aptamer was subtracted. The averaged background-subtracted data were fit to an equation that represents the ratio of bound to total receptors: (31). In this equation, Y is the averaged background-subtracted mean fluorescence intensity, Lt is the concentration of aptamer, Ymax is the maximum fluorescence intensity, and Kd is the dissociation constant. A derivation of this equation and verification that it is valid in our system is given in the Supporting Information. Ymax and Kd are determined by fitting the data to this equation using the Non-linear Curve-Fitting algorithm in Origin6.1 software (OriginLab Corp.). The mean and standard deviations of Kd obtained from this curve-fitting were used to calculate P-values using the normdist function in Excel (Microsoft). This function was used to estimate the probability that the mean of one Kd measurement could be generated by a normal distribution having the same mean and standard deviation of another Kd measurement.
RESULTS
General probe design
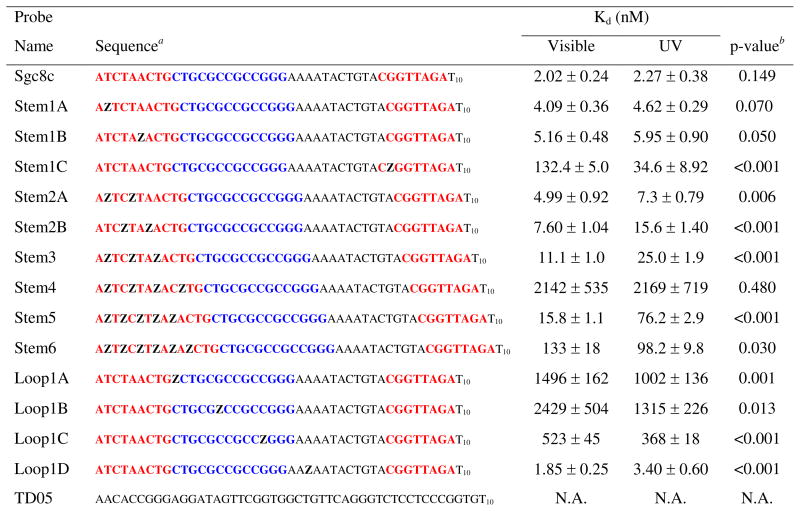
Our probe design consisted of the azobenzene modified aptamer sequence followed by a poly-T10 spacer and biotin moiety (see Schematic). Biotin was used to couple Streptavidin-PE, which is a sensitive fluorophore used frequently for flow cytometry measurements. Since azobenzene can quench fluorophores (30), a poly-T10 spacer was added to the 3′ end of Sgc8c to distance the Streptavidin-PE from the azobenzene moieties. Table 1 shows the specific azobenzene modified aptamer sequences used in this study. There are two basic groups of probes named Stem or Loop, depending on the position of the azobenzene moieties relative to the predicted stem. The number of azobenzene modifications is denoted after Stem or Loop and a capital letter A, B, C, or D denotes different permutations of azobenzene position for probes that contain the same number of azobenzene moieties.
Schematic.

Probe design depicting the melting of the stem region after UV illumination: Biotin (black polygon), trans-azobenzene (green polygon), cis-azobenzene (blue polygon), and DNA (colored cylinders/yellow ribbon).
Table 1.
Azobenzene modified Sgc8c sequences
|
Azobenzene modifications are signified using a bold faced capital Z, stem sequence is colored red, GC-rich loop region in blue, and sequences are written from 5′ to 3′.
P-value determined under the null hypothesis that the UV state is no different than the visible state.
Selectivity of azo-modified aptamers
To use azobenzene to probe the structure of an aptamer, the addition of azobenzene should not change the aptamer’s basic properties of selectivity and high affinity binding. Therefore, we first tested the selectivity of azo-Sgc8c probes by performing binding assays using both target (CEM) and control cells (Ramos), and the TD05 aptamer that binds only to control cells (39). These data are depicted as flow cytometry histograms in Figure 1A and 1B. We consistently observed increased fluorescence intensity of azo-modified probes compared to TD05 when bound to target cells and decreased fluorescence intensity when bound to control cells. Thus, the azo-modified probes retain the selectivity of Sgc8c towards target cells.
Figure 1.

Selectivity and photo-regulation of azo-modified aptamers using flow cytometry. A) Histograms of trans-probes binding to target cells: TD05 (black), Sgc8c (red), Stem2A (blue). B) Histograms of trans-probes binding to control cells: TD05 (red), Sgc8c (black), Stem2A (blue). A & B show that azo-modified aptamers had selectivity compared to unmodified aptamers. C) Histograms of Stem3 before and after 20 minutes of UV illumination show that cis-Stem3 binds less than trans-Stem3 at the same concentration: TD05 (black), Stem3-UV (blue), Stem3-Visible (red). D) Histograms of UV illuminated Stem3 before and after subsequent 5 minute visible illumination show that the reduced cell binding is reversible and not caused by DNA damage: TD05 (black), Stem3-UV (blue), Stem3-UV->Visible (red). All probes used in this figure were incubated with 106 cells at 200 nM for 20 minutes.
Photo-regulation of azo-modified aptamers
Having established that the selectivity of azo-Sgc8c probes was not affected, we next tested whether the azobenzene incorporation had endowed these probes with photo-regulated binding affinity. To show this, probes where first illuminated with either UV or visible light prior to cell binding. In general, the UV state of the probes had lower florescence signal compared to the visible state at the same concentration (Figure 1C). We also performed control experiments to test the photo-reversible property of azobenzene. In these experiments, probes were initially illuminated with UV light and subsequently illuminated with visible light for 5 minutes. Probes that had been cycled from the UV state and back to the visible state had higher fluorescence intensity compared to probes in the UV state (Figure 1D). These photo-regulation and photo-reversing experiments suggest that the azobenzene moieties are functioning in accord with published literature. Further, the photo-reversing experiments rule out DNA damage as a cause of lowered binding affinity in the UV state.
Dissociation constants of two-state probes
To quantify the difference in binding affinity between probes in the visible and UV states, dissociation constants with respect to target cells were measured using flow cytometry before and after UV illumination. Binding curves were generated for all probes in both UV and visible states and dissociation constants were extracted by fitting the curves to an equation that represents the ratio of bound receptors to total receptors, see Materials and Methods and Supporting Information for details. Representative flow cytometry histograms showing probes binding in both the UV and visible states at different concentrations are shown in Figure 2. An example of binding curves and fitted curves is displayed in Figure 3. The majority of probes were high affinity, Kd < 150 nM (Figure 4A), and four probes had Kd > 200 nM (Figure 4B). Since unmodified Sgc8c had Kd = 2.02 ± 0.24 nM, these experiments established that 7 out of 13 azo-Sgc8c probes bound with similar affinity compared to Sgc8c, i.e. < 20 nM. Further, when un-modified Sgc8c was illuminated with UV light, the dissociation constant was not significantly different (2.27 ± 0.38 nM, P > 0.1). This control measurement further proves that UV illumination has no deleterious effect on aptamer binding, and that any difference in binding affinity must be due to azobenzene and not DNA damage. When comparing all probes to Sgc8c, i.e. under the null hypothesis that the Kd is no different than that of Sgc8c, all probes had P < 0.05 except trans-Loop1D. When comparing the UV to visible state of each probe individually, i.e. under the null hypothesis that the UV state is no different than the visible state, all probes except for Sgc8c, Stem1A, and Stem4 had P < 0.05 (Table 1).
Figure 2.

Binding affinity of probes in both UV and visible state using flow cytometry. A–D) Histograms of probe Stem5 in the UV state (blue) and the visible state (red) at 200 nM, 10 nM, 5 nM, and 2 nM, respectively. TD05 is shown in black. Fluorescent means nearly saturate at 200 nM and cis-Stem5 has lower mean compared to trans-Stem5 at all concentrations.
Figure 3.

Representative binding curves for Stem5 showing average background-subtracted mean fluorescence signal for trans-Stem5 (square) and cis-Stem5 (triangle), and fitted curves (solid lines) for each binding curve. Data were fit to an equation that represents the ratio of bound receptors to total receptors, see Materials and Methods and Supporting Information for more details.
Figure 4.

Differences in dissociation constants of cis- and trans- states reveal important secondary structures associated with target binding. A) High affinity probes having Kd < 150 nM. B) Low affinity probes having Kd > 400 nM. Trans-form is shown in grey and cis-form is shown in black. Under the null hypothesis that the Kd of each probe is no different than that of Sgc8c, all probes had P < 0.05 except trans-Loop1D. Under the null hypothesis that the UV state is no different than the visible state for each probe, all probes except for Sgc8c, Stem1A, and Stem4 had P < 0.05 (*).
DISCUSSION
Verification of predicted stem structure
The first sub-set of probes that stands out are the probes that have azo-modifications placed before the sixth base in the stem, namely Stem1A, Stem1B, Stem2A, Stem2B, Stem3, and Stem5. As the number of azo-moieties within the stem increased, a larger decrease in binding affinity was observed after UV illumination (Figure 4A). Based on this data, we can make two conclusions. First, the initial 6 bases of the stem are double-stranded. In double-stranded regions, the UV state of azobenzene lowers the melting temperature or stability of the duplex, an effect that is proportional to the number of azobenzene modifications. Therefore, as the number of azobenzene modifications increases, the duplex becomes less stable in the UV state and, in the case of an aptamer, this translates into lower binding affinity in the UV state. Second, since the binding in the UV state was still high affinity, i.e. Kd < 80 nM, we can conclude that the first 6 bases in the stem do not represent the high-affinity binding site, but are needed to stabilize the overall structure of the aptamer.
T-G mismatch near stem-loop junction is sensitive to structural perturbation
A second set of probes could be grouped together based on the positioning of azobenzene near the stem-loop junction after the sixth base in the stem, namely Stem1C, Stem4, and Stem6. In this part of the stem, there is a T-G mismatch two bases before the end of the predicted stem. Stem4 had low binding affinity, Kd > 2 uM, having azo-moieties between C and T, whereas Stem6 and Stem1C had high affinity, Kd < 150 nM, having azo-moieties before C and after T, respectively. Therefore, the specific conformation of the C and T bases may be important for the high-affinity binding site and the T-G mismatch could coordinate the interaction with the protein or stabilize the aptamer secondary structure.
GC-rich portion of loop is important for protein binding
Control experiments with probes that had single azo-modifications in the loop were also performed. The loop is composed of a 13-base GC-rich region and a poly-A4 region. Probes that had modifications within the GC-rich portion of the loop, Loop1A, Loop1B, and Loop1C, showed low binding affinity, while Loop1D, which had a modification in the poly-A4 region, had high binding affinity. These data suggest that the GC-rich portion of the loop may be in direct contact with the protein or form an important secondary structure, whereas the poly-A4 region may be relatively unstructured. This interpretation is consistent with the fact that GC-rich regions are associated with more complex secondary structure when compared to poly-A regions.
CONCLUSION
Azobenzene can be used to determine the importance of predicted dsDNA within aptamer sequences. For the Sgc8c aptamer, the first 6 bases in the stem appear to be dsDNA. Further, bases in the GC-rich portion of the loop and the T-G mismatch near the stem-loop junction could have significant interaction with the target protein or stabilize the aptamer secondary structure. The modification of aptamers with moieties that can change their binding affinity on demand could have applications for targeted drug delivery. It may be possible to administer aptamer-drug conjugates in the non-binding state and target them to a tumor using visible light. This would reduce systemic side-effects in non-target organs. Future work will involve determining the efficacy of drug targeting using this type of light-regulated affinity modulation.
Supplementary Material
Acknowledgments
This study was supported by grants from the National Institutes for Health (NIH) and National Science Foundation (NSF).
Footnotes
Supporting Information Available: A schematic depicting the synthesis of azobenzene phosphoramidite monomer and a derivation of the formula used to fit binding curves and determine dissociation constants are contained in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
LITERATURE CITED
- 1.Jhaveri SD, Kirby R, Conrad R, Maglott EJ, Bowser M, Kennedy RT, Glick G, Ellington AD. Designed signaling aptamers that transduce molecular recognition to changes in fluorescence intensity. J Am Chem Soc. 2000;122:2469–2473. [Google Scholar]
- 2.Stojanovic MN, de Prada P, Landry DW. Aptamer-based folding fluorescent sensor for cocaine. J Am Chem Soc. 2001;123:4928–4931. doi: 10.1021/ja0038171. [DOI] [PubMed] [Google Scholar]
- 3.Ozaki H, Nishihira A, Wakabayashi M, Kuwahara M, Sawai H. Biomolecular sensor based on fluorescence-labeled aptamer. Bioorg Med Chem Lett. 2006;16:4381–4384. doi: 10.1016/j.bmcl.2006.05.054. [DOI] [PubMed] [Google Scholar]
- 4.Ferapontova EE, Olsen EM, Gothelf KV. An RNA Aptamer-based electrochemical biosensor for detection of theophylline in serum. J Am Chem Soc. 2008;130:4256–4258. doi: 10.1021/ja711326b. [DOI] [PubMed] [Google Scholar]
- 5.Hamaguchi N, Ellington A, Stanton M. Aptamer beacons for the direct detection of proteins. Anal Biochem. 2001;294:126–131. doi: 10.1006/abio.2001.5169. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto R, Kumar PKR. Molecular beacon aptamer fluoresces in the presence of Tat protein of HIV-1. Genes to Cells. 2000;5:389–396. doi: 10.1046/j.1365-2443.2000.00331.x. [DOI] [PubMed] [Google Scholar]
- 7.Jiang Y, Fang X, Bai C. Signaling aptamer/protein binding by a molecular light switch complex. Anal Chem. 2004;76:5230–5235. doi: 10.1021/ac049565u. [DOI] [PubMed] [Google Scholar]
- 8.Li W, Yang X, Wang K, Tan W, Li H, Ma C. FRET-based aptamer probe for rapid angiogenin detection. Talanta. 2008;75:770–774. doi: 10.1016/j.talanta.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 9.Patel DJ, Suri AK. Structure, recognition and discrimination in RNA aptamer complexes with cofactors, amino acids, drugs and aminoglycoside antibiotics. Rev Mol Biotechnol. 2000;74:39–60. doi: 10.1016/s1389-0352(99)00003-3. [DOI] [PubMed] [Google Scholar]
- 10.Schwalbe H, Buck J, Furtig B, Noeske J, Wohnert J. Structures of RNA switches: insight into molecular recognition and tertiary structure. Angew Chem Int Ed. 2007;46:1212–1219. doi: 10.1002/anie.200604163. [DOI] [PubMed] [Google Scholar]
- 11.Padmanabhan K, Padmanabhan KP, Ferrara JD, Sadler JE, Tulinsky A. The structure of α-thrombin inhibited by a 15-Mer single-stranded DNA aptamer. J Biol Chem. 1993;268:17651–17654. doi: 10.2210/pdb1hut/pdb. [DOI] [PubMed] [Google Scholar]
- 12.Marathias VM, Wang KY, Kumar S, Pham TQ, Swaminathan S, Bolton PH. Determination of the number and location of the manganese binding sites of DNA quadruplexes in solution by EPR and NMR in the presence and absence of thrombin. J Mol Biol. 1996;260:378–394. doi: 10.1006/jmbi.1996.0408. [DOI] [PubMed] [Google Scholar]
- 13.Padmanabhan K, Tulinsky A. An ambiguous structure of a DNA 15-mer thrombin complex. Acta Cryst. 1996;D52:272–282. doi: 10.1107/S0907444995013977. [DOI] [PubMed] [Google Scholar]
- 14.Long SB, Long MB, White RR, Sullenger BA. Crystal structure of an RNA aptamer bound to thrombin. RNA14. 2008:1–9. doi: 10.1261/rna.1239308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bouvet P, Allain FHT, Finger LD, Deickmann T, Feigon J. Recognition of pre-formed and flexible elements of an RNA stem-loop by nucleolin. J Mol Biol. 2001;309:763–775. doi: 10.1006/jmbi.2001.4691. [DOI] [PubMed] [Google Scholar]
- 16.Huang DB, Vu D, Cassiday LA, Zimmerman JM, Maher LJ, III, Ghosh G. Crystal structure of NF-κB (p50)2 complexed to a high-affinity RNA aptamer. Proc Natl Acad Sci USA. 2003;100:9268–9273. doi: 10.1073/pnas.1632011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yesodha SK, Pillai CKS, Tsutsumi N. Stable polymeric materials for nonlinear optics: a review based on azobenzene systems. Prog Polym Sci. 2004;29:45–74. [Google Scholar]
- 18.Renner C, Moroder L. Azobenzene as conformational switch in model peptides. Chembiochem. 2006;7:868–878. doi: 10.1002/cbic.200500531. [DOI] [PubMed] [Google Scholar]
- 19.Yager G, Barrett CJ. Novel photo-switching using azobenzene functional materials. J Photochem Photobiol A. 2006;182:250–261. [Google Scholar]
- 20.Barrett CJ, Mamiya J, Yager KG, Ikeda T. Photo-mechanical effects in azobenzene-containing soft materials. Soft Matter. 2007;3:1249–1261. doi: 10.1039/b705619b. [DOI] [PubMed] [Google Scholar]
- 21.Asanuma H, Ito T, Yoshida T, Liang X, Komiyama M. Photoregulation of the formation and dissociation of a DNA duplex by using the cis-trans isomerization of azobenzene. Angew Chem Int Ed. 1999;38:2393–2395. doi: 10.1002/(sici)1521-3773(19990816)38:16<2393::aid-anie2393>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 22.Liang X, Asanuma H, Komiyama M. Phenylazonaphthalene as a superb photo-regulator for DNA-triplex formation. Tetrahedron Lett. 2001;42:6723–6725. [Google Scholar]
- 23.Liang X, Asanuma H, Kashida H, Takasu A, Sakamoto T, Kawai G, Komiyama M. NMR study on the photoresponsive DNA tethering an azobenzene. assignment of the absolute configuration of two diastereomers and structure determination of their duplexes in the trans-form. J Am Chem Soc. 2003;125:16408–16415. doi: 10.1021/ja037248j. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi K, Yaegashi S, Asanuma H, Hagiya M. Photo- and Thermoregulation of DNA Nanomachines. Lect Notes Comput Sci. 2006;3892:336–346. [Google Scholar]
- 25.Liang X, Nishioka H, Takenaka N, Asanuma H. A DNA nanomachine powered by light irradiation. ChemBioChem. 2008;9:702–705. doi: 10.1002/cbic.200700649. [DOI] [PubMed] [Google Scholar]
- 26.Liang X, Takenaka N, Nishioka H, Asanuma H. Light driven open/close operation of an azobenzene-modified DNA nano-pincette. Nucleic Acids Symp Ser (Oxf) 2008;52:697–698. doi: 10.1093/nass/nrn352. [DOI] [PubMed] [Google Scholar]
- 27.Kim Y, Phillips JA, Liu HP, Kang HZ, Tan WH. Using photons to manipulate enzyme inhibition by an azobenzene-modified nucleic acid probe. Proc Natl Acad Sci USA. 2009;106:6489–6494. doi: 10.1073/pnas.0812402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogura Y, Nishimura T, Tanida J. Self-contained photonically-controlled DNA tweezers. Appl Phys Express. 2009;2:025004-1–025004-3. [Google Scholar]
- 29.Dohno C, Uno S, Sakai S, Oku M, Nakatani K. The effect of linker length on binding affinity of a photoswitchable molecular glue for DNA. Bioorg Med Chem. 2009;17:2536–2543. doi: 10.1016/j.bmc.2009.01.053. [DOI] [PubMed] [Google Scholar]
- 30.Kang H, Liu H, Phillips JA, Cao Z, Kim Y, Chen Y, Yang Z, Li J, Tan W. Single-DNA molecule nanomotor regulated by photons. Nano Lett. 2009;9:2690–2696. doi: 10.1021/nl9011694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shangguan D, Li Y, Tang Z, Cao ZC, Chen HW, Mallikaratchy P, Sefah K, Yang CJ, Tan W. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc Natl Acad Sci USA. 2006;103:11838–11843. doi: 10.1073/pnas.0602615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shangguan D, Tang Z, Mallikaratchy P, Xiao Z, Tan W. Optimization and modifications of aptamers selected from live cancer cell lines. ChemBioChem. 2007;8:603–606. doi: 10.1002/cbic.200600532. [DOI] [PubMed] [Google Scholar]
- 33.Shangguan D, Cao Z, Meng L, Mallikaratchy P, Sefah K, Wang H, Li Y, Tan W. Cell-specific aptamer probes for membrane protein elucidation in cancer cells. J Proteome Res. 2008;7:2133–2139. doi: 10.1021/pr700894d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang YF, Chang HT, Tan W. Cancer cell targeting using multiple aptamers conjugated on nanorods. Anal Chem. 2008;80:567–572. doi: 10.1021/ac702322j. [DOI] [PubMed] [Google Scholar]
- 35.Huang YF, Sefah K, Bamrungsap S, Chang HT, Tan W. Selective photothermal therapy for mixed cancer cells using aptamer-conjugated nanorods. Langmuir. 2008;24:11860–11865. doi: 10.1021/la801969c. [DOI] [PubMed] [Google Scholar]
- 36.Ma N, Sargent EH, Kelley SO. One-step DNA-programmed growth of luminescent and biofunctionalized nanocrystals. Nat Nanotechnol. 2008;4:121–125. doi: 10.1038/nnano.2008.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phillips JA, Xu Y, Xia Z, Fan ZH, Tan W. Enrichment of cancer cells using aptamers immobilized on a microfluidic channel. Anal Chem. 2009;81:1033–1039. doi: 10.1021/ac802092j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Asanuma H, Liang X, Nishioka H, Matsunaga D, Liu M, Komiyama M. Synthesis of azobenzene-tethered DNA for reversible photo-regulation of DNA functions: hybridization and transcription. Nature Protocols. 2007;2:203–212. doi: 10.1038/nprot.2006.465. [DOI] [PubMed] [Google Scholar]
- 39.Tang Z, Shangguan D, Wang K, Shi H, Sefah K, Mallikratchy P, Chen HW, Li Y, Tan W. Selection of aptamers for molecular recognition and characterization of cancer cells. Anal Chem. 2007;79:4900–4907. doi: 10.1021/ac070189y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.