

Abstract
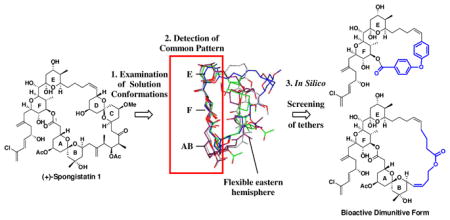
The design, synthesis, and biological evaluation of two diminutive forms of (+)-spongistatin 1, in conjunction with the development of a potentially general design strategy to simplify highly flexible macrocyclic molecules while maintaining biological activity, have been achieved. Examination of the solution conformations of (+)-spongistatin 1 revealed a common conformational preference along the western perimeter comprising the ABEF rings. Exploiting the hypothesis that the small molecule recognition/binding domains are likely to comprise the conformationally less mobile portions of a ligand led to the design of analogs, incorporating tethers (blue) in place of the CD and the ABCD components of the (+)-spongistatin 1 macrolide, such that the conformation of the retained (+)-spongistatin 1 skeleton would mimic the assigned solution conformations of the natural product. The observed nanomolar cytotoxicity and microtubule destabilizing activity for the ABEF analog provides support both for the assigned solution conformation of (+)-spongistatin 1 and the validity of the design strategy.
Introduction
The spongistatins (1–9; Table 1), independently reported by the Pettit,1 Fusetani2 and Kitagawa3 Laboratories in the early 1990’s, comprise a unique family of marine macrolides that embody remarkably complex structures possessing extraordinary antimitotic activity. Members of this family, in particular spongistatins 1 and 2 [(+)-1 and (+)-2, Table 1], rapidly attracted broad interest in the chemical and biological communities given their potential as cancer chemotherapeutic agents. Spongistatin 1, recognized at the time as one of the most selective cytotoxic agents known, has an average IC50 value of 0.12 nM against the NCI panel of 60 human cancer cell lines.1,4 The initially observed in vitro cell growth inhibition activity was subsequently explored by Hamel for tubulin polymerization, competitive microtubule assembly, and turbudity/aggregation.5 Their results revealed that (+)-spongistatin 1: (A) competitively inhibits tubulin binding of maytansine, rhizoxin, as well as GTP exchange;5,6 (B) non- competitively inhibits tubulin binding of dolastatin 10, halichondrin B and vinblastine;5,6 (C) does not induce formation of GTP-independent microtubule aggregates6 and (D) inhibits formation of the Cys-12-Cys-201/211 cross-linking on tubulin.7
Table 1.
Inhibition of cell growth of L1210 murine leukemia cells and average GI50 valuesa of 60 different cancer cell linesb
 | |||
|---|---|---|---|
| L1210 | Avg. | ||
| Spongistatin 1 [(+)-1] | R = Cl, R1 = R2 = Ac | 0.03 | 0.13 |
| Spongistatin 2 [(+)-2] | R = H, R1 = R2 = Ac | 2.0 | 0.85 |
| Spongistatin 3 (3) | R = Cl, R1 = H, R2 = Ac | 1.0 | 0.83 |
| Spongistatin 4 (4) | R = Cl, R1 = Ac, R2 = H | 0.10 | 0.10 |
| Spongistatin 6 (5) | R = R2 = H, R1 = Ac | 0.80 | 1.10 |
| Spongistatin 5 (6) | R = Cl, R1 = H | 0.20 | 0.12 |
| Spongistatin 7 (7) | R = H, R1 = H | 2.0 | 1.00 |
| Spongistatin 8 (8) | R = H, R1 = Ac | 3.0 | 0.23 |
| Spongistatin 9 (9) | R = Cl, R1 = Ac | 0.3 | 0.04 |
GI50 values are in nM.
Data is compiled from Reference 1
Based on these results, Hamel and coworkers proposed a “polyether” binding site on β-tubulin for the spongistatins located near the vinca domain,8 distinct from but in the vicinity of the “peptide” and “vinca” sites, where dolastatin 10/phomopsin A and vinblastine/vincristine respectively are known to bind.6 The development of the spongistatins as possible clinical agents however has not progressed given their low availability, notwithstanding seven total syntheses of (+)-spongistatin 19 and four total syntheses of (+)-spongistatin 2.10
Following completion of our first generation total syntheses of (+)-2 and (+)-1, respectively we focused on material advancement to deliver more than one gram of synthetic (+)-spongistatin 1 [(+)-1], via what in the end comprised an effective fourth generation synthetic route.9h, 11 In addition to defining the limitations and scalability issues along the synthetic route, a significant outcome of this synthetic campaign was the development of a one-pot, multicomponent dithiane union tactic,12 based on the earlier efforts of the Tietze Laboratories.13 This tactic has now evolved into a new synthetic paradigm termed Type I and II Anion Relay Chemistry (ARC)14 for the rapid construction of polyketide 15 and alkaloid 16 natural and unnatural products.17
With sufficient material for pre-clinical development in hand (ca. 1 g), we turned attention to identifying the architectural subunits of the spongipyran skeleton responsible for the subnanomolar cytotoxicity, with the goal of initiating structure activity studies. Notwithstanding the optimized (+)- spongistatin 1 route, construction of random analogs, appealed unattractive. We were, of course, well aware of the remarkable success of Halaven® (Eribulin) based on the marine natural product halichondrin B developed by the Kishi and Eisai Laboratories. We asked the question: Is the entire structure of the large, flexible polyketide skeleton of the spongistatins a prerequisite for biological activity?
In this, a full account, we report the design, synthesis and biological evaluation of two diminutive forms of (+)-spongistatin 1. Importantly, computational methods developed during this venture led to: (1) a general strategy to identify the binding regions of flexible small molecule macrolide natural products, and more specifically, (2) a general design strategy that permitted the simplification of the polyketide structure of the spongistatins, while maintaining biological activity.
Spongistatins Analogs: Early Congeners
Members of the spongistatin family (1–9, Table 1) possess an array of architecturally complex structural features including a 31-membered macrolactone ring endowed with 24 stereocenters, two [6,6] spiroketals (the AB and CD rings), one bis-tetrahydropyranylmethane moiety (E and F rings) complete with a diene side-chain differentiated by the acetate substitution pattern at C(5) and C(15), and in the case of (+)-spongistatins 6–9, by an additional tetrahydropyran ring (G ring) inscribed in the tether linking rings B and C. The cell growth inhibitory activity against the NCI-60 DTP human tumor panel for the natural congeners averaged at the sub-nanomolar level. Structural features for optimal activity comprise the chlorine substituent on the side-chain (1 vs. 2; 4 vs. 6; and 6 vs. 7) and an acetate at C(5) (1 vs. 3; and 6 vs. 9), while structural elements, not critical for maintaining activity include the C(15) acetate and ring G (1–5 vs. 6–9). Although these observations provided useful insights, our analog design criteria also benefited initially from synthetic intermediates and analogs that emanated from the Kishi,9a,b Paterson18 and Heathcock 19 Laboratories as well as our own.20
For example, in the Kishi synthesis of (+)-spongistatin 1,9b as well as in our synthesis of (+)- spongistatin 2,10b,e unexpected epimerizations that occurred at the C(23) center of the CD spiroketal led to congeners 10 and (−)-11 (Figure 1) that possessed modest tumor cell growth inhibition against several cancer cell lines [avg. GI50 of 0.2 μM for (−)-11].20c Subsequently, Paterson and coworkers constructed an analog lacking the diene side-chain (12) that significantly attenuated activity, highlighting the requirement for this structural unit.18 Later we prepared a series of glucose based mimetics of the F and EF rings possessing the (+)-spongistatin 2 side-chain [cf. (+)-13 and (+)-14a].20 Although modest tumor cell growth inhibition activity was observed with (+)-13, subsequent unpublished results suggested a different mode of action. 20a, 21 More recently (2008) Heathcock and coworkers reported a series of (+)- spongistatin 2 analogs, including (+)-14b which did not show any activity at the tested concentrations.19 The loss of activity led Heathcock to prepare macrolide congeners 15 and 16, employing polymethylene tethers to replace the ABCD and CD structural segments. Observation of only modest activity (cf. GI50 values of 4.8 and 4.6 μM against HCT116 human colon tumor cell lines, respectively) led Heathcock to conclude that the CD ring may comprise a structural moiety required to maintain the tumor cell growth inhibition.
Figure 1.

Spongistatin analogs
* Kishi et al. ref. 9a, 9b; # Paterson et al. ref. 18; † Smith et al. ref. 20; ‡ Heathcock et al. ref. 19
Analog Design: An EF Ring Congener Possessing a Rotationally Restricted ABCD Ring System
The lessons learned from analogs (+)-13–16, in conjunction with the activity observed for the C(23) epi-congeners [10 and (−)-11] led us to the hypothesis that the western perimeter of the spongistatins constituted a significant component of the recognition domain. We reasoned that the linker (cf. CD) was not important and might be replaced by a less complex unit, as long as effective control over the conformation of the western perimeter was maintained (i.e., the dihedral angles between rings E and F). Similar simplifying design tactics have been employed by the Kishi,22 Wender,23 Taylor24 and Romo25 Laboratories to construct biologically active halichondrin B, bryostatin and epothilone analogs, respectively.
To design a suitable tether that would orchestrate the C(37)–C(39) dihedral angles linking the spongistatin E and F rings, we turned to molecular modeling. We were cognizant of the solution conformation reported by Kitagawa26 during his elegant structural assignments of 5-desacetylaltohyrtin A (spongistatin 3, 3). To establish a baseline on the solution conformation of (+)-spongistatin 1 [(+)-1], we carried out repetitive Monte Carlo conformational searches on (+)-1 until no additional families of conformers were detected. Computations utilizing the MMFF force field27 with the GB/SA solvation model28 revealed that the energy ordering of the conformational populations changed significantly in different solvents.
According to our calculations, in chloroform, (+)-spongistatin 1 (1) adopts a “flat” conformation in which the surface area of the molecule is maximized by an increase in number of intramolecular hydrogen bonds (Figure 2).24d In water, a “twisted” conformer is preferred in which the oxygen atoms are oriented toward the solvent. When DMSO and acetonitrile solvent models are employed, the lowest energy conformer possesses a “saddle”-like shape. Interestingly, the orientation of the EF rings in the Kitagawa solution structure did not resemble any of these low energy conformers. We reasoned that the derived Kitagawa structure might well have comprised an virtual conformation, resulting from constrained conformational searches.
Figure 2.
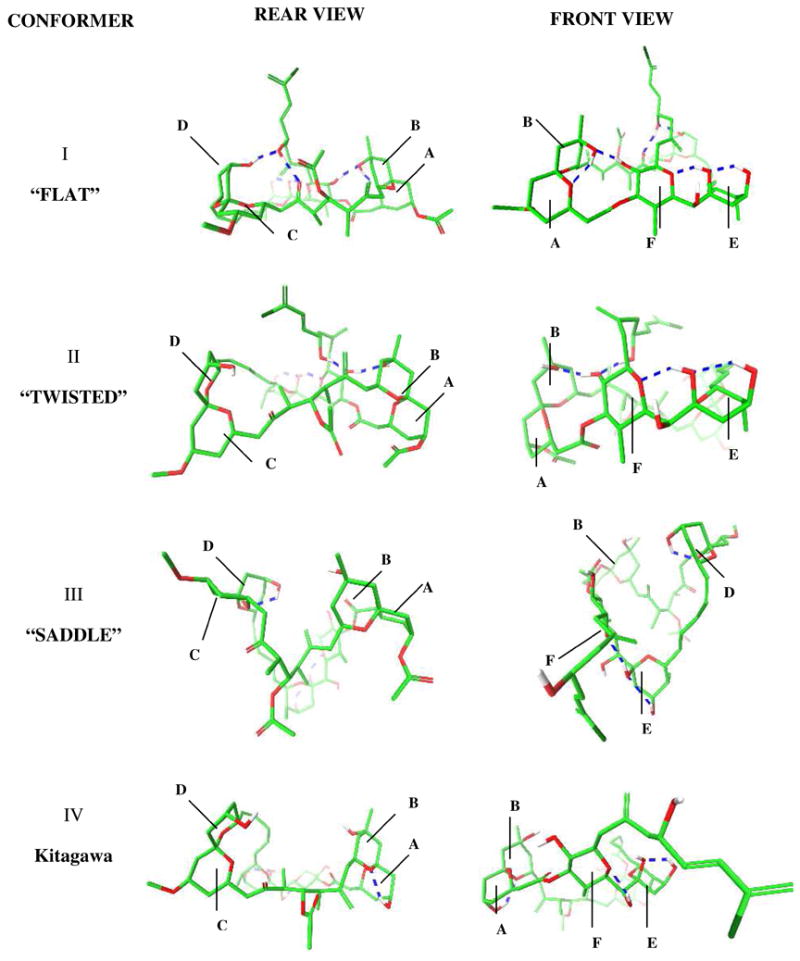
Front and rear 3D views (see supporting information section for stereo view images) of the lowest energy conformations (I–III) calculated by conformational searches employing different solvation models and Kitagawa solution structure (IV). Blue dashed lines represent hydrogen bonds. Rings A–F are shown in each conformation.
To understand further the solution behavior of (+)-spongistatin 1 (1), molecular dynamics (MD) simulations were performed using water as solvent, wherein changes in the dihedral angles of the macrocycle were analyzed. The flexible regions of (+)-1 were identified by examining: (1) the dihedral angle distributions of each Csp3–Csp3 bond (Figure 3A); and (2) the long range coupled torsional changes during the dynamics simulation. This analysis was performed exploiting polar coordinate graphs, in a fashion similar to the Taylor epothilone studies,24 wherein the coupled torsions were identified by analyzing the correlation in the torsional changes via principle component analysis. As illustrated in Figure 3A, red designated torsional bonds have rigid torsions, green labeled bonds have flexible torsions and blue labeled bonds have torsions with intermediate flexibility. In Figure 3B, the numbers indicate bonds where the torsions change together, corresponding to correlated movements. As seen in Figure 3B, tethers of the CD ring are flexible with the dihedral angle pairs labeled with the same number, when change occurs together, thereby permitting movement of the CD ring. Taken together, the results of this multivariate analysis reveal: (1) that the western perimeter, including ABEF rings has a preferred conformation, wherein each torsional angle has a preferred value; and (2) that reduction of a large, flexible molecule to a single solution structure derived by NMR constraints, in general is not feasible.
Figure 3.

Flexibility of macrocyclic dihedral angles in (+)-spongistatin 1. A. Red: rigid torsions, green: flexible torsions, blue: torsional angles with intermediate flexibility B. Numbers indicate bonds pairs where the torsions change together corresponding to long range movements during the simulations.
In support of these observations, molecular dynamics simulations constrained by NMR-derived distance and torsion values are known to generate average conformations; however the conformations generated are often of high energy due to the constraints applied. Equally important computational methods, solely relying on molecular mechanics force fields, suffer from inaccurate energy ordering of flexible organic molecules, due to the additive nature of parameterization errors. To circumvent these issues, we devised a computational hybrid strategy, wherein the NMR derived inter-proton distances and torsional angles are used to determine the most populated families of conformations. This effort led to the development and implementation of a software program termed Distribution of Solution Conformations (DISCON). The thrust of this software package is to define a combination of conformers that overall would provide the best match for the time averaged torsional angles and inter-proton distances obtained by NMR. Contrary to earlier methods developed,29 DISCON utilizes hierarchical clustering of conformations based on NMR variables for feature selection to avoid “overfitting”,30 as well as a genetic-algorithm based global optimization strategy to avoid local minima, two common problems often observed in NMR-based structure optimization. Details and availability of this software package for the deconvolution of NMR observables over an ensemble of solution conformations will be provided in a concurrent report.31
From this analysis, we deduce that the low energy conformations determined in water and chloroform (Figure 4a–d) define the major solution conformations of (+)-spongistatin 1. The two major calculated conformations comprise a twisted and a flat form (57% and 13% respectively) as illustrated in Figures 4a and 4b.
Figure 4.
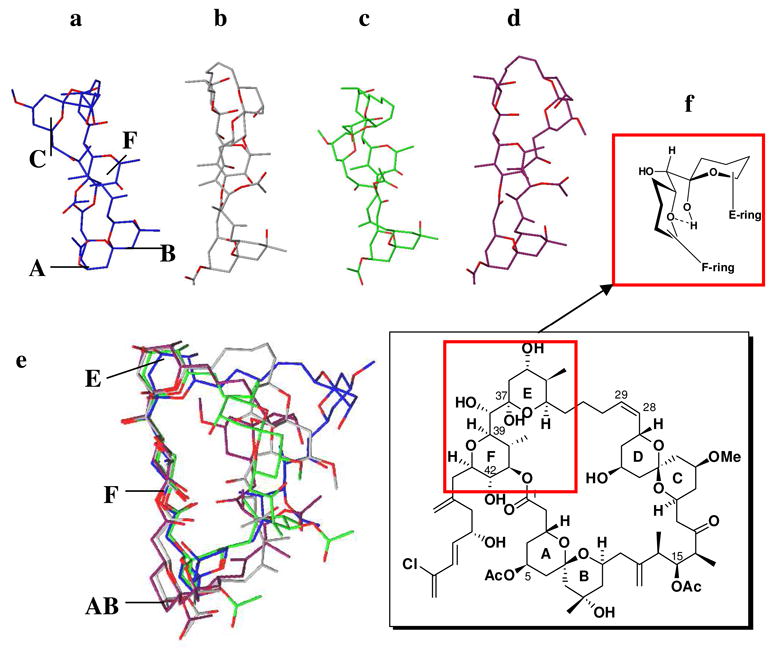
Calculated major solution conformers a–d. Overlay of these conformations (e) shows the conserved ABEF ring conformations where syn pentane interactions (f) are important in fixing the relative conformations of E and F rings.
The minor conformers (4c and 4d) are also similar to 4b, with the major difference being the orientation of the CD ring. Pleasingly, when the four major conformations are overlaid, a striking feature appeared: the western hemispheres, including the ABEF rings, have a common conformation (Figure 4e). This preference can be understood by earlier conformational studies of the bis-tetrahydropyranylmethane system reported by Hoffmann and coworkers32 (Figure 4f), wherein the preferred “skew” conformation minimizes the syn-pentane interactions between the E and F ring systems. This conformational arrangement of the E and F (+)-spongistatin 1 rings in turn facilitates an intramolecular hydrogen bond between the E ring hydroxyl and the F ring pyran oxygen (cf. the hydrogen bonds in Figure 2I and 2II).
The striking conformational similarities derived from the earlier MD simulations and subsequent DISCON analysis, in conjunction with the biological data of the epi-C(23) spongistatin congeners 10 and (−)-11, provided further support for the hypothesis that the ABEF portion of the spongistatin scaffold comprises the binding/small molecule recognition domain that interacts with β-tubulin.
To explore this scenario, we first turned to analogs possessing only the E and F ring system. In silico screening of a variety of linkers led to the biaryl ether tether in 17 (Scheme 1 in red), that would be suitable to orchestrate the relative conformational twist of the E and F ring system, to access the proposed conformation of the western perimeter.
Scheme 1.

Retrosynthetic analysis of EF ring analog 17.
To test our conformational design strategy we turned to the synthesis of 17. From the synthetic perspective, 17 was easily reduced to three fragments: phosphonium salt 18, allylstannane 19 and aldehyde 20 (Scheme 1). The route was designed to provide synthetic flexibility for SAR studies by a late-stage installation of the diene side-chain, exploiting intermediates in hand from our earlier spongistatins syntheses. One of the attributes of developing preparative scale syntheses of architecturally complex natural products possessing significant bio-regulatory properties is the subsequent readily availability of advanced intermediates for SAR studies. Fragments (+)-18 and aldehyde 20 were thus envisioned to be united via a Wittig reaction. Following macrolactonization, side-chain attachment would be achieved via the elegant glucal-epoxide union protocol developed by Evans et al.33 during the first total synthesis in the spongistatin area. Aldehyde 20 was particularly appealing given Ullmann coupling protocols.34
We began the synthesis of 17 with (+)-21, from our second generation (+)-spongistatin 1 synthesis.9f We envisioned that (+)-21 could be readily converted to the corresponding F-ring dihydropyran (+)-22 by treatment with trifluoromethanesulfonic anhydride (Tf2O), followed by elimination of TfOH (Table 2). In the event, use of lithium diisopropylamide (LDA) provided an inseparable mixture (ca. 1:1) of (+)- 22 and a compound whose structure was tentatively assigned as 23. Reasoning that a bulkier base might favor the desired compound, use of KHMDS resulted in a 3:1 mixture of (+)-22 and 23, albeit in modest yield (35%). Cooling the reaction mixture to −78 °C led to exclusive formation of (+)-22, although the reaction did not proceed to completion. In an effort to increase the efficiency, we examined the use of the less basic NaHMDS. Pleasingly, significant improvement in selectivity (cf. 12:1) was observed to provide (+)-22 in 72% overall yield. With reliable access to glucal (+)-22 in hand, removal of the benzyl protecting groups employing lithium 4,4′-di-tert-butylbiphenylide (LiDBB) proceeded smoothly to provide diol (+)-24 in good yield (Scheme 2). Selective conversion of the primary hydroxyl to furnish the 2,4,6-triisopropylbenzenesulfonate (trisylate) (+)-25 initially proved problematic, when the bulky base 2,6-di-tert-butyl-4-methylpyridine (DTBMP) was employed. The low reactivity of (+)-24 led us to screen a series of the bases. Triethylamine with DMAP at 0 °C proved optimal, selectively furnishing (+)-25 in 83% isolated yield, which in turn was treated with LiI, in the presence of 2,6- lutidine to generate iodide (+)-26. The secondary hydroxyl was then converted to the TES ether, followed by displacement of the iodide in (+)-27 with triphenylphosphine to furnish Wittig partner (+)- 18 in near quantitative yield.
Table 2.
Selectivity in the preparation of F ring pyran.
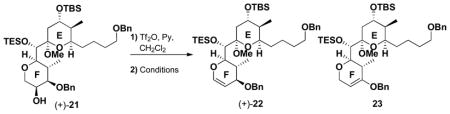 | ||||
|---|---|---|---|---|
| Entry | Base | Temperature (°C) | Yield | Ratio (+)-22: 23 |
| 1 | LDA | −45 to −20 | 56% | 1:1 |
| 2 | KHMDS | −45 to −20 | 35% | 3:1 |
| 3 | NaHMDS | −78 to −30 | 72% | 12:1 |
Scheme 2.

Preparation of (+)-18
The requisite biaryl ether tether 20 was constructed via condensation of 4-bromobenzoic acid with tert-butanol, mediated by 1,1′-carbonyl-diimidazole (CDI),35 followed the Ullmann coupling34 with 4- hydroxzybenzaldehyde to yield 20 (Scheme 3). While the efficiency of the last step was only modest, we moved forward to explore the viability of this tether.
Scheme 3.

Preparation of biarylether 20.
Wittig union36 of (+)-18 with 20 provided the Z olefin geometry, albeit in low yield. With studies to improve both the Ullmann and Wittig unions underway, we proceeded with removal of the tert-butyl ester. Unfortunately various conditions including treatment of (+)-30 with TMSOTf resulted only in decomposition, presumably due to the incompatibility of the enol ether with the required mild Lewis acidic conditions (Scheme 4).
Scheme 4.

Attempted removal of t-butyl group in (+)-30.
Concurrent with studies to access 17 from (+)-13, we also had access to EF Wittig salt (+)-31, again developed in conjunction with our gram-scale synthesis of (+)-spongistatin 1.10e A second generation approach to the spongistatin EF-analog 17 was thus initiated based on the availability of (+)-31 (Scheme 5). In the forward sense, we envisioned a biaryl linker now possessing a triisopropyl silyl group (TIPS) to protect the carboxylic acid, in view of our experience with the tert-butyl ester. Construction of this biaryl ether tether employed the Evans-modified Ullmann aryl ether synthesis,37 proved to be compatible with the labile TIPS ester (Scheme 6).
Scheme 5.

Second generation retrosynthesis of EF ring analog 17.
Scheme 6.

Preparation of 32.
Union of 32 with EF Wittig salt (+)-31 was achieved upon deprotonation with MeLi·LiBr. Addition of 32 provided 34 as in an inseparable mixture (4:1) of Z and E olefin isomers (Scheme 7). The lack of complete Z selectivity was surprising given that the identical conditions in the gram-scale synthesis of (+)-spongistatin 1, led exclusively the Z olefin. The mixture of olefin isomers, without separation, was then subjected to TBAF (3 equiv) to effect deprotection of both the TIPS ester, as well as the F-ring TES ethers; seco-acid 35 as a mixture of olefin Z:E isomers (4:1), was obtained in 67% yield. The macrocyclization conditions that had proven successful in the preparation of (+)-spongistatin 1 [cf. i-PrNEt2 (60 equiv); 2,4,6-TBCCl (20 equiv) toluene at room temperature; then DMAP, 90 °C, Scheme 8A] were explored first. A single product, containing a Z-olefin was generated, presumably due to the strain associated with incorporating an E-olefin into the macrocycle. The desired macrocycle (+)-37 however was not the product, but instead trichlorobenzoate 36, tentatively assigned based on NMR and MS analysis. Presumably, following macrocyclization, the molecule adopts a conformation which facilitates acylation at C(42). Reduction of the amount of each reagent, [cf. 9 equivalents of 2,4,6-trichlorobenzoyl chloride, 15 equivalents of i-Pr2NEt and 20 equivalents of DMAP (Scheme 8B)], furnished the desired macrocycle (+)-37, along with a minor inseparable impurity.
Scheme 7.

Preparation of macrolactonization precursor 35.
Scheme 8.

Macrolactonization of 35.
Global deprotection next yielded a mixture of products (11:1). Unfortunately, the major product was not the desired 17-lactol (Scheme 9), but instead identified as the E-ring-opened 17-ketone, assigned on the basis of a 13C NMR carbonyl resonance at 211.4 ppm. For comparison the hemiketal carbon resonance of (+)-37 appears at 100.5 ppm.
Scheme 9.

Global deprotection of (+)-37 under conditions followed in the gram-scale spongistatin 1 synthesis.
Isodesmic calculations,38 employing 17-ketone, (+)-spongistatin 1 [(+)-1] and the corresponding C(1) seco-acids, were performed in vacuum with the MMFF force field. The results revealed that the increase in strain energy upon macrocyclizations of (+)-spongistatin 1 [(+)-1] and 17-lactol to be 5.8 kJ/mol and 45.2 kJ/mol, respectively. In similar fashion calculations employing the 17-lactol and 17-ketone revealed the release in strain energy upon opening of the pyran ring of 17-ketone to be 11.2 kJ/mol. Although not definitive these calculations suggest upon the acidic global deprotection, the macrocyclic strain energy leads to opening of the E ring. We therefore decided to reinvestigate our design strategy.
Advanced Analog Design: Lessons Learned Lead to a (+)-Spongistatin Congener Possessing Nanomolar Tumor Cell Growth Inhibitory Activity
The design and synthesis of biarylether EF analog 17-lactol led to an important lesson: consideration of ring strain energy is critical in the design of macrocyclic analogs. Although a linker may fix the overall conformation, the induced ring strain associated with the designed macrocycle may not be compatible. To rectify this issue, we turned to more flexible polymethylene tethers. We also chose to incorporate an internal hydrogen bond acceptor in the form of a lactone carbonyl that might participate in a H-bond to the C(42)-hydroxyl on the F ring, in an effort to orchestrate the conformation of the (+)- spongistatin western binding/recognition domain. Our goal was to lower the overall flexibility observed both in our solution conformational studies of (+)-spongistatin 1 [(+)-1] and the Heathcock cyclic analogs (15 and 16; Figure 1). We also chose to include the AB spiro-ring moiety to expand the structural elements in an attempt to maximize tubulin binding.
Iterative in silico conformational searches, employing a series of tethers of different lengths with hydrogen bond acceptors, led to structure 38 (Scheme 10, in red), possessing a tether comprising a Z-olefin, in conjunction with a lactone moiety and a series of methylene units to mimic the span between ring B and the replaced CD spiroketal system. Importantly, the strain energy associated with the designed 29-membered diolide 38 was found to be nearly isoenergetic (9.1 kJ/mol) with the ring strain energy of (+)-spongistatin 1 (8.3 kJ/mol).
Scheme 10.

Retrosynthesis of ABEF ring analog.
From the synthetic perspective analog 38 was envisioned to arise from advanced AB and EF ring intermediates, again available from our gram scale synthesis of (+)-spongistatin 1 (1). That is, Wittig union involving EF phosphonium salt (+)-31 and now aldehyde 47, followed by macrolactonization and global deprotection would lead to 38 (Scheme 10). Tether 47, in turn, would be constructed from three fragments: known AB ring aldehyde 40,39 Wittig salt 41 and primary acid 44,40 the latter derived from δ-valerolactone.
Towards this end, Wittig union of (−)-40 with 41 led to (−)-42 in 51% yield, along with the deacetylated congener (−)-42a, which was readily reacetylated to furnish (−)-42 (Scheme 11); the overall yield was 63%. Given our earlier experience with (+)-30 (Scheme 4), the tert-butyl ester was converted to the TIPS-ester by a three step protocol (ca. 98% yield for the three steps). Known acid 44,38 the remaining component, was then appended to (−)-43 via esterification. Removal of the PMB group followed by oxidation41 delivered the desired tether (−)-47 in good yield.
Scheme 11.

Synthesis of (−)-47.
Union of (−)-47 with EF Wittig salt (+)-31 proceeded smoothly to furnish (−)-48 (Scheme 12) in 64% yield, in this case exclusively as the Z olefin. Removal of the TES and TIPS groups employing TBAF in THF next provided seco-acid (−)-49, which upon Yamaguchi lactonization,42 followed by global deprotection employing HF in CH3CN, conditions identical to those employed in our large scale (+)- spongistatin 1 synthesis,10e completed the construction of (−)-38, the ABEF analog. The yield for the final two steps was 48%.
Scheme 12.

Preparation ABEF ring analog (−)-38.
The solution conformations of (−)-38 was investigated by computational and NMR studies, similar in detail to those we employed to define the solution conformations of (+)-spongistatin 1. Three solution conformational families were found in CHCl3 (Figure 5A–C), wherein the western ABEF perimeters of all conformers effectively recapitulate the western ABEF perimeter conformation observed in (+)- spongistatin 1. The major solution conformer (cf. Figure 5A, 59%) incorporates two hydrogen bonds in addition to the H-bonds within the EF system. In accord with our design, the F ring hydroxyl makes a H-bond with the newly introduced lactone carbonyl, as well as the C(10) B ring hydroxyl, further fixing the conformation of the AB ring. The second major conformational family (cf. Figure 5B, 33%) incorporates three additional H-bonds, in this case with the F ring hydroxyl selecting the C(48) hydroxyl [(+)-spongistatin 1 numbering] group on the side-chain, rather than with the lactone carbonyl on the linker. Finally, the minor conformer family found (cf. Figure 5C, 8%) incorporates a H-bond between the linker carbonyl and the C(48) [(+)-spongistatin 1 numbering] side-chain hydroxyl. Overall, the conformations are quite rigid at the ABEF ring juncture, affording the side-chain with a more preferential alignment compared to the (+)-spongistatin 1 structures. Overlay of the three conformations of (−)-38 (Figure 5a-c) to the identified solution conformations of (+)-spongistatin 1 (Figure 4a–d) revealed they all possess a similar western conformation. (Figure 5D).
Figure 5.

Calculated major solution conformers a–c of ABEF analog (−)-38. Overlay of the major solution conformation a with (+)-spongistatin 1 conformer seen in Figure 4b.
Biological Studies
Not surprisingly, the first generation EF ring analog, isolated as a 11:1 mixture of structural isomers (17-ketone and 17-lactol), favoring the 17-ketone, revealed low biological activity in the cell-based growth inhibition assay (Table 1). Pleasingly however, the ABEF ring analog (−)-38, demonstrated by NMR to comprise a good conformational mimic of the western perimeter of (+)-spongistatin 1 [(+)-1], displayed nanomolar inhibitory activity (Table 3).43 The question that now arose, given that a substantial proportion of the natural product had been deleted, became: Does this analog have the same mechanism of action as (+)-spongistatin 1?
Table 3.
Cell growth inhibition (IC50, nM) with spongistatin 1 and the synthesized analogs
| MDA-MB-435 | HT-29 | H522-T1 | U937 | |
|---|---|---|---|---|
| (+)-Spongistatin 1 | 0.0225 | 0.058 | 0.16 | 0.059 |
| (−)-38 | 82.8 | 161.2 | 297.2 | 60.5 |
| 17 | >1000 | >1000 | >1000 | >1000 |
To answer this question, the effect of (+)-spongistatin 1 [(+)-1] and ABEF analog (−)-38 on cell cycle distribution was evaluated in a standard cell cycle analysis employing U937 lymphoma cells. As shown in Figure 6A, both (+)-1 and analog (−)-38 led to significant enrichment of cells in the G2/M phase of the cell cycle (G2/M peaks are indicated by arrows), consistent with the known mechanism of action of (+)-spongistatin 1 to target microtubule architecture. To ascertain further that analog (−)-38 retains the same mechanism of action as (+)-spongistatin 1, both compounds were evaluated in an in vitro tubulin polymerization assay. As shown in Figure 6B, analog (−)-38 inhibits tubulin polymerization in a dose dependent manner, similar to (+)-spongistatin 1 (1). The higher concentration (i.e., 30 μM) required for analog (−)-38 to inhibit tubulin polymerization reflects the lower potency of the analog as compared to the extraordinary potent (+)-spongistatin 1. Vinblastine, a microtubule destabilizing agent, and paclitaxel, a tubulin stabilization agent, were included as controls. Thus, like (+)-spongistatin 1, the designed analog (−)-38 possesses significant microtubule destabilizing activity.
Figure 6.

A. G2/M cell cycle effects of spongistatin 1 and (−)-38. U937 cells were treated with either spongistatin 1 or the (−)-38 analog for 18 h. Samples were subject to flow cytometric cell cycle analysis. Data shown represent relative number of cells (Y axis) as a function of fluorescence intensity representing DNA content (X axis). B. Inhibition of in vitro tubulin polymerization. An in vitro tubulin polymerization assay was carried out in the presence of indicated compounds and concentrations. Tubulin polymerization curves shown here represent the relative amount of tubulin polymer (OD340, Y axis) over time (X axis).
Summary
The feasibility of designing a diminutive congener of (+)-spongistatin 1 [cf. (−)-38], wherein approximately one third of the parent compound is replaced by a simplifying tether that retains significant microtubule destabilizing activity (ca. 60 nM for U937 cell lines) has been demonstrated. To achieve this goal, we introduced a new computational method (cf. DISCON) that led to a design strategy based on the hypothesis that the pharmacophoric elements of biologically active natural products are likely to reside on conformationally rigid portions of the molecule. Significantly, this synthetic effort led to the elimination of some 30 steps compared to that of (+)-spongistatin 1.11 Finally, a general strategy for the design of simplified bioactive analogs of large flexible natural products has been developed and validated.
Supplementary Material
Acknowledgments
Financial support was provided by the NIH (NCI) through Grant No. CA-70329. C.A.R. was supported by an American Cancer Society-Michael Schmidt Postdoctoral Fellowship.
Footnotes
Supporting Information Available: Spectroscopic and analytical data and selected experimental procedures. This material is available free of charge via Internet at http://pubs.acs.org.
LITERATURE CITED
- 1.(a) Pettit GR, Chicacz ZA, Gao F, Herald CL, Boyd MR, Schmidt JM, Hooper JNA. J Org Chem. 1993;58:1302–1304. [Google Scholar]; (b) Pettit GR, Cichacz ZA, Gao F, Herald CL, Boyd MR. J Chem Soc, Chem Commun. 1993:1166–1168. [Google Scholar]; (c) Pettit GR, Herald CL, Cichacz ZA, Gao F, Boyd MR, Christie ND, Schmidt JM. Nat Prod Lett. 1993;3:239–244. [Google Scholar]; (d) Pettit GR, Herald CL, Cichacz ZA, Gao F, Schmidt JM, Boyd MR, Christie ND, Boettner FE. J Chem Soc, Chem Commun. 1993:1805–1807. [Google Scholar]; (e) Pettit GR, Cichacz ZA, Herald CL, Gao F, Boyd MR, Schmidt JM, Hamel E, Bai R. J Chem Soc, Chem Commun. 1994:1605–1606. [Google Scholar]
- 2.Fusetani N, Shinoda K, Matsunaga S. J Am Chem Soc. 1993;115:3977–3981. [Google Scholar]
- 3.(a) Kobayashi M, Aoki S, Sakai H, Kawazoe K, Kihara N, Sasaki T, Kitagawa I. Tetrahedron Lett. 1993;34:2795–2798. [Google Scholar]; (b) Kobayashi M, Aoki S, Sakai H, Kihara N, Sasaki T, Kitagawa I. Chem Pharm Bull. 1993;41:989–991. doi: 10.1248/cpb.41.989. [DOI] [PubMed] [Google Scholar]; (c) Kobayashi, Aoki S, Kitagawa I. Tetrahedron Lett. 1994;35:1243–1246. [Google Scholar]
- 4.Boyd MR. In: Principles and Practices of Oncology Updates. 3. DeVita VT Jr, Hellman S, Rosenberg SA, editors. Vol. 10. 1994. pp. 1–12. [Google Scholar]
- 5.Bai R, Cichacz ZA, Herald CL, Pettit GR, Hamel E. Mol Pharmacol. 1993;44:757–766. [PubMed] [Google Scholar]
- 6.Bai R, Taylor GF, Cichacz ZA, Herald CL, Kepler JA, Pettit GR, Hamel E. Biochemistry. 1995;34:9714–9721. doi: 10.1021/bi00030a009. [DOI] [PubMed] [Google Scholar]
- 7.Luduena RF, Roach MC, Prasad V, Pettit GR, Cichacz ZA, Herald CL. Drug Dev Res. 1995;35:40–48. [Google Scholar]
- 8.Bai R, Pettit GR, Hamel E. J Biol Chem. 1990;265:17141–17149. [PubMed] [Google Scholar]
- 9.(a) Guo J, Duffy KJ, Stevens KL, Dalko PI, Roth RM, Hayward MM, Kishi Y. Angew Chem, Int Ed. 1998;37:187–192. [Google Scholar]; (b) Hayward MM, Roth RM, Duffy KJ, Dalko PI, Stevens KL, Guo J, Kishi Y. Angew Chem, Int Ed. 1998;37:192–196. [Google Scholar]; (c) Paterson I, Wallace DJ, Oballa RM. Tetrahedron Lett. 1998;39:8545–8548. [Google Scholar]; (d) Paterson I, Chen DYK, Coster MJ, Acena JL, Bach J, Gibson KR, Keown LE, Oballa RM, Trieselmann T, Wallace DJ, Hodgson AP, Norcross RD. Angew Chem, Int Ed. 2001;40:4055–4060. [PubMed] [Google Scholar]; (e) Smith AB, III, Doughty VA, Lin Q, Zhuang L, McBriar MD, Boldi AM, Moser WH, Murase N, Nakayama K, Sobukawa M. Angew Chem, Int Ed. 2001;40:191–195. doi: 10.1002/1521-3773(20010105)40:1<191::AID-ANIE191>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]; (f) Smith AB, III, Zhu W, Shirakami S, Sfouggatakis C, Doughty VA, Bennett CS, Sakamoto Y. Org Lett. 2003;5:761–764. doi: 10.1021/ol034037a. [DOI] [PubMed] [Google Scholar]; (g) Ball M, Gaunt MJ, Hook DF, Jessiman AS, Kawahara S, Orsini P, Scolaro A, Talbot AC, Tanner HR, Yamanoi S, Ley SV. Angew Chem, Int Ed. 2005;44:5433–5438. doi: 10.1002/anie.200502008. [DOI] [PubMed] [Google Scholar]; (h) Smith AB, III, Tomioka T, Risatti CA, Sperry JB, Sfouggatakis C. Org Lett. 2008;10:4359–4362. doi: 10.1021/ol801792k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Evans DA, Trotter BW, Cote B, Coleman PJ, Dias LC, Tyler AN. Angew Chem, Int Ed Engl. 1998;36:2744–2747. [Google Scholar]; (b) Smith AB, III, Lin Q, Doughty VA, Zhuang L, McBriar MD, Kerns JK, Brook CS, Murase N, Nakayama K. Angew Chem, Int Ed. 2001;40:196–199. [PubMed] [Google Scholar]; (c) Crimmins MT, Katz JD, Washburn DG, Allwein SP, McAtee LF. J Am Chem Soc. 2002;124:5661–5663. doi: 10.1021/ja0262683. [DOI] [PubMed] [Google Scholar]; (d) Heathcock CH, McLaughlin M, Medina J, Hubbs JL, Wallace GA, Scott R, Claffey MM, Hayes CJ, Ott GR. J Am Chem Soc. 2003;125:12844–12849. doi: 10.1021/ja030317+. [DOI] [PubMed] [Google Scholar]; (e) Smith AB, III, Lin Q, Doughty VA, Zhuang L, McBriar MD, Kerns JK, Boldi AM, Murase N, Moser WH, Brook CS. Tetrahedron. 2009;65:6470–6488. doi: 10.1016/j.tet.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith AB, Sfouggatakis C, Risatti CA, Sperry JB, Zhu W, Doughty VA, Tomioka T, Gotchev DB, Bennett CS, Sakamoto S, Atasoylu O, Shirakami S, Bauer D, Takeuchi M, Koyanagi J, Sakamoto Y. Tetrahedron. 2009;65:6489–6509. doi: 10.1016/j.tet.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith AB, III, Boldi AM. J Am Chem Soc. 1997;119:6925–6926. [Google Scholar]
- 13.Tietze LF, Geissler H, Gewert JA, Jakobi U. Synlett. 1994;7:511–512. [Google Scholar]
- 14.(a) Smith AB, III, Xian M. J Am Chem Soc. 2006;128:66–67. doi: 10.1021/ja057059w. [DOI] [PubMed] [Google Scholar]; (b) Smith AB, III, Wuest WM. Chem Commun. 2008:5883–5895. doi: 10.1039/b810394a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Smith AB, III, Xian M, Kim W-S, Kim D-S. J Am Chem Soc. 2006;128:12368–12369. doi: 10.1021/ja065033e. [DOI] [PubMed] [Google Scholar]; (d) Smith AB, III, Kim W-S, Wuest WM. Angew Chem, Int Ed. 2008;47:7082–7086. doi: 10.1002/anie.200802301. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Smith AB, III, Kim W-S, Tong R. Org Lett. 2010;12:588–591. doi: 10.1021/ol902784q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Smith AB, III, Tong R. Org Lett. 2010;12:1260–1263. doi: 10.1021/ol100130x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Smith AB, III, Kim D-S. Org Lett. 2007;9:3311–3314. doi: 10.1021/ol071282b. [DOI] [PubMed] [Google Scholar]; (b) Smith AB, III, Cox JM, Furuichi N, Kenesky CS, Zheng J, Atasoylu O, Wuest WM. Org Lett. 2008;10:5501–5504. doi: 10.1021/ol8019132. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Smith AB, III, Foley MA, Dong S, Orbin A. J Org Chem. 2009;74:5987–6001. doi: 10.1021/jo900765p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Smith AB, III, Smits H, Kim D-S. Tetrahedron. 2010;66:6597–6605. doi: 10.1016/j.tet.2010.01.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Smith AB, III, Kim D-S. Org Lett. 2005;7:3247–3250. doi: 10.1021/ol0510264. [DOI] [PubMed] [Google Scholar]; (b) Smith AB, III, Kim D-S. J Org Chem. 2006;71:2547–2557. doi: 10.1021/jo052314g. [DOI] [PubMed] [Google Scholar]
- 17.Smith AB, III, Kim W-S. Proc Nat Acad Sci. 2011;108:6787–6792. doi: 10.1073/pnas.1015265108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paterson I, Acena JL, Bach J, Chen DYK, Coster MJ. Chem Commun. 2003;4:462–463. [PubMed] [Google Scholar]
- 19.Wagner CE, Wang Q, Melamed A, Fairchild CR, Wild R, Heathcock CH. Tetrahedron. 2008;64:124–136. [Google Scholar]
- 20.(a) Smith AB, Lin Q. Bioorg Med Chem Lett. 1998;8:567–568. doi: 10.1016/s0960-894x(98)00050-x. [DOI] [PubMed] [Google Scholar]; (b) Smith AB, III, Corbett RM, Pettit GR, Chapuis JC, Schmidt JM, Hamel E, Jung MK. Bioorg Med Chem Lett. 2002;12:2039–2042. doi: 10.1016/s0960-894x(02)00305-0. [DOI] [PubMed] [Google Scholar]; (c) Sfouggatakis C. PhD Thesis. University Of Pennsylvania; Philadelphia: 2005. Toward the Gram-Scale Total Synthesis of (+)- Spongistatin 1. [Google Scholar]
- 21.Lin Q. PhD Thesis. University Of Pennsylvania; Philadelphia: 2000. The synthesis of spongistatin side-chain analogs: The total synthesis of spongistatin 2 and 23-epi-spongistatin 2. [Google Scholar]
- 22.Towle MJ, Salvato KA, Budrow J, Wels BF, Kuznetsov G, Aalfs KK, Welsh S, Zheng W, Seletsk BM, Palme MH, Habgood GJ, Singer LA, Dipietro LV, Wang Y, Chen JJ, Quincy DA, Davis A, Yoshimatsu K, Kishi Y, Yu MJ, Littlefield BA. Cancer Res. 2001;61:1013–1021. [PubMed] [Google Scholar]
- 23.(a) Wender PA, Cribbs CM, Koehler KF, Sharkey NA, Herald CL, Kamano Y, Pettit GR, Blumberg PM. Proc Natl Acad Sci U S A. 1988;85:7197–7201. doi: 10.1073/pnas.85.19.7197. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wender PA, Irie K, Miller BL. Proc Natl Acad Sci U S A. 1995;92:239–243. doi: 10.1073/pnas.92.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wender PA, De Brabander J, Harran PG, Jimenez JM, Koehler MFT, Lippa B, Park CM, Shiozaki M. Journal of the American Chemical Society. 1998;120:4534–4535. [Google Scholar]; (d) Wender PA, De Brabander J, Harran PG, Jimenez JM, Koehler MFT, Lippa B, Park CM, Siedenbiedel C, Pettit GR. Proc Natl Acad Sci U S A. 1998;95:6624–6629. doi: 10.1073/pnas.95.12.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wender PA, De Brabander J, Harran P, Hinkle KW, Lippa B, Pettit G. Tetrahedron Lett. 1998;39:8625–8628. [Google Scholar]; (f) Wender PA, Hinkle KW, Koehler MFT, Lippa B. Medicinal Research Reviews. 1999;19:388–407. doi: 10.1002/(sici)1098-1128(199909)19:5<388::aid-med6>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]; (g) Wender PA, Baryza JL, Bennett CE, Bi C, Brenner SE, Clarke MO, Horan JC, Kan C, Lacote E, Lippa B, Nell PG, Turner TM. J Am Chem Soc. 2002;124:13648–13649. doi: 10.1021/ja027509+. [DOI] [PubMed] [Google Scholar]; (h) Wender PA, Baryza JL, Bennett CE, Bi FC, Brenner SE, Clarke MO, Horan JC, Kan C, Lacôte E, Lippa B, Nell PG, Turner TM. Journal of the American Chemical Society. 2002;124:13648–13649. doi: 10.1021/ja027509+. [DOI] [PubMed] [Google Scholar]; (i) Baryza JL, Brenner SE, Craske ML, Meyer T, Wender PA. Chemistry & Biology. 2004;11:1261–1267. doi: 10.1016/j.chembiol.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 24.(a) Taylor RE, Zajicek J. Journal of Organic Chemistry. 1999;64:7224–7228. [Google Scholar]; (b) Taylor RE, Chen Y, Beatty A, Myles DC, Zhou YQ. J Am Chem Soc. 2003;125:26–27. doi: 10.1021/ja028196l. [DOI] [PubMed] [Google Scholar]; (c) Yoshimura F, Rivkin A, Gabarda AE, Chou TC, Dong HJ, Sukenick G, Morel FF, Taylor RE, Danishefsky SJ. Angew Chem. 2003;42:2518–2521. doi: 10.1002/anie.200351407. [DOI] [PubMed] [Google Scholar]; (d) Taylor RE, Chen Y, Galvin GM, Pabba PK. Org Biomol Chem. 2004;2:127–132. doi: 10.1039/b312213c. [DOI] [PubMed] [Google Scholar]
- 25.Romo D, Choi NS, Li S, Buchler I, Shi Z, Liu JO. J Amer Chem Soc. 2004;34:10582–10588. doi: 10.1021/ja040065s. [DOI] [PubMed] [Google Scholar]
- 26.Aoki S, Nemoto N, Kobayashi Y, Kobayashi M, Kitagawa I. Tetrahedron. 2001;57:2289–2292. [Google Scholar]
- 27.Halgren TA. Journal of Comp Chem. 1996;17:490–519. [Google Scholar]
- 28.Qiu D, Shenkin PS, Hollinger FP, Still WC. J Phys Chem A. 1997;101:3005–3014. [Google Scholar]
- 29.(a) Landis C, Allured VS. J Am Chem Soc. 1991;113:9493–9499. [Google Scholar]; (b) Mierke DF, Kurz M, Kessler H. J Am Chem Soc. 1994;116:1042–1049. [Google Scholar]; (c) Cicero DO, Barbato G, Bazzo R. J Am Chem Soc. 1995;117:1027–1033. [Google Scholar]; (d) Nikiforovich GV, Kover KE, Zhang WJ, Marshall GR. J Am Chem Soc. 2000;122:3262–3273. [Google Scholar]
- 30.Hawkins DM. J Chem Inf Comp Sci. 2004;44:1–12. doi: 10.1021/ci0342472. [DOI] [PubMed] [Google Scholar]
- 31.Information of DISCON software will be provided in a concurrent report.
- 32.(a) Hoffmann RW. Angew Chem Int Ed. 1992;31:1124–1134. [Google Scholar]; (b) Hoffmann RW, Sander T, Brumm M. Chem Ber. 1992;125:2319–2324. [Google Scholar]; (c) Gottlich R, Kahrs BC, Kruger J, Hoffmann RW. Chem Comm. 1997:247–251. [Google Scholar]; (d) Hoffmann RW, Stahl M, Schopfer U, Frenking G. Chem Eur J. 1998;4:559–566. [Google Scholar]; (e) Hoffmann RW. Angew Chem Int Ed. 2000;39:2054–2070. [PubMed] [Google Scholar]
- 33.Evans DA, Trotter BW, Coleman PJ, Cote B, Dias LC, Rajapakse HA, Tyler AN. Tetrahedron. 1999;55:8671–8726. [Google Scholar]
- 34.(a) Ullmann F, Bielecki J. Chem Ber. 1901;34:2174–2185. [Google Scholar]; (b) Lindley J. Tetrahedron. 1984;40:1433–1456. [Google Scholar]; (c) Ley SV, Thomas AW. Angew Chem. 2003;42:5400–5449. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]
- 35.Paul R, Anderson GW. Journal of the American Chemical Society. 1960;82:4596–4600. [Google Scholar]
- 36.Wittig G, Schöllkopf U. Chem Ber. 1954;87:1318–1330. [Google Scholar]
- 37.Evans DA, Katz JL, West TR. Tetrahedron Lett. 1998;39:2937–2940. [Google Scholar]
- 38.Term refers to “A reaction (actual or hypothetical) in which the types of bonds that are made in forming the products are the same as those which are broken in the reactants” Muller P, editor. Pure Appl Chem. 5. Vol. 66. 1994. pp. 1077–1184.
- 39.Hubbs JL, Heathcock CH. J Am Chem Soc. 2003;125:12836–12843. doi: 10.1021/ja030316h. [DOI] [PubMed] [Google Scholar]
- 40.(a) Hoye TR, Kurth MJ, Lo V. Tetrahedron Lett. 1981;22:815–818. [Google Scholar]; (b) Jacobi PA, Li Y. Org Lett. 2003;5:701–704. doi: 10.1021/ol0275116. [DOI] [PubMed] [Google Scholar]
- 41.Parikh JR, Doering WvE. J Am Chem Soc. 1967;89:5505–5507. [Google Scholar]
- 42.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989–1993. [Google Scholar]
- 43.Smith AB, III, Risatti CA, Atasoylu O, Bennett CS, TenDyke K, Xu Q. Org Lett. 2010;12:1792–1795. doi: 10.1021/ol100418n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.