

Abstract
Ricin is a potent toxin found in castor seeds. The A chain, RTA, enzymaticlly depurinates a specific adenosine in ribosomal RNA, inhibiting protein synthesis. Ricin is a known chemical weapons threat having no effective antidote. This makes the discovery of new inhibitors of great importance. We have previously used 6-substituted pterins, such as pteroic acid, as an inhibitor platform with moderate success. We now report the success of 7-carboxy pterin (7CP) as an RTA inhibitor; its binding has been monitored using both kinetic and temperature shift assays and by X-ray crystallography. We also discuss the synthesis of various derivatives of 7CP, and their binding affinity and inhibitory effects, as part of a program to make effective RTA inhibitors.
Keywords: Ricin inhibitors, pterin, 7-substituted derivatives, luciferase assay
1. Introduction
Ricin is an example of a type 2 ribosome inactivating protein (RIP). Such proteins are composed of two subunits, the A chain and B chain. The A chain (RTA) is involved in the inactivation of the ribosome itself while the B chain assists in the uptake of the protein into cells [1]. It is the B chain that results in the increased cytotoxicity of the type 2 ribosome inactivating protein, relative to type 1 RIPs. Once in the cytoplasm, RTA depurinates a specific adenosine on ribosomal RNA [2]. This inhibits protein synthesis and results in the death of the target cell.
Ricin can be toxic at doses as low as 0.1–1 μg/kg, depending on the mode of administration [3]. Ricin is extracted from castor seeds, and can be easily purified from the by-products of castor oil manufacturing [4]. Due to its ease of manufacture, as well as the very high toxicity, ricin represents a real threat as a chemical warfare agent. For example, one of the first well-known uses of ricin as a weapon was in the “umbrella tip” assassination of Georgi Markov by the KGB in 1978 [5]. This threat is compounded further due to the absence of a known antidote.
The X-ray structure of RTA has been solved and refined [6]. Site-directed mutagenesis and substrate binding in crystalline RTA provided key information leading to the accepted mechanism of depurination [7]. This information has in-turn assisted the design and development of small molecules that could potentially function as an antidote by binding to the RTA active site and inhibiting activity [3, 8].
The Schramm group has taken a different approach to inhibitor design. They constructed stem-loop oligonucleotides which were effective in the nM range, although this inhibition requires a low pH and appears nonfunctional at physiologic pH [9]. They have recently used locked, cyclic polynucleotides to inhibit RTA and the related RIP saporin, and have obtained a crystal structure of the complex [10]. In general, nucleic acid based compounds have limited cell uptake, are often subject to degradation, and therefore have limited “drugability”.
Computer based virtual screening of commercially available small molecule libraries has been used to expand the list of potential RTA inhibitors that can function at physiological pH. A number of chemical platforms have been identified for inhibitor design, including pterin, purine, and pyrimidine based inhibitors [3, 8b, 11]. Pteroic acid (PTA) was one such compound, showing modest inhibition in an in vitro translation assay IC50 of 600 μM. This assay involves translation of the protein luciferase, which gives a luminescent response with luciferin. Active RTA halts the protein synthesis, and the activity of potential inhibitors is determined by measuring the recovery of luciferase counts at different concentrations of the inhibitor. The crystal structure of the complex defined the active site of RTA. It showed that the protein had a large binding site for its ribosome target, but this cleft could be subdivided. Adenine binds in a well defined pocket called the specificity site. This pocket initially adopts a closed form and thus binding requires the Tyr80 residue of RTA to rotate 45° to the open form [7]. This site has evolved to bind aromatic rings with an exocyclic amine and a particular pattern of hydrogen bond donors and acceptors [8b]. The observed binding showed the feasibility of designing compounds that could bind in this pocket, but which if properly diversified, could make additional contacts that would provide drug-like specificity and affinity. Although the pterins have limited solubility, their ability to interact with a number of specific groups in the RTA active site makes them an attractive drug design platform. We therefore began an exploration of derivatized pterins with improved inhibitor characteristics. The X-ray structure suggests that pterin positions 6 and 7 could accommodate pendants that might reach the RTA surface and make favorable interactions.
An early effort centered on attaching a soluble carboxylate to the pterin platform. It was previously observed that 6-carboxypterin had little or no detectable inhibition and showed no electron density when soaked into RTA crystals [8b]. However, the newly synthesized, regioisomeric 7-carboxy pterin, 7CP, is one of the more potent small molecule inhibitors of RTA we have observed. This led to a paradigm shift in the direction of our pterin-based synthesis, as most pterin chemistry focuses on the 6-substituent due to similarities to naturally occurring folic acid [12]. We discuss herein the results of 7CP in an in vitro translation assay, a differential scanning fluorimetry (DSF) assay, as well as the X-ray structure of this new inhibitor bound into the active site of RTA. We also discuss the construction of new 7-substituted pterins and their subsequent results in these various assays, with many new compounds resulting in μM inhibition. We report two key X-ray structures in this paper; the full crystallographic description of all these structures will soon be released independently.
2. Chemistry
Many of the pterin based compounds evaluated were synthesized directly via a previously reported acyl radical insertion reaction (3–7, Scheme 1) [13]. The simple pterin core (2), which was prepared via condensation of glyoxal with 1, was taken up in aqueous sulfuric acid and treated with iron sulfate, tert-butyl hydroperoxide, and an acyl source to provide compounds 3–7 in yields ranging from 22–48%. Quantitative hydrolysis of 3 with NaOH at 80 °C provided 7CP (eg, 8, Scheme 1).
Scheme 1.

Synthesis of 7CP (8) and derivatives
N-Substituted carbamoyl structures (9–15) were synthesized via treatment of the methyl ester of 7CP (eg. 3) with the corresponding amine nucleophile in refluxing methanol in a sealed tube (Scheme 2). Compound 4 can also be constructed in this manner, as an alternative to the acyl radical reaction.
Scheme 2.

Synthesis of amide derivatives
3. Results and discussion
The active site of RTA contains various hydrogen bond donors and acceptors, aiding in the binding of the natural substrate adenine. It was previously seen that the pterin ring of pteroic acid and neopterin participate in a larger number and stronger interactions within the active site, compared to adenine [8b, 14]. As all current pterin-based inhibitors found from commercial libraries had relied on the 6-position for functionalization, we diversified our library by testing the regioisomeric 7-substituted derivatives, a task greatly assisted by our new synthetic means [13].
Beginning our study with 7-carboxy pterin (8), we immediately saw success with the in vitro luciferase-based assay, shown in Figure 1. As shown, the IC50 for 7CP is about 230 μM, superior to that of pteroic acid and neopterin, and vastly superior to 6-carboxy pterin [8b]. In general, protein-ligand complexes are more thermally stable, and will usually have a higher melting temperature, Tm. Therefore binding of 7CP to RTA can be observed using differential scanning fluorimetry, DSF [15]. As shown in Figure 2, the RTA•7CP complex has a Tm about 2 °C higher than RTA alone; the standard deviation of Tm measures is 0.1–0.3 °C. Experimental results for the inhibitor binding experiments are accumulated in Table 1.
Figure 1. RTA Inhibition by 7CP.

RTA activity is plotted as a function of 7CP concentration. The data were fitted to a hyperbolic decay function and the IC50 was determined to be 230 μM.
Figure 2. DSF profile for RTA and a 7CP-RTA complex.
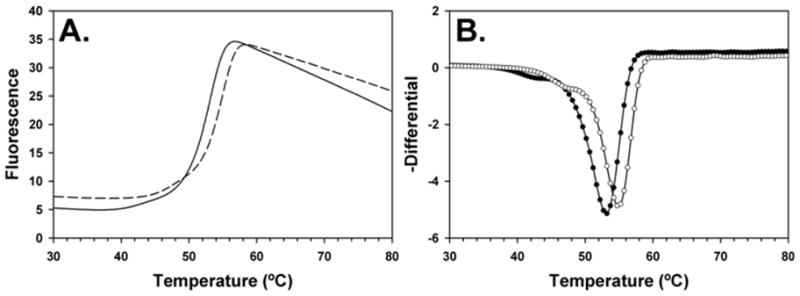
Panel A shows the fluorescence as a function of temperature for RTA (solid line) and with 7CP (dashed line). Panel B shows the negative differential of the data from panel A with RTA as black circles and the complex with open circles; the minimum of each curve marks the Tm for the unfolding.
Table 1.
Summary of Results
| Entry | Structure | Name | IC50a | ΔTm | Resolution |
|---|---|---|---|---|---|
| 8 |
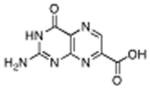
|
7-carboxy pterin (7CP) | 230 μM | 2°C | 1.29 Å |
| 4 |
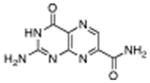
|
7-carbamoyl pterin (7AP) | No Inhibition | 1°C | 1.75 Å |
| 9 |
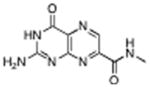
|
N-methyl-7-carbamoyl pterin | 1.6 mM | 0.4 °C | 1.26 Å |
| 10 |
|
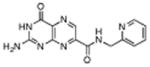
N-(methylamino pyridinyl)-7-carbamoyl pterin | 500 μM | 1°C | 1.26 Å |
| 11 |
|
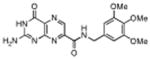
N-(3,4,5-trimethoxy benzyl)-7-carbamoyl pterin | No Inhibition | 1°C | NA |
| 12 |
|
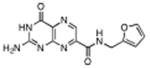
N-(furanylmethyl)-7-carbamoyl pterin | 380 μM | 2°C | 1.89 Å |
| 13 |
|
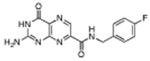
N-(4-fluorobenzyl)-7-carbamoyl pterin | 570 μM | 2°C | NA |
| 14 |
|
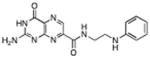
N-(2-(phenylamino) ethyl)-7-carbamoyl pterin | 210 μM | 3°C | 1.75 Å |
| 15 |
|
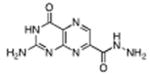
7-hydrazide pterin (7HP) | 500 μM (35%) | <1σ | 1.97 Å |
| 5 |
|
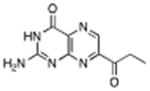
7-propionyl pterin | 700 μM | 2°C | 1.35 Å |
| 6 |
|
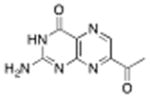
7-acetyl pterin | 500 μM (20%) | 1.3°C | 1.26 Å |
| 7 |
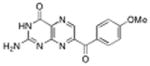
|
7-(p-methoxy benzoyl) pterin | No Inhibition | <1σ | NA |
= When 50% inhibition was not reached.
% inhibition given in parenthesis at maximum concentration
Compound 8 (7CP) was soaked into RTA crystals and X-ray data collected to 1.29 Å resolution. Figure 3 shows the electron density for the bound 7CP and Figure 4 shows details of the interaction with the protein. As seen in previous structures of RTA in complex with pterin derivatives, the 2-amino group of the pterin ring donates one hydrogen bond each to the carbonyl oxygen atoms of Val81 and Gly121, and the 4-oxo and N5 atoms accept hydrogen bonds from Arg180 [14]. The tautomeric form of the pterin with a proton on N1 is favored so that N1 can donate a hydrogen bond to a carboxyl oxygen of Gly121 and N3 can accept a hydrogen bond from the amido-N of Val81. An additional hydrogen bond, which was not seen in the previous 6-substituted pterins, is made between the amido-N of Tyr123 and the carbonyl oxygen of the 7-carboxy group. This extra hydrogen bond is retained in all subsequent derivatives based on this platform, likely contributing to the general increase in potency of this series of pterin-based inhibitors over previous ones.
Figure 3.
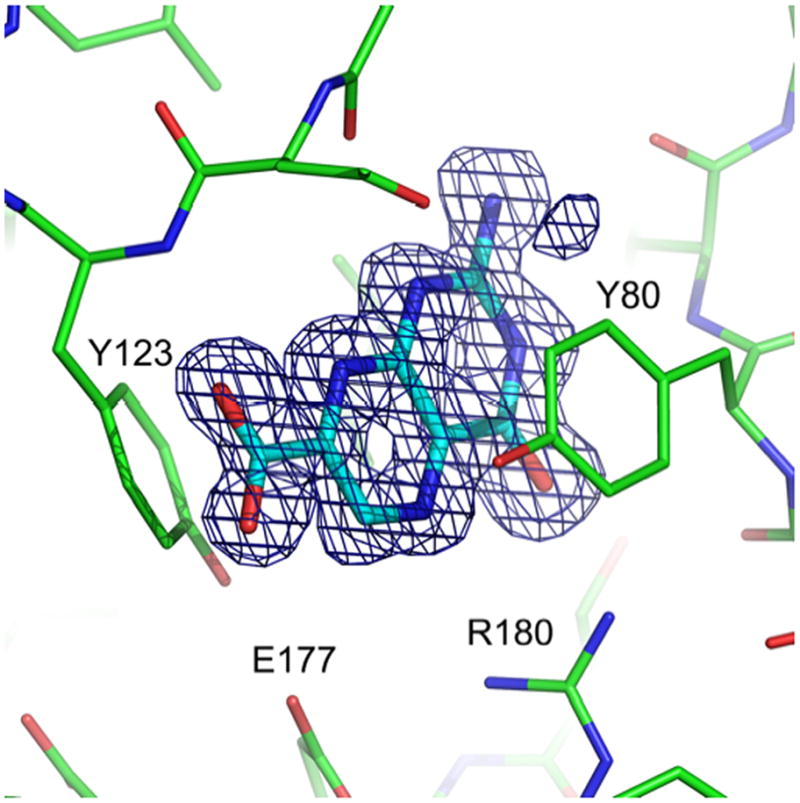
7-carboxy-pterin in complex with RTA. Density from the FO-FC map, shown as a blue mesh, is contoured at 3 σ around the ligand. Active site residues are labeled for orientation. (PDB ID 3PX8)
Figure 4.

Stereo view of 7CP bound to the active site of RTA. Hydrogen bonds are shown as black dashed lines.
Having seen success with the carboxylic acid at position 7, we turned our attention to amide derivatives, starting initially with the unfunctionalized 7-carbamoyl pterin (7AP, eg 4). Transitioning from the carboxylic acid, which is anionic at physiological pH, to the neutral amide reduced the solubility of the pterin. Pterins are notoriously insoluble in most any solvent, often requiring protection of the exocyclic amine, which would undoubtedly block binding to RTA due to the very specific interactions needed with this amine [7d, 8]. However, compound 4 exhibited a 1 °C Tm shift by DSF, and X-ray data of the RTA/7AP complex at 1.75 Å confirmed binding to the active site, as shown in Table 1. Unfortunately this amide did not show a strong response in the in vitro luciferase-based assay, which is attributed to its poor solubility in the HEPES buffer solution necessary to run the assay.
Despite the apparent decrease in activity of the simplest amide derivative of 7CP, we continued to explore larger amides that might make favorable contacts with RTA. Compound 9 was chosen to explore simple alkyl amides; this compound showed an IC50 of 1.6 mM in the in vitro assay. Although it is a poorer inhibitor than 7CP, it is still an improvement over the 6-substituted neopterin and previously reported guanine-based inhibitors [3, 8b]. Compound 9 was also crystallized with RTA, and a 1.26 Å X-ray structure confirmed it bound into the active site, with the pterin group making the expected interactions.
Compounds 11 and 13 were synthesized to investigate the effect of aromatic rings with electron rich and poor substituents, using 3,4,5-trimethoxybenzyl amine and 4-fluorobenzyl amine respectively. Compound 13 showed a marked improvement over the alkyl amide, giving an IC50 of 570 μM in the translation assay, and a Tm shift of 2 °C in the DSF experiment. Efforts to obtain a crystal structure for this compound have been unsuccessful to date, arising from an inability to find an appropriate cryoprotectant for this particular ligand. Compound 11 did not show any appreciable inhibition of RTA in the translation assay, and only shifted the Tm by 1 °C. This finding may suggest a binding preference towards electron deficient substituents, or that the size of the trimethoxyphenyl group may preclude favorable interaction with the protein.
Compounds 10 and 12 explore heterocycle pendants, using aminomethyl pyridine and furfuryl amine as the respective N-substituents. Compound 10 has an IC50 of 500 μM, and shows a Tm shift of 1 °C. Its binding was also observed in a 1.26 Å X-ray structure. The compound had diminished electron density around the pyridine ring, indicating the ring is not in a fixed position and appears to rotate about its long axis. Efforts are underway to diversify this pendant to provide more specific interactions within the active site, and thereby improve binding. Compound 12 showed an IC50 of 380 μM, and a Tm shift of 2 °C was observed in the DSF assay, indicating similar binding affinity to that of 7CP. This compound has been successfully seen in complex with RTA through X-ray data at 1.89 Å. In the crystal structure, the furan group is oriented such that its oxygen is facing the hydroxyl group on Tyr80, however the distance between the pair is too long, at approximately 4 Å, for a hydrogen bonding interaction. As seen in Figure 5, the furan is close to the so-called “second pocket” of RTA. This pocket is on the opposite side of the Tyr80 ring from the specificity pocket. The second pocket has been confirmed to be the binding site for guanine base in the GAGA tetraloop substrate adjacent to the adenine targeted for depurination [10]. Efforts are underway to create pendants extending from the 5 position on the furan ring that could potentially occupy this second pocket. An electrostatic surface map of the specificity pocket is shown in Figure 6 illustating possible requirements for these modifications.
Figure 5.

Surface representation of RTA in complex with furanomethyl carbamoyl pterin (Compound 12). While the pterin occupies the specificity pocket (A), the furan is in position to be derivatized at the 5 position to extend pendants around Tyrosine 80 and into the second pocket (B). In the surface representation of RTA, white represents hydrogen, green represents carbon, blue represents nitrogen, and red represents oxygen. (PDB ID 3PX9)
Figure 6.
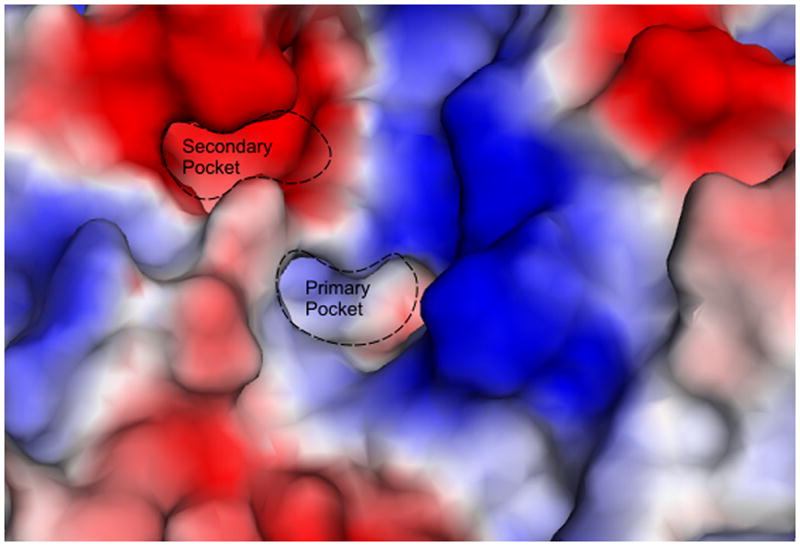
Electrostatic surface map of the RTA active site. Red represents electron rich regions, and blue indicates electron deficient or positive charge.
These results, along with those seen with 11 and 13, can again be taken to indicate a trend favoring the binding of electron deficient pendants to the 7CP platform. This conclusion is supported by the binding strength of the electron deficient benzoic acid moiety seen previously for pteroic acid [8b]. There may be important steric restraints on the inhibitors synthesized to this point, in that smaller amide derivatives gave better results than larger ones. Compound 14 was synthesized to determine if steric bulk could be better accommodated if located further from the relatively small specificity pocket. Indeed, this compound had an IC50 of 210 μM, and shifted the Tm of RTA by 3°C, even stronger than that observed for 7CP. The crystal structure of this complex shows that the aniline NH donates a hydrogen bond to the side-chain of Asn 209. This is one of the few polar interactions seen for the pendant groups of this inhibitor series. However, as observed for compound 10, only weak electron density was observed for the aromatic ring, suggesting it is mobile. We conclude from the results of the amide derivatives that smaller, electron deficient substituents are preferred when in close proximity to the pterin head-group, though larger groups can be successfully tolerated with proper spacer length.
Examination of the crystal structure of 7AP (eg. 4) indicated a water molecule forms a hydrogen bonded bridge from the amide NH2 to the carboxylate of Glu177 of RTA. With this in mind, we synthesized a hydrazide derivative (7HP, 15), postulating that the additional NH2 would replace this water molecule and enhance the binding compared with 7AP. 7HP was soaked into RTA crystals, and X-ray data collected to 1.97 Å, confirmed that this compound was bound into the RTA active site. However, it did not displace the water bound to Glu177. That water repositioned by about 0.4 Å and the water bridge to the hydrazide nitrogen remained intact. The 7HP compound has a maximum solubility of 500 μM in the HEPES buffer used for the in vitro translation assay. At this concentration, it inhibited RTA by 35%, indicating an IC50 below 1 mM range. Although not a strong inhibitor, this modification was a clear improvement over compound 4.
Finally, as part of our study we sought to examine how replacement of carboxylic acid derivatives with simple ketones would affect the binding of the pterin platform. Compounds 5–7 had been constructed previously, and allow for the investigation of van der Waals interactions as well as the effect of steric bulk. However, ketone derivatives suffer the same solubility problems seen when transitioning from carboxylic acids to amides. Compound 6, baring a simple methyl ketone, was soluble to 500 μM for the in vitro assay, however only 20% inhibition was observed in the translation assay. This compound was also seen in an X-ray crystal bound in the RTA active site in the same basic orientation as 7CP. Compound 5 was a better inhibitor, having an IC50 of 700 μM, similar to pteroic acid, and a Tm shift of 2 °C. Once again this pterin was observed in the X-ray analysis at 1.35 Å resolution. Compound 7 had a much bulkier pendant; it failed to give any detectable inhibition of RTA and could not be soaked into RTA crystals. Its insolubility and presumed poor binding by the restrained methoxybenzoyl made this a poor candidate for further development. While compounds such as 10, 13, and 14 all have similar sized pendants, they contain at least one sp3 carbon linking the amide to other chemical moieties. This methylene appears to allow for a more flexible binding geometry to the RTA active site. Compound 7 has a planar linking group, but lacks the flexible methylene in the linker. This restriction appears great enough to outweigh any favorable binding interactions by the pendant. The results from the ketone series are in agreement with the trend seen with the amides above; compounds with electron donating groups and bulky substituents in close proximity to the pterin core have diminished binding.
4. Summary
Pterins are able to bind in the RTA specificity pocket and make more, and stronger, hydrogen bonds to the protein than does the substrate adenine [8b]. This chemical specificity makes it an attractive inhibitor design platform despite its poor solubility. The platform can, in principle, be diversified to provide inhibitors with better solubility and specificity. In this paper we examined the utility of adding substituents at position 7 of the pterin. A carboxylate at position 6 had previously abolished binding and this site was not explored further [8b]. A carboxylate at position 7, in contrast, was a good inhibitor of RTA. A simple amide at position 7 renders the compound much less soluble and a poorer inhibitor. It is possible to make larger derivatives which are not necessarily more soluble, but where additional contacts with RTA provide comparable binding affinity. Amide derivatives with a methylene linker appear to be able to adjust to the RTA active site. For example, N-(furanylmethyl)-7-carbamoyl pterin has activity close to that of 7CP and places the furan ring in a position where further derivatization should be able to reach a second binding pocket on RTA. In principle, future compounds that use this potential may have both improved binding specificity and solubility. Nearly all of the new compounds showed a marked improvement in activity over neopterin, and a number of these are also superior to pteroic acid [8b]. This work highlights the importance of 7-substitued pterin analogs in the design of ricin inhibitors.
5. Experimental Section
5.1. Synthesis
All reagents used were of commercial quality and were obtained from Aldrich Chemical Co. and Fisher Scientific and were used as received. 1H (400 MHz) and 13C (125 MHz) NMR spectra were recorded in DMSO-d6 on a Varian spectrometer using the solvent as reference. Chemical shifts are given in parts per million (ppm). LC-MS data was recorded on an Agilent 6130 Quadrupole instrument. High resolution mass spectrometry was performed with a Varian 9.4T QFT-ESI ICR system. All solvents were removed by rotary evaporation under vacuum using a standard rotovap equipped with a dry ice condenser. All filtrations were performed with a vacuum. Purity of all final compounds was determined to be >95% on a Shimadzu HPLC with a 4.6 × 150 mm Phenomenex gemini 5 μm C18 column. The eluents were A, water, and B, acetonitrile. Gradient elution from 0% B to 30% B over 20 min with a final hold at 90% B for 5 min. Total run time was 25 min.
2-Aminopteridin-4-one (2), 7-methoxycarbonyl-pterin (3), 7-carbamoyl-pterin (4), 7-propionyl-pterin (5), 7-acetyl-pterin (6) and 7-(p-methoxybenzyl)-pterin (7) were all prepared through their literature procedures [12a, 13].
7-carbamoyl-pterin (7AP eg. 4) can also be prepared by suspending 3 in ammonia saturated methanol, in a sealed conical vial, and heating to 80 °C overnight with stirring. Addition of a small amount of 0.5M HCl, to neutralize, followed by filtration gave a yellow solid which, after washing with methanol and drying, was identical to the sample prepared through the previous method [13].
7-carboxy-pterin (7CP eg. 8) was prepared by hydrolysis of 3 in 0.5M NaOH at 80° C overnight. The pH of the homogeneous solution was then adjusted to 5 with 2M HCl, and the yellow precipitate filtered off and washed several times with water. This was dried under vacuum to provide 8 in quantitative yield. This compound had identical 1H and 13C NMR to those reported in the literature [16].
N-methyl-7-carbamoyl-pterin (9): To a suspension of 50 mg (0.23mmol) 3 in 2 mL MeOH in a sealed conical vial was added 0.3 mL methylamine. This was stirred overnight at 60 °C resulting in a homogeneous yellow solution, at which point the reaction was complete by LCMS. Addition of 1mL THF resulted in product precipitation. The yellow solid was filtered off and washed with cold methanol followed by THF, then dried under vacuum to give 42.3 mg (85% yield) 9. mp > 350 °C(dec) 1H NMR (d6-DMSO, 400M Hz) δ 8.82 (s, 1H), 2.81 (d, J=4.8, 3H); 13C NMR (d6-DMSO, 125M Hz) δ 162.1, 160.4, 155.3, 154.7, 145.2, 136.9, 131.9, 26.3; HRMS-ESI (m/z): [M+Na]+ calcd for (C8H8N6O2Na)+ 243.06009; found 243.05983.
N-(methyl pyridinyl)-7-carbamoyl-pterin (10): This compound was synthesized, using aminomethyl pyridine, through an analogous method as that used for 9, using 50 mg of 3. After the reaction was complete by LCMS analysis, it was cooled and added to 50 mL cold water and refrigerated, whereby a yellow solid formed. After filtration and drying, 40 mg (59% yield) of 10 was recovered. mp = 300 °C(dec) 1H NMR (d6-DMSO, 400M Hz) δ 9.52 (t, J=5.9, NH), 8.9 (s, 1H), 8.63 (d, J=5.2, 1H), 8.0 (t, J=7.7, 1H), 7.55 (d, J=7.9, 1H), 7.49 (t, J=7.3, 1H), 4.71 (d, J=5.5, 2H); 13C NMR (d6-DMSO, 125M Hz) δ 163.3, 160.3, 155.9, 155.5, 154.3, 147.2, 145.8, 140.8, 136.8, 131.9, 123.6, 123.0, 42.7; HRMS-ESI (m/z): [M+1]+ calcd for (C13H12N7O2)+ 298.10470; found 298.10461.
N-(3,4,5-trimethoxybenzyl)-7-carbamoyl-pterin (11): This compound was synthesized with 3,4,5-trimethoxybenzylamine through an analogous procedure as used for 9, using 30 mg of 3. After precipitation with THF, filtration, and drying, 38 mg (76% yield) of 11 was recovered. mp = 335 °C(dec) 1H NMR (d6-DMSO, 400M Hz) δ 9.33 (t, J = 6.2, NH) 8.84 (s, 1H), 6.66 (s, 2H), 4.4 (d, J = 6.3, 2H), 3.72 (s, 6H), 3.6 (s, 3H); 13C NMR (d6-DMSO, 125M Hz) δ 164.1, 160.5, 156.0, 154.5, 152.7 (2C), 147.8, 136.7, 136.4, 134.7, 131.6, 104.9 (2C), 59.9, 55.8 (2C), 42.7; HRMS-ESI (m/z): [M+H]+ calcd for (C17H19N6O5)+ 387.14114; found 387.14119.
N-(furanylmethyl)-7-carbamoyl-pterin (12): This compound was synthesized with furfurylamine through an analogous procedure as used for 9, using 30 mg of 3. After precipitation with THF, filtration, and drying, 34.5 mg (92% yield) of 12 was recovered. mp > 350 °C(dec) 1H NMR (d6-DMSO, 400M Hz) δ 9.26 (t, J =6.2, NH), 8.86 (s, 1H), 7.57 (d, J =2.8, 1H), 6.42–6.35 (m, 1H), 6.28 (d, J = 2.8, 1H), 4.50 (d, J = 6.4, 2H); 13C NMR (d6-DMSO, 125M Hz) δ 162.4, 159.5, 153.5, 152.0, 151.7, 147.4, 142.2, 138.2, 131.6, 110.5, 107.2, 35.9; HRMS-ESI (m/z): [M+H]+ calcd for (C12H11N6O3)+ 287.08871; found 287.08873.
N-(4-fluorobenzyl)-7-carbamoyl-pterin (13): This compound was synthesized with 4-fluorobenzylamine through an analogous procedure as used for 9, using 20 mg of 3. After precipitation with THF, filtration, and drying, 21 mg (74% yield) of 13 was recovered. mp > 350 °C(dec) 1H NMR (d6-DMSO, 400M Hz) δ 9.48 (t, J = 6.6, NH), 8.86 (s, 1H), 7.38 (dd, J = 5.3, 9.0, 2H), 7.14 (t, J = 8.9, 2H), 4.47 (d, J = 6.8, 2H); 13C NMR (d6-DMSO, 125M Hz) δ 162.9, 161.9, 160.3, 154.4, 147.7, 136.6, 135.4, 135.3, 131.6, 129.4, 129.3, 115.0, 114.8, 41.7; HRMS-ESI (m/z): [M+H]+ calcd for (C14H12N6O2F)+ 315.10003; found 315.10003.
N-(2-(phenylamino) ethyl)-7-carbamoyl pterin (14): This compound was synthesized with N-phenyl-ethylenediamine through an analogous procedure as used for 9, using 20 mg of 3. After precipitation with THF, filtration, and drying, 17.5 mg (60% yield) of 14 was recovered. mp = 330°C(dec) 1H NMR (d6-DMSO, 400M Hz) δ 8.95 (t, J = 6.1, NH), 8.81 (s, 1H), 7.00 (dd, J = 8.5, 7.3, 2H), 6.55 (d, J = 7.7, 2H), 6.45 (t, J = 7.2, 1H), 5.71 (t, J = 5.6, 1H) 3.43 (dd, J = 13.0, 6.5, 2H), 3.13 (dd, J = 12.6, 6.1, 2H); 13C NMR (d6-DMSO, 125M Hz) δ 162.9, 160.4, 156.0, 154.4, 148.6, 147.6, 136.6, 131.6, 128.9 (2C), 115.6, 111.9 (2C), 42.2, 38.3; HRMS-ESI (m/z): [M+Na]+ calcd for (C15H15N7O2Na)+ 348.11794; found 348.11795.
7-hydrazide-pterin (7HP eg. 15): This compound was synthesized with 98% hydrazine through an analogous procedure as used for 9, using 30 mg of 3. After precipitation with THF, filtration, and drying, 18 mg (60% yield) of 14 was recovered. mp = 290 °C(dec) 1H NMR (d6-DMSO, 400M Hz) δ 8.9 (s, 1H), 7.34 (s, NH2); 13C NMR (d6-DMSO, 125M Hz) δ 161.2, 160.1, 155.6, 154.5, 145.7, 136.6, 132.6; HRMS-ESI (m/z): [M+Na]+ calcd for (C7H7N7O2Na)+ 244.05534; found 244.05536.
5.2. Luciferase translation assay
An in vitro translation assay was adapted from the Promega original protocol for their Rabbit Reticulocyte Lysate System as previously described [8a]. The compounds to be tested were solubilized in 0.05 N KOH or 100% DMSO prior to their inclusion in the assay. After mixing RTA and various concentrations of compounds in the presence of BSA, translation reactions were initiated by mixing a portion of the RTA/compound mixture with reticulocyte lysate, and translation mix (amino acids, RNasin ribonuclease inhibitor, luciferase control mRNA). For reactions in which compounds were solubilized in DMSO, two distinct 30 minute room temperature incubations were performed. The first was done upon mixing the various concentrations of compounds with BSA, and the second when RTA was added. The reactions were incubated for 90 minutes at 30 °C, after which they were stopped by freezing them at −20 °C. After thawing at room temperature, 2.5 μl reaction mixture was mixed with 40 μl Luciferase Substrate Reagent (Promega), and their luminescence was measured on a Perkin Elmer Envision luminometer (Waltham MA).
For each concentration of inhibitor to be tested, reactions were run both in the presence and absence of RTA. To calculate the RTA activity, corrected for interference with the assay by the tested compounds, the percent differences in luminescence for the corresponding reaction pairs were divided by the percent difference for the controls with no inhibitor present. Values for IC50 were calculated by fitting the plot of RTA activity vs. inhibitor concentration to a hyperbolic decay function. An example plot is shown in Figure 1.
5.3. Differential Scanning Fluorimetry
Experiments were performed using a LightCycler 480 Real Time PCR instrument (Roche) in a 96 well plate format. The samples were prepared as 100 μl master mixes from which 30 μl triplicates were aliquotted into a LightCycler 480 multi-well plate. The samples contained RTA at a final concentration of 2 μM for both experimental and control wells, the inhibitor was at a nominal final concentration of 500 μM for the experimental wells, and both sets were buffered in 100 mM HEPES pH 7.5, 125 mM NaCl. After mixing the inhibitor with the RTA, a 30 minute room temperature incubation was done prior to the addition of fluorescent dye. SYPRO Orange (Invitrogen) was added to each sample well to a final concentration of 5X from a freshly prepared working stock of 100X. Samples were heated from 25 °C to 80 °C in a LightCycler with a ramp rate of 0.04 °C/sec. Fluorescence of SYPRO Orange (λex = 498 nm, λem = 610 nm) was measured at a frequency of 15 acquisitions per °C. The Tm for each sample was calculated from the first derivative plot of fluorescence versus temperature. An example plot is shown in Figure 2.
5.4. Crystallization of RTA-inhibitor complexes
Monoclinic RTA crystals were grown as described previously [17]. Briefly, RTA at a concentration of 3 mg/ml was mixed at a 1:1 ratio with mother liquor (75 mM Tris-HCl pH 8.9, 10 mM BME, 1 mM EDTA, 4.1% PEG MW 8000) containing a dilution of microseeds, and allowed to equilibrate in hanging drops of 10 μl over 500 μl reservoirs of mother liquor at 4 °C. Microseed stocks were prepared by adding an intact monoclinic RTA crystal to 500 μl of mother liquor and vortexing the solution with a Seed Bead (Hampton Research). Dilutions of the seed stock yielding optimum quality crystals were determined for each new seed stock.
The inhibitors were dissolved in modified mother liquor (75 mM BES pH 7.5, 4.1% PEG MW 8000) at an estimated final concentration of 1.5 mM. RTA crystals were transferred to hanging drops of modified mother liquor with inhibitor, and were allowed to soak for at least 48 hours prior to data collection. The crystals were transferred briefly to drops identical to the soaking condition with added cryoprotectant, mounted in a cryoloop (Hampton Research), and flash frozen in liquid nitrogen. In some cases, the presence of 1.5 mM concentrations of different inhibitors changes the interaction between the cryoprotectant and the crystal such that the crystal is destroyed or the suppression of ice crystal formation is no longer sufficient, therefore a cryoprotectant other than what was used for native RTA crystals must be used. For 7CP, 15% glycerol was used, and for compound 12, 10% PEG MW 400, 10% PEG MW 3350 was used. After flash freezing, crystals were either mounted immediately in a nitrogen cold stream for data collection on the home source, or they were stored in a dewar for shipping to the Advanced Light Source.
5.5. X-ray data collection and structure determination
Diffraction data for the RTA-Compound 12 complex were collected at 100 K on an R-AXIS IV++ image plate detector (Rigaku) with X-rays generated from a Rigaku MicroMax007 rotating anode generator operated at 40 kV, 30 mA. Data for the RTA-7CP complex were collected at 100 K on beamline 5.0.3 at the Advanced Light Source (Lawrence Berkeley National Laboratory). Diffraction images were processed and data reduced using HKL2000 [18]. For both structures, the unit cell parameters were isomorphous with the native monoclinic RTA crystals [17]. Data for the RTA-7CP complex were collected to 1.29 Å resolution, including 227,142 observations of 61,933 reflections, and the RTA-Compound 12 data were collected to 1.89 Å with 73,906 observations of 18,685 reflections. The data sets were scaled with Rmerge = 4.2 % and Rmerge = 5.0 % for RTA-7CP and RTA-Compound 12 respectively.
Models were generated by molecular replacement as described earlier [8a]. Briefly, the native RTA structure (PDB code 1RTC) was modified such that Tyrosine 80 was replaced by Alanine, and was used as the starting model for molecular replacement using MOLREP [19] from the CCP4 suite [20]. Refinement and electron density map generation were carried out by REFMAC [21] and manual model building and refinement, including the building of Tyrosine 80 and the ligand into the electron density, were done using COOT [22]. Omit maps with amplitudes Fo-Fc were generated by REFMAC using the refined complex structures, from which the ligand had been removed, for phasing. A summary of the processing and refinement statistics is shown in Table 2.
Table 2. X-ray Data.
Data Collection and Refinement Statistics for RTA/7CP and RTA/(12) Complexes
| Statistics | RTA·7CP | RTA·(12) |
|---|---|---|
| a (Å) | 42.7 | 42.4 |
| b (Å) | 67.6 | 67.3 |
| c (Å) | 49.4 | 49.5 |
| β(deg) | 112.8 | 112.5 |
| Space group | P21 | P21 |
| Resolution (Å) | 45.69–1.29 | 45.56–1.89 |
| No. observed | 227142 | 73906 |
| Rmerge (last shell) (%) | 4.2 (27.6) | 5.0 (43.2) |
| I/δ1,(last shell)(%) | 36.8 (3.2) | 44.9 (3.5) |
| Completeness (last shell) (%) | 95.3 (91.3) | 95.6 (89.5) |
| Redundancy | 3.7 | 3.7 |
| No. reflections | 61933 | 18685 |
| Rwork (%) | 20.6 | 19.5 |
| Rfree (%) | 23.7 | 23.9 |
| Rms deviation from ideality (Å, deg) | 0.028/2.24 | 0.022/1.96 |
| Estimated overall coordinate error (Å) | 0.060 | 0.157 |
| Correlation coefficient FO-FC | 0.953 | 0.961 |
| Correlation coefficient FO-FC FREE | 0.940 | 0.949 |
Supplementary Material
Acknowledgments
This work was supported by National Institute of Health (NIH) grant AI 075509, by Robert A. Welch Foundation grant F1225, and by the College of Natural Sciences support to the Center for Structural Biology.
Abbreviations
- RTA
ricin toxin A
- PTA
pteroic acid
- 7CP
7-carboxy pterin
- 7AP
7-carbamoyl pterin
- 7HP
7-hydrazide pterin
- DSF
differential scanning fluorometry
- BSA
bovine serum albumin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.a) Lord JM, Roberts LM, Robertus JD. Faseb J. 1994;8:201. [PubMed] [Google Scholar]; b) Robertus JDaM, AF . In: Protein toxin structure. Parker MW, editor. R.G. Landes Company; Austin: 1996. p. 253. [Google Scholar]
- 2.a) Endo Y, Tsurugi K. J Biol Chem. 1987;262:8128. [PubMed] [Google Scholar]; b) Ready MP, Kim Y, Robertus JD. Proteins. 1991;10:270. doi: 10.1002/prot.340100311. [DOI] [PubMed] [Google Scholar]
- 3.Miller DJ, Ravikumar K, Shen H, Suh JK, Kerwin SM, Robertus JD. J Med Chem. 2002;45:90. doi: 10.1021/jm010186s. [DOI] [PubMed] [Google Scholar]
- 4.Franz DRaJ, NK . Textbook of Military Medicine: Medical Aspects of Chemical and Biological Warfare. Office of the Surgeon General, Department of the Army, United States of America; 1997. [Google Scholar]
- 5.Loyd AaFM. London Times. 16. London, England: Nov, 2001. [Google Scholar]
- 6.a) Mlsna D, Monzingo AF, Katzin BJ, Ernst S, Robertus JD. Protein Sci. 1993;2:429. doi: 10.1002/pro.5560020315. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Montfort W, Villafranca JE, Monzingo AF, Ernst SR, Katzin B, Rutenber E, Xuong NH, Hamlin R, Robertus JD. J Biol Chem. 1987;262:5398. [PubMed] [Google Scholar]
- 7.a) Frankel A, Welsh P, Richardson J, Robertus JD. Mol Cell Biol. 1990;10:6257. doi: 10.1128/mcb.10.12.6257. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kim Y, Mlsna D, Monzingo AF, Ready MP, Frankel A, Robertus JD. Biochemistry. 1992;31:3294. doi: 10.1021/bi00127a035. [DOI] [PubMed] [Google Scholar]; c) Kim Y, Robertus JD. Protein Eng. 1992;5:775. doi: 10.1093/protein/5.8.775. [DOI] [PubMed] [Google Scholar]; d) Monzingo AF, Robertus JD. J Mol Biol. 1992;227:1136. doi: 10.1016/0022-2836(92)90526-p. [DOI] [PubMed] [Google Scholar]
- 8.a) Bai Y, Monzingo AF, Robertus JD. Arch Biochem Biophys. 2009;483:23. doi: 10.1016/j.abb.2008.12.013. [DOI] [PubMed] [Google Scholar]; b) Yan X, Hollis T, Svinth M, Day P, Monzingo AF, Milne GW, Robertus JD. J Mol Biol. 1997;266:1043. doi: 10.1006/jmbi.1996.0865. [DOI] [PubMed] [Google Scholar]
- 9.Chen XY, Berti PJ, Schramm VL. J Am Chem Soc. 2000;122:1609. [Google Scholar]
- 10.Ho MC, Sturm MB, Almo SC, Schramm VL. Proc Natl Acad Sci U S A. 2009;106:20276. doi: 10.1073/pnas.0911606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Bai Y, Watt B, Wahome PG, Mantis NJ, Robertus JD. Toxicon. 2010 doi: 10.1016/j.toxicon.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wahome PG, Bai Y, Neal LM, Robertus JD, Mantis NJ. Toxicon. 2010;56:313. doi: 10.1016/j.toxicon.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Waring P, Armarego WLF. Australian Journal of Chemistry. 1985;38:629. [Google Scholar]; b) Taylor EC, Henrie RN, Portnoy RC. Journal of Organic Chemistry. 1978;43:736. [Google Scholar]
- 13.Pruet JM, Robertus JD, Anslyn EV. Tetrahedron Lett. 2010;51:2539. doi: 10.1016/j.tetlet.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan X, Day P, Hollis T, Monzingo AF, Schelp E, Robertus JD, Milne GW, Wang S. Proteins. 1998;31:33. doi: 10.1002/(sici)1097-0134(19980401)31:1<33::aid-prot4>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 15.Niesen FH, Berglund H, Vedadi M. Nat Protoc. 2007;2:2212. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 16.Ewers U, Gunther H, Jaenicke L. Chemische Berichte-Recueil. 1973;106:3951. [Google Scholar]
- 17.Robertus JD, Piatak M, Ferris R, Houston LL. J Biol Chem. 1987;262:19. [PubMed] [Google Scholar]
- 18.Otwinowski Z, Minor W. Methods Enzymol. 1997;27:307. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 19.Vagin A, Teplyakov A. J Appl Cryst. 1997;30:1022. [Google Scholar]
- 20.CCP4. Acta Cryst. 1994;D5:760. [Google Scholar]
- 21.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr D Biol Crystallogr. 1997;53:240. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 22.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.