

Abstract
Background
Glutathione S-transferase Pi (GSTPi) is the predominant redox regulator in the lung. While evidence implicates an important role for GSTPi in asthma, the mechanism for this has remained elusive.
Objectives
To determine how GSTPi is regulated in asthma and to elucidate its role in maintaining redox homeostasis.
Methods
We elucidated the regulation of GSTPi in children with asthma and utilized murine models of asthma to determine the role of GSTPi in redox homeostasis.
Measurements and Main Results
Our findings demonstrate that GSTPi transcript levels are markedly down-regulated in allergen and IL-13 treated mouse models of asthma via STAT6 dependent and independent pathways. Nuclear factor-erythroid 2 related factor 2 (Nrf2) was also down-regulated in these models. The decrease in GSTPi expression was associated with decreased total GST activity in the lungs of mice. Examination of cystine intermediates uncovered a functional role for GSTPi in regulating Cys oxidation, whereby GSTPi-deficient mice exhibited increased oxidative stress (increase in % cystine) compared with wild-type mice following allergen challenge. GSTPi expression was similarly down-regulated in children with asthma.
Conclusions
These data collectively suggest that down-regulation of GSTPi following allergen challenge may contribute to the asthma phenotype due to disruption of redox homeostasis and increased oxidative stress. Furthermore, GSTPi may be an important therapeutic target for asthma, and evaluation of GSTPi expression may prove beneficial in identifying individuals who would benefit from therapy targeting this pathway.
Keywords: GSTPi, asthma, oxidative stress, redox homeostasis, gene
INTRODUTION
Asthma, a chronic inflammatory disorder of the airways, affects more than 9 million children (13%) in the U.S.1. A recent review of nearly 500 asthma gene association studies identified 25 genes associated with asthma phenotypes in six or more populations 2. Among those consistently associated with asthma is the glutathione S-transferase (GST) family of genes. GSTs comprise a family of phase II enzymes that catalyze the conjugation of reduced glutathione (GSH) via a sulfhydryl group to electrophilic sites on a wide variety of substrates found in air pollution3, cigarette smoke4, and mold5. Each of these environmental factors leads to the generation of reactive oxygen species (ROS) and has been implicated in the development of asthma6–8. The products of GST catalysis are more water-soluble promoting ROS detoxification and thereby protecting tissues from oxidative damage9.
In humans, GSTs are divided into eight families: Alpha, Kappa, Mu, Omega, Pi, Sigma, Theta, and Zeta10. A single gene in the Pi subfamily, glutathione S-transferase P1 (GST-P1), is the predominant cytosolic GST expressed in lung epithelium11. GST-P1 is a 2.8 kb gene located on chromosome 11q13, a known “hot spot” for asthma-related genes,12, 13 and studies by our group and others have demonstrated an association of mutations in this gene has been associated with asthma14–18. Many epidemiologic studies have implicated the GST-P1 Ile105Val polymorphism (rs1695) as a predictor for asthma. Although the contribution of the GST-P1 Val105 allele is not fully understood, the allele has been reported to have significantly lower GST enzyme activity19. In addition, we have previously shown that exposure to DEP, ETS, and mold each conferred an increased risk for wheezing in children that were carriers of the Val105 allele17.
GSTPi has been shown to play a unique, non-redundant role in total pulmonary GST activity4, 20. A recent study using mice deficient in both GSTPi genes (GSTP1/P2-deficient mice20) reported an increase in lung resistance compared to wild-type mice in response to ovalbumin treatment, further supporting a role for GSTPi in allergic airway disease21. Despite the compelling evidence supporting a strong role for GSTPi in asthma, very little is known about the regulation of GSTPi expression in asthma or the molecular mechanism underlying its role in asthma. The aim of this investigation was to determine how GSTPi expression and total GST activity are regulated in asthma and to determine the effect GSTPi on redox homeostasis in mouse models of asthma.
METHODS
Subjects
Recruitment, nasal mucosa sampling and RNA isolation from children with and without asthma were previously described22. Briefly, after IRB approval was obtained, healthy and asthmatic children (age 5–18 years old) presenting to Cincinnati Children’s Hospital Medical Center (CCHMC) were invited to participate in the study. Asthma was diagnosed in accordance with American Thoracic Society (ATS) criteria7, 8. All the children were positive by skin prick testing to at least one aeroallergen from an environmental panel that included dust mite, molds, cat, dog, feathers, weeds and ragweed, tree pollens and grass allergen extracts (Hollister-Stier Laboratories, Spokane, WA). Healthy control children had no history of chronic illnesses and were negative to the environmental skin test prick panel indicated above. Exclusion criteria to participate in the study included 18 years of age or older, the use of nasal or systemic steroids within the last 30 days, nasal malformations/tumors, and evidence of acute infectious disease. The use of inhaled steroids was not interrupted for this study. Skin prick testing was performed using DermaPiks (Greer Laboratories, Lenoir, NC). Histamine (1 mg/ml) and normal saline (0.9% NaCl) were used as positive and negative controls. Reactions were considered positive if there was an erythematous base with a wheal ≥ 3 mm in diameter. Nasal mucosa sampling and RNA isolation was performed using a CytoSoft Brush (Medical Packaging Corp., Camarillo, CA) and the sample was immediately taken to the laboratory for processing and RNA isolation as previously described22. The gender (male:female) and race (AfricanAmerican:Caucasian) ratios were 7:3 and 8:2 for the control group, and 7:3 and 7:3 for the asthma group. The children in the asthma group was predominantly African American males, which agrees with published data regarding the racial and gender distributions of childhood asthma in urban environments15–17.
Animals and care
Animals were maintained in a pathogen-free vivarium under institutional animal care using committee-approved procedures.
Allergen treatment of mice
Wild-type C57Bl/6 and Balb/c (Jackson Laboratory, Bar Harbor, ME), IL-13−/− mice kindly provided by Andrew McKenzie23 (Medical Research Council Laboratory of Molecular Biology, Cambridge, UK), STAT6−/− (Jackson Laboratory), and GSTPi−/− mice (kindly provided by Colin J. Henderson, University of Dundee)20 backcrossed for at least six generations with C57Bl/6 mice were immunized with house dust mite extract (HDM) (Dermatophagoides pteronyssinus) (Greer Laboratories, Lenoir, NC) as previously described24. For the kinetic experiments, i.t. challenges were performed at one-week increments using 100μg of HDM. One day after the last challenge, AHR to acetylcholine (50μg/kg) was measured as APTI 25. Blood, bronchoalveolar lavage fluid (BALF), and lung tissues were harvested.
Wild-type Balb/c mice were i.t. challenged with 100μg of Aspergillus fumigatus (Asp) crude protein extract (Greer Laboratories) as previously described26, 27.
IL-13 treatment of mice
Wild-type C57Bl/6 and Balb/c, STAT6−/−, and GSTPi−/− mice were i.t. challenged with 5μg of hIL-13 (Peprotech, Rocky Hill, NJ) on days 0, 3, and 6. On day 7, APTI was used to measure AHR; blood, BALF, and lung tissues were harvested.
Quantitative RT-PCR
Total lung RNA isolation and realtime PCR normalized to GAPDH or 18S rRNA28 were performed as previously described29. cDNA was generated using SuperScript® First-strand Synthesis System for RT-PCR (Invitrogen). Primers for mouse GSTPi (forward: 5′-ATCTTGAGACACCTTGGC-3′ and reverse: 5′-CCTTCACGTAGTCATTCTTACC-3′) were designed using LightCycler Probe Design Software 2.0 (Roche Diagnostics, GmbH, Mannheim, Germany). Remaining primers were previously published30–33.
Total GST activity in mouse lung
10μg of lung protein processed in 0.1M sodium phosphate buffer, pH=6.5, containing 2mM EDTA; quantified using Coomassie Plus™ Protein Assay Reagent (Thermo Scientific) were used to assess GST activity for 6 minutes using the GST assay kit (Sigma-Aldrich, linear range of detection was 0–0.25mg/ml GST). Experimental OD340 values fell within range of the manufacturer’s GST control.
Sample collection and analysis of cysteine (Cys) and cystine (CySS)
Cys and CySS were measured using high-performance liquid chromatography as previously described34.
GSTPi Immunohistochemistry
Slide-mounted paraffin sections were deparaffinized, rehydrated, and antigen retrieval was performed using high-pH target retrieval (Dako, Denmark). After hydrogen peroxide inactivation and serum blocking, slides were incubated at 4°C for 18 hours with a 1:150 dilution of rabbit anti-GSTPi antibodies (kindly provided by Colin J. Henderson). Sections were washed and incubated with biotinylated secondary antibody. Slides were developed using a peroxidase-labeled avidin detection system (Vector Labs, Burlingame, CA).
Statistical analysis
Statistical significance between two groups was determined by a two-tailed t-test and between multiple groups by one-way ANOVA followed by a Tukey-Kramer post-test. Significance was determined by a P ≤ 0.05 using PRISM software (GraphPad Software Inc., La Jolla, CA).
RESULTS
GSTPi expression and total GST activity are decreased in allergen challenged mice
We examined GSTPi expression and total GST activity in several different mouse models of asthma. Challenge with house dust mite (HDM) or Aspergillus fumigatus (Asp) induced a significant increase in airway hyperresponsiveness (AHR) as measured by airway pressure-time index (APTI) following allergen challenge as expected (data not shown). Contrary to expectation, we observed a marked decrease in the level of GSTPi mRNA expression in the lungs of mice following HDM or Asp challenge compared to control mice (Figure 1A, C). We also observed a concomitant decrease in total GST activity in the lungs following allergen challenge (Figure 1B, D). This was not due to a direct inhibitory effect of allergen on GST activity, because the addition of up to 100μg HDM to the assay had no effect on GST activity (data not shown).
Figure 1. Down-regulation of GSTPi expression and total GST activity in the lungs of mice following allergen challenge.

(A, B) Balb/c mice were sensitized i.p. twice and challenged i.t. twice with HDM or (C, D) challenged i.t. 9 times with Asp. (A, C) Total lung RNA was analyzed for GSTPi exression by real-time RT-PCR. (B, D) GST activity in total lung protein. * P<0.05, mean ± SD. Data are representative of at least 3 experiments.
Expression of GST family members (GSTK1, GSTM1, and GSTT1) is decreased in allergen challenged mice
Although it has been reported that GSTPi accounts for over 90% of the GST activity toward 1-chloro-2,4-dinitrobenzene (CDNB) in the lung 11, other GST family members may also contribute to the total activity. Thus, we evaluated the effect of allergen exposure on the expression of the other GST family members also expressed in the lung (GSTK1, GSTM1, and GSTT1). The mu and theta classes (GSTM1 and GSTT1), like the pi class (GSTPi), are cytosolic GSTs whereas the kappa GSTs (GSTK1) are mitochondrial35. Similar to GSTPi, lung transcript levels of all the GST family members examined were also significantly attenuated following HDM and Asp (Figure 2A–C).
Figure 2. HDM exposure attenuates mouse lung expression of GSTK1, GSTM1, and GSTT1.

(A-C) Balb/c mice were sensitized i.p. twice and challenged i.t. twice with HDM. (A) GSTK1, (B) GSTM1, and (C) GSTT1 RNA determined by real-time RT-PCR. * P<0.05. (D-E) C57BL/6 mice were sensitized i.p. once, twice, or sensitized i.p. twice and challenged i.t. 1, 2, 3, 5, 7, 9, or 11 times with HDM. Data is represented as fold changes in (D) GSTPi expression or (E) total GST activity in HDM treated mice relative to PBS treated mice. * p<0.05 comparing HDM vs. PBS treated mice. Using Bonferroni correction (P<0.004), there are significant differences between HDM and PBS treated mice in both GSTPi expression and total GST activity after 2 and 3 i.t. challenges. Values are the mean ± SD. N=3–5 mice per group. Data are representative of at least 3 experiments.
Kinetics of down-regulation of GSTPi expression and total GST activity following allergen challenge
In order to determine the kinetics of the observed decrease in GSTPi expression and total GST activity following acute and chronic allergen treatment, we analyzed the lungs of mice either following HDM sensitization alone or HDM sensitization followed by weekly intratracheal (i.t.) challenges for up to 11 weeks. There were no significant differences in either GSTPi expression or total GST activity following intraperitoneal (i.p.) sensitization. However, following the first i.t. challenge, there was a significant decrease in the GSTPi expression (Figure 2D). Total GST activity was significantly decreased after the second i.t. challenge (Figure 2E). Following only a single i.t. challenge, without prior sensitization, there was no significant difference in GSTPi expression between HDM and PBS treated mice (data not shown). GSTPi expression and total GST activity remained low up to 7 weeks of i.t. challenges and then returned to control levels. Collectively, these data suggest that systemic presence of HDM is insufficient to induce down-regulation of GSTPi expression and total GST activity. However, sensitization is required for the transient down-regulation of GSTPi expression and total GST activity.
IL-13 is sufficient to suppress GSTPi mRNA expression but is not required for allergen-induced down-regulation of GSTPi expression and total GST activity down-regulation
IL-13 is a key mediator in the pathogenesis of allergic asthma36. Similar to our observations following allergen treatment, IL-13 treatment resulted in marked down-regulation GSTPi expression and total GST activity (Figure 3A, B).
Figure 3. IL-13 is sufficient but not necessary for HDM-induced down-regulation of GSTPi expression and total GST activity.

(A-B) Balb/c mice were challenged i.t. 3 times with IL-13. (A) GSTPi RNA levels in total lung normalized to 18SrRNA. (B) Total GST activity. (C-D) IL-13+/+ and IL-13−/− mice were i.t. challenged 3 times with HDM and analyzed for: (C) GSTPi expression (D) total GST activity. Data are represented as fold changes in total GST activity in IL-13 treated mice relative to PBS treated mice. * P<0.05. mean ± sSD. Data are representative of 3 experiments.
In order to determine whether IL-13 was necessary for the observed decrease in GSTPi expression and GST activity, IL-13-deficient mice were treated with HDM. Even in the absence of IL-13, HDM treatment resulted in decreased GSTPi expression and total GST activity (Figure 3C, D) indicating that IL-13 is sufficient but not necessary for down-regulation of GSTPi expression.
Allergen-induced GSTPi mRNA expression and total GST activity down-regulation is STAT6 independent
To elucidate whether HDM down-regulation of GSTPi was dependent on STAT6, STAT6-deficient mice were treated with HDM. As expected, HDM exposed wild-type mice displayed increased AHR measured by APTI, whereas STAT6-deficient mice treated with HDM did not demonstrate an increase in AHR 37 (data not shown). GSTPi expression and total GST activity were similarly decreased after HDM treatment in wild type and STAT6-deficient mice (Figure 4A, B). Thus, STAT6 is not required for the observed decrease in GSTPi expression and total GST activity after HDM treatment. Interestingly, neither GSTPi expression nor total GST activity changed following IL-13 treatment in STAT6-deficient mice (Figure 4C, D). Thus, IL-13 induced down-regulation of GSTPi expression and total GST activity is STAT6-dependent in contrast to HDM induced GST down-regulation.
Figure 4. HDM-induced down-regulation of GSTPi expression and total GST activity is STAT6 independent, but IL-13-induced down-regulation is STAT6 dependent.

(A-B) STAT6+/+ and STAT6−/− mice were i.p. sensitized twice and i.t. challenged twice with HDM. (A) GSTPi RNA expression. (B) Total GST activity. (C-D) STAT6+/+ and STAT6−/− mice were i.t. challenged 3 times with IL-13 and analyzed for (C) GSTPi expression and (D) total GST activity. * P<0.05, mean ± SD.
Nuclear factor-erythroid 2 related factor 2 (Nrf2) expression is decreased in allergen and IL-13 challenged mice
Nrf2 is a transcription factor involved in the transcriptional regulation of many antioxidant genes, including those involved in the GST pathway38. In Nrf2-deficient mice, GSTPi expression is reduced, demonstrating the obligatory role that Nrf2 plays in GSTPi expression39. Since Nrf2 is upstream of GSTPi, we examined whether Nrf2 was also down-regulated in mouse models of asthma. HDM treatment in mice resulted in decreased Nrf2 mRNA levels (Figure 5A). Similar to GSTPi expression, the observed decreased in Nrf2 transcript levels was not allergen specific and was evident following HDM or Asp treatment in wild-type mice (Figure 5B). IL-13 treatment was sufficient to decrease Nrf2 expression levels (Figure 5C). Furthermore, Nrf2 expression levels were significantly depleted in both IL-13- and STAT6-deficient mice treated with HDM (Figure 5D, E). Together, the data suggest that the decrease in GSTPi expression is a consequence of decreased Nrf2 expression.
Figure 5. Expression of Nrf2 in mouse models of asthma.

Total lung RNA was isolated to determine expression levels of Nrf2 in Balb/c mice (A) sensitized i.p. twice and challenged i.t. twice with HDM; (B) challenged i.t. 9 times with Asp; or (C) challenged i.t. 3 times with IL-13. And in (D) IL-13+/+ and IL-13−/− mice or (E) STAT6+/+ and STAT6−/− mice sensitized i.p. twice and challenged i.t. twice with HDM. * P<0.05, mean ± SD. Data are representative of 3 experiments.
Lungs of GSTPi-deficient mice have increased oxidative stress following HDM challenge
Our data revealed that GSTPi expression and total GST activity were significantly down-regulated in a mouse model of asthma following allergen challenge. One mechanism by which environmental exposures can result in lung injury is by inducing inflammatory cells to generate ROS leading to oxidative injury40, 41. Thus, we next determined whether a decrease in GSTPi activity may contribute to increased oxidative stress in an asthma model. Glutathione (GSH), the substrate used by GSTs to catalyze detoxification reactions, is produced from cysteine (Cys), glycine, and glutamate. Cys availability is often a limiting factor for the rate of GSH synthesis42. Cys and its oxidized disulfide form, cystine (CySS), represent the major extracellular thiol/disulfide redox control system in mammals43. In models of lung injury, Cys/CySS was selectively oxidized early in inflammation supporting that it is a sensitive marker of redox homeostasis44. An increase in %CySS is indicative of oxidative stress. The %CySS (CySS/(Cys+CySS)) was analyzed in the lungs of wild-type and GSTPi-deficient mice following HDM challenge in an asthma model. Wild type mice did not exhibit any increase in %CySS following HDM challenge (Figure 6), supporting that in the presence of GSTPi, the mice were able to neutralize oxidative stress efficiently. In contrast, the lungs of GSTPi-deficient mice had significantly increased %CySS compared to wild-type mice following HDM challenge (P<0.05) (Figure 6). Thus, a loss in GSTPi activity significantly altered the ability of the lungs to handle the increased oxidative stress burden following allergen challenge.
Figure 6. Oxidative stress is increased in GSTPi−/− but not wild-type mice in an allergen induced asthma model.

Wild-type C57Bl/6 and GSTPi−/− mice were sensitized i.p. twice and challenged i.t. twice with HDM. Lung tissue was collected for high-performance liquid chromatography (HPLC) analysis of Cys and CySS. * P<0.05, mean ± SD. Cys=cysteine, CySS=cystine.
Localization of GSTPi in the lung
To determine the localization of GSTPi expression in the lung and the relevant cell type(s) in which it is regulated, GSTPi immunohistochemistry was performed on mouse lungs treated with PBS, HDM, and IL-13. GSTPi expression was expressed predominantly in epithelial cells including some expression in type II pneumocytes (Figure 7A). Interestingly, GSTPi expression appeared to be absent in goblet cells induced by either HDM or IL-13 in wild-type mice (Figure 7B, C). No staining was detected in GSTPi-deficient mice (Figure 7D–F). In contrast to wild-type mice, STAT6-deficient mice treated with HDM or IL-13 do not develop goblet cell hyperplasia. In these mice, GSTPi was mainly expressed in epithelial cells similar to wild-type mice treated with PBS (Figure 7G–I).
Figure 7. Localization of GSTPi in the lung.
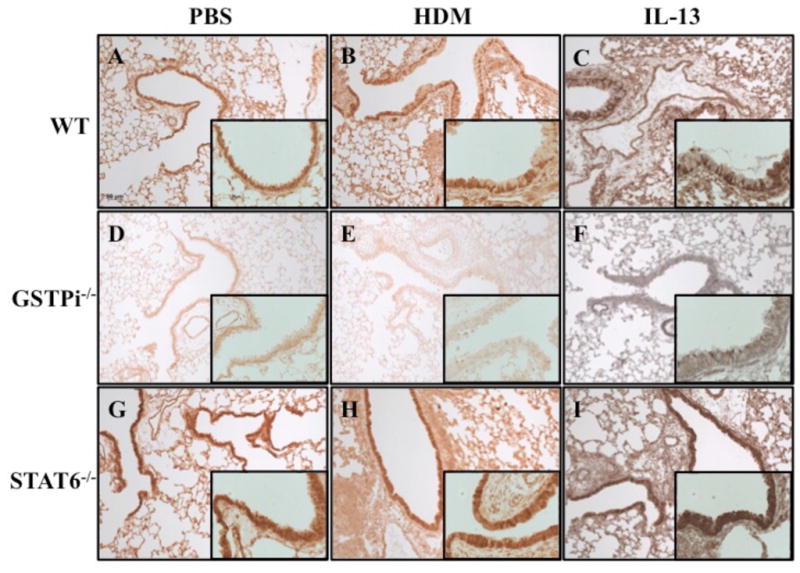
Wild-type, GSTPi−/−, and STAT6−/− mice were treated with PBS, HDM, or IL-13 and analyzed for GSTPi expression in the lung by immunohistochemistry. (A) Wild-type mice treated with PBS displayed positive GSTPi expression predominantly in epithelial cells. (B, C) GSTPi expression was not detected in goblet cells of wild-type mice treated with HDM or IL-13. (D-F) No staining was detected in GSTPi−/− mice treated with either PBS, HDM, or IL-13. (G-I) GSTPi expression in STAT6−/− mice treated with PBS, HDM, or IL-13 was mainly found in epithelial cells. All images were captured at 100X and inset images were captured at 400X.
GSTPi expression is decreased in nasal epithelial cells (NECs) of asthmatic children
Our data support a decrease in GSTPi expression following allergen challenge in a mouse model of asthma. To evaluate whether GSTPi was similarly down-regulated in children with asthma, we quantified GSTPi expression in RNA isolated from NECs of children with asthma as well as control (non-atopic, non-asthmatic) children. There were no significant differences in age or gender between the two groups (data not shown). Similar to the observed down-regulation of GSTPi expression in mouse models of asthma, GSTPi expression was also attenuated in asthmatic children compared to non-atopic, non-asthmatic controls (Figure 8).
Figure 8. GSTPi expression is decreased in nasal epithelial cells from children with asthma.

Children were divided into 2 groups: Control (non-atopic, non-asthmatic children) and children with asthma (Asthmatics). Values are the mean ± SD. *P<0.05
DISCUSSION
The contribution of GSTPi to asthma has been supported by epidemiologic, genetic, and animal studies by our group and others14–17, 21 although the mechanism for this has remained elusive. Our findings provide important novel insights into the contribution of GSTPi to asthma in humans and mice. Given that several environmental exposures associated with asthma result in enhanced oxidative stress, one might predict that GST enzymes would be induced in order to increase the capacity to handle the increased electrophilic load and restore redox homeostasis. However, we found that GSTPi transcript levels are markedly down-regulated in mouse models of asthma following allergen challenge via STAT6 dependent and independent pathways. The observed down-regulation was not unique to a specific allergen. The decrease in GSTPi expression was associated with decreased total GST activity in the lungs of mice. Levels of GSTPi mRNA have been shown to correlate to GST activity in other studies (R2=0.77, P<0.001)45. GSTPi was down-regulated in Balb/c and C57Bl/6 strains of mice supporting that the observation was not restricted to a given strain. Furthermore, GSTPi was similarly down-regulated in children with asthma. Our findings also uncovered a functional role for GSTPi in regulating Cys oxidation, revealing that GSTPi plays an important role in neutralizing oxidative stress in asthma. These data collectively suggest that down-regulation of GSTPi following allergen challenge may result in disruption of redox homeostasis and increased oxidative stress. Interestingly, patients with severe asthma have been found to have lower airway GSH with increased oxidized glutathione (GSSG), consistent with enhanced oxidative stress46.
Perturbations in the extracellular thiol/disulfide redox environment have been shown to correlate with the progression and severity of lung injury47. Cysteine (Cys) and its disulfide Cystine (CySS) constitute the most abundant, low-molecular-weight thiol/disulfide redox couple in the plasma, and Cys homeostasis is adversely affected during the inflammatory response. In models of lung injury, Cys/CySS was selectively oxidized early in inflammation supporting that it is a sensitive marker of redox homeostasis44. In the absence of GSTPi, the lungs of GSTPi-deficient mice had significantly increased %CySS following allergen challenge supporting that a loss in GSTPi activity significantly altered the ability of the lungs to handle the increased oxidative stress burden following allergen challenge. Key pathways relevant to allergic inflammation have been shown to be sensitive to Cys redox homestasis48, thus an increase in %CySS may effect on cytokine signaling in addition to the other effects of ROS.
Since GSTPi is an enzyme that plays a critical role in cellular detoxification of endogenous and xenobiotic substrates and protection against oxidative stress, it is not surprising that evidence exists supporting a substantial role for GSTPi in allergic asthma. We had predicted that following allergen exposure there would be an increased necessity for GSTPi-driven detoxification in the lung, marked by an increase in GSTPi expression and total GST activity. Therefore, it was surprising to observe a decrease in GSTPi expression and total GST activity following allergen exposure. One potential explanation for this apparent paradox is that the observed GSTPi down-regulation is transient. Down-regulation of GSTPi expression was evident after sensitization and a single i.t. challenge and remained low for several weeks. However, GSTPi levels and activity normalized. Since asthma is a chronic disease, the impact of the down-regulation may not be as evident in a more chronic model of exposure. The surprising decrease in GSTPi expression and total GST activity could also indicate that GSTPi is maladaptive in response to allergen exposure.
The mechanism by which down-regulation of GSTPi expression and total GST activity occurs may involve Nrf2. HDM and IL-13 treated mice displayed decreased Nrf2 expression in the lungs. IL-13-deficient and STAT6-deficient mice also display decreased Nrf2 expression following HDM exposure. Thus, although IL-13 contributes to HDM-induced Nrf2 down-regulation, neither IL-13 nor Stat6 is necessary for allergen-induced downregulation of Nrf2. There is an alternate pathway downstream of allergen treatment that results in the observed down-regulation in an IL-13 and STAT6 independent manner. Similarly, IL-13- but not HDM-induced GSTPi down-regulation was STAT6 dependent. Thus, HDM can induce down-regulation of GSTPi expression and total GST activity by IL-13 and STAT6 autonomous pathways that involve Nrf2. Furthermore, Nrf2 was found to be an important modulator for mounting an appropriate innate immune response during experimental sepsis49. Thus, transient down-regulation of Nrf2 and GSTPi following allergen exposure could signal the increase of proinflammatory cytokines and chemokines or the initiation of downstream detoxification pathways.
Our findings are consistent with previous studies that have demonstrated a correlation between GST activity and the level of GSTPi mRNA expression (R2=0.77, P<0.001)45. We observed a decrease in GSTPi gene expression following sensitization and one i.t. challenge whereas the total GST activity did not decrease until after two i.t. challenges. This suggests that the observed decrease in GSTPi gene expression precedes the decrease in total GST activity. Surprisingly, we did not observe a change in GSTPi protein expression analyzed by western blot (data not shown). One reason for this is may be that the assay used for measuring GSTPi protein expression in the lung may not be sensitive enough to detect modest changes in protein levels. Since the antibody does not distinguish between GSTP1 and GSTP2, it is also possible that one subtype is conserved or even upregulated relative to the other.
The observed decrease in GSTPi expression and total GST activity is not likely due to the dilutional effect of migratory cells infiltrating the lung in response to allergen exposure. STAT6-deficient mice have marked attenuation of lung inflammation, AHR, and mucus production 50, 51. Since STAT6-deficient mice have diminished airway inflammation, a decrease in GSTPi expression and total GST activity in STAT6-deficient mice treated with HDM indicates that this decrease is likely due to resident cells and not migratory cells.
In summary, GSTPi expression and total GST activity are dysregulated in mouse models of asthma. GSTPi is similarly dysregulated in human asthma. GSTPi plays an important role in neutralizing oxidative stress. The antioxidant and detoxification capacity of GSTPi and its potential role in GSH homeostasis suggest that the GSTPi pathway could be a critical therapeutic target for asthma, especially in response to environmental exposures. In fact, GSH has already been shown to alleviate IL-13 induced asthma in mice 52. Evaluation of GSTPi expression or GSH homeostasis may prove beneficial in identifying individuals who would benefit from therapy targeting this pathway.
KEY MESSAGES.
In children with asthma and in a murine model of asthma, GSTPi is markedly downregulated in asthma.
This downregulation of GSTPi may contribute directly to the asthma phenotype by disrupting of redox homeostasis resulting in increased oxidative stress.
Acknowledgments
This work was supported by National Institutes of Health grant U19A170235-01 (GKKH). We thank Jennifer Clark and Krista Dienger for assistance with AHR measurements, Mary Rolfes, and Amy Opoka for aid with the IHC, and Cynthia Chappell for help with preparation of the manuscript. We thank Dr. Nives Zimmermann for critical review of this manuscript.
ABBREVIATIONS
- AHR
airway hyperresponsiveness
- APTI
airway pressure time index
- Asp
Aspergillus fumigatus
- BALF
bronchoalveolar lavage fluid
- GSH
reduced glutathione
- GST
glutathione S-transferase
- HDM
house dust mite
- IP
intraperitoneal
- IT
intratracheal,
- ROS
reactive oxygen species
- SD
standard deviation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CAPSULE SUMMARY
Genetic and epidemiolic evidence supports a role for GSTPi in asthma. Our data reveal that down-regulation of GSTPi following allergen challenge may result in disruption of redox homeostasis and increased oxidative stress.
References
- 1.Bloom B, Dey AN. Summary health statistics for U.S. children: National Health Interview Survey, 2004. Vital Health Stat. 2006;10:1–85. [PubMed] [Google Scholar]
- 2.Ober C, Hoffjan S. Asthma genetics 2006: the long and winding road to gene discovery. Genes Immun. 2006;7:95–100. doi: 10.1038/sj.gene.6364284. [DOI] [PubMed] [Google Scholar]
- 3.Diaz-Sanchez D, Riedl M. Diesel effects on human health: a question of stress? Am J Physiol Lung Cell Mol Physiol. 2005;289:L722–3. doi: 10.1152/ajplung.00217.2005. [DOI] [PubMed] [Google Scholar]
- 4.Conklin DJ, Haberzettl P, Prough RA, Bhatnagar A. Glutathione-S-transferase P protects against endothelial dysfunction induced by exposure to tobacco smoke. Am J Physiol Heart Circ Physiol. 2009;296:H1586–97. doi: 10.1152/ajpheart.00867.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jussila J, Komulainen H, Huttunen K, Roponen M, Halinen A, Hyvarinen A, et al. Inflammatory responses in mice after intratracheal instillation of spores of Streptomyces californicus isolated from indoor air of a moldy building. Toxicol Appl Pharmacol. 2001;171:61–9. doi: 10.1006/taap.2000.9116. [DOI] [PubMed] [Google Scholar]
- 6.Peden DB. Development of atopy and asthma: candidate environmental influences and important periods of exposure. Environ Health Perspect. 2000;108 (Suppl 3):475–82. doi: 10.1289/ehp.00108s3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waegemaekers M, Van Wageningen N, Brunekreef B, Boleij JS. Respiratory symptoms in damp homes. A pilot study. Allergy. 1989;44:192–8. doi: 10.1111/j.1398-9995.1989.tb02261.x. [DOI] [PubMed] [Google Scholar]
- 8.Larsson ML, Frisk M, Hallstrom J, Kiviloog J, Lundback B. Environmental tobacco smoke exposure during childhood is associated with increased prevalence of asthma in adults. Chest. 2001;120:711–7. doi: 10.1378/chest.120.3.711. [DOI] [PubMed] [Google Scholar]
- 9.Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol. 1995;30:445–600. doi: 10.3109/10409239509083491. [DOI] [PubMed] [Google Scholar]
- 10.Mannervik B, Awasthi YC, Board PG, Hayes JD, Di Ilio C, Ketterer B, et al. Nomenclature for human glutathione transferases. Biochem J. 1992;282 ( Pt 1):305–6. doi: 10.1042/bj2820305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fryer AA, Hume R, Strange RC. The development of glutathione S-transferase and glutathione peroxidase activities in human lung. Biochim Biophys Acta. 1986;883:448–53. doi: 10.1016/0304-4165(86)90283-7. [DOI] [PubMed] [Google Scholar]
- 12.Kano T, Sakai M, Muramatsu M. Structure and expression of a human class pi glutathione S-transferase messenger RNA. Cancer Res. 1987;47:5626–30. [PubMed] [Google Scholar]
- 13.Doull IJ, Lawrence S, Watson M, Begishvili T, Beasley RW, Lampe F, et al. Allelic association of gene markers on chromosomes 5q and 11q with atopy and bronchial hyperresponsiveness. Am J Respir Crit Care Med. 1996;153:1280–4. doi: 10.1164/ajrccm.153.4.8616554. [DOI] [PubMed] [Google Scholar]
- 14.Tamer L, Calikoglu M, Ates NA, Yildirim H, Ercan B, Saritas E, et al. Glutathione-S-transferase gene polymorphisms (GSTT1, GSTM1, GSTP1) as increased risk factors for asthma. Respirology. 2004;9:493–8. doi: 10.1111/j.1440-1843.2004.00657.x. [DOI] [PubMed] [Google Scholar]
- 15.Gilliland FD, Gauderman WJ, Vora H, Rappaport E, Dubeau L. Effects of glutathione-S-transferase M1, T1, and P1 on childhood lung function growth. Am J Respir Crit Care Med. 2002;166:710–6. doi: 10.1164/rccm.2112065. [DOI] [PubMed] [Google Scholar]
- 16.Fryer AA, Bianco A, Hepple M, Jones PW, Strange RC, Spiteri MA. Polymorphism at the glutathione S-transferase GSTP1 locus. A new marker for bronchial hyperresponsiveness and asthma. Am J Respir Crit Care Med. 2000;161:1437–42. doi: 10.1164/ajrccm.161.5.9903006. [DOI] [PubMed] [Google Scholar]
- 17.Schroer KT, Myers JM, Ryan PH, Lemasters GK, Bernstein DI, Villareal M, et al. Associations between Multiple Environmental Exposures and Glutathione S- transferase P1 on Persistent Wheezing in a Birth Cohort. J Pediatr. 2008 doi: 10.1016/j.jpeds.2008.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Michel S, Liang L, Depner M, Klopp N, Ruether A, Kumar A, et al. Unifying candidate gene and GWAS Approaches in Asthma. PLoS One. 5:e13894. doi: 10.1371/journal.pone.0013894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watson MA, Stewart RK, Smith GB, Massey TE, Bell DA. Human glutathione S-transferase P1 polymorphisms: relationship to lung tissue enzyme activity and population frequency distribution. Carcinogenesis. 1998;19:275–80. doi: 10.1093/carcin/19.2.275. [DOI] [PubMed] [Google Scholar]
- 20.Henderson CJ, Smith AG, Ure J, Brown K, Bacon EJ, Wolf CR. Increased skin tumorigenesis in mice lacking pi class glutathione S-transferases. Proc Natl Acad Sci U S A. 1998;95:5275–80. doi: 10.1073/pnas.95.9.5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou J, Wolf CR, Henderson CJ, Cai Y, Board PG, Foster PS, et al. Glutathione transferase P1: an endogenous inhibitor of allergic responses in a mouse model of asthma. Am J Respir Crit Care Med. 2008;178:1202–10. doi: 10.1164/rccm.200801-178OC. [DOI] [PubMed] [Google Scholar]
- 22.Guajardo JR, Schleifer KW, Daines MO, Ruddy RM, Aronow BJ, Wills-Karp M, et al. Altered gene expression profiles in nasal respiratory epithelium reflect stable versus acute childhood asthma. J Allergy Clin Immunol. 2005;115:243–51. doi: 10.1016/j.jaci.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 23.McKenzie GJ, Emson CL, Bell SE, Anderson S, Fallon P, Zurawski G, et al. Impaired Development of Th2 Cells in IL-13-Deficient Mice. Immunity. 1998;9:423–32. doi: 10.1016/s1074-7613(00)80625-1. [DOI] [PubMed] [Google Scholar]
- 24.Tabata Y, Chen W, Warrier MR, Gibson AM, Daines MO, Hershey GK. Allergy-driven alternative splicing of IL-13 receptor alpha2 yields distinct membrane and soluble forms. J Immunol. 2006;177:7905–12. doi: 10.4049/jimmunol.177.11.7905. [DOI] [PubMed] [Google Scholar]
- 25.Walters DM, Breysse PN, Schofield B, Wills-Karp M. Complement factor 3 mediates particulate matter-induced airway hyperresponsiveness. Am J Respir Cell Mol Biol. 2002;27:413–8. doi: 10.1165/rcmb.4844. [DOI] [PubMed] [Google Scholar]
- 26.Mishra A, Hogan SP, Brandt EB, Rothenberg ME. An etiological role for aeroallergens and eosinophils in experimental esophagitis. J Clin Invest. 2001;107:83–90. doi: 10.1172/JCI10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zimmermann N, Doepker MP, Witte DP, Stringer KF, Fulkerson PC, Pope SM, et al. Expression and regulation of small proline-rich protein 2 in allergic inflammation. Am J Respir Cell Mol Biol. 2005;32:428–35. doi: 10.1165/rcmb.2004-0269OC. [DOI] [PubMed] [Google Scholar]
- 28.Schmittgen TD, Zakrajsek BA. Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J Biochem Biophys Methods. 2000;46:69–81. doi: 10.1016/s0165-022x(00)00129-9. [DOI] [PubMed] [Google Scholar]
- 29.Chen W, Sivaprasad U, Tabata Y, Gibson AM, Stier MT, Finkelman FD, et al. IL-13R alpha 2 membrane and soluble isoforms differ in humans and mice. J Immunol. 2009;183:7870–6. doi: 10.4049/jimmunol.0901028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watanabe T, Tanaka G, Hamada S, Namiki C, Suzuki T, Nakajima M, et al. Dose-dependent alterations in gene expression in mouse liver induced by diethylnitrosamine and ethylnitrosourea and determined by quantitative real-time PCR. Mutat Res. 2009;673:9–20. doi: 10.1016/j.mrgentox.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 31.Ruiz-Laguna J, Abril N, Garcia-Barrera T, Gomez-Ariza JL, Lopez-Barea J, Pueyo C. Absolute transcript expression signatures of Cyp and Gst genes in Mus spretus to detect environmental contamination. Environ Sci Technol. 2006;40:3646–52. doi: 10.1021/es060056e. [DOI] [PubMed] [Google Scholar]
- 32.Li N, Alam J, Venkatesan MI, Eiguren-Fernandez A, Schmitz D, Di Stefano E, et al. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J Immunol. 2004;173:3467–81. doi: 10.4049/jimmunol.173.5.3467. [DOI] [PubMed] [Google Scholar]
- 33.Rasmi Y, Allameh A, Nasseri-Moghaddam S, Gill P, Moghaddam MF, Hedayati M. Comparison of glutathione S-transferase-Pi expression at mRNA levels in oesophageal mucosa using RT-PCR-ELISA in individuals with reflux diseases, adenocarcinoma and squamous cell carcinoma. Clin Biochem. 2006;39:997–1001. doi: 10.1016/j.clinbiochem.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 34.Yeh MY, Burnham EL, Moss M, Brown LA. Non-invasive evaluation of pulmonary glutathione in the exhaled breath condensate of otherwise healthy alcoholics. Respir Med. 2008;102:248–55. doi: 10.1016/j.rmed.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knight TR, Choudhuri S, Klaassen CD. Constitutive mRNA expression of various glutathione S-transferase isoforms in different tissues of mice. Toxicol Sci. 2007;100:513–24. doi: 10.1093/toxsci/kfm233. [DOI] [PubMed] [Google Scholar]
- 36.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, et al. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–61. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 37.Kuperman D, Schofield B, Wills-Karp M, Grusby MJ. Signal transducer and activator of transcription factor 6 (Stat6)-deficient mice are protected from antigen-induced airway hyperresponsiveness and mucus production. J Exp Med. 1998;187:939–48. doi: 10.1084/jem.187.6.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ikeda H, Serria MS, Kakizaki I, Hatayama I, Satoh K, Tsuchida S, et al. Activation of mouse Pi-class glutathione S-transferase gene by Nrf2(NF-E2-related factor 2) and androgen. Biochem J. 2002;364:563–70. doi: 10.1042/BJ20011756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vachier I, Damon M, Le Doucen C, de Paulet AC, Chanez P, Michel FB, et al. Increased oxygen species generation in blood monocytes of asthmatic patients. Am Rev Respir Dis. 1992;146:1161–6. doi: 10.1164/ajrccm/146.5_Pt_1.1161. [DOI] [PubMed] [Google Scholar]
- 41.Owen S, Pearson D, Suarez-Mendez V, O’Driscoll R, Woodcock A. Evidence of free-radical activity in asthma. N Engl J Med. 1991;325:586–7. doi: 10.1056/NEJM199108223250816. [DOI] [PubMed] [Google Scholar]
- 42.Cotgreave IA, Gerdes RG. Recent trends in glutathione biochemistry--glutathione-protein interactions: a molecular link between oxidative stress and cell proliferation? Biochem Biophys Res Commun. 1998;242:1–9. doi: 10.1006/bbrc.1997.7812. [DOI] [PubMed] [Google Scholar]
- 43.Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8:1865–79. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 44.Iyer SS, Jones DP, Brigham KL, Rojas M. Oxidation of plasma cysteine/cystine redox state in endotoxin-induced lung injury. Am J Respir Cell Mol Biol. 2009;40:90–8. doi: 10.1165/rcmb.2007-0447OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bauer M, Herbarth O, Aust G, Hengstler JG, Dotzauer A, Graebsch C, et al. Expression patterns and novel splicing variants of glutathione-S-transferase isoenzymes of human lung and hepatocyte cell lines. Cell Tissue Res. 2006;324:423–32. doi: 10.1007/s00441-005-0150-8. [DOI] [PubMed] [Google Scholar]
- 46.Fitzpatrick AM, Teague WG, Holguin F, Yeh M, Brown LA. Airway glutathione homeostasis is altered in children with severe asthma: evidence for oxidant stress. J Allergy Clin Immunol. 2009;123:146–52. e8. doi: 10.1016/j.jaci.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iyer SS, Ramirez AM, Ritzenthaler JD, Torres-Gonzalez E, Roser-Page S, Mora AL, et al. Oxidation of extracellular cysteine/cystine redox state in bleomycin-induced lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2009;296:L37–45. doi: 10.1152/ajplung.90401.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharma P, Chakraborty R, Wang L, Min B, Tremblay ML, Kawahara T, et al. Redox regulation of interleukin-4 signaling. Immunity. 2008;29:551–64. doi: 10.1016/j.immuni.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–95. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akimoto T, Numata F, Tamura M, Takata Y, Higashida N, Takashi T, et al. Abrogation of bronchial eosinophilic inflammation and airway hyperreactivity in signal transducers and activators of transcription (STAT)6-deficient mice. J Exp Med. 1998;187:1537–42. doi: 10.1084/jem.187.9.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Malaviya R, Uckun FM. Role of STAT6 in IgE receptor/FcepsilonRI-mediated late phase allergic responses of mast cells. J Immunol. 2002;168:421–6. doi: 10.4049/jimmunol.168.1.421. [DOI] [PubMed] [Google Scholar]
- 52.Lowry MH, McAllister BP, Jean JC, Brown LA, Hughey RP, Cruikshank WW, et al. Lung lining fluid glutathione attenuates IL-13-induced asthma. Am J Respir Cell Mol Biol. 2008;38:509–16. doi: 10.1165/rcmb.2007-0128OC. [DOI] [PMC free article] [PubMed] [Google Scholar]