

Abstract
Aneuploidy, often preceded by tetraploidy, is one of the hallmarks of solid tumors. Indeed, both aneuploidy and tetraploidy are oncogenic occurrences that are sufficient to drive neoplastic transformation and cancer progression. True to form, the tumor suppressor p53 obstructs propagation of these dangerous chromosomal events by either instigating irreversible cell cycle arrest or apoptosis. The tumor suppressor Lats2, along with other tumor inhibitory proteins such as BRCA1/2 and BubR1, are central to p53‐dependent elimination of tetraploid cells. Not surprisingly, these proteins are frequently inactivated or downregulated in tumors, synergizing with p53 inactivation to establish an atmosphere of “tolerance” for a non‐diploid state.
Keywords: Aneuploidy, Tetraploidy, p53, Lats2, Genomic instability, Oncogenic H-Ras
Highlights
Aneuploidy is a tumorigenic process.
Spindle checkpoint, mitotic proteins and p53/p73 determine efficiency of checkpoint.
Inactive checkpoint, excess/deficit centrosomes or mis‐cytokinesis cause tetraploidy.
Tetraploid cells senesce, apoptosis or arrest, depending on the cellular context.
1. Aneuploidy as a tumorigenic process
Aneuploidy, numerical changes in whole chromosomes, is a frequent genetic characteristic of many cancers. It is a subclass of the more general phenomenon of chromosomal instability (CIN), which includes all types of chromosomal structural and numerical alterations.
Aneuploidy can be brought about by a myriad of defects, but one of the foremost precursors and principal instigators of aneuploidy is tetraploidy. It is generally speculated that preliminary polyploidization followed by loss or gain of certain chromosomes represents an important conduit en route to aneuploidy. In line with this, aneuploid cells frequently display modal chromosome numbers that are near‐tetraploid.
Tetraploidy, where the entire genome is duplicated and cells carry precisely twice the normal number of chromosomes, can lead to cellular transformation and tumor formation. Tetraploidy is observed in early stage cancers, preceding CIN and aneuploidy (Galipeau et al., 1996; Olaharski et al., 2006). Direct evidence of the oncogenic nature of tetraploidy comes from the observation that tetraploid mouse cells can initiate tumor formation when transplanted into immunocompromised mice, whereas isogenic diploid cells do not. Importantly, this can occur only in the background of nonfunctional p53 tumor suppressor protein, since in these conditions tetraploids expressing wild type p53 fail to propagate (Fujiwara et al., 2005).
Despite the distinct correlation between tetraploidy and aneuploidy, fusion of two diploid and genomically stable cells into one tetraploid cell does not routinely end up in aneuploidy (Lengauer et al., 1997; Stukenberg, 2004). In fact, these cells often arrest in the binucleated stage, through mechanisms that involve p53 and its major transcriptional target p21, as well as the tumor suppressors ARF and pRb (Andreassen et al., 2001; Margolis et al., 2003).
Oncoproteins of DNA tumor viruses have been instrumental in dissecting the distinct roles of p53 and another prominent tumor suppressor, pRb, in preventing aneuploidy. This is perhaps best exemplified by the E6 and E7 proteins of the human papillomavirus. E6 inactivates p53 (Scheffner et al., 1993, 1990) whereas E7 disrupts pRB signaling (Dyson et al., 1989; Funk et al., 1997; Gonzalez et al., 2001; Jones et al., 1997; Zerfass‐Thome et al., 1996). Expression of E6 oncoprotein renders cells prone to structural chromosomal changes, but is not sufficient in itself to cause aneuploidy (White et al., 1994). E7 itself induces a moderate level of aneuploidy (Duensing et al., 2000). Importantly, when the function of both tumor suppressors is neutralized together, the portion of aneuploid cells is dramatically elevated (Duensing et al., 2000). These data are in line with the notion that loss of p53 synergizes with pre‐existing “aneuploidy tolerance” to wreak genomic havoc.
Several mouse and human cancer models show that an increase in tetraploid and polyploid tumor cells is specifically correlated with loss of p53 function (Galipeau et al., 1996; Ramel et al., 1995). p53‐null mice proceed to develop tumors rapidly (Donehower et al., 1992; Jacks et al., 1994), in particular thymic lymphomas, which are usually aneuploid. Furthermore, lymphomas and sarcomas from p53+/− mice in which the remaining wild type p53 allele is lost, exhibit more chromosomal instability than tumors that retain the wild type p53 allele (Venkatachalam et al., 1998). Additionally, cells from p53−/− animals (Fukasawa et al., 1996) as well as cells deficient in p53 downstream transcriptional targets such as p21 (Mantel et al., 1999) or Gadd45 (Hollander et al., 1999) accumulate aberrant chromosomal numbers, even preceding any detectable malignant phenotype (Fukasawa et al., 1997).
How does aneuploidy facilitate neoplastic transformation? One accepted hypothesis is that aneuploidy entails the loss of chromosomes carrying tumor suppressor genes and/or the gain of chromosomes bearing oncogenes. Equally feasible, the aberrant number of chromosomes could result in a general imbalance of protein levels and/or transcription patterns within the cell, particularly in cases in which such events are under tight stoichiometric regulation (e.g. limiting amounts of a transcriptional repressor). This imbalance in itself could generate oncogenic signals. Furthermore, extra copies of chromosomes might buffer aneuploid cells against the deleterious effects of genomic alterations in one chromosome, extending their survival in a transitional state and allowing time for selection of cells with additional genetic or epigenetic alterations that stabilize growth‐promoting and transforming mutations.
2. Origins of tetraploidy
Aberrant tetraploidy can be the consequence of cell fusion, endoreduplication, cytokinesis failure or mitotic slippage. The mitotic checkpoint (often referred to as the spindle damage checkpoint) is one of the front‐line surveillance mechanisms against chromosome missegregation. This checkpoint delays completion of mitosis until all chromosomes have been properly aligned. Altered expression of mitotic checkpoint components has been documented in numerous human cancers including leukemia, breast, colorectal, ovarian and lung (Kops et al., 2005), implicating the importance of their function in preventing tumorigenesis. Despite this protective mechanism, cells that are exposed to a spindle damaging agent such as nocodazole, eventually exit from mitosis (“mitotic slippage”) without undergoing cytokinesis. Normal cells stably arrest in the subsequent G1 phase with 4N DNA content. In contrast, cells that lack functional p53 have an increased ability to re‐enter the cell cycle and initiate another round of DNA replication (Borel et al., 2002; Casenghi et al., 1999; Cross et al., 1995; Hirano and Kurimura, 1974; Khan and Wahl, 1998; Lanni and Jacks, 1998; Minn et al., 1996; Stewart et al., 1999). This p53‐dependent arrest of tetraploid cells is sometimes referred to as the “G1 tetraploidy checkpoint”. That being said, the existence of this “tetraploidy” checkpoint remains controversial, since some reports indicate that p53 activation might actually result from alterations of the cytoskeleton due to the use of the tetraploidy‐inducing agents rather than from tetraploidy itself (Stukenberg, 2004; Uetake and Sluder, 2004).
Aberrant centrosome dynamics can also result in tetraploidy. When centrosomes are experimentally removed (Hinchcliffe et al., 2001; Khodjakov and Rieder, 2001) or disrupted (Gromley et al., 2003), cytokinesis is impaired and cells arrest in the following G1 with tetraploid genomes, similarly to what one observes following the disruption of the mitotic spindle by nocodazole. For instance, targeted RNAi disruption of centrosome proteins in human diploid epithelial cells induces G1 arrest (Mikule et al., 2007). These experiments strengthen the notion that cell cycle regulatory mechanisms are sensitive to centrosome fidelity (Doxsey et al., 2005; Matsumoto and Maller, 2004). Of note, the nuclei of such centrosome‐depleted cells accumulate activated p53 protein that is phosphorylated on serine 33 (Ser33), rather than on serine 15 that is characteristic of DNA damage. Moreover, reduction of p53 levels or knockout of p21 suppresses the G1 arrest (Mikule et al., 2007). In line with the fact that Ser33 of p53 is known to be phosphorylated by p38 (Kishi et al., 2001), chemical inhibition or siRNA‐mediated silencing of p38 reduces the nuclear translocation of p53 under such conditions. Moreover, several reports suggest that p53 itself can localize to centrosomes (Brown et al., 1994; Ciciarello et al., 2001; Hollander et al., 1999; Mantel et al., 1999; Morris et al., 2000), which might imply a direct role of p53 in centrosome fidelity. In fact, activated p38 has been detected on centrosomes, and Ser33‐phosphorylated p53 can be found on centrosomes before the nuclear translocation of p53 (Minn et al., 1996). Interestingly, p53 requires both its transactivation function and its centrosome binding activity to control centrosome duplication (Shinmura et al., 2007). Together, these data suggest that impairment of centrosome integrity activates a p38–p53–p21 checkpoint akin to the “G1 tetraploidy” checkpoint induced by mitotic slippage.
On the other hand, also cells with extra centrosomes are highly correlated with loss or mutation of p53 (Tarapore, Horn, Tokuyama, Fukasawa, Oncogene, 2001). These cells are in danger of forming multipolar spindles, which allegedly generate aneuploid daughter cells. However, perhaps surprisingly, cell cycle progresses normally in cells with supernumerary centrosomes (Wong and Stearns, 2005). This is because multiple centrosomes tend to cluster into two functional poles (Kwon et al., 2008; Quintyne et al., 2005; see also Alwin Krämer, Bettina Maier and Jiri Bartek, this issue). Yet, despite this protective mechanism, clustered bipolar spindles in tetraploid cells do exhibit an increased occurrence of lagging chromosomes and segregation errors (Ganem et al., 2009; Silkworth et al., 2009), likely accelerating their transition toward an aneuploid state. Moreover, supernumerary centrosomes are frequently found in cancers and their presence often correlates with aneuploidy (Brinkley, 2001; Nigg and Raff, 2009).
Missegregation of chromosomes and aneuploidy can also be induced by directly inhibiting cytokinesis. In these cases, p53 is elevated only in the ensuing aneuploid cells – a daughter cell that has gained even a single chromosome as well as its sibling that has lost one chromosome (Thompson and Compton, 2010). Moreover, in this system, p53 deficiency is sufficient to enable the accumulation of an aneuploid population and to render cells genetically unstable with intrinsic CIN. Thus, conversion of diploid progenitor cells into cells that are aneuploid and manifest CIN involves two steps: reduction in chromosome segregation fidelity and acquisition of tolerance for a non‐diploid genome, which can arise through inactivation of the p53 pathway. Within this context, p53 activation is a critical impediment against the propagation of aneuploid cells. Therefore, linguistic definitions of “tetraploidy checkpoint” aside, it is clear that p53 plays a critical role in preventing the proliferation of these cells.
3. Effects of checkpoint activation
The G1 arrest of post‐slippage tetraploid cells is often long‐lasting, particularly in non‐cancerous cells, most likely due to the induction of cellular senescence (Rieder and Maiato, 2004). Nutlin is a drug presently in early clinical trials, which stabilizes p53 by blocking its interaction with its major negative regulator Mdm2. In a variety of cell types, Nutlin is capable of eliciting a permanent cell cycle arrest with distinct features of senescence; in some cases, however, this outcome is dependent on the duration of Nutlin exposure (Efeyan et al., 2007; Kumamoto et al., 2008). Recent studies imply that prolonged Nutlin treatment of cells expressing wild type p53, despite inducing senescent‐associated features such as acidic beta‐galactosidase staining, is largely if not entirely reversible upon Nutlin removal (Huang et al., 2009a; Korotchkina et al., 2009). In fact, Nutlin promotes a tetraploid G1 arrest in several normal and transformed cell lines that express wild type p53. These cell lines undergo DNA endoreduplication after Nutlin removal to become stable tetraploid clones that are more resistant to chemotherapy‐induced apoptosis (Shen and Maki, 2010). Interestingly, this is not observed in all wild type p53 cells, and seems to correlate with the ability of p53 to trigger persistent p21 expression in the tetraploid cells after Nutlin removal (Shen and Maki, 2010). These observations call for caution in combining Nutlin treatment with standard anti‐cancer chemotherapy, particularly when the tumor cells cannot be effectively and totally eradicated by the first round of treatment.
Besides withdrawing from the cell cycle, a large portion of tetraploid cells may undergo apoptosis (Castedo et al., 2006), suggesting that the “tetraploidy checkpoint” might serve as a means for dual removal of tetraploid cells, either via apoptosis or by irreversible cell cycle arrest. In support of this, depletion of the p53 transcriptional target and pro‐apoptotic protein BAX by RNAi mimics the effect of p53 deficiency on tetraploidization (Castedo et al., 2006). Similarly, overexpression of the anti‐apoptotic protein BCL‐XL facilitates polyploidization in the absence of p53 (Minn et al., 1996).
Moreover, the (rare) survival of p53‐proficient tetraploid cells strongly depends on the activity of the checkpoint kinase Chk1 (Vitale et al., 2007). Inhibition of Chk1 kills tetraploid cells via p38‐dependent activation of p53 (see also Ignacio Toledo, Matilde Murga and Oscar Fernandez‐Capetillo, in this issue) followed by p53‐mediated activation of Bax/Bak‐driven apoptosis (Senovilla et al., 2009). This may provide important clues toward the establishment of therapeutic regimens that eliminate p53‐proficient tetraploid cells more effectively, overcoming their inherent greater drug resistance and reducing the chance that they might eventually contribute to further tumor progression.
4. Upstream regulators of the G1 tetraploidy checkpoint
4.1. Spindle checkpoint
Growing evidence indicates that mitotic checkpoint function is required for efficient induction of a p53‐dependent G1 arrest of tetraploid cells (Andreassen et al., 2001; Vogel et al., 2004). This understanding emerged from the analysis of non‐transformed cells treated with spindle toxins, using time‐lapse microscopy. Cells that become only briefly arrested in prometaphase eventually continue to proliferate normally, whereas those undergoing a more prolonged prometaphase cell cycle arrest subsequently “freeze” as tetraploids in G1, in a p38‐, p53‐ and p21‐dependent manner (Uetake and Sluder, 2010). This and other studies (Gao et al., 2009; Vogel et al., 2004) suggest that a protracted spindle checkpoint‐mediated mitotic delay is required for proper p53‐driven G1 tetraploidy checkpoint function.
In line with this, haploinsufficiency of the mitotic checkpoint protein Mad2 has been shown to increase the frequency and number of aneuploid tumors in a p53+/− background (Chi et al., 2009). Interestingly, Mad2 and several other mitotic checkpoint genes are direct targets of the E2F family of transcription factors (Erez et al., 2008; Hernando et al., 2004; Iovino et al., 2006). Since the pRb tumor suppressor protein inhibits E2F, downregulation of mitotic checkpoint fidelity may account for the increased aneuploidy observed upon pRb inactivation (see above).
Intriguingly, similar to inactivation of the mitotic checkpoint, its hyperactivation may also promote tumorigenesis. Thus, despite the observation that Mad2 overexpression in normal fibroblasts has a marked negative effect on cellular viability, overexpression of Mad2 in mice leads to the generation of multiple tumors (Sotillo et al., 2007). Moreover, tumors that experience even transient overexpression of Mad2 recur at dramatically elevated rates and exhibit enhanced drug resistance (Sotillo et al., 2010), perhaps analogous to the “protective” aneuploidy observed following Nutlin removal (see above).
BubR1, another spindle checkpoint protein, operates in a positive regulatory loop with p53. BubR1 facilitates phosphorylation and stabilization of p53 after mitotic spindle damage (Ha et al., 2007) and is also a direct p53 transcriptional target (Oikawa et al., 2005). This positive feedback loop enhances apoptosis, chiefly of polyploid cells (Shin et al., 2003). Despite this seemingly epistatic relationship, inhibition of BubR1 in the presence of mitotic toxins accelerates polyploidy even in a p53‐null background, reinforcing the idea that reduction of chromosome segregation fidelity and acquisition of aneuploid tolerance work synergistically to promote cancer.
4.2. Mitotic proteins
BRCA1 and BRCA2, whose germline mutations predispose women to breast and ovarian cancers, are tumor suppressor proteins that, among other functions, regulate cytokinesis and centrosome cycle. Their absence induces polyploidization and allows the proliferation of tetraploid cells, in vitro as well as in vivo (Daniels et al., 2004). In mammary epithelial cells, deficient BRCA1 function leads to binucleation, abnormal centrosome amplification and tetraploidy (Schlegel et al., 2003). In line with this, overexpression of BRCA2 in breast cancer cell lines diminishes the proportion of polyploid cells. This correlates with downregulation of the Aurora A and Aurora B kinases as well as of E2F1, and an increase in p21 and pRb (Sagulenko et al., 2007). Moreover, BRCA2 mutations correlate with amplification of the Aurora A locus (Bodvarsdottir et al., 2007), indicating that BRCA2 might function upstream to Aurora A.
Amplification of regulators of mitotic progression, such as polo‐like kinase1 (PLK1), Aurora A and Aurora B, also generates polyploidy via mitotic dysfunction and cytokinesis failure (Meraldi et al., 2002). For instance, in ovarian cancer cells, RNAi‐mediated depletion of Aurora A limits genomic instability, centrosome amplification and in vivo tumorigenic potential (Yang et al., 2010). Correspondingly, in murine mammary epithelium, overexpression of Aurora A induces CIN and tetraploidy and consequent tumor formation (Wang et al., 2006). Thus, overexpression of Aurora A and loss of wild type p53 function induce similar phenotypes of centrosome amplification and aneuploidy. Consistent with the effects of excess Aurora A in experimental settings, Aurora A is frequently overexpressed in human cancers (Meraldi et al., 2002). Although hyperactivation of Aurora A can be augmented in tumors lacking functional p53 (Meraldi et al., 2002), the general observation is that tumors overexpressing Aurora A tend to retain a wild type p53 gene, supporting the notion that excessive Aurora A function may dampen the genome‐protective effects of wild type p53. These observations suggest that mitotic regulators act epistatically with p53. Indeed, both Aurora A and Plk1 physically bind and phosphorylate p53, leading to its reduced function. Aurora A achieves this outcome by phosphorylating p53 on Ser315, which promotes Mdm2‐mediated degradation (Katayama et al., 2004), whereas Plk does it by interacting with the DNA‐binding domain of p53 to inhibit its transcriptional function in apoptosis (Ando et al., 2004).
Aurora A also interacts with and antagonistically phosphorylates other components of the p53 pathway, including the tumor suppressor Lats2 (Toji et al., 2004). Lats2 is a serine/threonine kinase and a major component of the Hippo pathway. Importantly, Lats2 controls activation of the p53‐dependent tetraploidy checkpoint (Aylon et al., 2006). Thus, in cells exposed to nocodazole or other mitotic spindle poisons, Lats2 departs from its regular site of residence in the centrosomes and moves into the cell nucleus. There, it binds and inactivates Mdm2, resulting in an increase in p53 protein levels and induction of a p53‐driven transcriptional response (Aylon et al., 2006). It is tempting to speculate that the enhanced Mdm2‐mediated ubiquitination of p53, driven by phosphorylation by Aurora A (Katayama et al., 2004), might be mediated by the ability of Lats2 to bind all three players; Aurora A, Mdm2 and p53 (Aylon et al., 2006; Toji et al., 2004). Not surprisingly, Lats2‐null cells harbor a wide variety of CIN phenotypes, including centrosome fragmentation, chromosome misalignment, and cytokinesis defects with multinucleation (McPherson et al., 2004; Yabuta et al., 2007).
Within this process is also embedded a positive feedback loop, wherein the Lats2 gene itself is directly transcriptionally activated by p53, leading to a gradual and continuous increase in Lats2 protein levels (Aylon et al., 2006; Kostic and Shaw, 2000). Interestingly, p53 that has been phosphorylated by the spindle checkpoint kinase Mps1 is more efficient in transactivating the Lats2 gene in response to mitotic stress (Huang et al., 2009b). Together, these results suggest signal fortification and crosstalk between effective mitosis, centrosome integrity and p53 activation in preventing tetraploidy.
Oncogenic stimuli exacerbate the effect of p53 dysfunction by further impairing ploidy. For instance, a rapid onset of aneuploidy is observed when the adenovirus oncoprotein E1A or oncogenic H‐RasV12 are expressed either in p53‐null cells (Woo and Poon, 2004) or in cells in which p53 is inactivated by overexpression of Mdm2 (Seger et al., 2002). To combat these effects, transient expression of oncogenic H‐Ras (Aylon et al., 2009) or K‐Ras (Y. Aylon, unpublished) causes Lats2 nuclear translocation and p53 activation. Moreover, Lats2 has been shown to trigger apoptosis selectively in H‐RasV12 transformed polyploid cells (Aylon et al., 2010). In cells with activated oncogenes, the DNA replication stress checkpoint is orchestrated by an ATR‐Chk1 signaling cascade (Bartkova et al., 2006; Di Micco et al., 2006; Gorgoulis et al., 2005) (see also Ayguel Dereli, Gwennaelle Versini and Thanos Halazonetis, this issue). In turn, ATR‐Chk1‐activated Lats2 phosphorylates the p53‐associated ASPP1 protein to drive ASPP1 nuclear import simultaneously with that of Lats2 itself. In the nucleus, ASPP1 and Lats2 direct p53 to pro‐apoptotic target gene promoters, thus enhancing the p53‐mediated apoptotic response in tetraploid cells (Aylon et al., 2010). Following this wave of apoptosis, Lats2‐dependent oncogene‐induced senescence acts as a second line of defense against transformation by the H‐Ras oncoprotein. Cells surviving sustained oncogenic H‐Ras activity often do so by quenching Lats2 expression through hypermethylation of the Lats2 gene promoter; notably, such “escaper” cells are highly tetraploid (Aylon et al., 2009). These data further highlight two important points already mentioned previously: first, that the tetraploidy checkpoint can impinge on either cell cycle arrest or apoptosis, and second, that tumorigenic aneuploidy transpires as a two‐step process involving reduction of mitotic fidelity and augmentation of the tolerance of abnormal chromosome numbers.
Oncogenic H‐Ras also negatively regulates the activity of the Fbw7 protein (Minella et al., 2005). Fbw7 is a substrate recognition component of the SCF E3 ubiquitin ligase complex, and is mutated in cancers exhibiting enhanced genomic instability (Minella et al., 2007). More recently, Fbw7 has been shown to regulate both the mitotic and tetraploidy checkpoints. This occurs via Fbw7‐mediated degradation of mitotic signal transducers such as Aurora A, resulting in augmented activation of the Lats2‐p53 module (Finkin et al., 2008).
Additional members of the Hippo pathway also have been associated with maintenance of proper chromosome number. Recent studies have demonstrated that loss of RASSF1a and p53 synergistically accelerate progression to aneuploidy in vivo (Tommasi et al., 2011). Similarly, the Lats1 kinase is also implicated in the G1 tetraploidy checkpoint. Lats1, a paralog of Lats2, is a dynamic component of the mitotic apparatus (Hirota et al., 2000). Inhibition of Lats1 induces prolonged mitotic arrest followed by abortive cell division and polyploidy (Iida et al., 2004). Moreover, expression of kinase‐dead Lats1 is sufficient to override p53 checkpoint function, facilitating the survival and proliferation of tetraploid cells (Iida et al., 2004).
5. Aneuploidy and p73
As described above, p53 instigates the G1 tetraploidy checkpoint to prevent cells with a 4N DNA content from re‐entering the cell cycle and proliferating. Like p53, its paralog and family member p73 also aids in the battle against proliferation of aneuploid cells. Expression of dominant negative p73 in cells lacking p53 results in rapid accumulation of a tetraploid population. These cells, lacking both p53 and p73 function, exhibit diminished Chk1 levels and attenuated activating phosphorylation of Chk1 on Ser345 (Talos et al., 2007), indicative of checkpoint malfunction.
p73, in contrast to p53, is covalently modified specifically during mitosis. During mitotic arrest, hyperphosphorylated p73 is excluded from condensed, mitotic chromatin and suffers reduced transcriptional activity (Fulco et al., 2003). Instead, p73 interacts on the kinetochore with several components of the spindle checkpoint, including Bub1, Bub3 and BubR1 (Niikura et al., 2010; Tomasini et al., 2009; Vernole et al., 2009). Despite this, p73 is capable of activating a subset of transcriptional targets during mitosis, including the CDK inhibitor Kip2 (Merlo et al., 2005). The p73‐Kip2 axis, as well as the p73‐mitotic protein interactions, are required to control mitotic exit and prevent DNA endoreduplication in tetraploid cells.
Reminiscent of its inhibitory effect on p53, Plk1 binds and phosphorylates p73. This phosphorylation event both destabilizes p73 and reduces its transcriptional activity (Koida et al., 2008). Similarly, Aurora A also diminishes p73 transcriptional activity (Dar et al., 2008). Together, these data suggest that p73 plays an important and distinct role in maintaining genomic integrity. This genome safeguarding function of p73 may become particularly important in instances in which p53 function is compromised.
6. Conclusion
Aneuploidy is the byproduct of a multistep selection process that occurs during tumorigenesis. While p53 inactivation may not represent the primary cause of aneuploidy, it most probably facilitates chromosomal instability “tolerance” resulting from other various cellular insults. Damage during mitosis, either due to aberrant spindle checkpoint or deregulated mitotic kinase function, can “ramp up” the p53 response. Sensitized p53 preserves genomic stability on a multitude of levels; these include limiting the generation of tetraploid cells by instigating a G1/S cell cycle arrest, apoptotic removal of tetraploid cells, and inhibition of further genomic destabilization of illicitly generated tetraploid cells that might otherwise into more dangerous aneuploid cells. Failing this, p73 takes up arms to act as a backup guardian of the genome. When both p53 family members become defunct together with upstream mitotic regulators and p53 activators, all (genomic) hell breaks loose. Genomic instability gradually becomes more pronounced and more rampant, thereby contributing to the marked heterogeneity of the cancer cells residing within individual advanced tumors (Figure 1).
Figure 1.
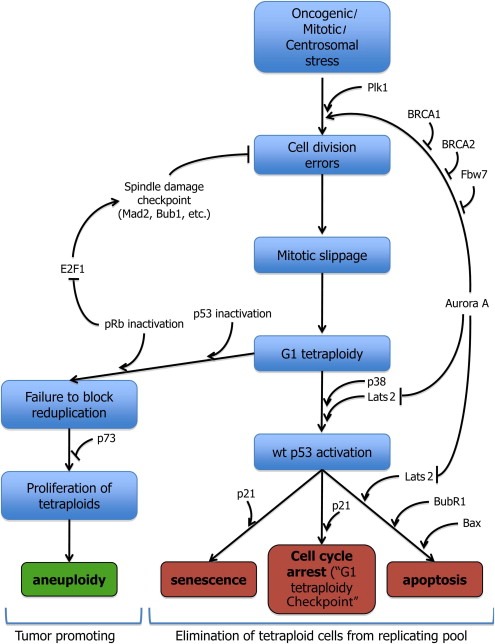
Schematic representation of the steps leading to, or avoiding, aneuploidy. Wild type p53 function is crucial for the elimination of tetraploid cells from the replicating pool. Loss of p53 and p73 tumor suppressors facilitates aneuploid tumor formation. Orange boxes represent proliferation “stops”, whereas green represents proliferation “go”.
Acknowledgments
Work in the authors' laboratory is supported in part by grant R37 CA40099 from the National Cancer Institute, EC FP7 funding (INFLACARE, agreement 223151 and ONCOMIRS, agreement 201102), a Center of Excellence grant from the Flight Attendant Medical Research Institute (FAMRI), the Dr. Miriam and Sheldon Adelson Medical Research Foundation, The Robert Bosch Stiftung, The Lower Saxony‐Israeli Association and the Estate of Harold Z. Novak. MO is incumbent of the Andre Lwoff chair in Molecular Biology. The EC is not liable for any use that may be made of the information contained herein.
Aylon Yael and Oren Moshe, (2011), p53: Guardian of ploidy, Molecular Oncology, 5, doi: 10.1016/j.molonc.2011.07.007.
References
- Ando, K. , Ozaki, T. , Yamamoto, H. , Furuya, K. , Hosoda, M. , Hayashi, S. , Fukuzawa, M. , Nakagawara, A. , 2004. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J. Biol. Chem.. 279, 25549–25561. [DOI] [PubMed] [Google Scholar]
- Andreassen, P.R. , Lohez, O.D. , Lacroix, F.B. , Margolis, R.L. , 2001. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol. Biol. Cell. 12, 1315–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon, Y. , Michael, D. , Shmueli, A. , Yabuta, N. , Nojima, H. , Oren, M. , 2006. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev.. 20, 2687–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon, Y. , Ofir-Rosenfeld, Y. , Yabuta, N. , Lapi, E. , Nojima, H. , Lu, X. , Oren, M. , 2010. The Lats2 tumor suppressor augments p53-mediated apoptosis by promoting the nuclear proapoptotic function of ASPP1. Genes Dev.. 24, 2420–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon, Y. , Yabuta, N. , Besserglick, H. , Buganim, Y. , Rotter, V. , Nojima, H. , Oren, M. , 2009. Silencing of the Lats2 tumor suppressor overrides a p53-dependent oncogenic stress checkpoint and enables mutant H-Ras-driven cell transformation. Oncogene. 28, 4469–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova, J. , Rezaei, N. , Liontos, M. , Karakaidos, P. , Kletsas, D. , Issaeva, N. , Vassiliou, L.V. , Kolettas, E. , Niforou, K. , Zoumpourlis, V.C. , Takaoka, M. , Nakagawa, H. , Tort, F. , Fugger, K. , Johansson, F. , Sehested, M. , Andersen, C.L. , Dyrskjot, L. , Orntoft, T. , Lukas, J. , Kittas, C. , Helleday, T. , Halazonetis, T.D. , Bartek, J. , Gorgoulis, V.G. , 2006. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 444, 633–637. [DOI] [PubMed] [Google Scholar]
- Bodvarsdottir, S.K. , Hilmarsdottir, H. , Birgisdottir, V. , Steinarsdottir, M. , Jonasson, J.G. , Eyfjord, J.E. , 2007. Aurora-A amplification associated with BRCA2 mutation in breast tumours. Cancer Lett.. 248, 96–102. [DOI] [PubMed] [Google Scholar]
- Borel, F. , Lohez, O.D. , Lacroix, F.B. , Margolis, R.L. , 2002. Multiple centrosomes arise from tetraploidy checkpoint failure and mitotic centrosome clusters in p53 and RB pocket protein-compromised cells. Proc. Natl. Acad. Sci. U S A. 99, 9819–9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkley, B.R. , 2001. Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol.. 11, 18–21. [DOI] [PubMed] [Google Scholar]
- Brown, C.R. , Doxsey, S.J. , White, E. , Welch, W.J. , 1994. Both viral (adenovirus E1B) and cellular (hsp 70, p53) components interact with centrosomes. J. Cell Physiol.. 160, 47–60. [DOI] [PubMed] [Google Scholar]
- Casenghi, M. , Mangiacasale, R. , Tuynder, M. , Caillet-Fauquet, P. , Elhajouji, A. , Lavia, P. , Mousset, S. , Kirsch-Volders, M. , Cundari, E. , 1999. p53-independent apoptosis and p53-dependent block of DNA rereplication following mitotic spindle inhibition in human cells. Exp. Cell Res.. 250, 339–350. [DOI] [PubMed] [Google Scholar]
- Castedo, M. , Coquelle, A. , Vivet, S. , Vitale, I. , Kauffmann, A. , Dessen, P. , Pequignot, M.O. , Casares, N. , Valent, A. , Mouhamad, S. , Schmitt, E. , Modjtahedi, N. , Vainchenker, W. , Zitvogel, L. , Lazar, V. , Garrido, C. , Kroemer, G. , 2006. Apoptosis regulation in tetraploid cancer cells. EMBO J.. 25, 2584–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi, Y.H. , Ward, J.M. , Cheng, L.I. , Yasunaga, J. , Jeang, K.T. , 2009. Spindle assembly checkpoint and p53 deficiencies cooperate for tumorigenesis in mice. Int. J. Cancer. 124, 1483–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciciarello, M. , Mangiacasale, R. , Casenghi, M. , Zaira Limongi, M. , D'Angelo, M. , Soddu, S. , Lavia, P. , Cundari, E. , 2001. p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J. Biol. Chem.. 276, 19205–19213. [DOI] [PubMed] [Google Scholar]
- Cross, S.M. , Sanchez, C.A. , Morgan, C.A. , Schimke, M.K. , Ramel, S. , Idzerda, R.L. , Raskind, W.H. , Reid, B.J. , 1995. A p53-dependent mouse spindle checkpoint. Science. 267, 1353–1356. [DOI] [PubMed] [Google Scholar]
- Daniels, M.J. , Wang, Y. , Lee, M. , Venkitaraman, A.R. , 2004. Abnormal cytokinesis in cells deficient in the breast cancer susceptibility protein BRCA2. Science. 306, 876–879. [DOI] [PubMed] [Google Scholar]
- Dar, A.A. , Belkhiri, A. , Ecsedy, J. , Zaika, A. , El-Rifai, W. , 2008. Aurora kinase A inhibition leads to p73-dependent apoptosis in p53-deficient cancer cells. Cancer Res.. 68, 8998–9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco, R. , Fumagalli, M. , Cicalese, A. , Piccinin, S. , Gasparini, P. , Luise, C. , Schurra, C. , Garre, M. , Nuciforo, P.G. , Bensimon, A. , Maestro, R. , Pelicci, P.G. , d'Adda di Fagagna, F. , 2006. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 444, 638–642. [DOI] [PubMed] [Google Scholar]
- Donehower, L.A. , Harvey, M. , Slagle, B.L. , McArthur, M.J. , Montgomery, C.A. , Butel, J.S. , Bradley, A. , 1992. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 356, 215–221. [DOI] [PubMed] [Google Scholar]
- Doxsey, S. , Zimmerman, W. , Mikule, K. , 2005. Centrosome control of the cell cycle. Trends Cell Biol.. 15, 303–311. [DOI] [PubMed] [Google Scholar]
- Duensing, S. , Lee, L.Y. , Duensing, A. , Basile, J. , Piboonniyom, S. , Gonzalez, S. , Crum, C.P. , Munger, K. , 2000. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc. Natl. Acad. Sci. U S A. 97, 10002–10007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson, N. , Howley, P.M. , Munger, K. , Harlow, E. , 1989. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 243, 934–937. [DOI] [PubMed] [Google Scholar]
- Efeyan, A. , Ortega-Molina, A. , Velasco-Miguel, S. , Herranz, D. , Vassilev, L.T. , Serrano, M. , 2007. Induction of p53-dependent senescence by the MDM2 antagonist nutlin-3a in mouse cells of fibroblast origin. Cancer Res.. 67, 7350–7357. [DOI] [PubMed] [Google Scholar]
- Erez, A. , Chaussepied, M. , Castiel, A. , Colaizzo-Anas, T. , Aplan, P.D. , Ginsberg, D. , Izraeli, S. , 2008. The mitotic checkpoint gene, SIL is regulated by E2F1. Int. J. Cancer. 123, 1721–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkin, S. , Aylon, Y. , Anzi, S. , Oren, M. , Shaulian, E. , 2008. Fbw7 regulates the activity of endoreduplication mediators and the p53 pathway to prevent drug-induced polyploidy. Oncogene. 27, 4411–4421. [DOI] [PubMed] [Google Scholar]
- Fujiwara, T. , Bandi, M. , Nitta, M. , Ivanova, E.V. , Bronson, R.T. , Pellman, D. , 2005. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 437, 1043–1047. [DOI] [PubMed] [Google Scholar]
- Fukasawa, K. , Choi, T. , Kuriyama, R. , Rulong, S. , Vande Woude, G.F. , 1996. Abnormal centrosome amplification in the absence of p53. Science. 271, 1744–1747. [DOI] [PubMed] [Google Scholar]
- Fukasawa, K. , Wiener, F. , Vande Woude, G.F. , Mai, S. , 1997. Genomic instability and apoptosis are frequent in p53 deficient young mice. Oncogene. 15, 1295–1302. [DOI] [PubMed] [Google Scholar]
- Fulco, M. , Costanzo, A. , Merlo, P. , Mangiacasale, R. , Strano, S. , Blandino, G. , Balsano, C. , Lavia, P. , Levrero, M. , 2003. p73 is regulated by phosphorylation at the G2/M transition. J. Biol. Chem.. 278, 49196–49202. [DOI] [PubMed] [Google Scholar]
- Funk, J.O. , Waga, S. , Harry, J.B. , Espling, E. , Stillman, B. , Galloway, D.A. , 1997. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev.. 11, 2090–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galipeau, P.C. , Cowan, D.S. , Sanchez, C.A. , Barrett, M.T. , Emond, M.J. , Levine, D.S. , Rabinovitch, P.S. , Reid, B.J. , 1996. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett's esophagus. Proc. Natl. Acad. Sci. U S A. 93, 7081–7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem, N.J. , Godinho, S.A. , Pellman, D. , 2009. A mechanism linking extra centrosomes to chromosomal instability. Nature. 460, 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, F. , Ponte, J.F. , Levy, M. , Papageorgis, P. , Cook, N.M. , Ozturk, S. , Lambert, A.W. , Thiagalingam, A. , Abdolmaleky, H.M. , Sullivan, B.A. , Thiagalingam, S. , 2009. hBub1 negatively regulates p53 mediated early cell death upon mitotic checkpoint activation. Cancer Biol. Ther.. 8, 548–556. [PubMed] [Google Scholar]
- Gonzalez, S.L. , Stremlau, M. , He, X. , Basile, J.R. , Munger, K. , 2001. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J. Virol.. 75, 7583–7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis, V.G. , Vassiliou, L.V. , Karakaidos, P. , Zacharatos, P. , Kotsinas, A. , Liloglou, T. , Venere, M. , Ditullio, R.A. , Kastrinakis, N.G. , Levy, B. , Kletsas, D. , Yoneta, A. , Herlyn, M. , Kittas, C. , Halazonetis, T.D. , 2005. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 434, 907–913. [DOI] [PubMed] [Google Scholar]
- Gromley, A. , Jurczyk, A. , Sillibourne, J. , Halilovic, E. , Mogensen, M. , Groisman, I. , Blomberg, M. , Doxsey, S. , 2003. A novel human protein of the maternal centriole is required for the final stages of cytokinesis and entry into S phase. J. Cell Biol.. 161, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha, G.H. , Baek, K.H. , Kim, H.S. , Jeong, S.J. , Kim, C.M. , McKeon, F. , Lee, C.W. , 2007. p53 activation in response to mitotic spindle damage requires signaling via BubR1-mediated phosphorylation. Cancer Res.. 67, 7155–7164. [DOI] [PubMed] [Google Scholar]
- Hernando, E. , Nahle, Z. , Juan, G. , Diaz-Rodriguez, E. , Alaminos, M. , Hemann, M. , Michel, L. , Mittal, V. , Gerald, W. , Benezra, R. , Lowe, S.W. , Cordon-Cardo, C. , 2004. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 430, 797–802. [DOI] [PubMed] [Google Scholar]
- Hinchcliffe, E.H. , Miller, F.J. , Cham, M. , Khodjakov, A. , Sluder, G. , 2001. Requirement of a centrosomal activity for cell cycle progression through G1 into S phase. Science. 291, 1547–1550. [DOI] [PubMed] [Google Scholar]
- Hirano, A. , Kurimura, T. , 1974. Virally transformed cells and cytochalasin B. I. The effect of cytochalasin B on cytokinesis, karyokinesis and DNA synthesis in cells. Exp. Cell Res.. 89, 111–120. [DOI] [PubMed] [Google Scholar]
- Hirota, T. , Morisaki, T. , Nishiyama, Y. , Marumoto, T. , Tada, K. , Hara, T. , Masuko, N. , Inagaki, M. , Hatakeyama, K. , Saya, H. , 2000. Zyxin, a regulator of actin filament assembly, targets the mitotic apparatus by interacting with h-warts/LATS1 tumor suppressor. J. Cell Biol.. 149, 1073–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander, M.C. , Sheikh, M.S. , Bulavin, D.V. , Lundgren, K. , Augeri-Henmueller, L. , Shehee, R. , Molinaro, T.A. , Kim, K.E. , Tolosa, E. , Ashwell, J.D. , Rosenberg, M.P. , Zhan, Q. , Fernandez-Salguero, P.M. , Morgan, W.F. , Deng, C.X. , Fornace, A.J. , 1999. Genomic instability in Gadd45a-deficient mice. Nat. Genet.. 23, 176–184. [DOI] [PubMed] [Google Scholar]
- Huang, B. , Deo, D. , Xia, M. , Vassilev, L.T. , 2009. Pharmacologic p53 activation blocks cell cycle progression but fails to induce senescence in epithelial cancer cells. Mol. Cancer Res.. 7, 1497–1509. [DOI] [PubMed] [Google Scholar]
- Huang, Y.F. , Chang, M.D. , Shieh, S.Y. , 2009. TTK/hMps1 mediates the p53-dependent postmitotic checkpoint by phosphorylating p53 at Thr18. Mol. Cell Biol.. 29, 2935–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida, S. , Hirota, T. , Morisaki, T. , Marumoto, T. , Hara, T. , Kuninaka, S. , Honda, S. , Kosai, K. , Kawasuji, M. , Pallas, D.C. , Saya, H. , 2004. Tumor suppressor WARTS ensures genomic integrity by regulating both mitotic progression and G1 tetraploidy checkpoint function. Oncogene. 23, 5266–5274. [DOI] [PubMed] [Google Scholar]
- Iovino, F. , Lentini, L. , Amato, A. , Di Leonardo, A. , 2006. RB acute loss induces centrosome amplification and aneuploidy in murine primary fibroblasts. Mol. Cancer. 5, 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks, T. , Remington, L. , Williams, B.O. , Schmitt, E.M. , Halachmi, S. , Bronson, R.T. , Weinberg, R.A. , 1994. Tumor spectrum analysis in p53-mutant mice. Curr. Biol.. 4, 1–7. [DOI] [PubMed] [Google Scholar]
- Jones, D.L. , Alani, R.M. , Munger, K. , 1997. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev.. 11, 2101–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama, H. , Sasai, K. , Kawai, H. , Yuan, Z.M. , Bondaruk, J. , Suzuki, F. , Fujii, S. , Arlinghaus, R.B. , Czerniak, B.A. , Sen, S. , 2004. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat. Genet.. 36, 55–62. [DOI] [PubMed] [Google Scholar]
- Khan, S.H. , Wahl, G.M. , 1998. p53 and pRb prevent rereplication in response to microtubule inhibitors by mediating a reversible G1 arrest. Cancer Res.. 58, 396–401. [PubMed] [Google Scholar]
- Khodjakov, A. , Rieder, C.L. , 2001. Centrosomes enhance the fidelity of cytokinesis in vertebrates and are required for cell cycle progression. J. Cell Biol.. 153, 237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi, H. , Nakagawa, K. , Matsumoto, M. , Suga, M. , Ando, M. , Taya, Y. , Yamaizumi, M. , 2001. Osmotic shock induces G1 arrest through p53 phosphorylation at Ser33 by activated p38MAPK without phosphorylation at Ser15 and Ser20. J. Biol. Chem.. 276, 39115–39122. [DOI] [PubMed] [Google Scholar]
- Koida, N. , Ozaki, T. , Yamamoto, H. , Ono, S. , Koda, T. , Ando, K. , Okoshi, R. , Kamijo, T. , Omura, K. , Nakagawara, A. , 2008. Inhibitory role of Plk1 in the regulation of p73-dependent apoptosis through physical interaction and phosphorylation. J. Biol. Chem.. 283, 8555–8563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops, G.J. , Weaver, B.A. , Cleveland, D.W. , 2005. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer. 5, 773–785. [DOI] [PubMed] [Google Scholar]
- Korotchkina, L.G. , Demidenko, Z.N. , Gudkov, A.V. , Blagosklonny, M.V. , 2009. Cellular quiescence caused by the Mdm2 inhibitor nutlin-3A. Cell Cycle. 8, 3777–3781. [DOI] [PubMed] [Google Scholar]
- Kostic, C. , Shaw, P.H. , 2000. Isolation and characterization of sixteen novel p53 response genes. Oncogene. 19, 3978–3987. [DOI] [PubMed] [Google Scholar]
- Kumamoto, K. , Spillare, E.A. , Fujita, K. , Horikawa, I. , Yamashita, T. , Appella, E. , Nagashima, M. , Takenoshita, S. , Yokota, J. , Harris, C.C. , 2008. Nutlin-3a activates p53 to both down-regulate inhibitor of growth 2 and up-regulate mir-34a, mir-34b, and mir-34c expression, and induce senescence. Cancer Res.. 68, 3193–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon, M. , Godinho, S.A. , Chandhok, N.S. , Ganem, N.J. , Azioune, A. , Thery, M. , Pellman, D. , 2008. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev.. 22, 2189–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanni, J.S. , Jacks, T. , 1998. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol. Cell Biol.. 18, 1055–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengauer, C. , Kinzler, K.W. , Vogelstein, B. , 1997. Genetic instability in colorectal cancers. Nature. 386, 623–627. [DOI] [PubMed] [Google Scholar]
- Mantel, C. , Braun, S.E. , Reid, S. , Henegariu, O. , Liu, L. , Hangoc, G. , Broxmeyer, H.E. , 1999. p21(cip-1/waf-1) deficiency causes deformed nuclear architecture, centriole overduplication, polyploidy, and relaxed microtubule damage checkpoints in human hematopoietic cells. Blood. 93, 1390–1398. [PubMed] [Google Scholar]
- Margolis, R.L. , Lohez, O.D. , Andreassen, P.R. , 2003. G1 tetraploidy checkpoint and the suppression of tumorigenesis. J. Cell Biochem.. 88, 673–683. [DOI] [PubMed] [Google Scholar]
- Matsumoto, Y. , Maller, J.L. , 2004. A centrosomal localization signal in cyclin E required for Cdk2-independent S phase entry. Science. 306, 885–888. [DOI] [PubMed] [Google Scholar]
- McPherson, J.P. , Tamblyn, L. , Elia, A. , Migon, E. , Shehabeldin, A. , Matysiak-Zablocki, E. , Lemmers, B. , Salmena, L. , Hakem, A. , Fish, J. , Kassam, F. , Squire, J. , Bruneau, B.G. , Hande, M.P. , Hakem, R. , 2004. Lats2/Kpm is required for embryonic development, proliferation control and genomic integrity. EMBO J.. 23, 3677–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi, P. , Honda, R. , Nigg, E.A. , 2002. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J.. 21, 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo, P. , Fulco, M. , Costanzo, A. , Mangiacasale, R. , Strano, S. , Blandino, G. , Taya, Y. , Lavia, P. , Levrero, M. , 2005. A role of p73 in mitotic exit. J. Biol. Chem.. 280, 30354–30360. [DOI] [PubMed] [Google Scholar]
- Mikule, K. , Delaval, B. , Kaldis, P. , Jurcyzk, A. , Hergert, P. , Doxsey, S. , 2007. Loss of centrosome integrity induces p38-p53-p21-dependent G1-S arrest. Nat. Cell Biol.. 9, 160–170. [DOI] [PubMed] [Google Scholar]
- Minella, A.C. , Grim, J.E. , Welcker, M. , Clurman, B.E. , 2007. p53 and SCFFbw7 cooperatively restrain cyclin E-associated genome instability. Oncogene. 26, 6948–6953. [DOI] [PubMed] [Google Scholar]
- Minella, A.C. , Welcker, M. , Clurman, B.E. , 2005. Ras activity regulates cyclin E degradation by the Fbw7 pathway. Proc. Natl. Acad. Sci. U S A. 102, 9649–9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn, A.J. , Boise, L.H. , Thompson, C.B. , 1996. Expression of Bcl-xL and loss of p53 can cooperate to overcome a cell cycle checkpoint induced by mitotic spindle damage. Genes Dev.. 10, 2621–2631. [DOI] [PubMed] [Google Scholar]
- Morris, V.B. , Brammall, J. , Noble, J. , Reddel, R. , 2000. p53 localizes to the centrosomes and spindles of mitotic cells in the embryonic chick epiblast, human cell lines, and a human primary culture: an immunofluorescence study. Exp. Cell Res.. 256, 122–130. [DOI] [PubMed] [Google Scholar]
- Nigg, E.A. , Raff, J.W. , 2009. Centrioles, centrosomes, and cilia in health and disease. Cell. 139, 663–678. [DOI] [PubMed] [Google Scholar]
- Niikura, Y. , Ogi, H. , Kikuchi, K. , Kitagawa, K. , 2010. BUB3 that dissociates from BUB1 activates caspase-independent mitotic death (CIMD). Cell Death Differ.. 17, 1011–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa, T. , Okuda, M. , Ma, Z. , Goorha, R. , Tsujimoto, H. , Inokuma, H. , Fukasawa, K. , 2005. Transcriptional control of BubR1 by p53 and suppression of centrosome amplification by BubR1. Mol. Cell Biol.. 25, 4046–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olaharski, A.J. , Sotelo, R. , Solorza-Luna, G. , Gonsebatt, M.E. , Guzman, P. , Mohar, A. , Eastmond, D.A. , 2006. Tetraploidy and chromosomal instability are early events during cervical carcinogenesis. Carcinogenesis. 27, 337–343. [DOI] [PubMed] [Google Scholar]
- Quintyne, N.J. , Reing, J.E. , Hoffelder, D.R. , Gollin, S.M. , Saunders, W.S. , 2005. Spindle multipolarity is prevented by centrosomal clustering. Science. 307, 127–129. [DOI] [PubMed] [Google Scholar]
- Ramel, S. , Sanchez, C.A. , Schimke, M.K. , Neshat, K. , Cross, S.M. , Raskind, W.H. , Reid, B.J. , 1995. Inactivation of p53 and the development of tetraploidy in the elastase-SV40 T antigen transgenic mouse pancreas. Pancreas. 11, 213–222. [DOI] [PubMed] [Google Scholar]
- Rieder, C.L. , Maiato, H. , 2004. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell. 7, 637–651. [DOI] [PubMed] [Google Scholar]
- Sagulenko, E. , Savelyeva, L. , Ehemann, V. , Sagulenko, V. , Hofmann, W. , Arnold, K. , Claas, A. , Scherneck, S. , Schwab, M. , 2007. Suppression of polyploidy by the BRCA2 protein. Cancer Lett.. 257, 65–72. [DOI] [PubMed] [Google Scholar]
- Scheffner, M. , Huibregtse, J.M. , Vierstra, R.D. , Howley, P.M. , 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 75, 495–505. [DOI] [PubMed] [Google Scholar]
- Scheffner, M. , Werness, B.A. , Huibregtse, J.M. , Levine, A.J. , Howley, P.M. , 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 63, 1129–1136. [DOI] [PubMed] [Google Scholar]
- Schlegel, B.P. , Starita, L.M. , Parvin, J.D. , 2003. Overexpression of a protein fragment of RNA helicase A causes inhibition of endogenous BRCA1 function and defects in ploidy and cytokinesis in mammary epithelial cells. Oncogene. 22, 983–991. [DOI] [PubMed] [Google Scholar]
- Seger, Y.R. , Garcia-Cao, M. , Piccinin, S. , Cunsolo, C.L. , Doglioni, C. , Blasco, M.A. , Hannon, G.J. , Maestro, R. , 2002. Transformation of normal human cells in the absence of telomerase activation. Cancer Cell. 2, 401–413. [DOI] [PubMed] [Google Scholar]
- Senovilla, L. , Vitale, I. , Galluzzi, L. , Vivet, S. , Joza, N. , Younes, A.B. , Rello-Varona, S. , Castedo, M. , Kroemer, G. , 2009. p53 represses the polyploidization of primary mammary epithelial cells by activating apoptosis. Cell Cycle. 8, 1380–1385. [DOI] [PubMed] [Google Scholar]
- Shen, H. , Maki, C.G. , 2010. Persistent p21 expression after Nutlin-3a removal is associated with senescence-like arrest in 4N cells. J. Biol. Chem.. 285, 23105–23114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, H.J. , Baek, K.H. , Jeon, A.H. , Park, M.T. , Lee, S.J. , Kang, C.M. , Lee, H.S. , Yoo, S.H. , Chung, D.H. , Sung, Y.C. , McKeon, F. , Lee, C.W. , 2003. Dual roles of human BubR1, a mitotic checkpoint kinase, in the monitoring of chromosomal instability. Cancer Cell. 4, 483–497. [DOI] [PubMed] [Google Scholar]
- Shinmura, K. , Bennett, R.A. , Tarapore, P. , Fukasawa, K. , 2007. Direct evidence for the role of centrosomally localized p53 in the regulation of centrosome duplication. Oncogene. 26, 2939–2944. [DOI] [PubMed] [Google Scholar]
- Silkworth, W.T. , Nardi, I.K. , Scholl, L.M. , Cimini, D. , 2009. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS One. 4, e6564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo, R. , Hernando, E. , Diaz-Rodriguez, E. , Teruya-Feldstein, J. , Cordon-Cardo, C. , Lowe, S.W. , Benezra, R. , 2007. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 11, 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo, R. , Schvartzman, J.M. , Socci, N.D. , Benezra, R. , 2010. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature. 464, 436–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, Z.A. , Leach, S.D. , Pietenpol, J.A. , 1999. p21(Waf1/Cip1) inhibition of cyclin E/Cdk2 activity prevents endoreduplication after mitotic spindle disruption. Mol. Cell Biol.. 19, 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukenberg, P.T. , 2004. Triggering p53 after cytokinesis failure. J. Cell Biol.. 165, 607–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talos, F. , Nemajerova, A. , Flores, E.R. , Petrenko, O. , Moll, U.M. , 2007. p73 suppresses polyploidy and aneuploidy in the absence of functional p53. Mol. Cell. 27, 647–659. [DOI] [PubMed] [Google Scholar]
- Thompson, S.L. , Compton, D.A. , 2010. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol.. 188, 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toji, S. , Yabuta, N. , Hosomi, T. , Nishihara, S. , Kobayashi, T. , Suzuki, S. , Tamai, K. , Nojima, H. , 2004. The centrosomal protein Lats2 is a phosphorylation target of Aurora-A kinase. Genes Cells. 9, 383–397. [DOI] [PubMed] [Google Scholar]
- Tomasini, R. , Tsuchihara, K. , Tsuda, C. , Lau, S.K. , Wilhelm, M. , Ruffini, A. , Tsao, M.S. , Iovanna, J.L. , Jurisicova, A. , Melino, G. , Mak, T.W. , 2009. TAp73 regulates the spindle assembly checkpoint by modulating BubR1 activity. Proc. Natl. Acad. Sci. U S A. 106, 797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tommasi, S. , Besaratinia, A. , Wilczynski, S.P. , Pfeifer, G.P. , 2011. Loss of Rassf1a enhances p53-mediated tumor predisposition and accelerates progression to aneuploidy. Oncogene. 30, 690–700. [DOI] [PubMed] [Google Scholar]
- Uetake, Y. , Sluder, G. , 2004. Cell cycle progression after cleavage failure: mammalian somatic cells do not possess a “tetraploidy checkpoint”. J. Cell Biol.. 165, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetake, Y. , Sluder, G. , 2010. Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr. Biol.. 20, 1666–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam, S. , Shi, Y.P. , Jones, S.N. , Vogel, H. , Bradley, A. , Pinkel, D. , Donehower, L.A. , 1998. Retention of wild-type p53 in tumors from p53 heterozygous mice: reduction of p53 dosage can promote cancer formation. EMBO J.. 17, 4657–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernole, P. , Neale, M.H. , Barcaroli, D. , Munarriz, E. , Knight, R.A. , Tomasini, R. , Mak, T.W. , Melino, G. , De Laurenzi, V. , 2009. TAp73alpha binds the kinetochore proteins Bub1 and Bub3 resulting in polyploidy. Cell Cycle. 8, 421–429. [DOI] [PubMed] [Google Scholar]
- Vitale, I. , Galluzzi, L. , Vivet, S. , Nanty, L. , Dessen, P. , Senovilla, L. , Olaussen, K.A. , Lazar, V. , Prudhomme, M. , Golsteyn, R.M. , Castedo, M. , Kroemer, G. , 2007. Inhibition of Chk1 kills tetraploid tumor cells through a p53-dependent pathway. PLoS One. 2, e1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel, C. , Kienitz, A. , Hofmann, I. , Muller, R. , Bastians, H. , 2004. Crosstalk of the mitotic spindle assembly checkpoint with p53 to prevent polyploidy. Oncogene. 23, 6845–6853. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Zhou, Y.X. , Qiao, W. , Tominaga, Y. , Ouchi, M. , Ouchi, T. , Deng, C.X. , 2006. Overexpression of aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene. 25, 7148–7158. [DOI] [PubMed] [Google Scholar]
- White, A.E. , Livanos, E.M. , Tlsty, T.D. , 1994. Differential disruption of genomic integrity and cell cycle regulation in normal human fibroblasts by the HPV oncoproteins. Genes Dev.. 8, 666–677. [DOI] [PubMed] [Google Scholar]
- Wong, C. , Stearns, T. , 2005. Mammalian cells lack checkpoints for tetraploidy, aberrant centrosome number, and cytokinesis failure. BMC Cell Biol.. 6, 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, R.A. , Poon, R.Y. , 2004. Activated oncogenes promote and cooperate with chromosomal instability for neoplastic transformation. Genes Dev.. 18, 1317–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabuta, N. , Okada, N. , Ito, A. , Hosomi, T. , Nishihara, S. , Sasayama, Y. , Fujimori, A. , Okuzaki, D. , Zhao, H. , Ikawa, M. , Okabe, M. , Nojima, H. , 2007. Lats2 is an essential mitotic regulator required for the coordination of cell division. J. Biol. Chem.. 282, 19259–19271. [DOI] [PubMed] [Google Scholar]
- Yang, G. , Chang, B. , Yang, F. , Guo, X. , Cai, K.Q. , Xiao, X.S. , Wang, H. , Sen, S. , Hung, M.C. , Mills, G.B. , Chang, S. , Multani, A.S. , Mercado-Uribe, I. , Liu, J. , 2010. Aurora kinase A promotes ovarian tumorigenesis through dysregulation of the cell cycle and suppression of BRCA2. Clin. Cancer Res.. 16, 3171–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerfass-Thome, K. , Zwerschke, W. , Mannhardt, B. , Tindle, R. , Botz, J.W. , Jansen-Durr, P. , 1996. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene. 13, 2323–2330. [PubMed] [Google Scholar]