

Abstract
Phosphorylation of IκB, an inhibitor of NF-κB, is an important step in the activation of the transcription factor NF-κB. Phosphorylation is mediated by the IκB kinase (IKK) complex, known to contain two catalytic subunits: IKKα and IKKβ. A novel, noncatalytic component of this kinase complex called NEMO (NF-κB essential modulator)/IKKγ was identified recently. We have generated NEMO/IKKγ-deficient mice by gene targeting. Mutant embryos die at E12.5–E13.0 from severe liver damage due to apoptosis. NEMO/IKKγ-deficient primary murine embryonic fibroblasts (MEFs) lack detectable NF-κB DNA-binding activity in response to TNFα, IL-1, LPS, and Poly(IC) and do not show stimulus-dependent IκB kinase activity, which correlates with a lack of phosphorylation and degradation of IκBα. Consistent with these data, mutant MEFs show increased sensitivity to TNFα-induced apoptosis. Our data provide in vivo evidence that NEMO/IKKγ is the first essential, noncatalytic component of the IKK complex.
Keywords: NEMO/IKKγ, IKK, NF-κB, liver apoptosis
The transcription factors of the Rel/NF-κB family are critical regulators of genes involved in inflammatory, immune and acute phase responses, proliferation, and apoptosis (Baeuerle and Henkel 1994). NF-κB activation is tightly controlled by IκB proteins. In resting cells, NF-κB is bound by IκB proteins, which mask the nuclear localization signal of NF-κB and prevent its nuclear translocation. Stimulation of the cell by a variety of stimuli, including proinflammatory cytokines such as tumor necrosis factor (TNFα) and interleukin-1 (IL-1), bacterial lipopolysaccharide (LPS) or viral infection, leads to the phosphorylation of IκB proteins on two conserved serine residues (Ser-32 and Ser-36 on IκBα and Ser-19 and Ser-23 on IκBβ) (DiDonato et al. 1996). The phosphorylated IκB proteins become targets for ubiquitination and proteasome-mediated degradation, consequently releasing NF-κB and allowing its translocation into the nucleus (Verma et al. 1995).
Recently, an IκB kinase activity was identified as a large complex, which contains two catalytic subunits, IKKα and IKKβ (DiDonato et al. 1997; Mercurio et al. 1997; Regnier et al. 1997; Woronicz et al. 1997; Zandi et al. 1997). These kinases are 52% identical and share several structural domains, including an amino-terminal kinase domain, a helix–loop–helix (HLH) domain and a leucine zipper (LZ) domain (Mercurio et al. 1997; Zandi et al. 1997). IKKα and IKKα form homo- and heterodimers in vitro, although only heterodimers are found in vivo (Woronicz et al. 1997). In vitro phosphorylation assays have shown that both kinases can phosphorylate IκBα on Ser-32 and Ser-36, but IKKβ seems to be more active in this regard (Woronicz et al. 1997). In addition, whereas IKKβ phosphorylates both critical serine residues of IκBα and IκBβ with equal efficiency (Woronicz et al. 1997), IKKα preferentially phosphorylates IκBβ on Ser-23 (Regnier et al. 1997). Catalytically inactive forms of both kinases inhibited TNFα and IL-1-induced NF-κB activation (Mercurio et al. 1997; Regnier et al. 1997; Woronicz et al. 1997; Zandi et al. 1997), suggesting that the kinases would have similar physiological functions. Surprisingly, recent gene targeting experiments have shown that, even though IKKα and IKKβ are part of the same IKK complex, share sequence homology and seem to have similar activities in vitro, IKKα-deficient mice and IKKβ-deficient mice have completely different phenotypes and the kinases seem to respond to different biological inducers. Although IKKβ mediates the response to proinflammatory cytokines such as TNFα and IL-1 (Li et al. 1999b; Z. Li et al. 1999; Tanaka et al. 1999), IKKα seems to respond to an unknown morphogenetic signal (Hu et al. 1999; Li et al. 1999a; Takeda et al. 1999). IKKβ−/− mice die at E12.5 to E14.5 from liver degeneration due to apoptosis (Li et al. 1999b; Z. Li et al. 1999; Tanaka et al. 1999), a phenotype similar to, but more severe than, that of RelA−/− mice (Beg et al. 1995). In contrast, IKKα−/− mice survive to term but die shortly after birth (Hu et al. 1999; Li et al. 1999a; Takeda et al. 1999). IKKα−/− mice display rudimentary limbs and a tail as well as severe craniofacial deformities and skeletal abnormalities. Lack of IKKα also affects skin development, because the mutant epidermis shows increased thickness due to hyperproliferation of cells at the basal layer and a block in keratinocyte differentiation.
Two additional subunits of the IκB kinase complex have now been identified, IKAP (IKK-complex-associated protein) (Cohen et al. 1998), a potential scaffold protein, and NEMO (NF-κB essential modulator) (Yamaoka et al. 1998), also called IKKγ (Rothwarf et al. 1998) or IKKAP1 (IκB kinase-associated protein 1) (Mercurio et al. 1999). NEMO/IKKγ is a glutamine-rich protein of 48 kD, which lacks a catalytic domain but contains two coiled–coil motifs and a LZ. NEMO/IKKγ seems to be essential for NF-κB activation. Introduction of a cDNA encoding NEMO/IKKγ was able to restore NF-κB DNA-binding activity in two cell lines with defective NF-κB activation (Yamaoka et al. 1998). Considering that both catalytic subunits of the IKK complex, IKKα and IKKβ, show similar in vitro activities but turned out to have very different physiological functions in vivo, we have generated NEMO/IKKγ-deficient mice to analyze the physiological relevance of this noncatalytic subunit of the IKK complex. In particular, we were curious to find out whether the phenotype of NEMO/IKKγ-deficient mice would be similar to the phenotype observed in IKKα- or IKKβ-deficient mice.
NEMO/IKKγ-deficient mice die at E12.5–E13.0 from severe liver degeneration due to apoptosis. NEMO/IKKγ-deficient primary murine embryonic fibroblasts (MEFs) show no detectable NF-κB DNA-binding activity in response to stimulation with TNFα, IL-1, LPS, or Poly(IC). Stimulus-dependent phosphorylation and degradation of IκBα is absent in mutant MEFs and IκB kinase activity is not detectable. Mutant MEFs are highly sensitive to TNFα and display reduced viability in response to TNFα treatment even in the absence of cycloheximide.
Results
Generation of NEMO/IKKγ-deficient mice
A targeting vector was designed to replace exon 1, encoding the translation start site, and exon 2 with a PGK–neo cassette (Fig. 1A). Correctly targeted ES cell clones were identified by Southern blot analysis and three ES clones were injected into C57BL/6 blastocysts to generate chimeric mice. Chimeric mice from all three clones transmitted the targeted NEMO/IKKγ-allele to their progeny. Genotypes were confirmed by Southern blot (Fig. 1B) and PCR analysis (Fig. 1C). NEMO/IKKγ has been mapped to the X chromosome (Jin and Jeang 1999; data not shown). Therefore, we could only generate heterozygous females from intercrosses of chimeric males with C57BL/6 females, which were then mated with C57BL/6 males to generate homozygous mutant mice. Although NEMO/IKKγ+/− females appeared normal and were fertile, viable homozygous mutant mice could not be generated from matings with C57BL/6 males, suggesting that a lack of NEMO/IKKγ results in embryonic lethality.
Figure 1.
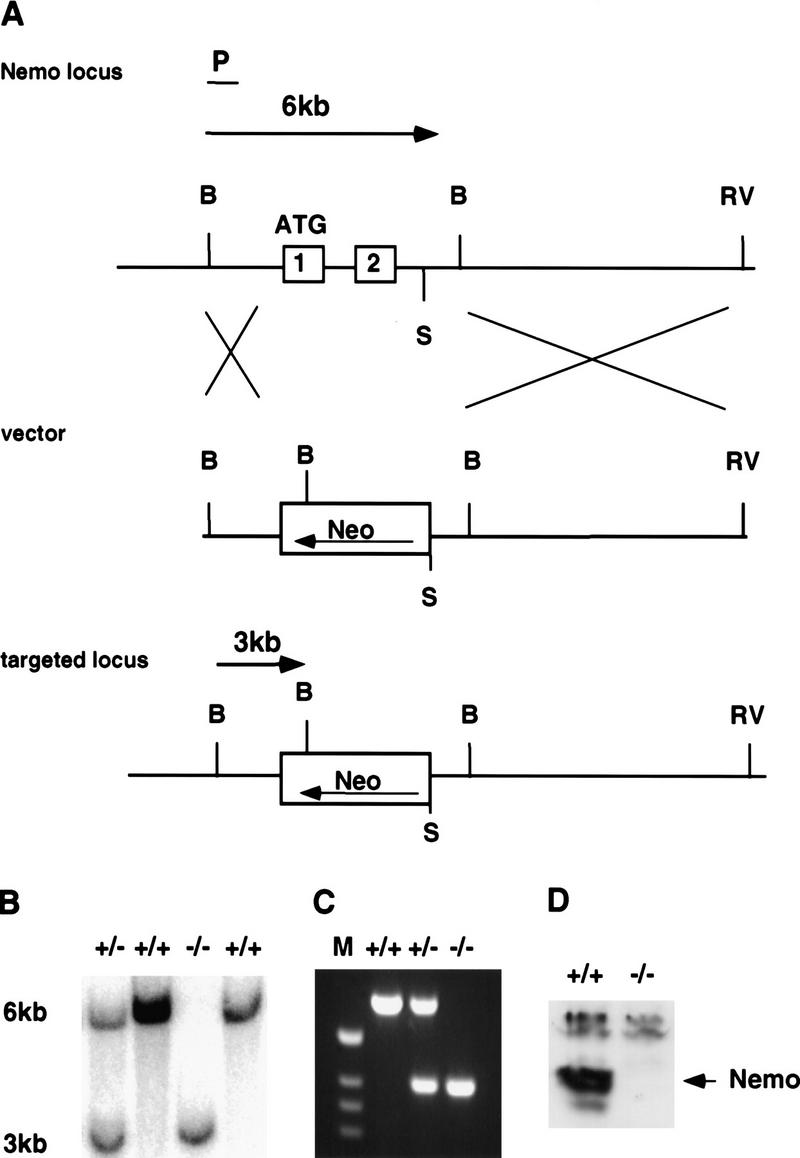
Generation of NEMO/IKKγ−/− mice. (A) Wild-type NEMO/IKKγ locus (top). Exon 1 and 2 are shown as open boxes and the ATG translation start site is indicated. (P) External flanking probe, (B) BamHI, (RV) EcoRV, (S) SpeI. The targeting vector (middle) was designed to replace exon 1 and exon 2 of the wild-type locus with a neomycin cassette in antisense orientation. Flanking probe P detects a 6-kb BamHI fragment corresponding to the wild-type allele and a 3-kb BamHI fragment for the targeted allele (bottom). (B) Genomic Southern blot of BamHI-digested DNA from E12.5 embryos of the indicated genotypes, showing the 6-kb wild-type band and the 3-kb mutant band detected by probe P. (C) Genotyping by PCR of DNA from E12.5 embryos using the primers described in Material and Methods. (D) Western blot analysis of NEMO/IKKγ protein expression in cytoplasmic extracts of MEFs of the indicated genotypes.
Western blot analysis of protein extracts from NEMO/IKKγ-deficient MEFs confirmed the absence of NEMO/IKKγ protein in mutant cells (Fig. 1D), indicating that we have generated a NEMO/IKKγ null mutation. We also did not detect changes in protein levels of IKKα, IKKβ, IκBα, or p65 (RelA) in mutant cells (data not shown).
Liver degeneration in NEMO/IKKγ-deficient embryos
Extensive timed matings were performed (Table 1). NEMO/IKKγ−/− embryos were normal at E11.5. On sections of E12.5−/− embryos, areas of hemorrhaging and tissue loss were apparent in parts of the liver (Fig. 2A,B; data not shown). At E13.0–E13.5, mutant embryos were dead and anemic in appearance. Mutant embryos at these stages showed massive liver hemorrhages, which are most likely due to the breakdown of the liver architecture at this developmental stage (Fig. 2D). To determine whether the observed cell death in the liver was due to apoptosis, TUNEL assays were performed on sections of E11.5 and E12.5 wild-type and mutant embryos. Only a few isolated TUNEL-positive cells could be detected on sections from wild-type embryos at both stages (Fig. 2G; data not shown). Mutant embryos at E11.5 showed only a slightly increased number of TUNEL-positive cells in small areas of the liver (data not shown). However, by E12.5, greatly increased numbers of TUNEL-positive cells in large areas throughout the liver were observed in mutant embryos (Fig. 2H), suggesting that apoptosis in the liver is a major cause of death of the mutant embryos. Enhanced apoptosis in the mutant embryos appeared to be restricted to the liver, as other parts of the NEMO/IKKγ−/− embryos did not show an increase in TUNEL-positive cells. Cell proliferation in E12.5 mutant embryos as detected by BrdU incorporation was generally comparable with wild-type embryos (data not shown) except in the liver (Fig. 2E,F). Of note, sections of dorsal skin at E12.5, which at this stage consists of a single layer of cells with underlying mesenchyme, showed similar proliferation in wild-type and mutant skin (data not shown).
Table 1.
Genotypes of embryos derived from matings of NEMO/IKKγ+/− females with C57BL/6 males
| Stage
|
+/+
|
+/−
|
−/−a
|
Total
|
|---|---|---|---|---|
| E11.5 | 12 | 8 | 2 | 22 |
| E12.5 | 54 | 29 | 20 | 103 |
| E13.0 | 3 | 1 | 3 | 7 |
| E13.5 | 13 | 5 | 2* | 20 |
| E14.5 | 6 | 2 | 1* | 9 |
(*) In resorption.
Figure 2.
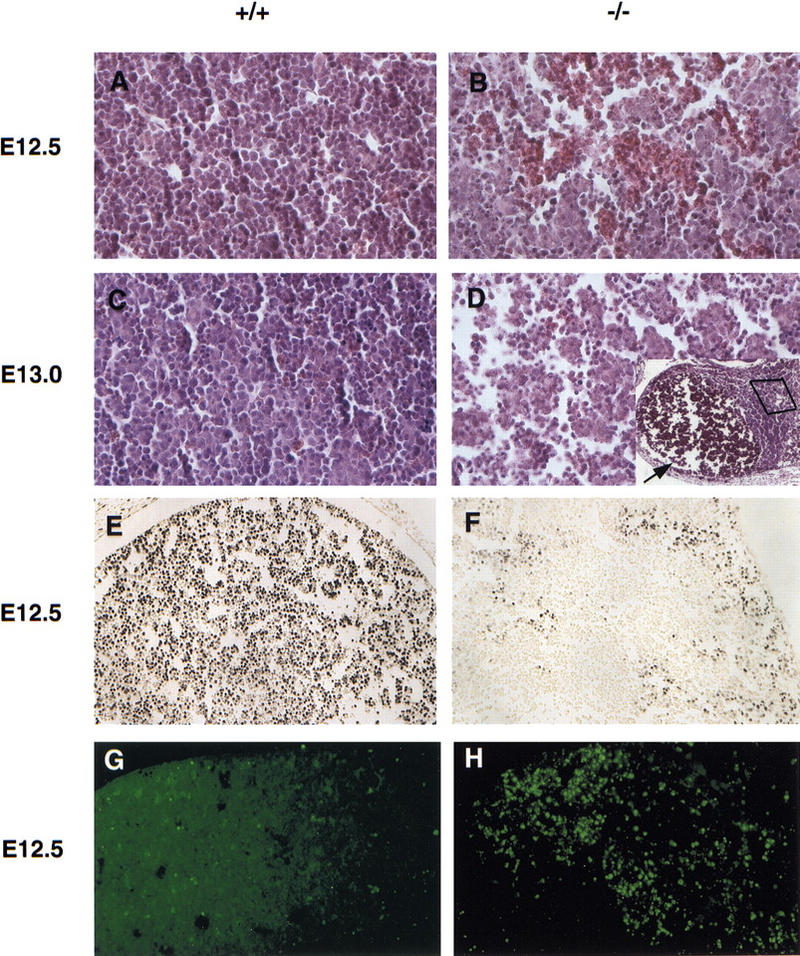
Massive liver apoptosis in the absence of NEMO/IKKγ. (A–D) Histological analysis of liver sections from E12.5 (A,B) and E13.0 (C,D) wild-type (A,C) and NEMO/IKKγ-deficient (B,D) embryos. Hematoxylin and eosin staining; high-power view. (D) The inset shows a low power view of the same region shown in D. The arrow points to a large area of tissue loss in which erythrocytes have accumulated. (E,F) Detection of cell proliferation as determined by BrdU incorporation. Pregnant females were injected IP with BrdU 60-min prior to sacrifice. Sections of E12.5 wild-type and mutant embryos were prepared and immunostained with anti-BrdU antibody. A high power view of the liver is shown. (G,H) Enhanced apoptosis in livers of E12.5 NEMO/IKKγ-deficient embryos. TUNEL assays were performed on sections of wild-type and mutant E12.5 embryos. A high power view of the liver is shown in both cases.
Lack of NF-κB activation in NEMO/IKKγ-deficient MEFs
The above-mentioned phenotype of NEMO/IKKγ−/− mice is similar to, albeit more severe than, that of RelA-deficient mice (Beg et al. 1995). RelA is a subunit of the most common form of NF-κB, the p50–RelA heterodimer. We therefore assessed whether the activation of NF-κB DNA-binding activity was impaired in NEMO/IKKγ-deficient MEFs. Wild-type and NEMO/IKKγ-deficient MEFs were treated with TNFα, IL-1, LPS, or poly(IC) to induce NF-κB DNA-binding activity and nuclear extracts were examined by gel shift assay. In contrast to wild-type MEFs, mutant MEFs showed no detectable NF-κB DNA-binding activity in response to any of these stimuli (Fig. 3; data not shown). Interestingly, IKKα−/− MEFs show normal NF-κB DNA-binding activity in response to these stimuli (Hu et al. 1999; Li et al. 1999a; Takeda et al. 1999), whereas NF-κB activation in IKKβ−/− MEFs is impaired, although it still occurred with delayed kinetics and reduced magnitude (Li et al. 1999b; Tanaka et al. 1999).
Figure 3.
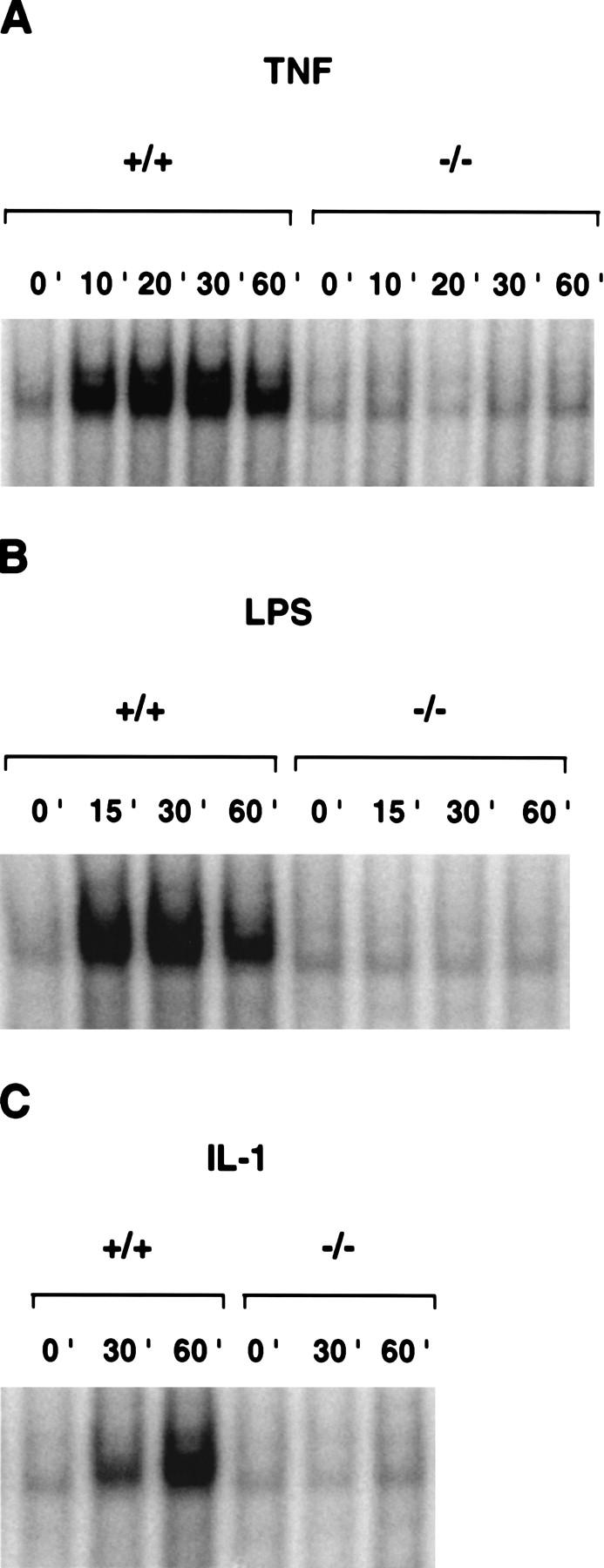
Impaired NF-κB activation in NEMO/IKKγ-deficient cells. Wild-type or NEMO/IKKγ−/− MEFs were incubated with mouse recombinant TNFα (10 ng/ml) (A), LPS (15 μg/ml) (B), or IL-1 (10 ng/ml) (C) for the indicated times. NF-κB activation in 10 μg of nuclear extract was determined by gel shift assay as described in Materials and Methods.
Lack of IκBα phosphorylation/degradation in NEMO/IKKγ-deficient MEFs
Next, we analyzed whether stimulus-dependent phosphorylation and degradation of IκBα was affected by the absence of NEMO/IKKγ. Wild-type and NEMO/IKKγ-deficient MEFs were stimulated with TNFα for 0, 6, and 10 min. Cytoplasmic extracts were prepared and used for Western blot analysis with an anti-phospho IκBα-specific antibody. Phosphorylation of IκBα could be detected in wild-type cells as soon as 6 min after TNFα stimulation (Fig. 4A, top left). However, no phosphorylation of IκBα could be detected in extracts of NEMO/IKKγ-deficient MEFs even after 10 min stimulation with TNFα (Fig. 4A, top right). An anti-IκBα specific antibody was used as a control to detect IκBα protein expression in −/− extracts. (Fig. 4A, bottom). As shown in Figure 4B, IκBα is degraded in wild-type cells and reappears 30–60 min after stimulation (Fig. 4B, top), whereas degradation of IκBα is completely absent in mutant cells (Fig. 4B, bottom).
Figure 4.
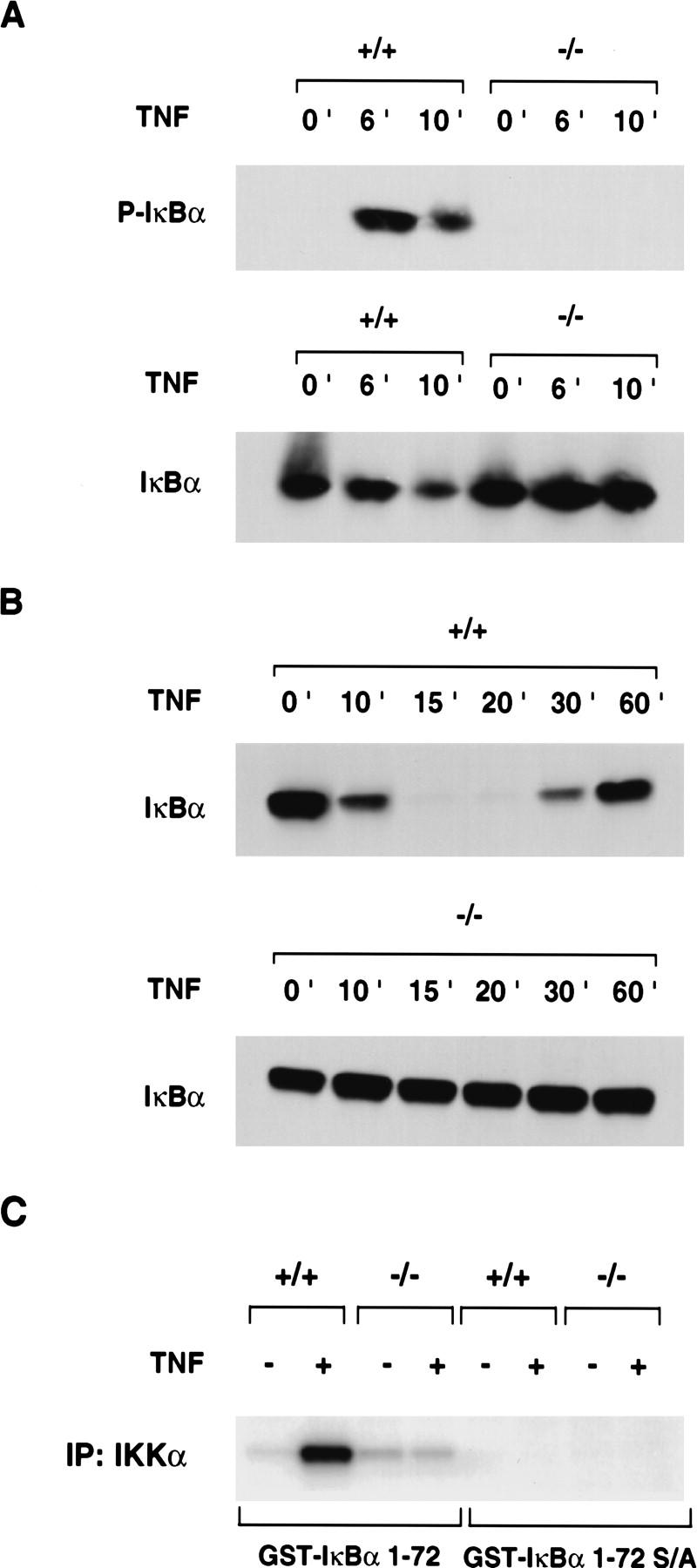
Defective TNFα-induced IκBα phosphorylation and degradation and IκB kinase activity in the absence of NEMO/IKKγ. (A) IκBα phosphorylation. (Top) Wild-type and NEMO/IKKγ-deficient MEFs were treated with TNFα (10 ng/ml) for the indicated times followed by Western blot analysis of cytoplasmic extracts with anti-phospho-IκBα-specific antibody. (Bottom) Western blot of the same extracts with anti-IκBα antibody as a control to show IκBα protein expression in mutant cells. (B) IκBα degradation. Wild-type (top) and NEMO/IKKγ-deficient (bottom) MEFs were treated with TNFα (10 ng/ml) for the indicated times followed by Western blot analysis with anti-IκBα specific antibody. (C) IκB kinase activity. Wild-type and NEMO/IKKγ-deficient MEFs were either left in medium alone or treated with TNFα (10 ng/ml) for 7 min. Whole cell extracts were immunoprecipitated with anti-IKKα antibody and in vitro kinase activity was determined as described in Materials and Methods using GST–IκBα (1–72) (left) or GST–IκBα (1–72) S/A (right) as a substrate. One representative experiment of four is shown.
Impaired IκB kinase activity in NEMO/IKKγ-deficient MEFs
The failure to phosphorylate IκBα in the absence of NEMO/IKKγ prompted us to examine the in vitro kinase activity of the IKK complex. Wild-type and NEMO/IKKγ-deficient MEFs were treated for 7 min with TNFα to induce kinase activity. Whole cell extracts were immunoprecipitated with anti-IKKα antibody and kinase activity was assessed in vitro using GST–IκBα (1–72) or GST–IκBα (1–72) S/A, containing a Ser-32/Ser-36 to Ala-32/Ala-36 mutation as a substrate. Extracts of wild-type cells were able to phosphorylate GST–IκBα (1–72) in vitro, whereas stimulus-dependent kinase activity in extracts of NEMO/IKKγ-deficient MEFs was severely impaired (Fig. 4C), indicating that NEMO/IKKγ is essential for IκB kinase activity. Similar results were obtained with a longer substrate [GST–IκBα (1–317)] (data not shown).
Expression of NF-κB-dependent target genes is impaired in NEMO/IKKγ-deficient MEFs
The above data showed that NF-κB activation is defective in the absence of NEMO/IKKγ. We therefore examined the expression of known NF-κB-dependent target genes. IκBα is one of the first genes to be up-regulated following NF-κB activation, establishing a feedback loop, which will eventually shut down the NF-κB response. This induction, but not basal IκBα mRNA expression, is regulated by RelA (Scott et al. 1993). To analyze IκBα mRNA expression in the absence of NEMO/IKKγ, RNA from wild-type and NEMO/IKKγ-deficient MEFs treated with TNFα for 0, 30, and 60 min was analyzed by Northern blot. As shown in Figure 5A, an IκBα transcript induced within 60 min of TNFα stimulation in wild-type cells is absent in NEMO/IKKγ-deficient MEFs. Interestingly, IKKβ−/− MEFs are still able to express the TNFα-induced IκBα transcript, although the level of transcription is reduced compared with wild-type cells (Li et al. 1999b).
Figure 5.
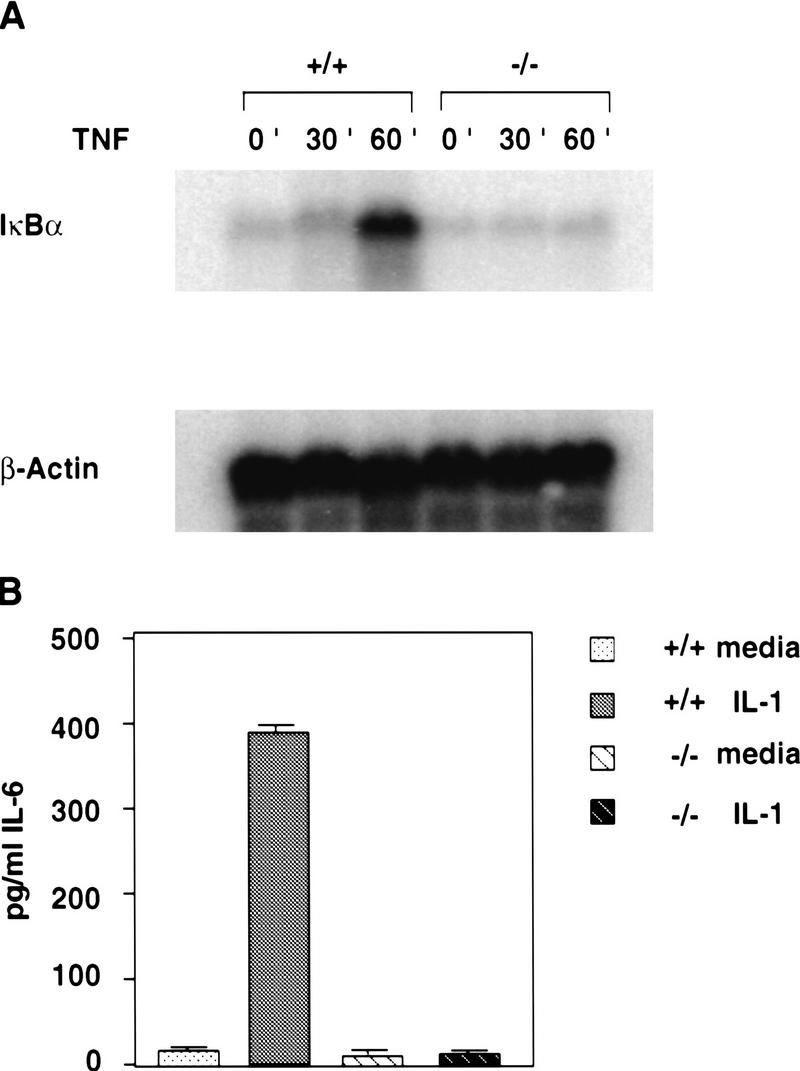
Impaired expression of NF-κB target genes in NEMO/IKKγ-deficient MEFs. (A) TNFα-induced IκBα mRNA synthesis. Wild-type and NEMO/IKKγ-deficient MEFs were stimulated with TNFα (10 ng/ml) for the indicated times and 20 μg of RNA was analyzed by Northern blot analysis using a 32P-labeled random priming probe corresponding to the full-length IκBα cDNA. β-actin, loading control. (B) IL-1 induced IL-6 production. Wild-type and NEMO/IKKγ-deficient MEFs (3 × 104) were left untreated or stimulated with IL-1 (10 ng/ml) for 16 hr. Levels of IL-6 (pg/ml) in culture supernatants were determined by ELISA. Values shown are the mean and standard deviation of triplicate samples.
Stimulation of cells with IL-1 leads to expression of various genes, including interleukin-6 (IL-6), an important mediator of acute phase reactions. IL-1 induces IL-6 expression through the coordinated action of NF-κB and the transcription factor NF-IL6 (Akira and Kishimoto 1992). We therefore treated wild-type and NEMO/IKKγ-deficient MEFs with IL-1 for 16 hr and measured the levels of IL-6 in the culture supernatant using an ELISA. Whereas wild-type MEFs showed stimulus-dependent production of IL-6, IL-1 treatment of mutant MEFs failed to up-regulate IL-6 expression (Fig. 5B). Levels of IL-6 in the stimulated NEMO/IKKγ-deficient cultures were equivalent to those in wild-type cultures treated with medium alone. These data demonstrate impaired induction of acute phase response proteins in the absence of NEMO/IKKγ.
Cells lacking NF-κB activity have been shown to undergo apoptosis in response to TNFα stimulation (Beg and Baltimore 1996; Van Antwerp et al. 1996; Wang et al. 1996), suggesting that NF-κB activation provides protection against TNFα-induced apoptosis. To determine whether sensitivity to TNFα-induced apoptosis was increased in the absence of NEMO/IKKγ, the viability of wild-type and NEMO/IKKγ-deficient MEFs after treatment with various concentrations of TNFα for 18 hr was assessed. Wild-type MEFs were resistant to TNFα-mediated cytotoxicity, but NEMO/IKKγ deficient MEFs showed a dramatic reduction in viability even in the absence of cycloheximide (Fig. 6A). TNFα-sensitivity of NEMO/IKKγ-deficient MEFs could be further increased by addition of very low concentrations of cycloheximide (30–100 ng/ml). Significantly higher concentrations of cycloheximide in combination with TNFα were required to affect viability of wild-type MEFs (Fig. 6B).
Figure 6.
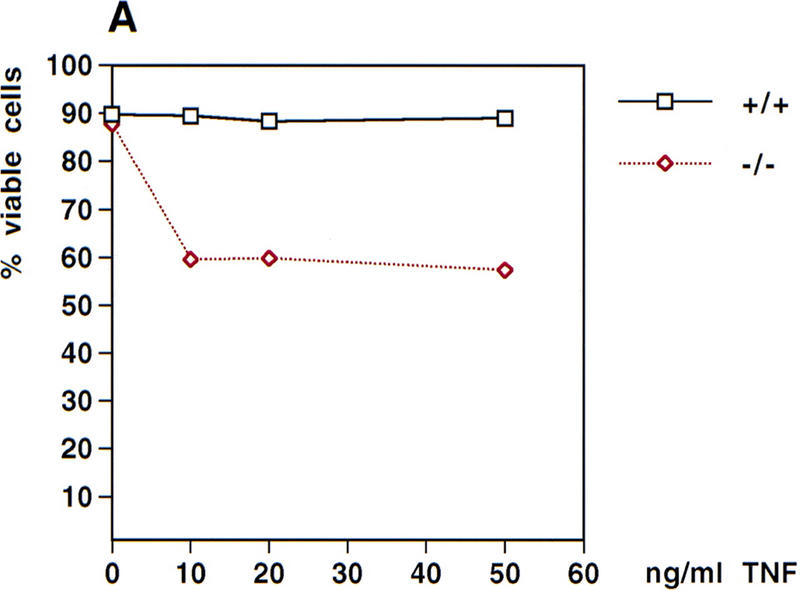
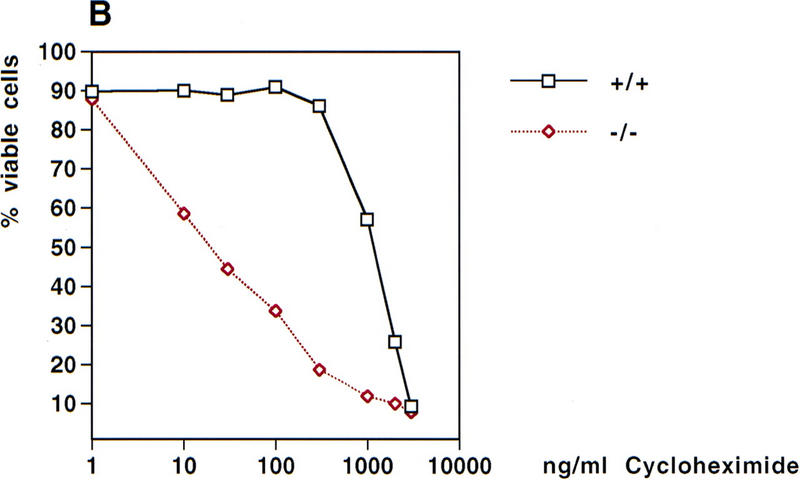
TNFα-induced cytotoxicity. (A) TNFα-induced cytotoxicity. Wild-type and NEMO/IKKγ-deficient (2 × 105) MEFs were left untreated or stimulated with the indicated concentrations of TNFα for 18 hr. Cells were then harvested and stained with PI/AnnexinV-FITC. Values shown are the percentage of viable cells after treatment relative to untreated controls. (B) Cytotoxicity induced by TNFα plus cycloheximide. Wild-type and NEMO/IKKγ-deficient MEFs (2 × 105) were left untreated or stimulated with TNFα (10 ng/ml) in combination with the indicated concentrations of cycloheximide for 18 hr. Cells were harvested and stained as described for A, and values shown were determined as for A.
Discussion
In this study, we have shown that NEMO/IKKγ, a component of the IKK complex, is required for the phosphorylation and degradation of IκBα. Lack of NEMO/IKKγ affects IκB kinase activity, in particular also IKKα kinase activity (Fig. 4C). We have demonstrated that NEMO/IKKγ is crucial for normal stimulus-dependent NF-κB activation and target gene expression. NEMO/IKKγ-deficient MEFs show increased sensitivity to TNFα treatment, implying that the liver apoptosis observed in NEMO/IKKγ-deficient mice could be due to a lack of NF-κB-mediated protection from TNFα cytotoxicity. RelA−/− and IKKβ−/− fibroblasts are also highly sensitized to TNFα in vitro (Beg et al. 1995; Li et al. 1999b; Tanaka et al. 1999). Interestingly, RelA/TNFα double-deficient mice (Doi et al. 1999) as well as IKKβ/TNFR1 double-deficient mice (Li et al. 1999b) are born alive, consistent with the hypothesis that enhanced sensitivity to endogenous TNFα is responsible for the liver apoptosis observed in these mice.
NEMO/IKKγ is a noncatalytic component of the IκB kinase complex. Precisely how it functions within the complex remains to be clarified. Analysis of a NEMO/IKKγ-deletion mutant series showed that the carboxyl terminus of the protein (amino acids 300–419) is required for TNFα-stimulated IκB kinase activity but not basal kinase activity (Rothwarf et al. 1998). Furthermore, this mutation did not interfere with the binding of NEMO/IKKγ to IKKα/β in cells, suggesting that the carboxyl terminus might be required to connect the IKK complex to upstream activators. Gene-targeting experiments have shown that IKKβ rather than IKKα mediates the response to proinflammatory cytokines and that IKKα seems to respond to an unknown morphogenetic signal (Hu et al. 1999; Li et al. 1999a,b; Z. Li et al. 1999; Takeda et al. 1999; Tanaka et al. 1999). At E18.5, IKKα−/− mice display rudimentary limbs and a tail as well as severe craniofacial deformities and skeletal abnormalities. Takeda et al. (1999) and Li et al. (1999a) show that IKKα−/− embryos at E12.5 already have mild alterations in the morphology of the limb buds (Li et al. 1999a; Takeda et al. 1999), whereas Hu et al. (1999) state that at E14.5, the forelimbs and hindlimbs of IKKα−/− embryos are not significantly shorter than those of wild-type littermates. At E12.5, limbbuds of NEMO/IKKγ-deficient mice are indistinguishable from those of wild-type littermates (data not shown), even though kinase assays (Fig. 4C) showed that IKKα activity is severely impaired in extracts of NEMO/IKKγ-deficient MEFs. However, this does not exclude the possibility that NEMO/IKKγ-deficient mice would show a phenotype similar to that observed in IKKα-deficient mice at E18.5, if they survived until this developmental stage. Lack of IKKα also affects skin development because the mutant epidermis at E18.5 shows increased thickness due to hyperproliferation of cells at the basal layer and a block in keratinocyte differentiation (Hu et al. 1999; Li et al. 1999a; Takeda et al. 1999). However, at E12.5, proliferation of the skin as detected by BrdU incorporation is comparable in wild-type and NEMO/IKKγ-deficient embryos (data not shown). At E12.5, the skin consists of a single layer of cells with underlying mesenchyme. Epidermal differentiation does not occur until later in development. Therefore, we cannot exclude the possibility that NEMO/IKKγ-deficient mice would show defects in epidermal differentiation similar to those observed in IKKα-deficient mice at E18.5, if they survived until this developmental stage, especially as IKKα kinase activity is severely impaired in NEMO/IKKγ-deficient MEFs (Fig. 4C).
Overall, the phenotype observed in NEMO/IKKγ-deficient mice is more severe than that of IKKβ−/− mice. In particular, the onset of the liver phenotype occurs at least 12 hr earlier in NEMO/IKKγ-deficient embryos than in IKKβ−/− embryos. Even though IKKβ−/− embryos show hemorrhages in the liver on E13.0, the overall liver architecture is still intact (Li et al. 1999b). In contrast, NEMO/IKKγ-deficient embryos show large areas of tissue loss at this stage and a massive accumulation of erythrocytes. Moreover, IKKβ−/− MEFs still show residual NF-κB DNA-binding activity in response to stimulation with TNFα or IL-1 (Z. Li et al. 1999; Tanaka et al. 1999), whereas NF-κB DNA-binding activity in response to these stimuli is completely abolished in NEMO/IKKγ-deficient MEFs. This suggests that the residual NF-κB DNA-binding activity observed in IKKβ−/− mice might provide limited protection to liver cells against apoptosis induced by endogenous TNFα. The earlier onset of the phenotype in NEMO/IKKγ-deficient mice might then be explained by the complete absence of NF-κB DNA-binding activity. As shown by Li et al. (1999b) the residual NF-κB DNA-binding activity observed in IKKβ−/− MEFs is still sufficient to induce a low level of expression of NF-κB-dependent target genes such as IκBα (Li et al. 1999b). Sixty minutes after TNFα stimulation of IKKβ−/− MEFs, about half the amount of IκBα mRNA observed in stimulated wild-type cells is detected in IKKβ mutant cells. However, IκBα mRNA is not induced at all in NEMO/IKKγ-deficient MEFs, which is consistent with the lack of detectable NF-κB DNA-binding activity in these cells.
In summary, the effects of the targeted disruption of the NEMO/IKKγ gene on NF-κB activation by a variety of stimuli are more dramatic than the deletion of either catalytic component of the IKK complex. We therefore speculate that the phenotype of IKKα/IKKβ double-deficient mice will be similar to that of NEMO/IKKγ-deficient mice. In any case, our data provide strong in vivo evidence that NEMO/IKKγ is essential for activation of NF-κB by various stimuli. Our study combined with previous reports clearly demonstrates that NEMO/IKKγ is the first essential, non-kinase component of the IKK complex.
Material and methods
Generation of NEMO/IKKγ-deficient mice
Phage clones containing the murine NEMO/IKKγ gene were isolated by screening a 129/J genomic library with a probe corresponding to the 5′ end of the NEMO cDNA (Yamaoka et al. 1998). A targeting vector was designed to replace exon 1 and exon 2 with a PGK–neo cassette in antisense orientation, leading to the replacement of the first 131 amino acids of the NEMO/IKKγ protein and a downstream frameshift. A total of 30 μg of the linearized targeting vector were used to electroporate 5 × 106 E14K ES cells (Bio-Rad Gene Pulser, 0.34 kV, 0.25 mF), which were subsequently cultured for 10 days in DMEM supplemented with 15% FBS, glutamine, pyruvate, β-mercaptoethanol, and leukemia inhibitory factor, containing 300 μg/ml G418 (Sigma). Recombinants were identified by PCR and confirmed by Southern blot analysis using BamHI-digested genomic DNA hybridized to an external flanking probe, which detects a 6-kb band for the wild-type locus and a 3-kb band for the mutant allele. Single integration was confirmed using a probe corresponding to the neo gene. Three correctly targeted ES clones were injected into C57BL/6 blastocysts and all three successfully generated germ-line chimeras. Genotyping of mice was performed by PCR using tail genomic DNA with the following primers: 5′-ACTTAAAGGTGTGTGGAGGAGGTAGG-3′, 5′-TGCATGTGACTCTTCACAGAGAACCC-3′, 5′-GGGTGGGATTAGATAAATGCCTGCTC-3′. These primers give rise to a 621-bp PCR product for the wild-type locus and a 354-bp PCR product for the mutant allele. The following PCR conditions were used: 1 min at 95°C, 30 sec at 64°C, 1.5 min at 72°C, for 35 cycles.
Animal husbandry
Mice were maintained in the animal facility of the Ontario Cancer Institute in accordance with its ethical guidelines.
Histology
Embryos were fixed with 4% buffered formalin at 4°C for 12 hr and embedded in paraffin. Sections were stained with hematoxylin and eosin according to standard protocols. For the detection of BrdU incorporation, immunostaining with anti-BrdU antibody was performed on paraffin sections according to standard procedures. Pregnant females were injected with 300 μl of BrdU (10 mg/ml) 60 min prior to sacrifice. For determination of apoptosis, TUNEL assays were performed using an in situ cell death detection kit (Boehringer Mannheim) according to the manufacturer's instructions.
Generation of primary MEFs
Primary MEFs were derived from E12.5 embryos. The head and liver were removed and the remainder of the embryo was cut into small pieces and trypsinized. Cells were washed in medium and plated onto 6-well plates. Embryonic fibroblasts were expanded in DMEM supplemented with 10% FBS and β-mercaptoethanol and frozen.
Western blot analysis
Cytoplasmic extracts were prepared from primary MEFs as described previously (Yamaoka et al. 1998). Briefly, cells were stimulated, washed with cold PBS, resuspended at 106/20 μl in hypotonic solution [10 mm HEPES at pH 7.8, 10 mm KCl, 2 mm MgCl2, 1 mm DTT, and 0.1 mm EDTA supplemented with a protease inhibitor cocktail (Boehringer Mannheim)] and kept on ice for 10 min. NP-40 was added to a final concentration of 1% and the cells were centrifuged at 14,000 rpm for 1 min. The supernatant, containing the cytoplasmic fraction, was recovered and the protein concentration was determined using a Bio-Rad protein assay. Protein samples were fractionated on a 10% SDS–polyacrylamide gel and blotted onto a PVDF membrane. Western blots were developed using an enhanced chemiluminescence system according to the manufacturer's instructions (Amersham). Anti-phospho-IκBα and anti-IκBα were from New England Biolabs. Anti-NEMO antiserum was kindly provided by Alain Israel (Institute Pasteur, Paris, France).
Gel mobility shift assay
MEFs (2 × 106) were lysed in 400 μl of hypotonic solution as described above and nuclear extracts were prepared for gel mobility shift assays as described previously (Yeh et al. 1997). A total of 10 μg of nuclear extract was incubated with an end-labeled, double-stranded oligo containing two NF-κB binding sites (5′-ATCAGGGACTTTCCGCTGGGGACTTTCCG-3′ and 5′-CGGAAAGTCCCCAGCGGAAAGTCCTGAT-3′). The reaction was performed in 20 μl of binding buffer (5 mm HEPES at pH 7.8, 50 mm KCl, 0.5 mm DTT, 1 μg of [poly d(I-C)], 10% glycerol] for 20 min at room temperature. Samples were fractionated on a 5% polyacrylamide gel and visualized by autoradiography.
Kinase assay
MEFs (5 × 106) were lysed as described previously (Tanaka et al. 1999). Briefly, cells were lysed with lysis buffer containing 50 mm HEPES (pH 7.6), 150 mm NaCl, 1.5 mm MgCl2, 1 mm EGTA, 10% glycerol, 1% Triton X-100, protease inhibitor cocktail (Boehringer Mannheim), 20 mm β-glycerophosphate, 1 mm sodium orthovanadate, and 10 mm NaF. After incubation on ice for 30 min, lysates were centrifuged for 10 min at 14,000 rpm and the protein concentration of the supernatant was determined using the Bio-Rad protein assay. A total of 500 μg of protein extract was precleared using protein G–Sepharose (Pharmacia) and immunoprecipitated with anti-IKKα (Santa Cruz). The immunoprecipitates were washed twice with lysis buffer and twice with kinase buffer [20 mm HEPES at pH 7.9, 10 mm MgCl2, 100 mm NaCl, protease inhibitor cocktail (Boehringer Mannheim), 20 mm β-glycerophospate, 1 mm sodium orthovanadate, 2 mm DTT] and assayed for kinase activity as described previously (Tanaka et al. 1999) using [γ32P]ATP and 3 μg of GST–IκBα (1–72) or GST–IκBα (1–72) S/A as a substrate. Reaction products were fractionated by 10% SDS-PAGE and phosphorylated IκBα was visualized by autoradiograhy.
Measurement of IL-6 production
MEFs (3 × 104) were plated onto 24-well plates in DMEM supplemented with 10% FBS. After 24 hr, cells were treated with IL-1 (10 ng/ml) (R&D Systems) for 16 hr. The concentration of IL-6 in the culture supernatants was determined by ELISA (R&D Systems).
Northern blot analysis
Total RNA was prepared from MEFs using Trizol (GIBCO BRL). RNA (20 μg) was fractionated on a 1% formaldehyde gel and transferred onto a Hybond N membrane (Amersham). Filters were hybridized at 65°C in 50% formamide, 5× SSC, 25 mm Na2HPO4 (pH 6.5), 8× Denhardt's solution, 0.5 mg/ml salmon sperm DNA, and 1% SDS with a random priming probe corresponding to the full-length murine IκBα cDNA or a β-actin cDNA as a control.
MEF cell death assay (PI/AnnexinV-FITC staining)
MEFs (2 × 105) were plated onto six-well plates 24 hr prior to induction with various concentrations of TNFα (R&D Systems) alone or TNFα (10 ng/ml) in combination with various concentrations of cycloheximide (Sigma). Cells were harvested and viability was determined by FACS analysis of PI/AnnexinV-FITC staining using an apoptosis detection kit (R&D Systems) according to the manufacturer's instructions.
Acknowledgments
We thank Alain Israel for providing the NEMO antiserum as well as GST–IκBα (1–72) and GST–IκBα (1–72) S/A expression plasmids, Mary Saunders for scientific editing, Malte Peters and Scott Pownall for critically reading the manuscript, Denis Bouchard for technical expertise, and Irene Ng for expert administrative assistance. D.R. was supported by a fellowship of the Deutsche Forschungsgemeinschaft.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL t.mak@oci.utoronto.ca; FAX (416) 204-2278.
References
- Akira S, Kishimoto T. IL-6 and NF-IL6 in acute-phase response and viral infection. Immunol Rev. 1992;127:25–50. doi: 10.1111/j.1600-065x.1992.tb01407.x. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- Cohen L, Henzel WJ, Baeuerle PA. IKAP is a scaffold protein of the IκB kinase complex. Nature. 1998;395:292–296. doi: 10.1038/26254. [DOI] [PubMed] [Google Scholar]
- DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M. Mapping of the inducible IκB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol. 1996;16:1295–1304. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- Doi TS, Marino MW, Takahashi T, Yoshida T, Sakakura T, Old LJ, Obata Y. Absence of tumor necrosis factor rescues RelA-deficient mice from embryonic lethality. Proc Natl Acad Sci. 1999;96:2994–2999. doi: 10.1073/pnas.96.6.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKα subunit of IκB kinase. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- Jin DY, Jeang KT. Isolation of full-length cDNA and chromosomal localization of human NF-κB modulator NEMO to Xq28. J Biomed Sci. 1999;6:115–120. doi: 10.1007/BF02256442. [DOI] [PubMed] [Google Scholar]
- Li Q, Lu Q, Hwang JY, Buscher D, Lee KF, Izpisua-Belmonte JC, Verma IM. IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes & Dev. 1999a;13:1322–1328. doi: 10.1101/gad.13.10.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Severe liver degeneration in mice lacking the IκB kinase 2 gene. Science. 1999b;284:321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. The IKKβ subunit of IκB kinase (IKK) is essential for NF-κB activation and prevention of apoptosis. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, et al. IKK-1 and IKK-2: Cytokine-activated IκB kinases essential for NF-κB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- Mercurio F, Murray BW, Shevchenko A, Bennett BL, Young DB, Li JW, Pascual G, Motiwala A, Zhu H, Mann M, et al. IκB kinase (IKK)-associated protein 1, a common component of the heterogeneous IKK complex. Mol Cell Biol. 1999;19:1526–1538. doi: 10.1128/mcb.19.2.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IκB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- Rothwarf DM, Zandi E, Natoli G, Karin M. IKKγ is an essential regulatory subunit of the IkappaB kinase complex. Nature. 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- Scott ML, Fujita T, Liou HC, Nolan GP, Baltimore D. The p65 subunit of NF-κB regulates IκB by two distinct mechanisms. Genes & Dev. 1993;7:1266–1276. doi: 10.1101/gad.7.7a.1266. [DOI] [PubMed] [Google Scholar]
- Takeda K, Takeuchi O, Tsujimura T, Itami S, Adachi O, Kawai T, Sanjo H, Yoshikawa K, Terada N, Akira S. Limb and skin abnormalities in mice lacking IKKα. Science. 1999;284:313–316. doi: 10.1126/science.284.5412.313. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL, Goeddel DV. Embryonic lethality, liver degeneration, and impaired NF-κB activation in IKKβ-deficient mice. Immunity. 1999;10:421–429. doi: 10.1016/s1074-7613(00)80042-4. [DOI] [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNFα-induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NFκB/IκB family: Intimate tales of association and dissociation. Genes & Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Baldwin AS., Jr TNF- and cancer therapy-induced apoptosis: Potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- Woronicz JD, Gao X, Cao Z, Rothe M, Goeddel DV. IκB kinase-β: NF-κB activation and complex formation with IκB kinase-α and NIK. Science. 1997;278:866–869. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, Kirk HE, Kay RJ, Israel A. Complementation cloning of NEMO, a component of the IκB kinase complex essential for NF-κB activation. Cell. 1998;93:1231–1240. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]
- Yeh WC, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A, de la Pompa JL, Ferrick D, Hum B, Iscove N, et al. Early lethality, functional NF-κB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997;7:715–725. doi: 10.1016/s1074-7613(00)80391-x. [DOI] [PubMed] [Google Scholar]
- Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]