

Abstract
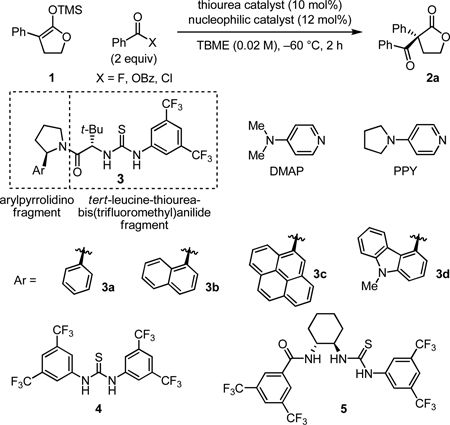
A highly-enantioselective acylation of silyl ketene acetals with acyl fluorides has been developed to generate useful α, α-disubstituted butyrolactone products. This reaction is promoted by a new thiourea catalyst and 4-pyrrolidinopyridine and represents the first example of enanti-oselective thiourea anion-binding catalysis with fluoride.
Pyridine derivatives induce significant rate accelerations in acyl transfer reactions through the generation of electrophilic N-acylpyridinium ion intermediates.1 While pyridine itself is a competent nucleophilic catalyst, analogs bearing strongly electron-donating substituents such as 4-(dimethylamino)pyridine (DMAP)2 and 4-pyrrolidinopyridine (PPY)3 are up to four orders of magnitude more reactive in representative acyl transfer reactions.4 The amplified reactivity of aminopyridine derivatives is ascribable both to a greater equilibrium concentration of the acylpyridinium ion intermediate and to increased electrophilicity of that intermediate as a result of looser ion pairing.5 Both effects can be enhanced, in principle, through stabilization of the counteranion by a specific hydrogen-bond donor (Scheme 1).6 We have shown that chiral urea and thiourea derivatives can catalyze enantioselective reactions via mechanisms involving anion binding, 7 and Seidel has demonstrated recently the successful application of this concept in the context of chiral thiourea-DMAP co-catalyzed acylative kinetic resolutions of primary amines.8 This strategy is fundamentally different from classical approaches to catalytic asymmetric acyl transfer reactions that rely on construction of chiral nucleophilic catalysts.9
Scheme 1.

Anion-Binding/Nucleophilic Co-Catalysis of Acyl Transfer
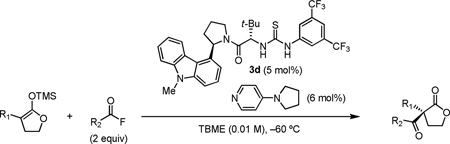
We sought to apply this reactivity principle to the C-acylation of enolate equivalents, a transformation that affords versatile β-dicarbonyl derivatives with generation of a quaternary stereocenter. We report here the highly enantioselective acylation of silyl ketene acetals with acyl fluorides promoted by the new thiourea catalyst 3d and PPY.10 This method provides a preparatively useful route to α,α-disubstituted butyrolactones11 and represents the first example of enantioselective thiourea anion-binding catalysis with fluoride.12
Benzoylation of silyl ketene acetal 1 was selected as a model reaction. A lead result afforded lactone 2a in 19% yield and 41% ee by acylation of 1 with benzoic anhydride in the presence of phenylpyrrolidine-derived thiourea 3a and DMAP (Table 1, entry 1). Slightly improved enantioselectivity and yield were obtained by replacing DMAP with PPY (entry 2). However, a more striking dependence on the identity of the acylating agent was observed, with benzoyl chloride being completely unreactive (entry 3) and benzoyl fluoride affording 2a in 84% yield and 81% ee (entry 4). Accordingly, chiral catalyst optimization studies were carried out on the PPY-catalyzed acylation of 1 with benzoyl fluoride.
Table 1.
Catalyst and Reaction Optimization
 | |||||
|---|---|---|---|---|---|
| entrya | thiourea catalyst |
nucleophilic catalyst |
acylating agent (X =) |
yieldb (%) |
eec (%) |
| 1 | 3a | DMAP | OBz | 19 | 41 |
| 2 | 3a | PPY | OBz | 21 | 52 |
| 3 | 3a | PPY | Cl | 0 | - |
| 4 | 3a | PPY | F | 84 | 81 |
| 5 | 3b | PPY | F | 88 | 87 |
| 6 | 3c | PPY | F | 29 | 75 |
| 7 | 3d | PPY | F | 80 | 92 |
| 8 | 4 | PPY | F | 9 | - |
| 9 | 5 | PPY | F | 20 | <5 |
| 10d | 3d | PPY | F | 86 | 93 |
Reactions run on a 0.08 mmol scale.
Yields determined by 1H NMR analysis relative to p-xylene as an internal standard.
Enantiomeric excess determined by HPLC analysis on commercial chiral columns.
Reaction run using 5 mol% thiourea catalyst and 6 mol% PPY at −60 °C in TBME (0.01 M) for 8 h.
Catalysts with the general structure of 3 have been demonstrated recently to be broadly effective in a wide range of enantioselective transformations7 and also proved optimal in the present study. Notably, Schreiner’s thiourea13 (4) and Seidel’s optimal catalyst for the acylative kinetic resolution of primary amines8b,c (5) were considerably less reactive than 3. The right-hand tert-leucine-thiourea-3,5-bis(trifluoromethyl)anilide fragment of 3 is a common feature in most of these systems, and it was found to be strictly superior to other amino acid-H-bond donor-anilide combinations in the model acylation reaction. Enantioselectivity also proved strongly responsive to the structure of the left-hand 2-arylpyrrolidino fragment. Systematic variation of the aryl group of the pyrrolidine led to the identification of 3d as the most enantioselective catalyst. Although elucidation of the mechanism of stereoinduction must await detailed mechanistic investigation, the important influence of the arylpyrrolidino component of the thiourea catalyst on both reaction rate and enantioselectivity14 is suggestive of differential stabilizing non-covalent interactions with the cationic electrophile in the transition states leading to the major and minor enantiomers of product.15
Under the optimized reaction conditions, silyl ketene acetal 1 underwent acylation with a variety of substituted benzoyl fluorides (Table 2).16 Substrates containing both electron-donating and electron-withdrawing groups, as well as 2-naphthoyl fluoride, provided acylation products in high yields and enantioselectivities. Substitution at the meta- and para-positions was well tolerated, however substitution at the ortho-position resulted in a complete loss of reactivity. This observation supports the notion of an acylpyridinium intermediate, as the rates of reactions that proceed through such species are known to be severely affected by ortho-substitution on the acylating agent.17 A number of silyl ketene acetals were also viable substrates, with derivatives bearing electron-rich and electron-poor arene substituents, as well as heteroaromatic functionality, participating in acylation with benzoyl fluoride in good yields and enantioselectivities.18 The efficiency of this protocol is illustrated in the acylation of 1 with 2-naphthoyl fluoride on a preparative scale using only 0.5 mol% thiourea catalyst (Scheme 2). The product of this reaction was purified by crystallization from the crude reaction mixture without the need for chromatography. A single recrystallization provided pure acylation product 2b in 70% overall yield and >99% ee.
Table 2.
Substrate Scope
 | ||||
|---|---|---|---|---|
| entrya | Product | time | yieldb (%) |
eec (%) |
| 1 |  |
6 h | 88 | 92 |
| 2d |  |
6 h | 85 | 95 |
| 3 | 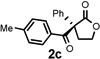 |
24 h | 91 | 95 |
| 4 | 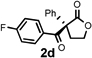 |
6 h | 90 | 91 |
| 5 | 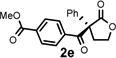 |
4 h | 95 | 86 |
| 6 | 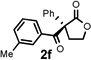 |
16 h | 77 | 92 |
| 7 | 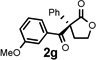 |
6 h | 95 | 92 |
| 8 | 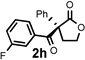 |
6 h | 78 | 87 |
| 9 | 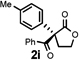 |
6 h | 75 | 93 |
| 10 | 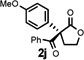 |
6 h | 80 | 88 |
| 11 | 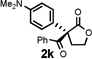 |
24 h | 76 | 89 |
| 12 | 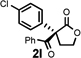 |
6 h | 76 | 89 |
| 13 | 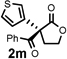 |
6 h | 77 | 89 |
Reactions run on a 0.20 mmol scale.
Isolated yield of purified product.
Enantiomeric excess determined by HPLC analysis on commercial chiral columns.
The structure and absolute configuration of 2b was established by X-ray crystallography and the stereochemistry of all other products was assigned by analogy.
Scheme 2.

Preparative-Scale Reaction
No reaction between silyl ketene acetal 1 and benzoyl fluoride is observed in the absence of either thiourea catalyst or nucleophilic catalyst. This observation indicates the thiourea is playing a role not only in the enantioselectivity-determining acylation event, but also in the generation of the key acylpyridinium ion intermediate. It is likely that the outstanding hydrogen-bond accepting ability of the fluoride anion is important in this regard.19 Furthermore, activation of the silyl ketene acetal by fluoride or benzoate seems necessary for acylation to occur, particularly given the complete absence of reactivity observed with benzoyl chloride under the standard reaction conditions.20 Consistent with this hypothesis, variation of the silyl group of the silyl ketene acetal has a measurable influence on the rate of the reaction, with larger silyl groups leading to diminished reaction rates. However, the identity of the silyl group has a negligible effect on reaction enantioselectivity, indicating that it plays no significant role in the organization of the stereoselectivity-determining step.21 This result raises the possibility of a thiourea-bound enolate as a key intermediate.22
A proposed catalytic cycle that is consistent with these observations is presented in Scheme 3. As noted, the thiourea catalyst activates benzoyl fluoride for reaction with PPY, presumably via initial complexation to the carbonyl group of the acyl fluoride (A). A thiourea-bound N-acylpyridinium/fluoride intermediate (B) is then proposed, in which the thiourea is associated to the fluoride anion and the catalyst arene substituent is engaged in a stabilizing interaction with the N-acylpyridinium cation.15 Reaction of B with the silyl ketene acetal likely proceeds via a pentavalent silicate intermediate23 and is proposed to be rate-determining based on the observed dependence of the overall rate on the identity of the silyl group. However, the independence of reaction enantioselectivity on the identity of the silyl group points to a thiourea-bound enolate such as C as the intermediate involved in enantiodetermining acylation.
Scheme 3.

Proposed Catalytic Cycle
In conclusion, a highly enantioselective acylation of silyl ketene acetals with acyl fluorides has been developed to generate useful α,α-disubstituted butyrolactone products. The remarkable hydrogen-bond acceptor properties and silaphilicity of the fluoride anion facilitate an efficient reaction protocol with low catalyst loadings and high yields and selectivities. A more-complete mechanistic elucidation of this acylation reaction that includes analysis of the basis for enantioinduction is the focus of ongoing studies.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM-43214), a predoctoral fellowship to J. A. B. from Eli Lilly, and a postdoctoral fellowship to J.-N. D. from NSERC (PDF). We thank Dr. Shao-Liang Zheng for crystal structure determination and Dr. Alan Hyde for catalyst development and synthesis.
Footnotes
Supporting Information Available: Complete experimental procedures and characterization data for acylation products and all isolated intermediates, 1H and 13C NMR spectra of acylation products, HPLC traces of racemic and enantioenriched acylation products, catalyst and silyl ketene acetal optimization data, and crystallographic information for compound 2b. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Fersht AR, Jencks WP. J. Am. Chem. Soc. 1969;91:2125–2126. [Google Scholar]; (b) Fersht AR, Jencks WP. J. Am. Chem. Coc. 1970;92:5432–5442. [Google Scholar]
- 2.(a) Litvinenko LM, Kirichenko AI. Dokl. Akad. Nauk. SSSR, Ser. Khim. 1967;176:97–100. [Google Scholar]; (b) Steglich W, Höfle G. Angew. Chem., Int. Ed. 1969;8:981. [Google Scholar]
- 3.Hassner A, Krepski LR, Alexanian V. Tetrahedron. 1978;34:2069–2076. [Google Scholar]
- 4.For reviews on DMAP catalysis, see ref 3 and the following: Höfle G, Steglich W, Vorbrüggen H. Angew. Chem., Int. Ed. 1978;17:569–583. Scriven EFV. Chem. Soc. Rev. 1983;12:129–161. Murugan R, Scriven EFV. Aldrichimica Acta. 2003;36:21–27.
- 5.For discussions on the mechanism of DMAP catalysis, see ref 4a and the following: Spivey AC, Arseniyadis S. Angew. Chem., Int. Ed. 2004;43:5436–5441. doi: 10.1002/anie.200460373. Xu S, Held I, Kempf B, Mayr H, Steglich W, Zipse H. Chem. Eur. J. 2005;11:4751–4757. doi: 10.1002/chem.200500398. Held I, Villinger A, Zipse H. Synthesis. 2005;9:1425–1430. Lutz V, Glatthaar J, Würtele C, Serafin M, Hausmann H, Schreiner PR. Chem. Eur. J. 2009;15:8548–8557. doi: 10.1002/chem.200901379.
- 6.For a review on thiourea anion-binding catalysis, see: Zhang Z, Schreiner PR. Chem. Soc. Rev. 2009;38:1187–1198. doi: 10.1039/b801793j.
- 7.(a) Reisman SE, Doyle AG, Jacobsen EN. J. Am. Chem. Soc. 2008;130:7198–7199. doi: 10.1021/ja801514m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Klausen RS, Jacobsen EN. Org. Lett. 2009;11:887–890. doi: 10.1021/ol802887h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zuend SJ, Coughlin MP, Lalonde MP, Jacobsen EN. Nature. 2009;461:968–970. doi: 10.1038/nature08484. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Knowles RR, Lin S, Jacobsen EN. J. Am. Chem. Soc. 2010;132:5030–5032. doi: 10.1021/ja101256v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) De CK, Klauber EG, Seidel D. J. Am. Chem. Soc. 2009;131:17060–17061. doi: 10.1021/ja9079435. [DOI] [PubMed] [Google Scholar]; (b) Klauber EG, De CK, Shah TK, Seidel D. J. Am. Chem. Soc. 2010;132:13624–13626. doi: 10.1021/ja105337h. [DOI] [PubMed] [Google Scholar]; (c) Klauber EG, Mittal N, Shah TK, Seidel D. Org. Lett. 2011;13:2464–2467. doi: 10.1021/ol200712b. [DOI] [PubMed] [Google Scholar]
- 9.For reviews on catalytic asymmetric acyl transfer reactions, see: Fu GC. Acc. Chem. Res. 2004;37:542–547. doi: 10.1021/ar030051b. Wurz RP. Chem. Rev. 2007;107:5570–5595. doi: 10.1021/cr068370e. Spivey AC, Arseniyadis S. Top. Curr. Chem. 2010;291:233–280. doi: 10.1007/978-3-642-02815-1_25. Müller CE, Schreiner PR. Angew. Chem. Int. Ed. 2011;50:6012–6042. doi: 10.1002/anie.201006128.
- 10.For related racemic and diastereoselective methods, see: Poisson T, Dalla V, Papamicaël C, Dupas G, Marsais F, Levacher V. Synlett. 2007:381–386. Woods PA, Morrill LC, Lebl T, Slawin AMZ, Bragg RA, Smith AD. Org. Lett. 2010;12:2660–2663. doi: 10.1021/ol1008747.
- 11.For catalytic asymmetric methods that form α,α-disubstituted butyrolactones, see: Spielvogel DJ, Buchwald SL. J. Am. Chem. Soc. 2002;124:3500–3501. doi: 10.1021/ja017545t. Mermerian AH, Fu GC. J. Am. Chem. Soc. 2003;125:4050–4051. doi: 10.1021/ja028554k. Ooi T, Miki T, Fukumoto K, Maruoka K. Adv. Synth. Catal. 2006;348:1539–1542.
- 12.For reviews on quaternary ammonium fluoride catalysis, see: Ooi K, Maruoka K. Acc. Chem. Res. 2004;37:526–533. doi: 10.1021/ar030060k. Shirakawa S, Ooi T, Maruoka K. Chiral Quaternary Ammonium Fluorides for Asymmetric Synthesis. Chapter 9. In: Maruoka K, editor. Asymmetric Phase Transfer Catalysis. Weinheim: Wiley-VCH; 2008. pp. 189–206. for a desilylative kinetic resolution of silyl ethers using a chiral bis(hydroxy) polyether catalyst/KF complex, see: Yan H, Jang HB, Lee J-W, Kim HK, Lee SW, Yang JW, Song CE. Angew. Chem. Int. Ed. 2010;49:8915–8917. doi: 10.1002/anie.201004777.
- 13.Schreiner PR, Wittkopp A. Org. Lett. 2002;4:217–220. doi: 10.1021/ol017117s. [DOI] [PubMed] [Google Scholar]
- 14.For a more comprehensive summary of catalyst optimization studies, see the Supporting Information.
- 15.For examples of stabilizing non-covalent interactions involving pyridinium ions in asymmetric catalysis, see: Kawabata T, Nagato M, Takasu K, Fuji K. J. Am. Chem. Soc. 1997;119:3169–3170. Birman VB, Uffman EW, Jiang H, Kilbane CJ. J. Am. Chem. Soc. 2004;126:12226–12227. doi: 10.1021/ja0491477. Wei Y, Held I, Zipse H. Org. Biomol. Chem. 2006;4:4223–4230. doi: 10.1039/b610140b. Li X, Houk KN, Birman VB. J. Am. Chem. Soc. 2008;130:13836–13837. doi: 10.1021/ja805275s. Hu B, Meng M, Du W, Fossey JS, Hu X, Deng WP. J. Am. Chem. Soc. 2010;132:17041–17044. doi: 10.1021/ja108238a.
- 16.Acyl fluorides were prepared in a single step from the corresponding benzoic acids upon treatment with cyanuric fluoride: Olah GA, Nojima M, Kerekes I. Synthesis. 1973;8:487–488.
- 17.Gold V, Jefferson EG. J. Chem. Soc. 1953:1409–1415. [Google Scholar]
- 18.α-alkyl-substituted, acyclic, and six-membered silyl ketene acetal analogs provided no desired C-acylation product using this reaction protocol. For a summary of silyl ketene acetal substrates examined, see the Supporting Information.
- 19.(a) Hibbert F, Emsley J. Adv. Phys. Org. Chem. 1990;26:255–379. [Google Scholar]; (b) Jeffrey GA. An Introduction to Hydrogen Bonding. New York: Oxford University Press; 1997. Chapter 3. [Google Scholar]
- 20.For other examples where acyl fluorides react with silylprotected nucleophiles but acyl chlorides are unreactive, see ref 10a and the following: Bappert E, Müller P, Fu GC. Chem. Commun. 2006:2604–2606. doi: 10.1039/b603172b. Ryan SJ, Candish L, Lupton DW. J. Am. Chem. Soc. 2011;133:4694–4697. doi: 10.1021/ja111067j.
- 21.Silyl groups that were examined include SiMe3, SiEt3, and SiMe2tBu. For experiments evaluating the influence of the silyl group on reactivity and enantioselectivity, see the Supporting Information. For an example of another reaction where variation of the silyl group of a silyl ketene acetal substrate does not influence enantioselectivity, see ref 11b.
- 22.For another catalytic asymmetric reaction where a hydrogen-bonded enolate may be a reactive intermediate, see: Ohmatsu K, Kiyokawa M, Ooi T. J. Am. Chem. Soc. 2011;133:1307–1309. doi: 10.1021/ja1102844.
- 23.For discussions on hypervalent silicon species in bond-forming processes, see: Rendler S, Oestreich M. Synthesis. 2005;11:1727–1747. Denmark SE, Beutner GL. Angew. Chem. Int. Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.