

Abstract
Gene therapy has shown a tremendous potential to benefit patients in a variety of disease conditions. However, finding a safe and effective systemic delivery system is the major obstacle in this area. Although viral vectors showed promise for high transfection rate, the immunogenicity associated with these systems has hindered further development. As an alternative to viral gene delivery, this review focuses on application of novel safe and effective non-condensing polymeric systems that have shown high transgene expression when administered systemically or by the oral route. Type B gelatin-based engineered nanocarriers were evaluated for passive and active tumor-targeted delivery and transfection using both reporter and therapeutic plasmid DNA. Additionally, we have shown that nanoparticles-in-microsphere oral system (NiMOS) can efficiently deliver reporter and therapeutic gene constructs in the gastrointestinal tract. Additionally, there has been a significant recent interest in the use small interfering RNA (siRNA) as a therapeutic system for gene silencing. Both gelatin nanoparticles and NiMOS have shown activity in systemic and oral delivery of siRNA, respectively.
Keywords: Non-viral gene delivery, non-condensing polymers, type B gelatin, nanoparticles, nanoparticles-in-microsphere oral system (NiMOS)
1. INTRODUCTION
Nucleic acid therapy holds significant promise in improving clinical outcomes for many acute and chronic diseases. This strategy is based on two different paradigms: (1) introducing target genes in the form of oligonucleotides or plasmids to recover or induce the expression of therapeutic proteins and (2) providing antisense oligonucleotides or small interfering RNA (siRNA) to interrupt the function of target genes, and trigger silencing. Although there have been many clinical trials of gene therapy for diseases such as cancer, inflammation, and diabetes, there is still no approved product in the United States at the present time. Development of safe and effective vectors for delivery of nucleic acid to the tissue and cell of interest has been a major barrier to clinical translation of this very exciting experimental concept [1].
Unlike most small molecule therapeutics, nucleic acid constructs are large, hydrophilic, and negatively charged molecules. In order to achieve therapeutic effect, these molecules need to overcome the physical and biological barriers in the systemic circulation, reach the tissue and cell of interest, and survive the harsh intracellular environment. As of June 2010, there have been over 1,640 gene therapy clinical trials and among them, viral vectors were used in up to 68% of cases [2]. Viral vectors usually consist of viral capsids and viral genome. The therapeutic gene cassette could be inserted into the viral genome to replace the native genes by recombination or introduced into the viral vector as episomal genes [3]. During the treatment of viral vectors, transduction happens, which indicate the infectious process that functional genetic information could be introduced and expressed into the target cells [3]. Viral vectors usually present high efficiency for infection and have broad tropism, which make them to be the most prevailing vectors in gene therapy [3]. However, many viral vectors that have a history of immunotoxicity and gene insertion into the host chromosome. In September of 1999, University of Pennsylvania researchers and clinicians carried out an experimental gene therapy study using adenovirus-based treatment for replacement of ornithine transcarbamylase deficiency [4]. The patient, an 18-year old, Jesse Gelsinger had a fatal reaction due to acute respiratory distress syndrome and an overwhelming systemic inflammatory response, which was diagnosed to relate to the immunogenic reaction to the large dose of adenoviral vector [4]. Since this tragic incident, the safety of viral vectors has been a major limiting concern for their use in routine gene therapy protocols. Additionally, viral vectors possess constraints of transgene size limits and quality-assured reproducible large-scale production for clinical translation [5].
Non-viral gene delivery systems have received a lot of attention based on improved safety profile, but the efficiency of transfection is significantly less than that of viral counterparts. Non-viral methods generally include naked nucleic acid delivery, physical methods for nucleic acid delivery, condensing vector-based constructs, and non-condensing vector-based constructs. Recently, the use of condensing and non-condensing delivery vehicles have received significant interest at the preclinical level [6]. Cationic polymer and lipid molecules have the ability to neutralize the negative charges of nucleic acids and form a condensed electrostatic complex, which are called polyplexes and lipoplexes, respectively [7]. With the electrostatic forces between the polymer and the nucleic acid, the complex could maintain a stable and condensed nano-size structure, promote cellular endocytosis, and possibly enhance transfection efficiency of therapeutic genes [7]. Although the condensing vectors seem to be an excellent substitute for viral vectors, some drawbacks inherent in the condensing system limit their application for systemic delivery. These include toxicity of the cationic polymer or lipid, rapid clearance by the reticulo-endothelial (RES) system, inability of the complex to escape from the endosome/lysosome compartments in the cells, and lack of intracellular unpacking of the nucleic acid construct from the electrostatic complex [8].
We and others have hypothesized that non-condensing lipid and polymeric nano-sized vectors can be engineered for tissue and cell specific delivery and allow for enhanced transfection efficiency with significantly less toxicity concerns (Table 1). Non-condensing lipids and polymers posses either a neutral or net negative charge. Plasmid DNA, siRNA, and oligonucleotide payload is encapsulated within the system (e.g., liposomes or nanoparticles) either by physical entrapment of nucleic acid constructs within the matrix or through hydrogen bonds between polymer and nucleic acid bases [9, 10]. Physical encapsulation enables protection from the enzymes and other plasma proteins during its transit from blood to the site of action. Cellular uptake is facilitated since masking negative charge of DNA prevents electrostatic repulsion between negatively charged DNA and negatively charged cell surface. In contrast to condensing lipids and polymers, absence of positive charges on non-condensing systems limits its recognition by the mononuclear phagocyte system and hence limits its early clearance.[11, 12] In addition, opsonization by IgM and innate immune response is not favored in case of non-condensing polymers because of their neutral or slightly negatively charged state [11, 12]. This review paper will focus on non-condensing polymeric nanoparticles and microparticles that are used as vectors for gene therapy.
Table 1.
Non-Condensing polymers applied in gene delivery.
| Polymer Name | Structure | Charasteristics | Applications | Refs |
|---|---|---|---|---|
| Gelatin | 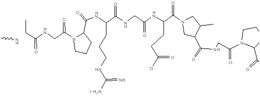 |
Biodegradable polymers obtained from hydrolysis of collagen. Gelatin is considered as a (Generally recognized as safe) GRAS excipient by FDA. Based on the pH and isoelectric point of different types of gelatin, they could be either negative or positive charge. | Neutral or negative charged gelatin can physically encapsulate plasmid DNA or siRNA inside. By desolvation method, gelatin nanoparticles could be formed and used for therapeutic gene delivery or gene silencing therapy. | [28, 32, 57, 58, 67, 68] |
| Hydroxyproryl-methacrylate copolymers (HPMA) | 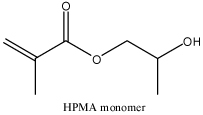 |
HPMA copolymers are hydrophilic, non-immunogenic and not toxic. During the synthesis, stimuli-sensitive bond or different electrolyte could be included to fulfill the purpose of the delivery system. | Multivalent HPMA copolymers were used to stabilize DNA complex and circulation of this system has been improved. siRNA could be conjugated to HPMA copolymers to for the delivery system. | [62, 69–71] |
| Poly(ε-caprolactone) (PCL) | 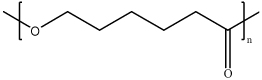 |
PCL is biodegradable polyester, could interact with nucleic acids physically. | PCL polymers are usually conjugated with other polymers such as polyethylenimine to form copolymer and enhance the efficacy during gene delivery. | [72–74] |
| Poly (ethylene glycol) (PEG) | 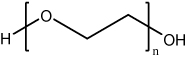 |
PEG and derivatives are neutral and hydrophilic molecules with flexible polymer chains. With two ends susceptible to modification, PEG could be synthesized as mono, homo- or heterofunctional polymers. | PEG is usually conjugated on the surface of gene delivery vectors to enhance the accumulation for anti-cancer treatment due to EPR effect. Copolymers could also be formed such as poloxamer or poly(lactic acid) (PLA) – PEG and further used as a more efficient gene delivery system. | [17, 75–79] [64, 65] |
| Poly(D,L-Lactide-co-glycolide) (PLG) | 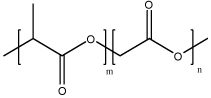 |
Copolymer synthesized by polymerization of lactic acid and glycolic acid. PLG, as a hydrophobic, biodegradable and biocompatible polymer, could encapsulate with nucleic acids physically. | PLG could form microspheres or nanoparticles with emulsion method or spray drying techniques. With encapsulation of DNA, PLG particles could be used as DNA vaccines with intravenous, intraperitoneal or oral administration. These polymers could also serve as therapeutic gene delivery vectors. | [35, 60, 80–85] |
| Poloxamer (Pluronic®) | 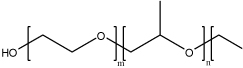 |
Pluronic® block copolymers are amphiphilic molecules, consisted of hydrophilic ethylene oxide (EO) and hydrophobic propylene oxide (PO) with PEO-PPO-PEO structure. These polymers could self assemble into micelles. | By mixture of different block copolymers, Pluronic® could encapsulate nucleic acids into the micellar structure by physical interaction and protect them from the enzyme digestion. | [10, 86–88] |
| Poly (N-vinyl pyrrolidone) (PVP) | 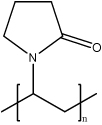 |
PVP is synthetic hydrophilic homopolymer with mild adhesive properties. Although not biodegradable, they are biocompatible and proved as a safe excipient. These polymers could not form either micelles or particles. By binding to the base pairs in the major groove of DNA, PVP could associate with nucleic acids by hydrogen bonds at pH 4–6. | PVP is used to formulate with plasmid DNA and use as local and systemic therapeutic gene delivery vector or DNA vaccine vector. Since these polymers do not particulate, this system is susceptible to extracellular degradation. Usually, PVP is applied as a stabilizer in other formulations to enhance the incorporation with nucleic acids. | [59, 89–92] |
2. NANOPARTICLES FOR TUMOR-TARGETED GENE DELIVERY
2.1 Passive and Active Tumor Targeting
Tumor formation is related to abnormalities in the genetic sequences and expression or suppression of oncogene or tumor suppressor genes, which result from inherited or environment-induced mutations. As cancer is a genetic disease, gene therapy represents a promising treatment for tumor, with localized, sustained gene expression in the target and low cytotoxicity to the host. In order to deliver genes to tumor, the delivery system need to recognize the host cells, avoid nonspecific binding and uptake, resist degradation during the systemic circulation and after reaching to the target cells, it should cross the cell membrane, afford escape from endosomal/lysosomal compartment, release genes from the complex and let the cargo to get into the nucleus or accomplish its function in the cytoplasm (e.g., for siRNA) [13]. For targeting to specific tumor cells, delivery system is usually designed based on passive or active targeting mechanism (Figure 1).
Figure 1.

Passive targeting and active targeting strategies for anti-cancer gene therapy. For passive targeting, the following strategies are usually applied. a. Pegylated nanoparticles accumulate in the tumor due to leaky vasculature and EPR effect. b. Nanovectors sensitive to low pH response to the acidic tumor microenvironment and release nucleic acids. c. Nanovectors with disulfide bond or other chemical structure react with high level of glutathione or redox enzymes in tumor and release the cargo. For active targeting, the following strategies are usually applied. d. With antibody conjugated on the surface of nanovectors, they could recognize tumor antigen and internalize by facilitated endocytosis and release cargo after endosome escape. e. Receptor mediated endocytosis help transport of nanovectors and transfer nucleic acids.
Solid tumors are characterized with heterogeneous vasculature, which has different size and distribution from the periphery to the core region. Generally, due to the rapid growth of the tumor mass, the vascular system usually has big gap junctions between endothelial cells and lack of lymphatic drainage [51]. The pore size of endothelial junctions in tumor neovasculature is between 100 nm to 780 nm, which is significantly larger than those on normal blood vessels [14, 15]. Nanoparticles with size of up to 200 nm in diameter and having hydrophilic surface, such as with poly(ethylene glycol) (PEG) modification, tend to have a longer duration of circulation in the blood stream and are able to reach the tumor mass through exravasation at higher concentrations [16] (Shown in Figure 1a). This unique pathophysiologic feature is termed the “enhanced permeability and retention” (EPR) effect and it was first shown by Maeda et al. [17] in preclinical models with polymer-conjugated anticancer therapeutics. Meanwhile, the microenvironment of tumor cells also present different properties compared to normal tissue. In order to proliferate, cancer cells usually have a high metabolic rate, which uses aerobic glycolysis to obtain energy and resulting lactate produces an acidic environment [18]. pH responsive polymers could help to stabilize the complex during normal physiological pH and release genes in the slightly acidic tumor microenvironment [19]. Additional cancer cells express different levels of enzymes and proteins, such as redox enzymes and glutathione [20, 21]. Polymeric system designed specifically to form disulfide crosslinks are susceptible to glutathione-induced intracellular delivery of the encapsulated payload in cancer cells [19].
Even with passive targeting, most gene therapy vectors still face intrinsic limitation of this mechanism: lack of specificity. To solve this problem, a variety of delivery systems are modified with targeting moieties to improve active targeting. In order to achieve tumor-targeted delivery, we should explore several unique properties of cell surface to differentiate target cells population with normal cells. Based on the expression of antigen or receptors on the tumor cells surface, antibodies or receptor’s substrate could be conjugated on the delivery system, and help to target specifically on the tumor cells (Shown in Figure 1d and 1e). Ideally, after targeting moiety interacting with cell surface, they should trigger the internalization and endocytosis process [16]. While selecting the targeting moiety, all the above properties need to be taken into consideration.
2.2 Passive Tumor Targeting with PEG-Modified Gelatin Nanoparticles
For passive targeting, there are several ways to achieve, such as modification on the surface of polymeric nanoparticle with hydrophilic polymer chain or cooperating environment sensitive polymers into the nanoparticles. PEG or poly(ethylene oxide) (PEO), with a structure of HO-(CH2CH2O)n-CH2CH2-OH is a commonly used polymer for surface modification of long-circulating vesicles [22]. With coating of PEG, it could form a dense and hydrophilic shell of long chains and protect the core from interacting with different solutes, especially on physiologic level, non-specific hydrophobic interaction with the reticuloendothelial system (RES) [22]. This polymeric protection for solid particles is termed as “steric stabilization” [22]. Meanwhile, terminal hydroxyl group of PEG could be modified into different derivatives, which provides monofunctional, homo- or hetero- bifunctional and even multi-arm PEG, capable for further conjugation of selected ligands [9]. To sum up, PEG modification of nanoparticles presents the following advantages: increasing the circulation time, stabilizing the therapeutic payload during transportation, decreasing RES accumulation and providing potential for conjugation of targeting moieties.
Gelatin is one of the most versatile natural biopolymer derived from collagen, and it has been widely used in food products and medicines. With solvent displacement, type B gelatin, derived from alkaline hydrolysis of collagen, which has an isoelectric point at around 4.5–5.5, could physical encapsulate nucleic acid construct at neutral pH. Furthermore, the physical encapsulation in gelatin nanoparticles preserves the supercoiled structure of the plasmid DNA and improves the transfection efficiency upon intracellular delivery [23].
Kaul and Amiji [20] were the first to develop type B gelatin-based nanoparticles as non-condensing gene delivery systems for tumor-targeting by passive accumulation due to the EPR effect. They prepared the unmodified and PEG-modified gelatin nanoparticles by ethanol precipitation method, which gave them size in the range of 200 to 500 nm. Tetramethylrhodamine (TMR)-dextran, a hydrophilic fluorescently-labeled molecule, was first used as model drug for in vitro cell uptake studies. The results showed that control and PEG-modified type B gelatin nanoparticles could be taken up by cells through non-specific endocytosis.[24] Confocal fluorescence microscopy studies showed time-dependent intracellular localization of the nanoparticles and the results illustrated that PEG-modified gelatin nanoparticles were internalized in NIH-3T3 murine fibroblast cells by non-specific endocytosis, and within 12 hours, the payload could be released and accumulated around the perinuclear region. Plasmid DNA, encoding for enhanced green fluorescence protein (EGFP-N1), was encapsulated into the particles during the precipitation process. After digestion with protease, gel electrophoresis analysis proved that the plasmid was stably encapsulated at a high loading efficiency (>95% at 0.5% (w/v)). In vitro transfection studies in NIH-3T3 cells confirmed the long-lasting transgene expression potential of PEG-modified type B gelatin nanoparticles as compared to other controls, including Lipofectin®, a cationic lipid transfection reagent that is commercially available. Additionally, neither gelatin non PEG-modified gelatin nanoparticles showed any toxicity to the NIH-3T3 cells even after several days [21].
Following in vitro evaluations, the authors further examined the biodistribution profiles of unmodified and PEG-modified 125Iodine (125I)-labeled gelatin nanoparticles following intravenous administration through the tail vein in Lewis Lung carcinoma (LLC)-bearing C57BL/6J mice. Periodically, the tumor-bearing mice were sacrificed and the radioactivity levels in blood, tumor, and other highly perfusing organs were measured. PEG-modified nanoparticles showed long circulating properties in the blood and preferentially accumulated in the tumor for up to 24 hours post-administration. There was also significant accumulation of the PEG-modified nanoparticles in the liver. Conversely, unmodified nanoparticles were rapidly cleaned from the circulation and remained mostly in the liver and spleen. These results showed that PEG-modified gelatin nanoparticles could be passively targeted to the tumor mass following systemic administration and they could be an effective vector for anti-cancer gene therapy [25].
Reporter plasmid DNA encoding β-galactosidase (pCMV-β) was encapsulated into nanoparticles and used to validate the potential of PEG-modified system as a systemic gene delivery vector. In vitro studies with LLC cells first illustrated that PEG-modified gelatin nanoparticles are preferable vector for gene delivery, which is on the basis of qualitative and quantitative analysis after transfection with reporter genes. The in vivo studies further demonstrated that PEG-modified nanoparticles have the potential to be systemic gene delivery vector, since it showed higher transfection with PEG-modified system after intravenous administration than that after intratumoral administration [26]. The unique physical, chemical, and biological properties of type B gelatin and the “steric stabilization” property of PEG modification made these nanoparticles a highly promising system for systemic gene delivery to tumor mass.
2.3 Passive Tumor Targeting with PEG-Modified Thiolated Gelatin Nanoparticles
Tumor cells usually present different physiological properties as compared to normal cells. On the basis of these differences, one could design delivery system to target the tumor mass and afford greater cellular delivery. Glutathione (GSH) is a tripeptide, generally expressed in the cell cytoplasm and functions as an antioxidant to prevent damage related to the reactive oxygen species [27]. The intracellular GSH concentration (5–10 mM) is generally higher than the extracellular concentrations (1–10 µM). While during active proliferation of tumor cells, GSH and peroxide levels are even higher in the cytoplasm [27]. Introduction of thiol (i.e., SH) groups is a common modification that can allow for intracellular delivery through the reduction disulfide crosslinks.
Based on this delivery rationale, Kommareddy and Amiji designed thiolated type B gelatin nanoparticles (SHGel) for systemic gene delivery [28]. Gelatin was first thiolated with different concentration of 2-iminothiolane and after lyophilization, the thiolated gelatins were used to prepare nanoparticles by the solvent displacement using ethanol. In cytotoxicity study, SHGel-20 (1 g of gelatin modified with 20 mg 2-iminothiolane) with 6.08 mM/g thiol groups, showed the highest cell viability out of all the thiolated gelatin nanoparticles and was selected for additional studies. Release profiles in GSH containing phosphate buffer (pH 7.4) proved that the thiolated gelatin nanoparticles showed a greater percent release of the payload (fluorescein isothiocyanate-dextran) as compared to the unmodified nanoparticles. Green fluorescent protein (GFP) expressing plasmid DNA was encapsulated into the nanoparticles and glyoxal was used to crosslink the system. Both unmodified and thiolated type B gelatin nanoparticles were internalized by NIH-3T3 cells and the expression profile of GFP was analyzed by fluorescence microscopy and by flow cytometry. Crosslinked SHGel-20 was found to have greater transfection efficiency as compared to all the other nanoparticle systems, including Lipofectin®-complexed DNA. With this study, thiolation of gelatin and formation of nanoparticles was found to be an effective strategy for intracellular GSH-induced DNA delivery [28].
Furthermore, Kommareddy and Amiji evaluated methoxy-PEG-succinimidyl glutarate (mPEG-SG, MW 2 kDa) modified preformed gelatin and thiolated gelatin nanoparticles. Surface modification with PEG chains was confirmed with 2,4,6-trinitrobenzene sulfonic acid (TNBS) assay and electron spectroscopy for chemical analysis (ESCA). Encapsulated plasmid DNA stability and release studies were performed in control and PEG modified nanoparticles and the results showed that surface PEG modification of preformed nanoparticles did not affect the stability or release properties of the payload. In vitro transfection studies with EGFP-N1 plasmid DNA encapsulated in gelatin (Gel), PEG-modified gelatin (PEG-Gel), thiolated gelatin (SHGel), and PEG-modified thiolated gelatin (PEG-SHGel) nanoparticle formulations was evaluated in NIH-3T3 cells. Of all the different formulations tested, including Lipofectin®-complexed DNA, PEG-SHGel nanoparticles showed the highest GFP expression, which proved the ability of this system to efficiently transfect target cells in vitro and could be used as a promising gene delivery vector in vivo [29].
Before using the control and PEG modified nanoparticle systems for in vivo gene therapy, biodistribution and pharmacokinetic analysis were performed in orthotopic estrogen negative MDA-MB-435 human breast adenocarcinoma-bearing female nu/nu (athymic) mice. In this study, 2 µCi dose of 111Indium- (111In)-labeled PEG-Gel and PEG-SHGel nanoparticles were injected intravenously through the tail vein in the tumor-bearing mice. At pre-determined time points, blood, tumor and highly perfusing organs were collected from mice and analyzed for the radioactivity levels using a gamma counter. The concentration of nanoparticle accumulation was based on percent administered dose per gram of fluid or tissue. Non-linear pharmacokinetic analysis was performed with plasma and tumor concentrations as a function of time to show the variability in biodistribution profiles. Overall, PEG modified nanoparticles (i.e., PEG-Gel and PEG-SHGel) showed longer circulation in blood and higher accumulation in the tumor as compared to the unmodified nanoparticles (i.e., Gel and SHGel). Although a certain portion of radioactivity was found in the liver and spleen for all treatments, surface modification with PEG significantly decreased this non-specific uptake. Meanwhile, the pharmacokinetics analysis supported the above statement and also showed that PEG-SHGel nanoparticles had a higher tumor concentration and much longer half-life, which means that thiolation enhances nanoparticles’ sensitivity to the reducing environment of the tumor and may enhance delivery efficiency in tumor cells [30].
Finally, to confirm the transfection of a therapeutic gene construct in Gel, SHGel, PEG-Gel and PEG-SHGel nanoparticle formulations, plasmid DNA encoding for the soluble form of the extracellular domain of vascular endothelial growth factor receptor-1 (VEGF-R1 or sFlt-1) was chosen for anti-angiogenesis therapy. After obtaining a sample of sFlt-1 encoding plasmid DNA from Professor Kensuke Egashira at Kyushu University in Fukuoka, Japan, it was amplified in E. coli, purified, and encapsulated in the nanoparticle formulations. After confirmation of DNA encapsulation, MDA-MB-435 breast adenocarcinoma cells were transfected at a dose of 20 µg per 200,000 cells. The expressed sFlt-1 that was obtained from the cell culture media over a period of up to 8 days was concentrated and measured by ELISA and western blot analysis. Of all the formulations tested, PEG-SHGel proved to be the superior system for transfection of sFlt-1 expressing plasmid in MDA-MB-435 tumor cells. Following confirmation of in vitro transfection potential with sFlt-1 expressing plasmid DNA, an orthotopic MDA-MB-435 breast adenocarcinoma model was established in the mammary fat pad of female nu/nu mice. Once the MDA-MB-435 tumor mass had reached approximately 50 mm3, the nude mice received a total of 60 µg dose divided in three intervals at day 1, 3, and 5 by intravenous administration in PEG-Gel and PEG-SHGel nanoparticle formulations. Only PEG modified nanoparticles were used for in vivo transfection studies since the previous biodistribution analysis clearly showed that unmodified Gel and SHGel nanoparticles accumulated primarily in organs of the RES and did not reach the tumor mass. Naked and Lipofectin®-condensed plasmid DNA were used as controls. Periodically, the changes in tumor volume were measured for up to 40 days post-administration (Figure 2). The tumor growth suppression analysis also confirmed that PEG-SHGel nanoparticles were superior relative to all other formulations. After 40 days, tumor-bearing mice were sacrificed and the tumor, liver, and other tissues were analysed for sFlt-1 expression and the excised tumor tissues were also immunostained with CD-31 antibodies for microvessel density analysis. ELISA and western blot have shown the similar expression rates as the in vitro studies with PEG-SHGel nanoparticles having the highest in vivo transfection potential relative to other formulations. CD-31 immunostaining of tumor cryosection showed that PEG-Gel and PEG-SHGel nanoparticles significantly reduced the tumor microvessel density (Figure 3) [28].
Figure 2.

In vivo antitumor efficacy studies of expressed sFlt-1 in orthotopic MDA-MB-435 human breast adenocarcinoma-bearing female Nu/Nu mice. PEG-Gel and PEG-SHGel nanoparticles, with sFlt-1 encoding plasmid DNA, were administered intravenously to tumor-bearing mice at a plasmid DNA dose of 20 µg three times every other day. Untreated animals and those receiving naked plasmid DNA served as controls. Tumor volume changes were measured daily following administration of the plasmid DNA. The naked plasmid DNA-treated and –untreated animals were used as controls. N=6, mean±s.d. *P<0.01 as compared with naked plasmid DNA treated, #P<0.01 compared with PEG-Gel treated, (non-parametric t-test). At the time of killing (40 days post-therapy), the tumor masses from control and test animals were surgically excised. (Reprinted with permission of Macmillan Publishers Ltd: Cancer Gene Therapy[30], copyright (2007))
Figure 3.

Antiangiogenic effect of expressed sFlt-1 in orthotopic MDA-MB-435 human breast adenocarcinoma-bearing female Nu/Nu mice. Tumor microvessels were detected and quantified by CD-31 (PECAM-1) antibody staining of tumor cryosections after harvesting the tissue at the time of killing (40 days post-therapy). (a) Quantitative analysis of microvessel density in tumor cryosections from animals receiving 20 µg of plasmid DNA dose every other day for 3 days in PEG-Gel and PEG-SHGel nanoparticles. Untreated animals and those receiving naked plasmid DNA served as controls. The results represent an average of total number of blood vessels counted in at least three fields per tissue section in three different animals. The naked plasmid DNA-treated and -untreated animals are used as control. N=9 fields of observation, mean ± S.D. P<0.01 (ANOVA). (b) CD-31 immunostaining images of tumor cryosections from the control and treated animals. (Reprinted with permission of Macmillan Publishers Ltd: Cancer Gene Therapy[30], copyright (2007))
On the basis of these results, it is evident that non-condensing type B gelatin-based DNA delivery vehicle could be used as a safe and effective vector for systemic administration of therapeutic genes to solid tumor. According to these studies, chemical modification of polymers could significantly change the biological properties of nanoparticles. Based on those passive targeting mechanism, one could design similar strategies, such as PEG modification and thiolation, with other nanoparticles formulation, to enhance passive targeting to tumor mass.
2.4 Active Targeting with EGFR Targeting Peptide-Modified Gelatin Nanoparticles
Although passive tumor targeting with PEG surface modification and stimuli-responsive mechanisms can provide some preferential accumulation in tumor mass and allow for intracellular delivery, there are certain tumors that do not have adequate vascularity or these nanoparticles may not penetrate deep into the tumor mass. Passively targeted PEG-Gel and PEG-SHGel nanoparticles also were found to accumulate in the liver, which resulted in high levels of sFl-1 transfection in this organ [31]. Actively targeted delivery, based on surface functionalization with a specific bio-recognizable molecule, can further facilitate accumulation in the tumor mass and also reduce non-specific accumulation in RES organs.
One example of active targeting non-condensing gene delivery system is based on human epidermal receptor (HER) targeting peptide functionalized type B gelatin nanoparticles system developed by Magadala and Amiji [32]. In this system, a heterobifunctional PEG derivative, maleimide-PEG-succinimidyl carboxymethyl (MAL-PEG-SCM, MW 2kDa), was first anchored on the surface of gelatin nanoparticles through the amine reactive SCM functionality and the maleimide functional group was available to react with four HER targeting peptides that had a terminal cysteine residue. This approach allowed for surface modification of gelatin nanoparticles with HER peptides through a flexible PEG spacer (Figure 4). Following confirmation of epidermal growth factor receptor (EGFR, erbB-1) over-expression in a several pancreatic adenocarcinoma cell lines, which is correlated to metastasis and resistance [33], Panc-1 cells were used to evaluate in vitro delivery efficiency and transfection using reported plasmid expressing GFP. One of the four peptides tested with the sequence YHWYGYTPQNVI was found to be the best for EGFR-specific delivery of nanoparticles in Panc-1 cells.
Figure 4.

(a) Chemical reaction scheme illustrating surface modification of type B gelatin nanoparticles with epidermal growth factor receptor (EGFR) binding peptide through a poly(ethylene glycol) (PEG) spacer and (b) scanning electron microscopy of control, PEG-modified, and EGFR-targeted gelatin nanoparticles. (Reprinted with permission of The AAPS Journal [32], copyright (2008)).
Following confirmation of lack of toxicity of control and EGFR peptide-modified type B nanoparticles, the efficacy of this system as gene delivery vehicle was evaluated with GFP-expressing plasmid in Panc-1 cells. The cells were transfected with 20 µg plasmid DNA dose per 200,000 cells for up to 96 hours. The qualitative fluorescence microscopy and quantitative flow cytometric as well as GFP-specific ELISA results showed that the EGFR targeted nanoparticle formulations showed highest transgene expression relative to all the other controls, including Lipofectin®-condensed DNA (Figure 5) [32]. The enhanced transgene expression was attributed to the surface presence of EGFR peptide, which triggers rapid internalization by facilitated endocytosis and then quickly releases the cargo in the cells. EGFR-targeted gelatin nanoparticles showed superior DNA delivery in pancreatic cancer cells in vitro, which makes this system a potential treatment for gene therapy in vivo. Further in vivo study needs to be done to show the safety and efficacy especially with a therapeutic gene construct in the treatment of pancreatic cancer.
Figure 5.

Quantitative and qualitative in vitro enhanced green fluorescence protein (GFP) transgene expression efficiency studies in control (unmodified) type B gelatin nanoparticles, poly(ethylene glycol)-modified gelatin nanoparticles (PEG-Gel NP), and epidermal growth factor receptor (EGFR)-targeted gelatin nanoparticles (Peptide-Gel NP) in Panc-1 human pancreatic adenocarcinoma cells. Quantitative analysis was performed by (a) flow cytometry and (b) enzyme-linked immunoassay for GFP at different time points from 24 to 96 h post-transfection. (c) Qualitative analysis of GFP transfection was performed by epifluorescence microscopy after 48 h. The Panc-1 cells were treated with the plasmid DNA dose of 20 µg per 200,000 cells for a period of 4 h, followed by washing with sterile phosphate-buffered saline (pH 7.4) and replacement of regular cell culture medium. Epifluorescence microscopy images were obtained at ×40 original magnification. Cells treated with blank gelatin nanoparticles served as a negative control, while the commercial cationic lipid-based DNA transfection reagent, Lipofectin®, was used as a positive control. (Reprinted with permission of The AAPS Journal [32], copyright (2008)).
3. NANOPARTICLES-IN-MICROSPHERE ORAL SYSTEM (NiMOS) for GENE DELIVERY
3.1 Oral Gene Therapy
Gene delivery through oral route seems very attractive as compared to other more invasive routes due to the ease of administration and very high patient compliance [12]. Efficient oral delivery can provide sustained production of therapeutic proteins at the disease site in the gastrointestinal (GI) tract. Large surface area of GI tract and a large number of stem cells in the intestinal crypts often help to improve the DNA uptake and transgene expression resulting in sustained local production of therapeutic proteins. This has significant promise for treatment of local diseases such as infections, gastric and duodenal ulcers, inflammatory bowel disease, and GI cancer. In addition, oral DNA administration also allows access to the luminal side of the intestine for treatment of regional disorders. The efficient oral delivery has significant potential for administration of DNA vaccines that can provide both mucosal and systemic immunity. The mucosal immune system is geared not only to protect against antigenic entry into the systemic immune system, but also to be unresponsive to food antigens [34]. Lastly, oral DNA delivery can also provide region-specific transfection and protein expression that can be absorbed for systemic therapy.
Although there are many potential applications of oral gene therapy, the delivery of gene construct in the GI tract is extremely challenging due extracellular and intracellular barriers. The extracellular barriers include various anatomical (mucus and epithelial layer) and physiological constraints (varying pH, degrading enzymes, etc) exhibited throughout by the tract. In addition, once the DNA reaches the intended cell of interest, uptake, endosomal/lyosomal escape, and efficient nuclear entry for protein expression remain an additional challenge to successful gene therapy.
To overcome the delivery barrier, a number of investigators have relied on polymeric microparticle or nanoparticle systems to carry the plasmid DNA. Poly(D,L-lactide-co-glycolid) (PLGA) and chitosan are examples of synthetic and natural polymers, respectively, that have received significant attention in oral DNA delivery [35]. Both of these polymers in microparticulate formulations have been used predominantly for oral vaccination that targets the M-cells in the Peyer’s patch region of the small intestine.
3.2 NiMOS for Oral Gene Delivery
Bhavsar and Amiji [36] developed a unique multicompartmental oral DNA delivery system based on encapsulation of type B gelatin nanoparticles in poly(epsilon-caprolactone) (PCL) microsphere. This delivery system was termed “nanoparticles-in-microsphere oral system” or NiMOS. Based on the success of using non-condensing type B gelatin nanoparticles for systemic gene therapy, it was envisioned that this formulation would also be suitable for oral gene delivery if the payload can efficiently reach the cell of interest. As such, DNA-encapsulated gelatin nanoparticles were further encapsulated in PCL microspheres to protect against premature pH- or enzyme-induced degradation of the matrix and the payload in the GI tract (Figure 6) [35]. PCL is a biocompatible and biodegradable polymer which has been used in various medical and pharmaceutical applications. Additionally, PCL is known be degraded by lipases, which are abundantly present in the small and large intestine. Using a statistical factorial design optimization approach, NiMOS of 1–5 µm in diameter were formulated by a “double emulsion-like” technique with encapsulated type B gelatin nanoparticles of 200 nm diameter [36, 37]. As such, NiMOs were believed to be able to protect orally-administered DNA during transit from the stomach and release the nanoparticles in the small and large intestine. These DNA-containing gelatin nanoparticles then get internalized by enterocytes or other cells of the GI lumen for transfection of the encoded protein.
Figure 6.

Schematic illustration showing the cross-sectional view of nanoparticles-in-microsphere oral system (NiMOS). On the left is the scanning electron microscopy (SEM) image of gelatin nanoparticles, which are less than 200 nm in diameter, and can physically encapsulate plasmid DNA at a loading efficiency of >93%. On the right is the SEM image of 2–5 µm NiMOS with the overall DNA encapsulation efficiency of >46%. (Reprinted with permission of Macmillan Publishers Ltd: Gene Therapy [38], copyright (2008)).
Following oral administration of reporter plasmid DNA encoding GFP or beta-galactosidase in NiMOS to fasted Sprague-Dawley rats, there was significant GFP and beta-galactosidase expression in the small and large intestine as area compared to controls including DNA-encapsulated type B gelatin nanoparticles [36]. The oral biodistribution studies with 111In-labeled gelatin nanoparticles and NiMOS showed clear accumulation in the small and large intestines at later time points (after 6 hours), which was believed to be important for optimization DNA transfection in the lower part of the GI tract. Following confirmation of transfection with reporter plasmid in both naïve and 1,4,6, trinitrobenezene sulfonic acid (TNBS)-induced colitis model established in Balb/c mice, the colitis group was treated with anti-inflammatory murine IL-10 (mIL-10) expressing plasmid DNA in NiMOS [38].
3.3 IL-10 Gene Therapy for Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) is a chronic condition that involves the inflammation of mucosal layer of the GI tract. Evidence supports that it could be mainly coming from genetically determined dysregulation of the mucosal immune response to luminal antigens. The normal mucosal layer of intestine generally shows a balance between endogenous pro- and anti-inflammatory cytokines. Pathogenesis of IBD seem to cause excess production of pro-inflammatory and deficiency in anti-inflammatory cytokines. Evidence suggests that the IL-10 is known to play an important role in the immunological balance of mucosal immune system and its expression seems to inhibit the production of pro-inflammatory cytokines and chemokines, thus inhibiting antigen presentation [39–41]. In order to tilt the balance towards an anti-inflammatory state, mIL-10 expressing plasmid was encapsulated in NiMOS. Following characterization of the formulation for DNA loading, release, and stability, a dose of 100 µg per animal was administered in NiMOS orally to TNBS-induced acute colitis bearing Balb/c mice. Control animals received no treatment, naked mIL-10 expressing plasmid, or mIL-10 plasmid encapsulated in gelatin nanoparticles.
When administered orally, the mice that received mIL-10 expressing plasmid DNA in NiMOS showed significantly higher mRNA and protein levels as compared to control groups including the group that received mIL-10 expressing plasmid DNA in gelatin nanoparticles [38]. The therapeutic benefits were exhibited by significantly reduced levels of pro-inflammatory cytokines such as IL-1a, IL-1b, IFN-gamma, TNF-alpha, and IL-12 [38]. Along with the reduction of these, there was also significant reduction of chemokines demonstrated (MCP-1, MIP-1a). Additional more dramatic effect of transfected mIL-10 activity was evident from the gain in body weight in the animals to near baseline levels in naïve (no colitis) animals. In contrast, the control animals with TNBS colitis lost almost 30% of their body weight a few days and had to be sacrificed. The NiMOS treated group also had a restoration of colon length and corresponding colon weight back to the baseline levels in the absence of acute colitis (Figure 7). Lastly, the excess IL-10 production also reduced the cellular infiltration demonstrated by colonic tissue myeloperoxidase activity and histology. The results of this study showed, for the first time, the potential of oral therapeutic gene delivery for treatment of acute colitis using biocompatible and biodegradable NiMOS.
Figure 7.

Changes in body weight, clinical activity score, and the lengths and weights of colonic tissue upon oral administration of murine interleukin (IL)-10-expressing plasmid DNA in nanoparticles-in-microsphere oral system (NiMOS). The body weight change was used as a marker of therapeutic efficacy achieved with locally expressed IL-10 over the course of 8 days (a). The clinical activity scores in control and treatment animals as measured using an aggregate of body weight changes, rectal bleeding and stool consistency (b). Additionally, the colon length (c) and colon weights (d) were also measured. Each conscious animal received a 100 µg oral dose of pORF5-mIL-10 in gelatin nanoparticles or NiMOS. Mean ± S.D. (n=4). (Reprinted with permission of Macmillan Publishers Ltd: Gene Therapy [38], copyright (2008)).
4. NANOPARTICLES FOR SYSTEMIC siRNA DELIVERY
4.1. Extracellular and Intracellular Barriers to siRNA Delivery
First discovered by Fire and Mello, RNA interference has emerged as a powerful post-transcriptional gene silencing approach. This strategy is especially appealing for therapeutic targets that are difficult for development of small molecule drugs. However, like other nucleic acid therapies, delivery of siRNA duplexes to the target tissue and subsequently cellular internationalization and availability in the cytosol for effective binding to RISC for mRNA degradation is a major hurdle before this experimental approach can be translated for routine clinical use [42].
Systemically administered siRNA faces multiple challenges in the extracellular environment and various barriers for the intracellular uptake before it reaches the site of action from its site of administration [43]. Following intravenous administration, large hydrophilic siRNA molecules are unstable in serum and rapidly degraded by nucleases and cleared from the body predominantly by the renal excretion route [44]. There is also non-specific distribution of these siRNAs throughout the body, which decreases to some extent the local concentration in the disease area. To reach the target cell, these siRNAs also need to overcome the blood vessel endothelial wall and multiple tissue barriers [45–47]. The negatively-charged siRNA molecules also do not efficiently cross biological membranes. For the small fraction of administered dose that reaches the cell of interest, intact siRNA molecules need to efficiently escape from the endosome/lysosome compartments for binding with RISC and exert biological function [45]. Naked siRNA duplexes can be administered locally in select areas of the body (e.g., eyes) [48]; however, they are not effective when administered systemically. As such, it is critical to develop safe and effective siRNA delivery vehicle in order to realize the tremendous clinical promise of RNA interference therapy.
Unlike plasmid DNA, siRNA duplexes need to be delivered to the cytosol for therapeutic effect. Due to the fact that one antisense strand can bind with multiple mRNA molecules, there is tremendous potency and nanomolar doses are effective for in vitro and in vivo silencing effects [42]. An ideal siRNA delivery system, therefore, should protect the labile payload from degradation in the systemic circulation and afford tissue- and cell-specific targeted delivery. Phagocytic cells, such as macrophages and monocytes, generally act as a significant immunological barrier as they are highly efficient in removing any foreign material including certain therapeutic nanocomplexes and macromolecules. The cells of the mononuclear phagocytic system also non-specifically bind to the unwanted negatively charged cells to ultimately cause toxicity [49, 50]. As previously described, the PEG modification minimizes RES uptake and help the particle to accumulate at tumor site [51].
Once the siRNA molecules reach the cell of interest, the carrier should be efficiently internalized and escape the endosome/lyososome compartments. If the carrier/siRNA system is positively charged, it interacts with the negatively charged cell membrane to form endocytic vesicle, although it involves additional non-specific interaction with other cells leading to toxicity. Alternatively, ligands or antibodies can be attached on the surface of the carriers to promote a specific receptor mediated endocytosis. If this complex unable to escape the endosome, it ultimately traffics through the compartments and fuses with lysosomes, where it is subjected to the low pH and enzyme-induced degradative conditions. In those cases, extra help is provided to disrupt the endosome membrane by having fusogenic peptide or pH-sensitive polymer/lipid backbone. Additionally, the delivery vehicle should afford release of intact siRNA duplex for binding to RISC [16, 42]. Above all, the delivery system should be safe to administer to patients on a chronic basis and allow for reproducible quality-assured large-scale manufacturing [52].
4.2 Gelatin Nanoparticles for HIF-1α Gene Silencing
Shah and Amiji used the unmodified or PEG-modified gelatin nanoparticles that were previously developed in their lab [24], for successful encapsulation and intracellular delivery of siRNA. Surface PEG modification was used to impart long-circulating properties and efficient delivery following non-specific endocytosis to HIF-1α activated tumor cells. As previously stated, at neutral pH, the uncharged gelatin particles trap siRNA by physical encapsulation [24]. The encapsulated siRNA was shown to be stable even in RNAse rich environment. Following encapsulation of HIF-1α siRNA in these particles, these were used to down-regulate the overly expressed HIF-1α in cancer cells. HIF-1α is known to activate the transcription of many genes that involve in propagation and progression of cancerous cells under hypoxic conditions. Several studies confirm that HIF-1α is present in many late stage aggressive carcinomas and over-expression of HIF-1α is correlated with poor prognosis and decreased survival [53]. Meanwhile, expression level of HIF-1α is also found to be related with expression of angiogenic markers such as VEGF and metastatic markers such as MMPs and Ki67 [53–55]. It was also observed that the expression levels of HIF-1α, VEGF, MMP2 and 9 were increased significantly under hypoxic conditions compared to their levels under normal conditions [56]. Following treatment of HIF-1α loaded Gelatin or PEG-modified gelatin particles in HIF-1α over expressed SKOV3 or MDA-MB-231 cells, it has demonstrated significant down regulation of HIF-1α [57]. With the decreased levels of HIF-1α, the downstream markers VEGF, MMP2 and MMP9 were significantly reduced [57] which may indicate the reversal of the aggressive phenotype of the tumors with HIF-1α knockdown, thus this treatment could offer a great potential for the therapy of aggressive tumors.
4.3 NiMOS for TNF-α Gene Silencing in Inflammatory Bowel Disease
Kriegel and Amiji [36] have recently extended the application of NiMOS for oral delivery siRNA duplexes for treatment of inflammatory bowel disease (IBD). For these studies, siRNA was encapsulated in the type B gelatin nanoparticles, which were further encased in PCL microspheres. Specifically, the gene for pro-inflammatory cytokine, tumor necrosis factor (TNF-α), which is known to be up-regulated in the IBD, was selected as a target for siRNA delivery. Unlike plain gelatin particles, the NiMOS resided longer time in small and large intestine and efficiently accumulated and released the siRNA at the inflamed site [36]. Acute colitis was established using dextran sulfate sodium (DSS) exposure to the Balb/c mice in their drinking water. NiMOS with TNF-α silencing siRNA was orally administered along with blank NiMOS and NiMOS with a scrambled siRNA duplexes as controls. Successful delivery was shown by efficient gene silencing in the large intestine, demonstrated by decreased colonic levels of TNF-α and other pro-inflammatory cytokines such as IL-1β, IFN-γ and chemokines (MCP-1) in mice treated with TNF-α /NiMOS compared to the other groups [58]. While the group that had DSS treatment, blank NiMOS or scramble siRNA/NiMOS treatment exhibited clear signs of inflammation by histological analysis, the tissues collected from the NiMOS/TNF-α treated group showed no signs of any inflammation or disruption of healthy tissue morphology [58]. The mice that had NiMOS/TNF-α also demonstrated Increase in body weights and colon length along with reduced myeloperoxidase activity compared to a significant reduction in colon length reported with DSS induction [58]. Mice treated with blank NiMOS and scramble siRNA/NiMOS, also exhibited statistically significant shortening just like the DSS treatment [58]. While the mice in all the treatment groups lost body weight until the last dose of treatment, the recovery was significantly faster in mice that had NiMOS/TNF-α treatment compared to all the other groups. Myeloperoxidases are the index markers of inflammation and the down regulation of these markers in NiMOS/TNF-α treated group compared to all the other groups further support less inflammation in mice in this group. These results all together suggest the potential of NiMOS as an oral therapeutic option for treatment of IBD.
5. CONCLUSIONS
Tremendous progress has been achieved in the development of non-condensing delivery systems during the last decade. More polymers have been utilized as non-condensing systems for gene delivery based on “physical encapsulation”. Besides, for both natural and synthetic polymers, safety, biodegradability and biocompatibility are the obligatory requirements in the selection of non-condensing polymers. Mainly, “physical encapsulation” relates to the following criteria. Firstly, hydrophilic polymers, such as gelatin could be used as a non-condensing system. During desolvation step, gelatin polymers would particulate to avoid hydrophobic interaction and nucleic acids tend to escape from the organic solvent to hydrophilic compartment, which helps them to get into gelatin nanoparticles [23]. Secondly, polymers with hydrogen donors and acceptors could form non-condensing system. For example, poly (N-vinyl pyrrolidone) could bind to the base pairs in the major groove of DNA and associate with nucleic acids by hydrogen bonds at pH 4–6 [59]. Lastly, for hydrophobic polymers, if they could form emulsions, they would also have the potential to form non-condensing system. For poly(D,L-Lactide-co-glycolide), polymers and nucleic acids were dissolved into different phases. With emulsion method, nucleic acids in aqueous phase could embed in the organic phase polymer and form microspheres or nanoparticles.[60] For polymers with above characteristics, further researches could be done to investigate the ability of these polymers as gene delivery vectors.
Although non-condensing polymers have shown certain encapsulation with nucleic acids, compared to viral vectors and cationic polymers, they still need to improve transfection efficiency and efficacy. Two main strategies have been applied during the development: passive targeting and active targeting. Pegylation is a commonly used passive targeting strategy, which conjugate PEG on the surface or out-layer of nanoparticles. Besides this, stimuli-responsive chemical modification is also generally applied. Lin and El-Sayed et al. have developed a cationic copolymer system for nucleic acids delivery, with pH-sensitive ethyl acrylic acid (EAA) monomers. [61] With this system, they have shown that this system is stable at physical pH and could successfully silence target after administration of nanoparticles into cells. Similar pH-sensitive monomers could also be conjugated into non-condensing copolymers. Meanwhile, disulfide crosslinks have been modified to response to high level of glutathione in cytoplasm [27]. Regarding to active targeting, antibodies and receptor specific peptides are conjugated on the surface of vectors. By association to the specific antigens or receptors on the target cells, nanoparticles could specifically enter cells through receptor mediated pathway and release the cargo into the target cell by endocytosis. In order to further improve the efficacy of gene delivery systems, more recent studies combine different strategies into the same system and produce multi-targeting delivery system. York and McCormick et al. have developed a HPMA copolymer system with multiconjugation of folate ligand, a cancer cell targeting moiety and siRNA. [62] With similar design, 3 or even more strategies could be incorporated together into the same system for develop a more efficient non-condensing system.
Furthermore, oral administration of non-condensing system has shown a strong promise. Properties of previous delivery systems have limited gene therapy to systemic and parenteral administration. However, NiMOS system successfully delivered the plasmid DNA and siRNA into intestinal cells and accomplished gene delivery by oral administration. NiMOS system is based on blend of two polymers.[36, 58] By using PCL polymer as outer layer, nucleic acids loaded gelatin nanoparticles could get protected from the gastric environment and transported to intestines. With mixture of different polymers, better encapsulation could be achieved [63], dual or multiple protections could be secured to ensure the stabilization of nucleic acids during delivery and new administration type such as oral dosage form could also be established.
Vila and Alonso et al. synthesized a copolymer poly(lactic acid)(PLA)-PEG and used this copolymer for intranasal gene delivery. [64, 65] By incorporating the advantages of these two non-condensing polymers, the new copolymer system could efficiently encapsulate the plasmid DNA and circulate for longer time to trigger the systemic immune response. Another group, Mao and Wand et al. have produced a triblock copolymer system, consisting of monomethoxy poly(ethylene glycol), poly(ε-caprolactone) and poly(2-aminoethyl ethylene phosphate).[66] They used these positive charged copolymers to self-assemble and encapsulate siRNA, successfully delivered them to tumor and, down-regulated acid ceramidase genes to ultimately inhibit tumor growth. [66] In their system, they enhanced the siRNA loading with positive charged portion, poly(2-aminoethyl ethylene phosphate) and prevent recognition by the mononuclear phagocyte system and early clearance by the shield of PEG chain. By synthesizing new copolymers, advantages of having different polymers could be collected and better delivery systems could be further established.
Although non-condensing polymers are not efficient enough to replace other gene delivery system for now, multiple techniques could be applied to improve these systems. The research on non-condensing gene therapy will move on with increased knowledge and innovative delivery strategies. With continuous development, non-condensing gene therapy will eventually lead toward better treatments for many diseases.
ACKNOWLEDGEMENTS
Our studies with systemically-administered type B gelatin nanoparticles for tumor-targeted gene delivery and transfection were supported by a grant (R01-CA95522) from the National Cancer Institute. The studies with nanoparticles-in-microsphere oral system (NIMOS) for gene and siRNA delivery in inflammatory bowel disease is funded by a grant (R01-DK080477) from the National Institute of Diabetes, and Digestive Diseases, and Kidney Diseases. The work described in this review was originally conducted by many postdoctoral associates and graduate students in the lab. Their tremendous effort in furthering the development of novel gene therapy strategies is greatly appreciated.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Hoag H. Gene therapy rising? Nature. 2005;435(7041):530–531. doi: 10.1038/nj7041-530a. [DOI] [PubMed] [Google Scholar]
- 2.gene. Gene Therapy Clinical Trials Worldwide. Journal of Gene Medicine. 2010 June [Google Scholar]
- 3.Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4(5):346–358. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- 4.Hollon T. Researchers and regulators reflect on first gene therapy death. Am J Ophthalmol. 2000;129(5):701. doi: 10.1016/s0002-9394(00)00442-6. [DOI] [PubMed] [Google Scholar]
- 5.Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med. 2001;7(1):33–40. doi: 10.1038/83324. [DOI] [PubMed] [Google Scholar]
- 6.Li S, Huang L. Nonviral gene therapy: promises and challenges. Gene Ther. 2000;7(1):31–34. doi: 10.1038/sj.gt.3301110. [DOI] [PubMed] [Google Scholar]
- 7.Kabanov AV, Felgner PL, Seymour LW. Self-assembling complexes for gene delivery: from laboratory to clinical trial. xvii. Chichester; New York: Wiley; 1998. 442 pp. [Google Scholar]
- 8.Merdan T, Kopecek J, Kissel T. Prospects for cationic polymers in gene and oligonucleotide therapy against cancer. Adv Drug Deliv Rev. 2002;54(5):715–758. doi: 10.1016/s0169-409x(02)00046-7. [DOI] [PubMed] [Google Scholar]
- 9.Kommareddy S, Tiwari SB, Amiji MM. Long-circulating polymeric nanovectors for tumor-selective gene delivery. Technol Cancer Res Treat. 2005;4(6):615–625. doi: 10.1177/153303460500400605. [DOI] [PubMed] [Google Scholar]
- 10.Lemieux P, et al. A combination of poloxamers increases gene expression of plasmid DNA in skeletal muscle. Gene Ther. 2000;7(11):986–991. doi: 10.1038/sj.gt.3301189. [DOI] [PubMed] [Google Scholar]
- 11.Morille M, et al. Progress in developing cationic vectors for non-viral systemic gene therapy against cancer. Biomaterials. 2008;29(24–25):3477–3496. doi: 10.1016/j.biomaterials.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 12.Bhavsar MD, Amiji MM. Polymeric nano- and microparticle technologies for oral gene delivery. Expert Opin Drug Deliv. 2007;4(3):197–213. doi: 10.1517/17425247.4.3.197. [DOI] [PubMed] [Google Scholar]
- 13.Amiji MM. Polymeric gene delivery: principles and applications. Boca Raton, Fla.: CRC Press; 2005. pp. 29–37. [Google Scholar]
- 14.Yuan F, et al. Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. Cancer Res. 1994;54(17):4564–4568. [PubMed] [Google Scholar]
- 15.Maeda H, et al. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 16.Cho K, et al. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14(5):1310–1316. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- 17.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392. [PubMed] [Google Scholar]
- 18.Pelicano H, et al. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25(34):4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 19.Bulmus V, et al. A new pH-responsive and glutathione-reactive, endosomal membrane-disruptive polymeric carrier for intracellular delivery of biomolecular drugs. J Control Release. 2003;93(2):105–120. doi: 10.1016/j.jconrel.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30(11):1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 21.Dong L, et al. A pH/enzyme-responsive tumor-specific delivery system for doxorubicin. Biomaterials. 2010;31(24):6309–6316. doi: 10.1016/j.biomaterials.2010.04.049. [DOI] [PubMed] [Google Scholar]
- 22.Napper DH. Colloid science. xvi. London; New York: Academic Press; 1983. Polymeric stabilization of colloidal dispersions; 428 pp. [Google Scholar]
- 23.Young S, et al. Gelatin as a delivery vehicle for the controlled release of bioactive molecules. J Control Release. 2005;109(1–3):256–274. doi: 10.1016/j.jconrel.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 24.Kaul G, Amiji M. Long-circulating poly(ethylene glycol)-modified gelatin nanoparticles for intracellular delivery. Pharm Res. 2002;19(7):1061–1067. doi: 10.1023/a:1016486910719. [DOI] [PubMed] [Google Scholar]
- 25.Kaul G, Amiji M. Biodistribution and targeting potential of poly(ethylene glycol)-modified gelatin nanoparticles in subcutaneous murine tumor model. J Drug Target. 2004;12(9–10):585–591. doi: 10.1080/10611860400013451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaul G, Amiji M. Tumor-targeted gene delivery using poly(ethylene glycol)-modified gelatin nanoparticles: in vitro and in vivo studies. Pharm Res. 2005;22(6):951–961. doi: 10.1007/s11095-005-4590-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Navarro J, et al. Changes in glutathione status and the antioxidant system in blood and in cancer cells associate with tumour growth in vivo. Free Radic Biol Med. 1999;26(3–4):410–418. doi: 10.1016/s0891-5849(98)00213-5. [DOI] [PubMed] [Google Scholar]
- 28.Kommareddy S, Amiji M. Preparation and evaluation of thiol-modified gelatin nanoparticles for intracellular DNA delivery in response to glutathione. Bioconjug Chem. 2005;16(6):1423–1432. doi: 10.1021/bc050146t. [DOI] [PubMed] [Google Scholar]
- 29.Kommareddy S, Amiji M. Poly(ethylene glycol)-modified thiolated gelatin nanoparticles for glutathione-responsive intracellular DNA delivery. Nanomedicine. 2007;3(1):32–42. doi: 10.1016/j.nano.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kommareddy S, Amiji M. Biodistribution and pharmacokinetic analysis of long-circulating thiolated gelatin nanoparticles following systemic administration in breast cancer-bearing mice. J Pharm Sci. 2007;96(2):397–407. doi: 10.1002/jps.20813. [DOI] [PubMed] [Google Scholar]
- 31.Kommareddy S, Amiji M. Antiangiogenic gene therapy with systemically administered sFlt-1 plasmid DNA in engineered gelatin-based nanovectors. Cancer Gene Ther. 2007;14(5):488–498. doi: 10.1038/sj.cgt.7701041. [DOI] [PubMed] [Google Scholar]
- 32.Magadala P, Amiji M. Epidermal growth factor receptor-targeted gelatin-based engineered nanocarriers for DNA delivery and transfection in human pancreatic cancer cells. AAPS J. 2008;10(4):565–576. doi: 10.1208/s12248-008-9065-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tobita K, et al. Epidermal growth factor receptor expression in human pancreatic cancer: Significance for liver metastasis. Int J Mol Med. 2003;11(3):305–309. [PubMed] [Google Scholar]
- 34.Roy K, et al. Oral gene delivery with chitosan--DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat Med. 1999;5(4):387–391. doi: 10.1038/7385. [DOI] [PubMed] [Google Scholar]
- 35.Jones DH, et al. Poly(DL-lactide-co-glycolide)-encapsulated plasmid DNA elicits systemic and mucosal antibody responses to encoded protein after oral administration. Vaccine. 1997;15(8):814–817. doi: 10.1016/s0264-410x(96)00266-6. [DOI] [PubMed] [Google Scholar]
- 36.Bhavsar MD, Amiji MM. Gastrointestinal distribution and in vivo gene transfection studies with nanoparticles-in-microsphere oral system (NiMOS) J Control Release. 2007;119(3):339–348. doi: 10.1016/j.jconrel.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 37.Bhavsar MD, Tiwari SB, Amiji MM. Formulation optimization for the nanoparticles-in-microsphere hybrid oral delivery system using factorial design. J Control Release. 2006;110(2):422–430. doi: 10.1016/j.jconrel.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 38.Bhavsar MD, Amiji MM. Oral IL-10 gene delivery in a microsphere-based formulation for local transfection and therapeutic efficacy in inflammatory bowel disease. Gene Ther. 2008;15(17):1200–1209. doi: 10.1038/gt.2008.67. [DOI] [PubMed] [Google Scholar]
- 39.Li MC, He SH. IL-10 and its related cytokines for treatment of inflammatory bowel disease. World J Gastroenterol. 2004;10(5):620–625. doi: 10.3748/wjg.v10.i5.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindsay J, et al. IL-10 gene therapy prevents TNBS-induced colitis. Gene Ther. 2002;9(24):1715–1721. doi: 10.1038/sj.gt.3301841. [DOI] [PubMed] [Google Scholar]
- 41.Lindsay JO, et al. IL-10 gene therapy is therapeutic for dextran sodium sulfate-induced murine colitis. Dig Dis Sci. 2004;49(7–8):1327–1334. doi: 10.1023/b:ddas.0000037830.22065.71. [DOI] [PubMed] [Google Scholar]
- 42.Dominska M, Dykxhoorn DM. Breaking down the barriers: siRNA delivery and endosome escape. J Cell Sci. 123(Pt 8):1183–1189. doi: 10.1242/jcs.066399. [DOI] [PubMed] [Google Scholar]
- 43.Xie FY, Woodle MC, Lu PY. Harnessing in vivo siRNA delivery for drug discovery and therapeutic development. Drug Discov Today. 2006;11(1–2):67–73. doi: 10.1016/S1359-6446(05)03668-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Layzer JM, et al. In vivo activity of nuclease-resistant siRNAs. Rna. 2004;10(5):766–771. doi: 10.1261/rna.5239604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Au JL, et al. Determinants of drug delivery and transport to solid tumors. J Control Release. 2001;74(1–3):31–46. doi: 10.1016/s0168-3659(01)00308-x. [DOI] [PubMed] [Google Scholar]
- 46.Jang SH, et al. Drug delivery and transport to solid tumors. Pharm Res. 2003;20(9):1337–1350. doi: 10.1023/a:1025785505977. [DOI] [PubMed] [Google Scholar]
- 47.Wang J, et al. Delivery of siRNA Therapeutics: Barriers and Carriers. Aaps J. 12(4):492–503. doi: 10.1208/s12248-010-9210-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Fougerolles A, et al. Interfering with disease: a progress report on siRNA-based therapeutics. Nat Rev Drug Discov. 2007;6(6):443–453. doi: 10.1038/nrd2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Omidi Y, Barar J, Akhtar S. Toxicogenomics of cationic lipid-based vectors for gene therapy: impact of microarray technology. Curr Drug Deliv. 2005;2(4):429–441. doi: 10.2174/156720105774370249. [DOI] [PubMed] [Google Scholar]
- 50.Pecot CV, et al. RNA interference in the clinic: challenges and future directions. Nat Rev Cancer. 11(1):59–67. doi: 10.1038/nrc2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8(2):129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leng Q, et al. Advances in Systemic siRNA Delivery. Drugs Future. 2009;34(9):721. doi: 10.1358/dof.2009.034.09.1413267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koukourakis MI, et al. Hypoxia inducible factor 1 alpha and 2 alpha expression is independent of anemia in patients with stage I endometrial cancer. Anticancer Res. 2002;22(6C):4137–4140. [PubMed] [Google Scholar]
- 54.Harris AL. Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2(1):38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 55.Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. Embo J. 1998;17(11):3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carmeliet P, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394(6692):485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 57.Shah S, A.M . Master's thesis. 2010. Hypoxia in Tumor Angiogenesis and Metastasis: Evaluation of VEGF and MMP Over-expression and Down-regulation of HIF-1a with RNAi in Hypoxic tumor cells. [Google Scholar]
- 58.Kriegel C, Amiji M. Oral TNF-alpha gene silencing using a polymeric microsphere-based delivery system for the treatment of inflammatory bowel disease. J Control Release. doi: 10.1016/j.jconrel.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mumper RJ, et al. Polyvinyl derivatives as novel interactive polymers for controlled gene delivery to muscle. Pharm Res. 1996;13(5):701–709. doi: 10.1023/a:1016039330870. [DOI] [PubMed] [Google Scholar]
- 60.Mundargi RC, et al. Nano/micro technologies for delivering macromolecular therapeutics using poly(D,L-lactide-co-glycolide) and its derivatives. J Control Release. 2008;125(3):193–209. doi: 10.1016/j.jconrel.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 61.Lin YL, et al. Degradable, pH-sensitive, membrane-destabilizing, comb-like polymers for intracellular delivery of nucleic acids. Biomaterials. 2010;31(27):7150–7166. doi: 10.1016/j.biomaterials.2010.05.048. [DOI] [PubMed] [Google Scholar]
- 62.York AW, Huang F, McCormick CL. Rational design of targeted cancer therapeutics through the multiconjugation of folate and cleavable siRNA to RAFT-synthesized (HPMA-s-APMA) copolymers. Biomacromolecules. 2010;11(2):505–514. doi: 10.1021/bm901249n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Csaba N, et al. PLGA:poloxamer and PLGA:poloxamine blend nanoparticles: new carriers for gene delivery. Biomacromolecules. 2005;6(1):271–278. doi: 10.1021/bm049577p. [DOI] [PubMed] [Google Scholar]
- 64.Vila A, et al. PLA-PEG particles as nasal protein carriers: the influence of the particle size. Int J Pharm. 2005;292(1–2):43–52. doi: 10.1016/j.ijpharm.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 65.Vila A, et al. Transport of PLA-PEG particles across the nasal mucosa: effect of particle size and PEG coating density. J Control Release. 2004;98(2):231–244. doi: 10.1016/j.jconrel.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 66.Mao CQ, et al. A biodegradable amphiphilic and cationic triblock copolymer for the delivery of siRNA targeting the acid ceramidase gene for cancer therapy. Biomaterials. 2011;32(11):3124–3133. doi: 10.1016/j.biomaterials.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 67.Administration, U.S.F.a.D. Database of Select Committee on GRAS Substances (SCOGS) Reviews. 2006 Report No. 58.
- 68.Ward AG, Courts A. Food science and technology. xvi. London; New York: Academic Press; 1977. The science and technology of gelatin; 564 pp. [Google Scholar]
- 69.Oupicky D, et al. Importance of lateral and steric stabilization of polyelectrolyte gene delivery vectors for extended systemic circulation. Mol Ther. 2002;5(4):463–472. doi: 10.1006/mthe.2002.0568. [DOI] [PubMed] [Google Scholar]
- 70.Oupicky D, Parker AL, Seymour LW. Laterally stabilized complexes of DNA with linear reducible polycations: strategy for triggered intracellular activation of DNA delivery vectors. J Am Chem Soc. 2002;124(1):8–9. doi: 10.1021/ja016440n. [DOI] [PubMed] [Google Scholar]
- 71.York AW, Kirkland SE, McCormick CL. Advances in the synthesis of amphiphilic block copolymers via RAFT polymerization: stimuli-responsive drug and gene delivery. Adv Drug Deliv Rev. 2008;60(9):1018–1036. doi: 10.1016/j.addr.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 72.Liu Y, et al. A new synthesis method and degradation of hyper-branched polyethylenimine grafted polycaprolactone block mono-methoxyl poly (ethylene glycol) copolymers (hy-PEI-g-PCL-b-mPEG) as potential DNA delivery vectors. Polymer. 2009;50(16):3895–3904. [Google Scholar]
- 73.Shuai X, et al. Supramolecular gene delivery vectors showing enhanced transgene expression and good biocompatibility. Bioconjug Chem. 2005;16(2):322–329. doi: 10.1021/bc0498471. [DOI] [PubMed] [Google Scholar]
- 74.Arote R, et al. A biodegradable poly(ester amine) based on polycaprolactone and polyethylenimine as a gene carrier. Biomaterials. 2007;28(4):735–744. doi: 10.1016/j.biomaterials.2006.09.028. [DOI] [PubMed] [Google Scholar]
- 75.Jung S, et al. Gene silencing efficiency of siRNA-PEG conjugates: effect of PEGylation site and PEG molecular weight. J Control Release. 2010;144(3):306–313. doi: 10.1016/j.jconrel.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 76.Kim HK, et al. Enhanced siRNA delivery using cationic liposomes with new polyarginine-conjugated PEG-lipid. Int J Pharm. 2010;392(1–2):141–147. doi: 10.1016/j.ijpharm.2010.03.047. [DOI] [PubMed] [Google Scholar]
- 77.Kunath K, et al. The structure of PEG-modified poly(ethylene imines) influences biodistribution and pharmacokinetics of their complexes with NF-kappaB decoy in mice. Pharm Res. 2002;19(6):810–817. doi: 10.1023/a:1016152831963. [DOI] [PubMed] [Google Scholar]
- 78.Vittaz M, et al. Effect of PEO surface density on long-circulating PLA-PEO nanoparticles which are very low complement activators. Biomaterials. 1996;17(16):1575–1581. doi: 10.1016/0142-9612(95)00322-3. [DOI] [PubMed] [Google Scholar]
- 79.Choi SW, et al. Multifunctional siRNA delivery system: polyelectrolyte complex micelles of six-arm PEG conjugate of siRNA and cell penetrating peptide with crosslinked fusogenic peptide. Biotechnol Prog. 2010;26(1):57–63. doi: 10.1002/btpr.310. [DOI] [PubMed] [Google Scholar]
- 80.Okada H, Toguchi H. Biodegradable microspheres in drug delivery. Crit Rev Ther Drug Carrier Syst. 1995;12(1):1–99. doi: 10.1615/critrevtherdrugcarriersyst.v12.i1.10. [DOI] [PubMed] [Google Scholar]
- 81.Tahara K, et al. Chitosan-modified poly(D,L-lactide-co-glycolide) nanospheres for improving siRNA delivery and gene-silencing effects. Eur J Pharm Biopharm. 2010;74(3):421–426. doi: 10.1016/j.ejpb.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Walter E, et al. Microencapsulation of DNA using poly(DL-lactide-co-glycolide): stability issues and release characteristics. J Control Release. 1999;61(3):361–374. doi: 10.1016/s0168-3659(99)00151-0. [DOI] [PubMed] [Google Scholar]
- 83.Newman KD, et al. Uptake of poly(D,L-lactic-co-glycolic acid) microspheres by antigen-presenting cells in vivo. J Biomed Mater Res. 2002;60(3):480–486. doi: 10.1002/jbm.10019. [DOI] [PubMed] [Google Scholar]
- 84.Lima KM, et al. Single dose of a vaccine based on DNA encoding mycobacterial hsp65 protein plus TDM-loaded PLGA microspheres protects mice against a virulent strain of Mycobacterium tuberculosis. Gene Ther. 2003;10(8):678–685. doi: 10.1038/sj.gt.3301908. [DOI] [PubMed] [Google Scholar]
- 85.Abbas AO, Donovan MD, Salem AK. Formulating poly(lactide-co-glycolide) particles for plasmid DNA delivery. J Pharm Sci. 2008;97(7):2448–2461. doi: 10.1002/jps.21215. [DOI] [PubMed] [Google Scholar]
- 86.Alakhov V, et al. Block copolymeric biotransport carriers as versatile vehicles for drug delivery. Expert Opin Biol Ther. 2001;1(4):583–602. doi: 10.1517/14712598.1.4.583. [DOI] [PubMed] [Google Scholar]
- 87.Kabanov AV, Alakhov VY. Pluronic block copolymers in drug delivery: from micellar nanocontainers to biological response modifiers. Crit Rev Ther Drug Carrier Syst. 2002;19(1):1–72. doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.10. [DOI] [PubMed] [Google Scholar]
- 88.Batrakova EV, Kabanov AV. Pluronic block copolymers: evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J Control Release. 2008;130(2):98–106. doi: 10.1016/j.jconrel.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mumper RJ, et al. Protective interactive noncondensing (PINC) polymers for enhanced plasmid distribution and expression in rat skeletal muscle. J Control Release. 1998;52(1–2):191–203. doi: 10.1016/s0168-3659(97)00215-0. [DOI] [PubMed] [Google Scholar]
- 90.Anwer K, et al. Systemic effect of human growth hormone after intramuscular injection of a single dose of a muscle-specific gene medicine. Hum Gene Ther. 1998;9(5):659–670. doi: 10.1089/hum.1998.9.5-659. [DOI] [PubMed] [Google Scholar]
- 91.Jin X, et al. A novel HIV T helper epitope-based vaccine elicits cytokine-secreting HIV-specific CD4+ T cells in a Phase I clinical trial in HIV-uninfected adults. Vaccine. 2009;27(50):7080–7086. doi: 10.1016/j.vaccine.2009.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Quezada A, et al. Safety toxicity study of plasmid-based IL-12 therapy in Cynomolgus monkeys. J Pharm Pharmacol. 2002;54(2):241–248. doi: 10.1211/0022357021778439. [DOI] [PubMed] [Google Scholar]