

Abstract
Cell line models have been widely used to investigate glioblastoma multiforme (GBM) pathobiology and in the development of targeted therapies. However, GBM tumours are molecularly heterogeneous and how cell lines can best model that diversity is unknown. In this report, we investigated gene expression profiles of three preclinical growth models of glioma cell lines, in vitro and in vivo as subcutaneous and intracerebral xenografts to examine which cell line model most resembles the clinical samples. Whole genome DNA microarrays were used to profile gene expression in a collection of 25 high-grade glioblastomas, and comparisons were made to profiles of cell lines under three different growth models. Hierarchical clustering revealed three molecular subtypes of the glioblastoma patient samples. Supervised learning algorithm, trained on glioma subtypes predicted the intracerebral cell line model with one glioma subtype (r = 0.68; 95% bootstrap CI –0.41, 0.46). Survival analysis of enriched gene sets (P < 0.05) revealed 19 biological categories (146 genes) belonging to neuronal, signal transduction, apoptosis- and glutamate-mediated neurotransmitter activation signals that are associated with poor prognosis in this glioma subclass. We validated the expression profiles of these gene categories in an independent cohort of patients from ‘The Cancer Genome Atlas’ project (r = 0.62, 95% bootstrap CI: –0.42, 0.43). We then used these data to select and inhibit a novel target (glutamate receptor) and showed that LY341595, a glutamate receptor specific antagonist, could prolong survival in intracerebral tumour-implanted mice in combination with irradiation, providing an in vivo cell line system of preclinical studies.
Keywords: supervised model, glioblastoma, orthotopic model, cell lines, xenograft
Introduction
Despite advances in neurooncology treatment regimens, the overall prognosis of patients with glioblastoma multiforme (GBM) tumours remains dismal. Aimed at developing more effective GBM treatment strategies, most preclinical studies have relied on glioma cell lines grown and maintained as in vitro (iv) monolayer cultures. This experimental model has served as an essential system for cancer drug development and has proven invaluable in identifying cytotoxic agents [1, 2]. However, its application is based on the critical assumption that the iv cells simulate the cancer phenotype in situ. Whereas this may be valid for some tumour types, for GBM this assumption remains questionable.
An obvious deficiency of the iv culture model is the failure to account for the microenvironment, which can significantly influence tumour cell biology and may thus affect the molecular determinants of treatment response. To account for the microenvironment, the subcutaneously (sc) grown tumour xenograft is frequently applied for therapy development and preclinical testing. However, with respect to GBMs, the sc microenvironment may not sufficiently mimic the unique circumstances of the growth within the brain. Along these lines, using gene expression profiles to compare tumour phenotypes, we have previously shown that the gene expression patterns of glioma cell lines U87 and U251 grown in vitro, subcutaneously and intracerebrally are significantly different [3]. These results also showed that although the two glioma lines have disparate gene expression profiles when evaluated in vitro or as subcutaneous tumours, under intracerebral conditions their profiles are very similar. Finally, as compared to the other growth conditions, the glioma cells grown intracerebrally (ic) expressed genes related to central nervous system development and function. Whereas these data suggested a profound effect of the orthotopic microenvironment on the phenotype of the glioma cell lines, whether this model system better simulates GBM in situ was unclear.
Thus, in the present work we sought to determine if the gene expression profile of the ic model replicates the gene expression profile of clinical samples implying a similar biology. Moreover, if the ic model represented the clinical situation then the ic gene expression profile could be used to select novel targeted agents to be used alone or in combination with irradiation. The data presented indicate that the ic gene expression profile is similar to the gene expression profile of a subset of clinical GBM specimens. Moreover, the gene expression profile and relevant gene ontology (GO) pathway analysis lead to a new molecular pathway whose inhibition in combination with irradiation inhibited ic tumour growth in our xenograft model.
Materials and methods
Cell lines
The human glioma cell lines U251 and U87 grown in vitro and in vivo as sc or ic implants were subjected to RNA extraction procedures and processed on microarray with 7680 human clones as described [3].
Study populations
Gene expression profiling data [4] from 25 grade IV glial brain tumours (GBM) and four normal brains using 42,000-feature cDNA microarrays (from total RNA) was used (GEO accession number GSE2223). All glioma specimens were subjected to standard WHO classification. The normal brain samples were used as a negative validation set. For positive validation, the RMA pre-processed Affymetrix U133A chip transcriptomic profiles of 255 glioblastomas generated by Broad Institute, were retrieved from Controlled-Access Data Tiers Portal (http://tcga-data.nci.nih.gov/tcga/findArchives.htm) of The Cancer Genome Atlas (TCGA) pilot project [5] upon National Human Genome Research Institute approval.
Gene expression profiling
For both cell line and GBM study sets, Universal Common Reference was used as reference RNA. For cell line data, each sample had a biological replicate and each replicate was run on duplicate arrays. Amplified RNA was labelled with Cy5-dUTP (experimental RNA) or Cy3-dUTP (Stratagene) and hybridized to 7680 human cDNA clone microarrays in duplicate. For glioma tissue samples, tumour samples and universal human reference total RNA were reverse transcribed using Cy5- and Cy3-specific primers and hybridized to 42,000-feature cDNA microarray. Ratios (sample/reference RNA) of background corrected and filtered raw data from cell lines (downloaded from mAdb repository available at http://madb.nci.nih.gov/) and GBM tissue samples were normalized using locally weighted scatterplot smoothing (LOWESS) algorithm. Datasets were exported to R bioconductor package (available at http://www.r-project.org) and all further statistical and computational analyses were performed in R (http://www.R-project.org) [6]. Raw data were further subjected to filtering if each row on the array contained greater than 60% missing values. The rest of the arrays were imputed for the missing values using k-nearest neighbour method and duplicate samples in cell line data were averaged. Because of different number of features for each sample set, only common features between the two arrays were used giving a total of 3322 genes. Finally, the gene expression data are log2 transformed and standardized to z-scores that will allow direct comparison between the datasets independent of original hybridization intensities.
Statistical methods: unsupervised analysis
Unsupervised analysis employs unbiased searches for the patterns of expression that can be used to develop hypothesis regarding the mechanistic associations between genotype and phenotype. These approaches are useful for identifying expression patterns within the set of differentially regulated genes that may have phenotypic significance. An unsupervised average linkage hierarchical clustering was used to separate GBM tissue samples into genotypic groups. Three major clusters were identified and these sets were used for supervised class prediction method. To visualize the data, we used classical multidimensional scaling, which takes as an input, a distance matrix between samples and returns a set of points in a lower dimensional space such that the distances between the points are approximately equivalent to original distances.
Class prediction
Supervised methods employ known phenotypic relationships between samples as well as information about molecular signatures that can be used for tumour classification. For supervised learning model, prediction analysis for microarrays (PAM) [7] method was employed to identify 10-fold cross-validated gene expression predictor for the hierarchical cluster defined classes. Cell line data were used to test the prediction model using the centroids found in training set after cross validation. To ascertain the relative degree of similarity between training and test classes, we employed Pearson correlation measure corrected by bootstrap confidence limits after 1000 randomizations.
Gene set enrichment analysis
To screen for differentially expressed groups of genes between computationally defined sample clusters we used the gene set analysis (GSA) methods proposed previously [8]. GSA was chosen because it uses a stringent max-mean algorithm to identify significantly differentially regulated genesets. The cutoff P value was adjusted to 0.05. We used precompiled module of gene lists from KEGG (Kyoto Encyclopaedia ofGenes and Genomes, http://www.genome.ad.jp/kegg) categories available at (MolSigDB2, Stanford repository).
Survival analysis
To discover clinically relevant signatures from the list of enriched gene sets, we employed Kaplan–Meier survival analysis. To determine the prognostic relevance we assessed the significance (P < 0.05) using log-rank test. For function level analysis, an overall multivariate survival profile was built using the genes belonging to each gene set and it was tested whether this signature added to the predictive significance. The significance of each gene was measured based on a univariate Cox proportional hazards regression of survival time versus the log expression level.
Clonogenic survival following radiation
Cultures were trypsinized to generate a single cell suspension and a specified number of cells were seeded into each well of a 6-well tissue culture plate. After allowing cells time to attach (4 hrs), cultures received LY341495 (1 μM) or dimethyl sulfoxide (DMSO) (vehicle control) for 16 hrs prior to irradiation: media were then removed and replaced with drug-free media. Ten to 14 days after seeding, colonies were stained with crystal violet; the number of colonies containing at least 50 cells was determined and surviving fractions calculated. Survival curves were then generated after normalizing for the amount of LY341495-induced cell death. Data presented are representative of three independent experiments.
Subcutaneous tumour model and tumour growth delay assay
Four- to 6-week-old female nude mice were caged in groups of five or less, and all animals were fed a diet of animal chow and water ad libitum. U251 tumour cells (5 × 106 cells) were injected sc into the right hind leg. When tumours grew to a mean volume of ∼172 mm3, mice were randomized into four groups: vehicle alone, LY351495 (10 mg/kg) alone, IR (4 Gy) alone or LY341495 + IR. The mice were given a single dose of LY341495 (10 mg/kg) by intraperitoneal injection 1 hr before local tumour irradiation (4 Gy). Irradiation was performed with a Pantak irradiator with animals restrained in a custom jig. To obtain tumour growth curves, perpendicular diameter measurements of each tumour were made every 2 days with digital callipers, and volumes were calculated using formula (L× W× W)/2. Tumours were followed until the group’s tumours reached a mean size of 2000 mm3. Specific tumour growth delay was calculated for each individual animal in a treatment group as the number of days for the mean of the treated tumours to grow to 2000 mm3 minus the number of days for the mean of the control group to reach the same size. Standard errors (S.E.) in days were calculated about the mean of the treated groups. Each experimental group contained 6 mice; 10 mice were included in the control group. All animal studies were conducted in accordance with the principles and procedures outlined in the NIH Guide for the Care and Use of Animals.
Intracerebral xenograft survival model
Four- to 6-week-old mice were anesthetized by i.p. injection of ketamine (83 mg/kg) and rompun (8.3 mg/kg) dissolved in saline. Using a blank syringe, 1 × 106 of U251 tumour cells suspended in 5 μl of phosphate-buffered saline were injected into the caudate nucleus at a depth of 3 mm from the dura over 10 min. The needle was left in place for 2 min. and then withdrawn slowly. The scalp wound was closed with 5–0 synthetic polydioxanone suture (PDS) suture. Surgery was performed with sterile technique. Mice were placed on a heating pad in sterile cages and allowed to awaken from anaesthesia. Three days after implantation, animals were randomized into one of four treatment groups: vehicle alone, LY341495 alone, IR alone or LY341495 + IR. The mice were given a single dose of LY341495 (10 mg/kg) by intraperitoneal injection 1 hr before local tumour irradiation (4 Gy). Each experimental group contained five mice. The day of tumour implantation was assigned as day 0.
Results
Selection of a representative subset of genes
To maximize cross platform compatibility all datasets were standardized as described in the methods. Initially, common genes were identified by matching Human Genome Organization nomenclature gene symbols between the training and test sets. To compare our previously published glioma cell line gene expression data (test set) with that derived from clinical samples (training set) and to reduce platform dependent variability, we used cDNA microarray datasets for the initial analysis. The test set was derived from glioma cell lines grown under three experimental conditions iv, sc and ic [3] and the training set was derived from clinical tumour samples from patients with GBM [9]. The 3322 genes common between these two datasets were used for further analysis. To determine whether this common gene set divided the patient samples into categories, as reported for other clinical datasets, we performed an unsupervised agglomerative hierarchical cluster analysis showing that the clinical samples sub-grouped into three major classes (Fig. 1A): these were arbitrarily named GBM1, GBM2 and GBM3 for further analysis. To determine whether this classification was influenced by the subset of genes selected, the clustering was verified by normalizing the initial data before selecting the common genes, which lead to a similar clustering pattern (data not shown).
Fig 1.

GBM sample classification and comparison of cell line models using a subset of common genes between two array platforms (n = 3322). (A) Identification of three major cluster groups of GBM patient samples by hierarchical agglomerative cluster analysis. The dendrogram was created using 1-correlation distance metric and average linkage measure. (B–D) Scatterplots comparing U251 (x-axis) and U87 (y-axis) gene expression of cell grown in vitro (B) and in vivo as sc (C) and ic (D) tumours in mice. Pearson correlation coefficients were calculated for each comparison and listed in each plot.
We have previously shown that the microarray profiles for glioma cells grown as orthotopic tumours are more similar to each other than they are when grown as subcutaneous tumours or as in vitro cultures [3]. To determine if this pattern was maintained when using the 3322 gene subset we used scatterplots to directly compare the expression levels of the selected genes in the U251 and U87 cell lines. For each growth condition, the gene expression profile of U251 cells was plotted against the corresponding profile of U87 cells (Fig. 1B–D). As a measure of similarity of gene expression profiles, Pearson correlation coefficients were calculated. As shown in Figure 1D, the gene expression profiles for U251 and U87 cells grown as orthotopic tumours were highly concordant (r = 0.98) in comparison to cells grown as subcutaneous tumours (r = 0.56) or cells grown as monolayer cultures (r = 0.31) indicating that the 3322 gene subset used for this analysis did not deviate significantly from our earlier reported results.
Supervised analysis of GBM classes predicts the orthotopic model to subset GBM1
Having established that this gene list divides the clinical GBM specimens into three categories and accurately reproduces the delineation of glioma cell lines as a function of growth condition, we determined whether there were any similarities between GBM subgroups and cell line expression profiles. Towards this end, a robust subset of genes was selected that defines each GBM subgroup using the PAM method [7] with a 10-fold cross validation to build a class prediction model. This analysis identified 1215 genes (Table S1) (GBM1 = 614; GBM2 = 723; GBM3 = 577 genes) that accurately predicted the three GBM classes with zero misclassifications (Fig. 2A). As shown in Table 1A, by using the classifier model, the in vitro cell lines were predicted into two different GBM sets (GBM2 and GBM3) consistent with disparate expression profiles for those cell lines grown in monolayer culture. The cell lines grown as sc tumours were predicted with the GBM3 subset and the cell lines grown as orthotopic ic tumours were predicted with the GBM1 subset. To quantitate the degree of similarity between the sample classes from each of the datasets, we determined the Pearson correlation coefficient followed by bootstrap analysis between each of the GBM subgroups and each of the cell line growth conditions for the 1215 gene subset selected by the PAM model. As shown in Table 1B, there was a high correlation between the tumours grown ic and the clinical GBM1 subgroup, a nominal correlation between sc model cell lines and GBM3 subgroup, whereas the rest of paired sample measurements had correlations that overlapped with the bootstrap confidence intervals signifying that the correlations were no better than random associations. As an additional verification of the similarity between the cell lines grown as ic tumours and GBM1, the correlation between the cell line growth environment and the GBM subgroups was analysed by heatmap (Fig. 2B), and multidimensional analysis (Fig. 2C). Taken together, these three analyses show a strong association of the gene expression profile derived from cell lines grown as orthotopic ic tumours with the GBM1 subgroup of clinical samples.
Fig 2.
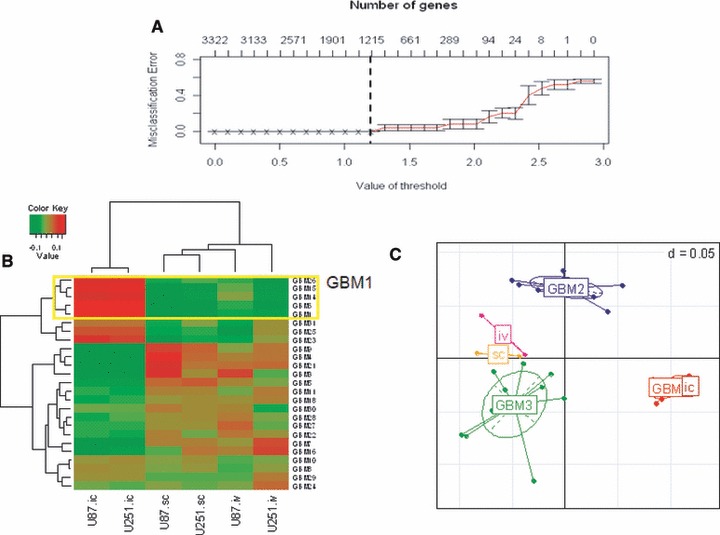
Supervised model of GBM sub class and association of cell line models using the subset of genes selected by the model (n = 1215). (A) Cross-validated error curves from the nearest shrunken centroid classifier with the three subclasses of GBM patient samples. (B) Heatmap of correlation between GBM and cell line sample data showing a close cluster between GBM1 with ic model (C) Principle component analysis showing the association of ic cell lines with GBM1 in comparison with other samples in the dataset.
Table 1.
Classification and prediction of training and test data using supervised PAM model
| (a) Prediction of sample classes using gene profiles from supervised learning model. | |||
|---|---|---|---|
| S. no. | True | Predicted | |
| 1 | U251 (iv) | GBM2 | |
| 2 | U87 (iv) | GBM3 | |
| 3 | U251 (sc) | GBM3 | |
| 4 | U87 (sc) | GBM3 | |
| 5 | U251 (ic) | GBM1 | |
| 6 | U87 (ic) | GBM1 | |
| (b) Pearson correlation measures between GBM classes and the average cell line data for each growth condition. 95% C.I. after bootstrap permutation analysis are shown as numbers within parenthesis next to r-value. | |||
| ic | sc | iv | |
| GBM1 | 0.73 (−0.37, 0.44) | −0.54 (−0.35, 0.29) | −0.49 (−0.32, 0.28) |
| GBM2 | −0.12 (−0.38, 0.43) | 0.03 (−0.34, 0.33) | 0.15 (−0.31, 0.29) |
| GBM3 | −0.47 (−0.38, 0.44) | 0.4 (−0.35, 0.31) | 0.26 (−0.31, 0.27) |
Gene set analysis similarities and survival significance of ic model and GBM1 tumours
To identify the underlying pathways or gene modules that are similar between the ic tumours and the GBM1 subgroup we used the gene set analysis (GSA) algorithm. GSA uses lists of genes that are empirically related to the transcriptional profiles between two diverse datasets [8]. As shown in Figure 3, GSA computed 36 unique, statistically significant, KEGG-defined gene sets. The corresponding heatmap, plotted using the feature scores, indicated that the ic tumours had profiles that clustered with the profiles from the tumours in the GBM1 subclass (yellow box). To assess if the pathways had potential clinical significance, the associated survival times for each clinical subgroup was estimated by Kaplan–Meier product-limit method, and the survival distributions between groups were compared using log-rank test statistic. To view the gene functional organization, we plotted this output next to the hierarchically clustered KEGG categories. Only KEGG categories with a significant difference in survival between the groups of GBM would result in significant P values (P ≤ 0.05); thus, this approach highlights large sets of co-regulated KEGG categories of the GBM1 subgroup whose expression is associated with patient survival. A representative Kaplan–Meier analysis using four KEGG categories among the three GBM subgroups is available as Figure S2A–D. We identified a total of 19 KEGG categories with a significant survival difference between the GBM1–3 subgroups (P < 0.05). Sixteen of these 19 categories were up regulated in the GBM1 subgroup and the ic model. For comparison, we have computed the log-rank test statistic for all the genes associated with the 36 KEGG categories shown in Figure 3, and these data are available in Table S2. The pathways associated with survival included many of the previously confirmed signatures such as neurogenesis [10], growth factor-induced signal transduction [11, 12], processes that control cell cycle progression such as p53 and apoptosis pathways [12, 13]. Additional top pathways from our analysis that are less well described included long-term depression, long-term potentiation and glutamate-mediated neurotransmitter activation signals.
Fig 3.
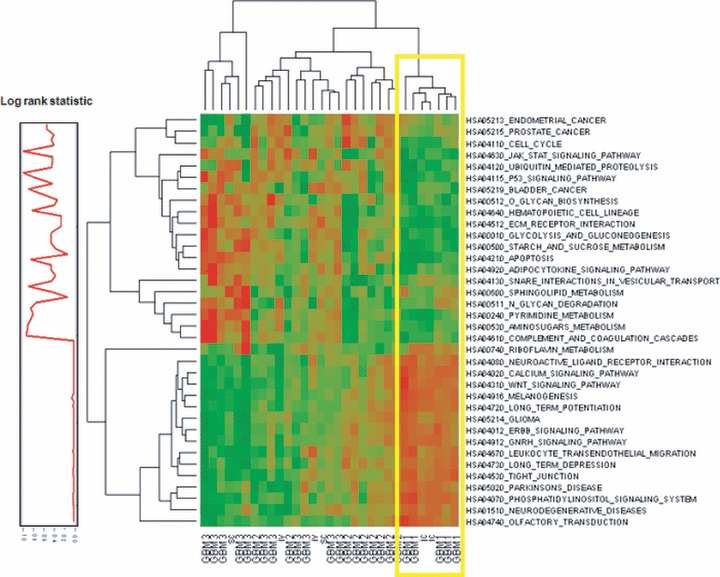
Heatmap of co-regulated categories that correlate with patient survival. Gene sets were enriched for functional categories using GSA method. The resulting feature score for each of the categories is used to build the heatmap. The log-rank statistic was calculated for each category using the 25 GBMs for which we could obtain survival data. These scores were plotted adjacent to the heatmap of functional categories.
Independent validation of GBM subtype classifiers
To determine if the PAM model classifiers of GBM subclasses is independent of the dataset used and to determine if the classifiers correlate with the most comprehensive malignant glioma tumour data, we selected 255 GBM expression profiles from the TCGA portal (Affymetrix platform) for conformation [5]. We used our classifiers to stratify TCGA sample data using the subset of 3322 matching genes used to train our model. Interestingly, the proportion of samples predicted into the three GBM subtypes 1 to 3 is 0.2:0.4:0.4 that is the same distribution of samples assigned in our training model. Next, Pearson correlations and bootstrap 95% confidence intervals were calculated measuring the similarity in the subset of gene expression patterns corresponding to the 19 clinically significant categories. As shown in Table 2, comparisons were made between the cell line growth models, the GBM test dataset, the normal brain as negative validation dataset and the TCGA GBM as independent validation dataset. There was a strong correlation between the ic cell line model with both GBM1 (r = 0.68) and TCGA1 (r = 0.62). A similar comparison with the normal brain dataset showed no correlation between any of the cell line growth conditions and the normal brain (correlation values near zero). As expected, when we tested for similarity at the pathway level, a similar trend in correlations was observed between cell line ic model with both GBM1 and TCGA1 whereas the correlations with normal brain sample data either had a negative or near zero values (Fig. S1 and Table S3). Taken together these results suggest that the ic model of cell line growth has the most similar profile to a subset of patient samples irrespective of source or platform type.
Table 2.
Validation of gene functional categories enriched in GBM classes and ic model with independent datasets. 95% C.I. after bootstrap permutation are shown as numbers within parenthesis next to r-value.
| ic | Sc | iv | |
|---|---|---|---|
| GBM1 | 0.68 (−0.41,0.46) | −0.6 (−0.47,0.41) | −0.34 (−0.32,0.3) |
| GBM2 | −0.02 (−0.4,0.46) | −0.04 (−0.47,0.41) | 0.07 (−0.29,0.28) |
| GBM3 | −0.47 (−0.45,0.45) | 0.47 (−0.42,0.44) | 0.19 (−0.28,0.29) |
| BRAIN | −0.1 (−0.43,0.43) | 0.01 (−0.46,0.38) | 0.13 (−0.31,0.29) |
| TCGA1 | 0.62 (−0.42,0.43) | −0.61 (−0.44,0.4) | −0.25 (−0.28,0.3) |
| TCGA2 | 0.16 (−0.44,0.45) | −0.16 (−0.44,0.42) | −0.06 (−0.31,0.31) |
| TCGA3 | −0.51 (−0.43,0.44) | 0.51 (−0.44,0.43) | 0.20 (−0.29,0.31) |
Evaluation of a glutamate receptor antagonist as a radiation modifier
If the ic growth of glioma cells provides a more biologically accurate model of GBM in situ, as suggested by the data presented above, then the gene expression profiles generated from the ic xenografts may provide unique insights into the development of clinically relevant treatment strategies. To illustrate this potential with respect to radiosensitization we focused on the glutamate receptor. As shown in Figure 3, multiple KEGG pathways contain glutamate receptor (neuroactive ligand receptor interaction, long-term potentiation and neurodegenerative diseases) as well as signalling molecules downstream of the glutamate receptor (calcium signalling pathway and phosphatidylinositol signalling system) implying that inhibition of glutamate receptor may lead to growth inhibition. When glutamate receptor is stimulated by ligand, the signal is propagated through both the MAPK and PI3K pathways [14]. As inhibition of MAPK and PI3K has been shown to enhance tumour radiosensitivity [15] we suggested that inhibition of glutamate receptor signalling may sensitize ic tumours to irradiation. To test whether inhibiting the activity of glutamate receptor enhances radiosensitivity under each growth condition we used LY341495, which has previously been shown to inhibit glutamate receptor activity in vitro and to inhibit the growth of glioma sc xenografts [16]. The effect of LY341495 on radiosensitivity was first defined in vitro according to clonogenic survival analysis. Exposure of U87 and U251 cells to LY341495 (1 μM), a dose previously shown to inhibit downstream signalling of mGlu2/3 [17], for 16 hrs resulted in surviving fractions of 67% and 95%, respectively, which is an appropriate range for evaluating clonogenic survival in combination with irradiation. For the combination studies, cells were exposed to drug for 16 hrs, irradiated and colonies counted 12 days later. As shown in Figure 4A and B, there was no enhancement of radiosensitivity in either cell line when grown as in vitro mono-layer cultures. To determine whether LY341495 enhances tumour cell radiosensitivity under in vivo growth conditions we focused on U251 cells. Initially, the effect of the glutamate receptor inhibitor on U251 cells grown as sc xenografts was determined using a tumour growth delay assay. Mice bearing U251 sc xenografts (172 mm3) were randomized into one of four groups: vehicle; LY341495 only (10 mg/kg); irradiation only (4 Gy) and LY341495 (10 mg/kg) given as an i.p. injection 1 hr prior to irradiation (4 Gy). As shown in Figure 4C, LY351495 had no significant effect on the radiosensitivity of U251 cells grown sc as determined by tumour growth delay. The combination of LY351495 was then evaluated against U251 cell grown as ic xenografts following the same treatment protocol used for sc tumours. Specifically, 5 days after undergoing intracerebral implantation, groups of five mice were randomized into control, LY341495 only (10 mg/kg), irradiation only (4 Gy) or LY341495 (10 mg/kg) given as an i.p. injection 1 hr prior to irradiation (4 Gy). As shown in the Kaplan–Meier plot in Figure 4D, there was a significant prolongation of survival in the combination group consistent with LY341495-induced radiosensitization. Thus, these results suggest that the glutamate receptor provides a novel target for GBM radiosensitization, information that would not be generated from the more standard glioma models of in vitro monolayer culture and sc xenografts.
Fig 4.

Effect of glutamate receptor antagonist LY341495 on tumour cell radiosensitivity. (A, B) Cells were exposed to the designated concentrations of drug for 16 hrs before irradiation. Colony-forming efficiency was determined 10–14 days later and survival curves were generated after normalizing for the cytotoxicity induced by drug alone. PE: plating efficiency; IR: irradiation. (C) Tumour volume in mice after treatment with LY341495 ± irradiation. When tumours reached 172 mm2, mice were randomized into four groups: vehicle, LY341495 (10 mg/kg), irradiation (4 Gy) or drug/irradiation. To obtain tumour growth curve, perpendicular diameter measurements of each tumour were measured every two days. (D) Kaplan–Meier survival curve showing survival of mice with implanted ic with U251 cells and randomized to either vehicle, LY341495 (10 mg/kg), irradiation (4 Gy) or drug/irradiation. Log-rank statistic was computed comparing the survival curves and is shown in the figure.
Discussion
Gene expression profiling of human GBM specimens has recently led to the identification of molecular subgroups prognostic of patient survival, independent of traditional factors such as age, resection status and mental status [18-20]. This categorization of GBMs into distinct molecular entities has suggested that a given subgroup may preferentially respond to a specific targeted therapy [21, 22]. Although it is possible to test suggested targets through clinical trials, the availability of a model system that simulates a given GBM subgroup would be of considerable benefit in testing novel therapeutic strategies. Towards this end, the data presented here indicate that the gene expression profiles generated for the established glioma cell lines U87 and U251 grown as orthotopic xenografts in nude mice recapitulate those of a specific GBM subgroup.
Our classification system of GBM clinical samples was based entirely on gene expression profiles without prior knowledge of pathological, genetic or clinical subgroups, allowing the unbiased selection of GBM subtype classifiers. Several studies attempted to classify gliomas to identify differentially expressed genes among morphologically defined glioma subsets [23]. These studies confirmed that morphological differences among gliomas are reflected at the mRNA transcript level and that differentially expressed genes could be utilized to distinguish among morphologically defined subtypes. Recently, two large transcriptome studies examined specimens from patients with high-grade gliomas. Phillips et al., using only WHO grade III and IV astrocytomas, showed that the gene expression profiles divided the tumours into three molecular subclasses (proneural, proliferation and mesenchymal) with a significantly longer survival in the proneural subgroup [21]. However, the majority of the grade 3 astrocytomas were in the proneural subgroup consistent with the longer survival in this subgroup. The second study by Li et al., using both oligodendrogliomas and astrocytomas, showed that transcriptome profiles divided into two oligodendroglioma subgroups (OA and OB) and four glioma subgroups (GA1, GA2, GB1 and GB2) with the GA1 subgroup having a significantly shorter survival [24]. In the GA1–GB2 subgroups there was a range in the number of grade IV astrocytomas in each group (53–73%). Although we only evaluated WHO grade IV astrocytomas, there are similarities between our dataset and the larger glioma classes from Li et al. as well as their glioma subclasses (GA1–GB2). Gene set analysis derived pathways separating the larger glioma class included DNA damage, Cell cycle regulation and Proliferation and for the GB1 subclass Neuronal function and G-protein signalling, which were similar to our IPA-derived GBM1 pathways including Nervous system development, DNA repair/recombination and Cellular assembly. Determining whether these similarities between GBM1 and GB1 can be extended to the level of gene expression will require additional analyses.
Given that similar gene expression profiles imply similar biology, if the ic xenograft transcriptome recapitulates that of the GBM1 clinical subset, then the orthotopic growth of glioma cells may also provide a preclinical model for testing targeted therapies. As an initial investigation into this potential application we focused on the glutamate receptor. Metabotropic glutamate receptors have previously been suggested as possible therapeutic targets for GBMs [16, 17]. The data presented here indicate that the glutamate receptor antagonist LY341495 enhanced the radiosensitivity of a glioma cell line grown as an ic xenograft, yet had no effect on the radiosensitivity of glioma cells grown in vitro or as subcutaneous leg xenografts. These results are consistent with the unique biology of the ic xenografts as compared to the more standard experimental growth conditions and provide support for the use of the orthotopic model in the development of novel therapies for GBM1 subgroup.
The training model used in this study was based on the limited number of genes (3322), i.e. those common to the two experimental datasets. It would appear that further analyses using microarray platforms encompassing larger gene sets would allow for the identification of additional genes/pathways that may be of significance. Moreover, whereas U87/U251 xenografts were found to model a single subgroup of GBMs, it would seem that extending these studies to ic xenografts initiated from additional cell lines including lines derived from GBM tumour stem cells may provide experimental models simulating other subgroups. Clearly, the goal would be to establish preclinical models with which to develop targeted therapies against each GBM subtype.
Acknowledgments
This research was supported by Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Supporting Information
Heatmap of pairwise correlations of ic cell line model and tissue samples.
Pairwise Pearson correlation coefficients were estimated between all samples under study using the GSA scores. The correlation measures were plotted as heatmap to show the relative association of ic cell line model with GBM1 and TCGA1 subsets of clinical samples.
Survival curves of gene set functions showingdifferences between three GBM subtypes. Kaplan–Meier analyseswere conducted on four representative pathways with statisticallysignificant differences (a–b) and insignificant differences(c–d) between the GBM subsets. Differences in survivalbetween the three GBM subsets are summarized by log-rank teststatistic (insert in legend title) in all comparisons.
List of significant genes identified by PAM analysis. The scores are the shrunken centroid measures from the model for each group.
Results of gene set enrichment analysis.Log-rank test statistic for genes associated with each of thesignificant pathway identified in Fig. 3. The ‘N’ column is the number of genes from the KEGG category found in the study.
Summary of pairwise correlations showing similarity between i.c. model with subset of GBM samples from test and validation sets.
References
- 1.Alley MC, Scudiero DA, Monks A, et al. Feasibility of drug screening with panels of human tumour cell lines using a microculture tetrazolium assay. Cancer Res. 1988;48:589–601. [PubMed] [Google Scholar]
- 2.Deschavanne PJ, Fertil B. A review of human cell radiosensitivity in vitro. Int J Radiat Oncol Biol Phys. 1996;34:251–66. doi: 10.1016/0360-3016(95)02029-2. [DOI] [PubMed] [Google Scholar]
- 3.Camphausen K, Purow B, Sproull M, et al. Influence of in vivo growth on human glioma cell line gene expression: convergent profiles under orthotopic conditions. Proc Natl Acad Sci USA. 2005;102:8287–92. doi: 10.1073/pnas.0502887102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bredel M, Bredel C, Juric D, et al. Functional network analysis reveals extended gliomagenesis pathway maps and three novel MYC-interacting genes in human gliomas. Cancer Res. 2005;65:8679–89. doi: 10.1158/0008-5472.CAN-05-1204. [DOI] [PubMed] [Google Scholar]
- 5.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.R Development Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2011. [Google Scholar]
- 7.Tibshirani R, Hastie T, Narasimhan B, et al. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci USA. 2002;99:6567–72. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Efron B, Tibshirani R. On testing the significance of sets of genes. Ann Appl Stat. 2007:107–29. [Google Scholar]
- 9.Bredel M, Bredel C, Juric D, et al. High-resolution genome-wide mapping of genetic alterations in human glial brain tumours. Cancer Res. 2005;65:4088–96. doi: 10.1158/0008-5472.CAN-04-4229. [DOI] [PubMed] [Google Scholar]
- 10.Freije WA, Castro-Vargas FE, Fang Z, et al. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004;64:6503–10. doi: 10.1158/0008-5472.CAN-04-0452. [DOI] [PubMed] [Google Scholar]
- 11.Liang Y, Diehn M, Watson N, et al. Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme. Proc Natl Acad Sci USA. 2005;102:5814–9. doi: 10.1073/pnas.0402870102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohgaki H, Dessen P, Jourde B, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64:6892–9. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe K, Sato K, Biernat W, et al. Incidence and timing of p53 mutations during astrocytoma progression in patients with multiple biopsies. Clin Cancer Res. 1997;3:523–30. [PubMed] [Google Scholar]
- 14.Bruno V, Battaglia G, Copani A, et al. Metabotropic glutamate receptor subtypes as targets for neuroprotective drugs. J Cereb Blood Flow Metab. 2001;21:1013–33. doi: 10.1097/00004647-200109000-00001. [DOI] [PubMed] [Google Scholar]
- 15.Chung EJ, Brown AP, Asano H, et al. In vitro and in vivo radiosensitization with AZD6244 (ARRY-142886), an inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2 kinase. Clin Cancer Res. 2009;15:3050–7. doi: 10.1158/1078-0432.CCR-08-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arcella A, Carpinelli G, Battaglia G, et al. Pharmacological blockade of group II metabotropic glutamate receptors reduces the growth of glioma cells in vivo. Neuro Oncol. 2005;7:236–45. doi: 10.1215/S1152851704000961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.D’Onofrio M, Arcella A, Bruno V, et al. Pharmacological blockade of mGlu2/3 metabotropic glutamate receptors reduces cell proliferation in cultured human glioma cells. J Neurochem. 2003;84:1288–95. doi: 10.1046/j.1471-4159.2003.01633.x. [DOI] [PubMed] [Google Scholar]
- 18.Liang Q, Xiong H, Gao G, et al. Inhibition of basigin expression in glioblastoma cell line via antisense RNA reduces tumour cell invasion and angiogenesis. Cancer Biol Ther. 2005;4:759–62. doi: 10.4161/cbt.4.7.1828. [DOI] [PubMed] [Google Scholar]
- 19.Mischel PS, Shai R, Shi T, et al. Identification of molecular subtypes of glioblastoma by gene expression profiling. Oncogene. 2003;22:2361–73. doi: 10.1038/sj.onc.1206344. [DOI] [PubMed] [Google Scholar]
- 20.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–73. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 22.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–73. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vitucci M, Hayes DN, Miller CR. Gene expression profiling of gliomas: merging genomic and histopathological classification for personalised therapy. Br J Cancer. 104:545–53. doi: 10.1038/sj.bjc.6606031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li A, Walling J, Ahn S, et al. Unsupervised analysis of transcriptomic profiles reveals six glioma subtypes. Cancer Res. 2009;69:2091–9. doi: 10.1158/0008-5472.CAN-08-2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Heatmap of pairwise correlations of ic cell line model and tissue samples.
Pairwise Pearson correlation coefficients were estimated between all samples under study using the GSA scores. The correlation measures were plotted as heatmap to show the relative association of ic cell line model with GBM1 and TCGA1 subsets of clinical samples.
Survival curves of gene set functions showingdifferences between three GBM subtypes. Kaplan–Meier analyseswere conducted on four representative pathways with statisticallysignificant differences (a–b) and insignificant differences(c–d) between the GBM subsets. Differences in survivalbetween the three GBM subsets are summarized by log-rank teststatistic (insert in legend title) in all comparisons.
List of significant genes identified by PAM analysis. The scores are the shrunken centroid measures from the model for each group.
Results of gene set enrichment analysis.Log-rank test statistic for genes associated with each of thesignificant pathway identified in Fig. 3. The ‘N’ column is the number of genes from the KEGG category found in the study.
Summary of pairwise correlations showing similarity between i.c. model with subset of GBM samples from test and validation sets.