

Abstract
A mechanistic model is formulated to account for the high reactivity of chelating azides (organic azides capable of chelation-assisted metal coordination at the alkylated azido nitrogen position) and copper(II) acetate (Cu(OAc)2) in copper(II)-mediated azide-alkyne cycloaddition (AAC) reactions. Fluorescence and 1H NMR assays are developed for monitoring the reaction progress in two different solvents – methanol and acetonitrile. Solvent kinetic isotopic effect and pre-mixing experiments give credence to the proposed different induction reactions for converting copper(II) to catalytic copper(I) species in methanol (methanol oxidation) and acetonitrile (alkyne oxidative homocoupling), respectively. The kinetic orders of individual components in a chelation-assisted, copper(II)-accelerated AAC reaction are determined in both methanol and acetonitrile. Key conclusions resulting from the kinetic studies include (1) the interaction between copper ion (either in +1 or +2 oxidation state) and a chelating azide occurs in a fast, pre-equilibrium step prior to the formation of the in-cycle copper(I)-acetylide, (2) alkyne deprotonation is involved in several kinetically significant steps, and (3) consistent with prior experimental and computational results by other groups, two copper centers are involved in the catalysis. The X-ray crystal structures of chelating azides with Cu(OAc)2 suggest a mechanistic synergy between alkyne oxidative homocoupling and copper(II)-accelerated AAC reactions, in which both a bimetallic catalytic pathway and a base are involved. The different roles of the two copper centers (a Lewis acid to enhance the electrophilicity of the azido group and a two-electron reducing agent in oxidative metallacycle formation, respectively) in the proposed catalytic cycle suggest that a mixed valency (+2 and +1) dinuclear copper species be a highly efficient catalyst. This proposition is supported by the higher activity of the partially reduced Cu(OAc)2 in mediating a 2-picolylazide-involved AAC reaction than the fully reduced Cu(OAc)2. Finally, the discontinuous kinetic behavior that has been observed by us and others in copper(I/II)-mediated AAC reactions is explained by the likely catalyst disintegration during the course of a relatively slow reaction. Complementing the prior mechanistic conclusions drawn by other investigators which primarily focus on the copper(I)/alkyne interactions, we emphasize the kinetic significance of copper(I/II)/azide interaction. This work not only provides a mechanism accounting for the fast Cu(OAc)2-mediated AAC reactions involving chelating azides, which has apparent practical implications, but suggests the significance of mixed-valency dinuclear copper species in catalytic reactions where two copper centers carry different functions.
Keywords: Copper-catalyzed azide-alkyne cycloaddition, click chemistry, mixed-valency dinuclear copper complex, copper(II) acetate, alkyne oxidative homocoupling, Glaser reaction
Introduction
The discovery of copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) reaction by Fokin/Sharpless1 and Meldal2 groups has stimulated advances across many scientific disciplines.3–6 It is a simple yet powerful tool to put molecular pieces together, for instance, in applications of bioconjugation,7 surface modification,8 and syntheses of new polymeric materials.9 One of the most astonishing facts of the CuAAC reaction is that it proceeds under a large number of conditions that are developed,5 not necessarily to make the reaction itself faster, but to accommodate various stringent requirements rarely faced in a laboratory of traditional synthetic organic chemistry.10,11 The ability of the CuAAC reaction to proceed under a vast array of different conditions suggests that a high degree of mechanistic redundancy exist, by which the azide and alkyne substrates can be channeled via various pathways to 1,4-substituted-1,2,3-triazoles over the potential energy surface despite of the tremendous variations in substrate structures and reaction conditions. Such a situation renders mechanistic investigations challenging, because the sequence of the events on the reaction coordinate and the structures of the intermediates and catalytic species vary when the reaction parameters are altered as needed in kinetic investigations.
Two significant characteristics in the prevailing mechanistic model of the CuAAC reaction12–16 are the requirements of a terminal alkyne substrate and a copper catalyst in the +1 oxidation state. There are two ramifications of these two observations that are relevant to this report: first, the recognition of the roles of terminal alkyne and copper(I) understandably led to the accepted mechanistic significance of copper(I)-acetylide.16 The importance of copper(I/II)-azide interaction, on the other hand, has been relatively understated.17 Second, anecdotal reports did appear where copper(II) salts apparently “catalyze” the CuAAC reactions.18–24 These earlier observations were suspected at times as arising from using a copper(II) salt that was contaminated with a small amount but potent copper(I) catalytic species.5 Consequently, the likelihood of the active engagement of copper(II) in CuAAC reactions was largely dismissed.5,25 In this paper, we describe the mechanistic significance of copper(I/II)-azide interaction and the possible beneficial effect of a direct participation of copper(II) in CuAAC reactions.
Mizuno et al. reported that a dicopper(II)-substituted silicotungstate accelerates the CuAAC reaction.26 A preceding alkyne oxidative homocoupling (OHC) reaction was postulated to generate the needed copper(I) catalyst for the subsequent CuAAC reaction. Gratifyingly, a kinetic study was conducted to support the hypothesis of the copper(I)-generating induction process,26 which provides an appropriate foundation for the discussion of the findings reported herein.
Our group showed that organic azides that are capable of chelating copper(II) at the alkylated azido nitrogen (chelating azides) have high reactivity in CuAAC reactions in the presence of an air-stable copper(II) salt without the addition of a reducing agent as usually required for in situ copper(I) generation.27 In the current work, we present kinetic and structural data to map out a mechanistic pathway of this variant of the CuAAC reaction. The discussion of our mechanistic investigation is conducted under the context of the currently known mechanistic models.12–15,26 The newly discovered features in our model emphasize the dependence of mechanism on substrate structures (e.g. chelating azide vs. non-chelating azide) and counter ion of the copper(II) salt. In addition to advancing the understanding in the multifaceted nature of the mechanisms of CuAAC reactions,16 this work may offer insights into other copper(I/II)-involved catalytic processes.
Before delving into our findings, a summary of the current mechanistic understanding of the CuAAC reaction is warranted. As illustrated in the simplified model in Figure 1, the in-cycle copper(I) catalyst may be generated in an induction period (i.p., step A) by reducing an air stable copper(II) salt. The formation of the copper(I) acetylide (step B) follows, which binds the azide (step C) to set up an intramolecular nucleophilic attack step. The rate-determining ring closure (step D), as supported by computational studies,12,15 involves the oxidation of copper(I) to copper(III) upon the formation of a six-membered (or larger if multiple copper centers are involved28) metallacycle.29,30 The metallacycle readily converts to copper(I) triazolide (step E)31 which exits the catalytic cycle upon protonation (step F). The protonation step has been suggested to be kinetically significant when the overall proton donating ability of the reaction mixture is low (e.g. when an aprotic solvent is used).31 Therefore, it is likely that more than one elementary step in the catalytic cycle may affect the overall rate of the reaction. The formations of copper(I) acetylide32,33 and copper(I) triazolide31 have been directly observed during CuAAC reactions. Many other lines of experimental and computational evidence are consistent with this model.16 The formation and retention of unaggregated copper(I) species, the accessibility of the copper(I) acetylide by an azide, and the involvement of two copper centers in the rate-determining step,34 are the subtle issues unveiled in the mechanistic studies which have profound effects on the efficiency of the reaction.16
Figure 1.

A mechanistic proposal by Fokin et al.1,16 L: a ligand or a counter ion associated with copper(I/II). i.p.: induction period; r.d.s.: rate-determining step. The numbering of azide and acetylide are marked blue. The brackets around “CuLn” indicate that bi- or polynuclear copper(I) species may be involved.
Our group recently found that Cu(OAc)2 in alcoholic solvents mediates the CuAAC reaction without the deliberate addition of a reducing agent.27,35 Based on the mechanistic model in Figure 1, the catalytic copper(I) species was postulated to emerge in an induction period via alcohol oxidation and/or alkyne oxidative homocoupling (OHC) reaction.27 This hypothesis is consistent with experimental observations of other groups.26,36 In particular, the OHC-enabled copper(I) generation in a dicopper(II)-substituted silicotungstate-accelerated CuAAC process has been invoked by Mizuno et al. and substantiated by their kinetic studies.26
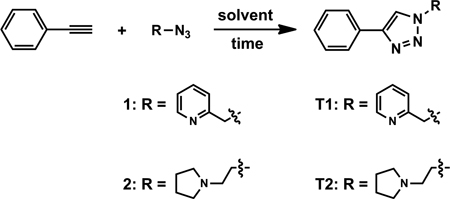
The key motivations that propelled us to conduct the mechanistic investigation of the Cu(OAc)2-accelerated CuAAC reaction are the following two observations. First, chelating azides, such as compounds 1–6, are superior substrates under the Cu(OAc)2-accelerated conditions.27,35,37 Second, Cu(OAc)2 is the most effective copper(II) salt that drives the CuAAC reaction in non-oxidizable solvents (see next section) in which other counter ions are largely ineffective. Herein, we reveal how chelating azides and Cu(OAc)2 change the potential energy landscape of the CuAAC reaction.

Results and Discussion
This section is organized as the following: first, various solvents, alkynes, and counter ions are screened for their effects on the efficiency of CuAAC reactions involving a chelating azide as represented by the time required for a complete conversion. The appropriate conditions are subsequently selected for kinetic experiments using fluorescence and 1H NMR assays to gain mechanistic pictures in two different concentration regimes.38 Following the kinetic studies, preliminary mechanistic models are proposed that account for the reaction orders of individual components. The structural studies focus on the interactions between Cu(OAc)2 and chelating azide or terminal alkyne, based on which the final mechanistic model is described to present our proposition on the unique role of Cu(OAc)2 in the CuAAC reactions.
1. Solvent screening
The reactions between azides 1 or 2 and phenylacetylene are selected for screening for solvent effects. The solvents listed in Table 1 are arranged into two groups. The first group includes aprotic organic solvents CH2Cl2, CH3CN, THF, and toluene (Table 1, entries 1–4). The heterogeneous reaction mixture shows green-blue color throughout the reaction in most cases, while the colors of the others change to yellow (e.g., see Figure S1). The fading of the bluish color of the reaction mixture over the course of the reaction is an indication that the copper(II) precatalyst is transformed into copper(I) catalyst(s).26,27 The reactions generally complete within 1–5 h. In some cases, an initial rapid conversion is followed by a much slower phase to reach completion. Such a discontinuous kinetic profile is indicative of the inactivation of the catalyst over the course of the reaction.17,39
Table 1.
Solvent effect on the Cu(OAc)2-accelerated CuAAC reactions.a
 | |||
|---|---|---|---|
| Entry | Solvent | Timeb (T1) | Timeb (T2) |
| 1 | CH2Cl2 | 1 h | 2.8 h |
| 2 | CH3CN | 55 min | 3.5 h |
| 3 | THF | 5 h | 3.5 h |
| 4 | toluene | 3 h | 8 h (58%)c |
| 5 | tBuOH | < 5 min | < 5 min |
| 6 | CH3OH | < 5 min | < 5 min |
| 7 | iPrOH | < 5 min | < 5 min |
| 8 | water ([HEPES] = 0.5 M, pH 7.0) |
< 5 min | 2 h (89%)c |
Reaction conditions: azide 1 or 2 (0.2 mmol), phenylacetylene (0.22 mmol), Cu(OAc)2·H2O (5 mol%), solvent (0.5 mL), rt.
Time for the azide to disappear on TLC, followed by the confirmation of a full conversion (> 95%) by 1H NMR.
Incomplete conversion with percentage yield in parenthesis.
Protic solvents (Table 1, entries 5–8) constitute the second group. In alcoholic solvents (entries 5–7) the reaction mixture is more homogeneous than those in the first group of aprotic solvents. Sharp color transitions from blue/greenish to yellow are observed in most cases (Figure S2). The reactions reach completion in 5 min or less without any apparent difference in reactivity between azides 1 and 2. These are some of the fastest solution-phase CuAAC reactions reported, yet without the direct addition of a copper(I) catalyst. The shorter time frames required for full conversions in alcoholic solvents than that in aprotic organic solvents are consistent and striking. Three factors might contribute to the “faster” reactions in alcoholic solvents: (1) the induction period required for copper(I) generation in an aprotic solvent, presumably via the OHC reaction, may be relatively long. Whereas in most alcoholic solvents alcohol oxidation provides an additional pathway to copper(I). (2) The protonation of copper(I)-triazolide, which may affect the overall rate,17,31 is rapid in an alcoholic solvent. In an aprotic solvent, however, the only proton sources are the terminal alkyne, the protonated azide 1 or 2 (resulting from deprotonation of alkyne), and the minimal amount of water introduced with Cu(OAc)2. Therefore, the protonation step may be relatively slow due to the paucity of proton donors.40 (3) The dynamic exchange between the counter ion and azide or alkyne substrate to the catalytic copper centers, which has been emphasized as important in CuAAC reactions,17,41 may be faster in a protic solvent than that in an aprotic solvent.
The reactions proceed well under the aqueous conditions (entry 8). Once again, similar color transition from blue/green to yellow is observed (Figure S3). The reactions are reasonably rapid; much faster than the CuAAC reactions under similar conditions reported by Reddy et al. (a 20-h reaction time was reported).18 The difference reflects the high reactivity of the chelating azides 1. The relatively inefficient reaction involving azide 2 under the conditions of entry 8 may be attributed to the protonation of the tertiary amino group at neutral pH, which hampers the chelation between 2 and the copper centers. The effectiveness of Cu(OAc)2 in enabling rapid CuAAC reactions involving chelating azides offers promises in developing CuAAC-based bioconjugation protocols without the sensitivity to molecular oxygen.
Based on the solvent screening data, CH3OH and CH3CN are chosen as representative solvents for kinetic studies. In either solvent, all species involved in the reactions have satisfactory solubilities, which allow spectroscopic interrogation without complication due to aggregation. In addition, the induction processes during which copper(I) catalytic species is generated are expected to differ in these two solvents – alcohol oxidation in CH3OH and OHC reaction in CH3CN. It should be noted that copper(II) is a stronger oxidant in CH3CN than in most other solvents.42 Therefore, a solvent environment of CH3CN provides a thermodynamic driving force for copper(II) reduction.
2. Alkyne screening
The reactivities of various alkynes against azide 1–3 are studied in CH3OH (Table 2) and CH3CN (Table S1), respectively. In either solvents no apparent correlation between the structure of alkyne and the efficiency of the reaction under the preparative, heterogeneous conditions is observed, although under homogenous reaction conditions where the kinetic experiments are conducted it is revealed otherwise (see section 8). One exception is 3,3-dimethylbutyne (entry 7) whose slow reactions can be attributed to the steric effect imposed by the t-butyl group. The pyridyl-containing azide 1 consistently takes shorter time to finish the reaction than the tertiary amino-containing 2 and carboxylate-containing 3, particularly in CH3CN (see Table S1). In reactions involving 2 and 3, a very rapid reaction is routinely observed via TLC during the early phase of the reaction (5–30 min). However the reaction slows down subsequently to result in an overall less efficient reaction than that involving 1. The two-phase observation may be caused by the reorganization of the catalyst structure during the course of the reaction, which alters, and often lowers, the catalytic activities (see section 11).39 The transformation of Cu(OAc)2 structure in the presence of 3-azidopropionate 3 is evident in the crystal structure shown in Figure 15C. Due to such complications involving azides 2 and 3, azide 1 is used in the majority of the kinetic experiments described herein.
Table 2.
Effect of alkyne on the Cu(OAc)2-accelerated reactions involving chelating azides 1–3.a
| Entry | Timeb | Timeb | Timeb | |
|---|---|---|---|---|
 |
||||
| 1 | < 5 min | < 5 min | 8 h (84%)c | |
| 2 | < 5 min | 5–6 min | 2.5 h | |
| 3 | < 5 min | < 5 min | 8 h | |
| 4 | < 5 min | < 5 min | 30 min | |
| 5 | < 5 min | 8 h (93%)c | 8 h (75%)c | |
| 6 | 2.5 h | 25 min | 8 h (94%)c | |
| 7 | 8 h (84%)c | 8 h (96%)c | 2 h |
Reaction conditions: azide (0.2 mmol), alkyne (0.22 mmol), Cu(OAc)2 (5 mol%), in CH3OH (0.5 mL), rt. For reactions involving azide 3, TEA (0.2 mmol) was included.
Time for azide to disappear on TLC, followed by the confirmation of a full conversion (> 95%) by 1H NMR.
Incomplete conversion with percentage yield in parenthesis.
Figure 15.

ORTEP views (50% probability ellipsoids) of the asymmetric units of (A) [Cu2(1)2(OAc)4]. Selected distances (Å): Cu1-N1 2.236, Cu1-Cu1i 2.644, Cu1-O1 1.975, Cu1-O2 1.977, Cu1-O3 1.960, Cu1-O4 1.977. (B) [Cu2(6)2(OAc)4]. Selected distances (Å): Cu1-N1 2.239, Cu1-Cu2 2.641, Cu1-O1 1.979, Cu1-O2 1.962, Cu1-O3 1.973, Cu1-O4 1.954. (C) [Cu2(3)6]2−. Selected distances (Å): Cu1-N4 2.219, Cu1-Cu1i 2.587, Cu1-O1 1.957, Cu1-O2 1.965, Cu1-O3 1.967, Cu1-O4 1.958.
3. Counter ion screening
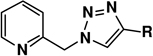
The effect of counter ion is evaluated using the reaction between 2-azidomethylquinoline 4, as an example of chelating azides,43 and phenylacetylene (Table 3). In the non-oxidizable solvent CH3CN, acetate stands out as the only viable counter ion that affords a rapid conversion (within an hour). All other counter ions tested are far inferior. The coordinating strength of a counter ion (e.g., strongly coordinating chloride vs. non-coordinating perchlorate or triflate) does not appear to be a significant factor. The reactivity difference between Cu(OAc)2 and Cu(CF3CO2)2 will be addressed in Section 10–2, note #78. When an oxidizable solvent is used (e.g. CH3OH, middle column in Table 3), or other means of copper(II) reduction is provided (e.g. via treating with sodium ascorbate, right column in Table 3), the dependence of reaction efficiency on counter ion diminishes. These observations suggest the requirement of acetate in generating the catalytic copper(I) species in a non-oxidizable solvent during the induction period, presumably involving the OHC process.
Table 3.
Effect of counter ion on the copper(II)-accelerated reactions involving azide 4.a
 | ||||
|---|---|---|---|---|
| Entry | CuII salt | Time (conversion)b (CH3CN) |
Time (conversion)b (CH3OH) |
Time (conversion)b (tBuOH/NaAsc)c |
| 1 | Cu(OAc)2 | 25 min (> 95%) | 1 h (> 95%) | < 5 min (>95%) |
| 2 | CuCl2 | 16 h (14%) | 1 h (60%) | 50 min (> 95%) |
| 3 | CuSO4 | 16 h (33%) | 1 h (27%) | < 5 min (>95%) |
| 4 | Cu(NO3)2 | 18 h (27%) | 1 h (14%) | < 5 min (>95%) |
| 5 | Cu(ClO4)2 | 16 h (8%) | 1 h (14%) | 60 min (> 95%) |
| 6 | Cu(OTf)2 | 14 h (86%); 1 h (10%) |
1 h (40%) | 45 min (> 95%) |
| 7 | Cu(CF3CO2)2 | 16 h (89%); 1 h (2%) |
1 h (26%) | 30 min (> 95%) |
Reaction conditions: 2-azidomethylquinoline 4 (0.2 mmol), phenylacetylene (0.22 mmol), copper(II) salt (5 mol%), rt. The 1H NMR of the triazole product is consistent with the reported data in ref. #27.
The conversion values in parentheses were determined by 1H NMR.
NaAsc: sodium ascorbate.
4. Fluorescence and 1H NMR assays
In a previous kinetic study conducted by Finn, Fokin et al.,13 aliquots of the reaction mixture were taken and quenched before subjecting to LC-MS analysis. We aim to develop an operationally simple kinetic assay that allows real-time monitoring of the reaction.44 Fahrni et al. reported that an analogue of alkyne 7 (Scheme 1) underwent fluorescence enhancement upon forming the triazole product with an organic azide.11,45 Upon transforming 7 to triazole 8, the fluorescence quantum yield is indeed increased by 10-fold in CH3OH (Scheme 1). Therefore, the reaction in CH3OH shown in Scheme 1 is monitored by the growth of fluorescence intensity at 400 nm.
Scheme 1.

(a) 5 mol% Cu(OAc)2·H2O, CH3OH or CD3CN. The fluorescence quantum yields (ϕ) were measured in CH3OH.
In the fluorescence assay in CH3OH (Figure 2A), the alkyne concentration ([7]) is in most cases kept under 10 µM so that the fluorescence intensity can be approximated to have a linear relationship with the triazole product concentration ([8]).46 Azide 1 is used in large excesses (0.5–3.5 mM) to manage reasonable reaction rates. The concentration of Cu(OAc)2 is varied within 0.8–2.5 µM during its reaction order determination, which is catalytic with respect to the azide and alkyne components. A control experiment shows that in the presence of Cu(OAc)2 up to 15 µM, the fluorescence of the triazole product 8 (15 µM) is unaffected (Figure S4). In addition, the formation of fluorescent diyne side product 9 (Scheme 1) is negligible in CH3OH (Figure S5). Therefore, the observed fluorescence enhancement (e.g. in Figure 2A) can be correlated to the formation of fluorescent triazole product 8.
Figure 2.

(A) Fluorescence spectral changes (λex 320 nm) during the fluorogenic CuAAC reaction between alkyne 7 and azide 1. Reaction conditions: 7 (10 µM), 1 (5 mM), Cu(OAc)2·H2O (10 µM) in CH3OH. (B) 1H NMR spectral evolution during the CuAAC reaction between 7 and 1. Conditions: 7 (10 mM), 1 (10 mM), Cu(OAc)2·H2O (1 mM) in CD3CN.
The reaction (Scheme 1) in CH3CN requires millimolar concentration of alkyne 7 to proceed within a reasonable time frame.47 However, the linearity between fluorescence intensity and [8] no longer applies in this concentration regime.46 Furthermore, the fluorescence of the diyne side product 9 produced in CH3CN is difficult, if not impossible, to be separated from that of the triazole product 8. Therefore, the reaction in CD3CN (solvent deuteration does not affect the reaction progress) is analyzed using 1H NMR.
As shown in Figure 2B, a clean transition from alkyne 7 to triazole 8 could be monitored by 1H NMR spectroscopy. No buildup of intermediates could be detected in the experiment. The presence of catalytic amount of Cu(OAc)2 slightly blurs the signals of the pyridyl portions of 1 and 8 due to the paramagnetic nature of a copper(II) salt (see the difference between the first and second spectra from the bottom of Figure 2B) in otherwise well-resolved NMR spectra. The product formation is quantified via integration of the CH2 signal of triazole 8 at 5.7 ppm or the C5-H triazole hydrogen at 8.3 ppm.
5. The induction period
The reaction in Scheme 1 is monitored by fluorescence at 400 nm immediately after mixing 2-picolylazide (1), alkyne 7, and Cu(OAc)2·H2O in CH3OH. In most cases, an induction period precedes the triazole product formation (e.g., blue trace in Figure 3A). The length of the induction period is inversely correlated to the concentration of azide 1 ([1]). When [1] is 3.5 mM, the induction period is barely noticeable (Figure 7A in Section 6). We hypothesize that copper(I) catalytic species is generated during the induction period via the reduction of Cu(OAc)2 by CH3OH. The reduction process appears to be aided by the presence of 2-picolylazide 1 as a base in the oxidation of CH3OH.48 Consistent with a 2-picolylazide-dependent copper(II) reduction process, by premixing 2-picolylazide 1 and Cu(OAc)2 up to 2 h, the induction period is no longer observed in CH3OH (see the red trace in Figure 3A).
Figure 3.

(A) Growth of fluorescence intensity at 400 nm of the mixture of 1 (1 mM), 7 (10 µM), and Cu(OAc)2 (10 µM) in CH3OH over time with (red) or without (blue) pre-mixing azide 1 and Cu(OAc)2 for 2 h. (B) Product (8) generation over time monitored by 1H NMR with (red) or without (blue) pre-mixing azide 1 and Cu(OAc)2. Conditions: [1] = 10 mM, [7] = 10 mM, [Cu(OAc)2] = 1 mM in CD3CN.
Figure 7.

(A) The dependence of fluorescence time course (λex 320 nm, λem 400 nm) on [1]. Conditions: [7] = 10 µM, [Cu(OAc)2·H2O] = 10 µM, and [1] = 0.5–3.5 mM in CH3OH. (B) Plot of lnVint vs. ln[1]. The slope yields the kinetic order of 2-picolylazide 1. Vint: initial observed rate = d[8]/dt (M·s−1).
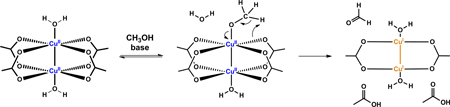
The occurrence of methanol oxidation during the induction period is supported by the solvent kinetic isotope effect (SKIE) experiment when the reaction is run in CH3OH, CH3OD, or CD3OD (Figure 4). The deuterium substitution on the methyl group in methanol slows down the reaction, whereas the deuteration of the hydroxyl group only does not significantly alter the reaction rate. The SKIE is consistent with the hypothesis that in the induction reaction methanol is oxidized to formaldehyde during which the C-H/D bond cleavage takes place in the rate-determining step.
Figure 4.

(A) Growth of fluorescence intensity at 400 nm of the mixture of 1 (10 mM), 7 (10 µM), and Cu(OAc)2·H2O (10 µM) in CH3OH (cornflower squares), CH3OD (garnet diamonds), and CD3OD (lime triangles), respectively.
In CD3CN, on the contrary, premixing 2-picolylazide 1 and Cu(OAc)2 has little effect on the induction period (Figure 3B). We postulate that the alkyne OHC reaction provides the required copper(I) species in CD3CN, which would not take place until the entrance of alkyne into the reaction mixture. The high reactivity of 2-picolylazide 1 under Cu(OAc)2-involved conditions can be attributed, in part, to its acceleration of the initial OHC induction reaction.
Based on the absorption spectral difference between diyne 9 and alkyne 7 (Figure 5A), the generation of diyne 9 in an OHC reaction can be monitored at 374 nm. In a control experiment, the effect of pyridine as an additive on the efficiency of the Cu(OAc)2-mediated OHC reaction is investigated.49 The diyne (9) formation is very slow in the absence of pyridine as shown by the absorption at 374 nm (Figure 5B, cornflower trace). The addition of pyridine accelerates the reaction (Figure 5B, garnet trace). Pyridine, however, cannot initiate the OHC reaction using CuCl2 (resulting in unidentified aggregates) or CuSO4 (Figure 5B, lime trace), suggesting a productive combination of Cu(OAc)2 and pyridine in the OHC reaction in CH3CN. 2-Picolylazide 1 may have an analogous effect as pyridine, thus offering an explanation on the rapid generation of copper(I) catalytic species via an accelerated OHC induction reaction.50 Little absorption change at 374 nm is observed, however, during the CuAAC reaction in CH3OH under the conditions shown in the caption of Figure 2A (Figure S5), suggesting that the OHC reaction is at best marginal.
Figure 5.

(A) Absorption spectra of alkyne 7 (cornflower), triazole 8 (garnet), and diyne 9 (lime) in CH3CN at 2 µM each. (B) The growth of absorption at 374 nm in CH3CN of the mixture of 7 (10 mM) and Cu(OAc)2·H2O (1 mM) in the presence (garnet) and absence (cornflower) of pyridine, and of the mixture of 7 (10 mM) and CuSO4·5H2O (1 mM) in the presence of pyridine (lime), respectively. The absorbance values at time = 0 are zeroed.
6. Reaction orders in CH3OH
The product formation is monitored as the fluorescence enhancement at 400 nm immediately after mixing the fluorogenic alkyne 7, 2-picolylazide 1, and Cu(OAc)2·H2O. The reaction orders (Table 4, entries 1–3) of the three components – alkyne 7, 2-picolylazide 1, and Cu(OAc)2·H2O – are determined under initial kinetics conditions (Figures 6–8).51 The time course data are processed based on the method reported by Vallee, et al. (see derivations in the Supporting Information).52 Alkyne 7 shows a first-order behavior (Figure 6). 2-Picolylazide 1, in excess amounts, has fractional order (0.4), which suggests saturation kinetics (Figure 7).53 The reaction is second-order in Cu(OAc)2 when it is maintained at catalytic levels (< 5 mol%, Figure 8). A second-order dependence on Cu(OAc)2 is consistent with the currently accepted kinetic model which favors the participation of two copper(I) centers in the metallacycle formation step based on both kinetic data13 and computation.14,15 The reaction orders of CuAAC reactions determined by Finn, Fokin, et al.14 and Mizuno, et al.27 are included in Table S2 for comparison.
Table 4.
Kinetic orders in various components of CuAAC reactions under different conditions.
| entry | component | alkyne | azide | Cu(OAc)2 | solvent | order |
|---|---|---|---|---|---|---|
| 1 (FL)a | alkyne 7 | 1–10 µM | 5 mM | 10 µM | CH3OH | 0.9 |
| 2 (FL) | azide 1 | 10 µM | 0.5–3.5 mM | 10 µM | CH3OH | 0.4 |
| 3 (FL) | Cu(OAc)2 | 50 µM | 5 mM | 0.8–2.5 µM | CH3OH | 2.2 |
| 4 (NMR)b | alkyne 7 | 3–10 mM | 10 mM | 1 mM | CD3CN | 2.5 |
| 5 (NMR) | azide 1 | 10 mM | 1–10 mM | 1 mM | CD3CN | 0.072 |
| 6 (NMR) | Cu(OAc)2 | 25 mM | 25 mM | 0.05–0.12 mM |
CD3CN | 2.1 |
| 7 (NMR) | 1-ethynyl-4- nitrobenzene |
4–12 mM | 20 mM | 1 mM | CD3CN | 2.3 |
| 8 (NMR) | azide 2 | 10 mM | 10–20 mM | 1 mM | CD3CN | −0.13 |
| 9 (NMR) | Cu(OAc)2 | 20 mM | 20 mM | 0.05–0.12 mM |
CD3CN | 1.7 |
FL: kinetic orders are determined in a fluorescence assay;
NMR: kinetic orders are determined in a 1H NMR assay.
Figure 6.

(A) The dependence of fluorescence time course (λex 320 nm, λem 400 nm) on [7]. Conditions: [1] = 5 mM, [Cu(OAc)2·H2O] = 10 µM, and [7] = 1–10 µM in CH3OH at 25 °C. (B) Plot of lnVint vs. ln[7]. The slope yields the kinetic order of 7. Vint: initial observed rate = d[8]/dt (M·s−1). The spectra taken after 48 h, assuming full conversion, are included in Figure S6.
Figure 8.

(A) The dependence of fluorescence time course (λex 320 nm, λem 400 nm) on [Cu(OAc)2·H2O]. Conditions: [7] = 50 µM, [Cu(OAc)2·H2O] = 0.8–2.5 µM, and [1] = 5 mM in CH3OH. (B) Plot of lnVint vs. ln[Cu(OAc)2·H2O]. The slope yields the kinetic order of Cu(OAc)2·H2O. Vint: initial observed rate = d[8]/dt (M·s−1).
7. Reaction orders in CD3CN.54
The Cu(OAc)2-accelerated CuAAC reaction between alkyne 7 and 2-picolylazide 1 proceeds smoothly in CD3CN (Figure 2B). The kinetic orders of various components are determined via integrating the 1H NMR signals which are sharp enough in the presence of up to 10 mol% Cu(OAc)2·H2O. Invariably, an induction period is observed (e.g., in Figure 9A). The slope of the immediate linear portion following the induction period, which is defined as the “reaction phase”, is considered as the initial rate of each reaction. The kinetic order of alkyne 7 is 2.5 (Figure 9B), as opposed to 0.9 in CH3OH. Three possibilities may account for a kinetic order of or over two: (1) two alkyne molecules take part in the rate-determining step, (2) two or more alkyne molecules are involved in separate kinetically significant steps, and (3) an autoinductive process55 is operating. The last possibility is not consistent with the observation that addition of the triazole product 8 barely affects the reaction (Figure S9). The mechanistic model that we propose in Section 9 is consistent with the second scenario.
Figure 9.

(A) The dependence of reaction time course on [7]. Conditions: [1] = 10 mM, [Cu(OAc)2·H2O] = 1 mM, and [7] = 3–10 mM in CD3CN at 25 °C. Full conversions of each reaction would afford 8 at 3, 4, 6, 8, and 10 mM, respectively. (B) Plot of lnVint vs. ln[7]. The slope yields the kinetic order of 7. Vint: initial observed rate = d[8]/dt (M·s−1).
When the concentration of 2-picolylazide ([1]) is lowered, the rate of the reaction phase is largely unchanged following a progressively longer induction period (Figure 10A). The observed zero order in 2-picolylazide 1 (Figure 10B) supports a pre-equilibrium process in which the copper(II) center is saturated by 1 prior to its reduction to copper(I) and the entry to the catalytic cycle. The faster interaction of 2-picolylazide 1 with copper(II) than that of alkyne 7 is also evident from the 1H NMR spectra of the initial mixture (Figure 2B), where the signals of azide 1 is significantly blurred due to copper(II) binding during the first 20 min of the reaction. The peaks of alkyne 7 are barely affected.
Figure 10.

(A) The dependence of reaction time course on [1]. Conditions: [1] = 4–15 mM, [Cu(OAc)2·H2O] = 1 mM, and [7] = 10 mM in CD3CN at 25 °C. Full conversions of each reaction would afford 8 at 4, 6, 8, 10, and 10 mM, respectively. (B) Plot of lnVint vs. ln[1]. The slope yields the kinetic order of 1. Vint: initial observed rate = d[8]/dt (M·s−1).
The second order in Cu(OAc)2·H2O at a true catalytic, less than 0.5% loading level (Figure 11) is in agreement with that reported by Fokin and Finn under the typical copper(I)-catalyzed conditions.13 Two copper ions acting in concert are considered to best activate alkyne and/or azide toward the formation of the key C-N bond in the metallacycle intermediate.
Figure 11.

(A) The dependence of reaction time course on [Cu(OAc)2·H2O]. Conditions: [1] = 25 mM, [Cu(OAc)2·H2O] = 0.05–0.12 mM, and [7] = 25 mM in CD3CN at 25 °C. Full conversions of each reaction would afford 8 at 25 mM. (B) Plot of ln Vint vs. ln[Cu(OAc)2·H2O]. The slope yields the kinetic order of Cu(OAc)2·H2O. Vint: initial observed rate = d[8]/dt (M·s−1).
As shown in Figures 9–10, the length of the induction period is dependent on the concentrations of both azide 1 and alkyne 7, suggesting the participation of these two components outside the catalytic cycle. Both reaction order and induction period data contribute to the following mechanistic picture of the reaction: (1) the interaction of copper(II) and 2-picolylazide 1 is fast and occurs prior to the entry of copper(I) into the catalytic cycle; (2) the association between the copper(I)/azide complex and alkyne, which leads to the formation of the triazole product, is rate-determining; (3) the most effective catalyst may contain a dinuclear copper core; (4) the induction period results in the reduction of copper(II) to copper(I) via the alkyne OHC reaction, the rate of which is dependent on both 2-picolylazide 1 (as a base or other active roles in enhancing the rate of the homocoupling reaction) and alkyne.
The reactivity of azide 2 is lower than that of azide 1. Discontinuous kinetic traces of the reaction between azide 2 and alkyne 7 are collected which challenge reaction order determination (e.g., see Figure S10). Therefore, a more reactive alkyne partner, 1-ethynyl-4-nitrobenzene, is used in the 1H NMR assay (CD3CN) at an elevated temperature of 313 K for the reactions to proceed within reasonable time frames to minimize the probability of change in mechanism. The kinetic orders (Table 4, entries 7–9) of both 1-ethynyl-4-nitrobenzene and Cu(OAc)2 are close to 2 (2.3 and 1.7, respectively, see Figures S11, S13), whereas azide 2 carries a zero order (−0.17, Figure S12). These numbers are similar to those recorded in the reaction between 2-picolylazide (1) and 7 in CD3CN, suggesting similarity in mechanism.
8. Deuterium kinetic isotope effect (KIE) and substituent effect on phenylacetylene
The kinetic significance of alkyne deprotonation revealed in the kinetic order determinations is further investigated using deuterium KIE experiments and effect of substitution on the reactivity of phenylacetylene. A primary deuterium KIE (kH/kD) of 2.3 is observed in the reaction shown in Scheme 3 (R = H) in CD3CN (Figure 12A). This result is consistent with the positive kinetic order on alkyne, and suggests that deprotonation of the alkyne component is rate-determining. The reaction involving phenylacetylene-d also sustains a longer induction period than that of the protonated alkyne (Figure 12A), indicating that the rate-determining step of the alkyne OHC reaction to generate copper(I) is also the deprotonation of the terminal alkyne. The reaction in Scheme 3 also shows a normal KIE in CD3OD (Figure S14) which is consistent with alkyne deprotonation being kinetically significant. Barely any induction period is observed in CD3OD, echoing the observations made in fluorescence assays.
Scheme 3.

Conditions: (a) 5 mol% Cu(OAc)2·H2O, CD3CN. R = -NO2, -F, -H, -OCH3, and –N(CH3)2.
Figure 12.

The time courses of triazole product formation determined by integrating 1H NMR signals. Conditions: [1] = 20 mM, [alkyne] = 20 mM, and [Cu(OAc)2·H2O] = 1 mM in CD3CN at 40 °C. (A) phenylacetylene (cornflower diamonds); phenylacetylene-d (garnet squares). (B) 1-ethynyl-4-nitrobenzene (cornflower diamonds); 1-ethynyl-4-fluorobenzene (garnet squares); phenylacetylene (lime triangles); 1-ethynyl-4-methoxybenzene (purple crosses); 1-ethynyl-4-dimethylaminobenzene (turquoise crosses).
The time courses of various para-substituted phenylacetylene are collected to study the effect of substitution on the reactivity of alkyne (Scheme 3). As an electron-withdrawing group (e.g., -F, -NO2) replaces hydrogen at the para position, the induction period is significantly reduced accompanying a higher rate in the reaction phase (Figure 12B). For 1-ethynyl-4-nitrobenzene, the reaction completes within 10 min after mixing the reactants with a barely noticeable induction period. The electronic effect reported herein echoes the results by Bohlman et al. in their mechanistic studies of the OHC reaction almost half a century ago.56 The substituent effect provides another piece of experimental evidence for the kinetic significance of alkyne deprotonation in both OHC reaction in the induction period and the CuAAC reaction in the reaction phase under the acceleration of Cu(OAc)2 in an aprotic solvent.
9. Mechanistic models based on the kinetic measurements
The kinetic profile of the CuAAC reaction between benzylazide and phenylacetylene mediated by a dicopper(II)-substituted silicotungstate catalyst was described by Mizuno et al.26 A comparison of their results and ours reveals subtle mechanistic differences between the two cases. In Mizuno’s work, the rate had a zero order dependence on phenylacetylene and first order in benzylazide (Table S2, entries 4–6). Furthermore, no deuterium KIE was found when using phenylacetylene-d. This result suggests that a fast copper(I) acetylide formation occur prior to the azide/copper(I) interaction. The following azide binding and cycloaddition are slower, kinetically significant steps. This conclusion is in line with the accepted ease of copper(I)-acetylide formation16,57 when a non-chelating azide is used as the reaction partner.
The kinetic orders of chelating azide 1, alkyne 7, and Cu(OAc)2 reported herein offer a different scenario. In CD3CN (Figure 13A), the zero order in chelating azide 1 indicates that in contrary to Mizuno’s system, the binding between copper(II) and chelating azide 1 (step A) occurs prior to the rate-determining step(s) in a pre-equilibrium. This conclusion is also supported by 1H NMR experiment where only the interaction between copper(II) with azide 1 was detected in the initial reaction mixture (Figure 2B). Therefore, the significance of chelation is in facilitating the interaction between azide and copper center to the extent that the usually rapid copper(I)-acetylide formation becomes rate-determining. Consequently, an overall faster reaction than those involving non-chelating azides is observed.35
Figure 13.

Postulated catalytic cycles accounting for the results of the kinetic studies on the CuAAC reactions in Scheme 1 in CD3CN (A) and CH3OH (B). L: copper-binding ligand or the counter ion. py: 2-pyridyl. Blue, orange, and purple represents the +2, +1, and +3 oxidation states of copper, respectively. The steps of copper(I) triazolide protonation are in green.
Copper(II) is subsequently reduced to copper(I) via the alkyne OHC reaction aided by azide 1 in step B (Figure 13A). Steps B and C which involve alkyne are kinetically significant as shown in both kinetic order and deuterium KIE experiments. The intramolecular steps D and E are likely very rapid, whereas the protonation step F could also be kinetically significant in an aprotic solvent.
In the sequence depicted in Figure 13A, azide 1 acts as a base in step B in aiding the formation of copper(II) acetylide prior to diyne 9 formation. This explains the dependence of the induction period on [1]. Azide 1 does not appear to be the base in deprotonating alkyne in step C because of its zero order in producing triazole 8.58 In the aprotic solvent CD3CN, alkyne 7 likely is the primary proton source in step F, which releases the triazole product 8 and supplies the acetylide for the next cycle. Therefore, as soon as the catalytic cycle commences, an external base is no longer needed. Alkyne participates in steps B, C, and F, all of which might be kinetically significant in the aprotic CD3CN, which is consistent with the larger than second-order dependence on alkyne.
In CH3OH (Figure 13B), a slightly different mechanistic picture emerges. The induction period (step B’) entails the oxidation of CH3OH which is base (azide 1) dependent. Copper(I) triazolide may no longer have long enough lifetime to deprotonate alkyne because it could easily acquire proton from the solvent CH3OH, or solvent-mediated proton transfer from protonated azide 1 (1H+) as depicted in step F’. In step C’, azide 1 helps deprotonate alkyne which explains the fractional positive order of azide 1 observed in CH3OH.59 Alkyne is only needed in step C’, thus accounting for the first-order dependence.
10. The role of acetate
With a rough sketch of the mechanism of the chelation-assisted, Cu(OAc)2-accelerated CuAAC reaction completed (Figure 13), we turn our attention to the structural details of the individual components involved in the postulated mechanism and look for answers for the extraordinarily fast reactions and the counter ion specificity that we have observed. There are two clues that direct our structural studies. First, among all copper(II) salts tested, Cu(OAc)2 is the most effective by a large margin that accelerates the reaction between azide 4 and phenylacetylene (Table 3) in the non-oxidizable solvent CH3CN. This observation suggests that in the presence of a base, Cu(OAc)2 mediates the alkyne oxidative homocoupling (OHC) reaction much more efficiently than other copper(II) salts. The observations shown in Figure 5 support this conclusion. Second, the rate of the reaction shows a second-order dependence on Cu(OAc)2. The acceleratory effect of acetate on copper(I)-catalyzed CuAAC reactions has been observed anecdotally,11,60,61 and in a recent case by Hu et al.62 been recognized to arise from the dinuclear copper core of copper(I) acetate (CuOAc) which bears a Cu-Cu distance (2.556 Å)63 favoring the CuAAC reaction as shown in computational studies.15
Herein, we offer a structural model that accounts for the extraordinary reactivity and selectivity of the acetate counter ion in the CuAAC reactions involving chelating azides under apparently non-reducing conditions. We will start by (1) examining the interactions between chelating azides and Cu(OAc)2 and (2) summarizing the key mechanistic features that have been discovered by other groups. Our proposition on the unique reactivity of Cu(OAc)2 will be subsequently presented to demonstrate that how various pieces of experimental observations from us and others come together.
10-1. Interactions between chelating azides and copper(II) salts
We have demonstrated that azides 1, 5, and 6 act as chelating ligands toward copper(II) ion (Figure 14) in single crystal structures.27,35,37 An earlier case of coordination between a chelating azide and copper(II) was reported by Thiel, et al.64 Chelation-assisted binding between the Nα of the azido group and copper(I/II) center was proposed to enhance the electrophilicity of the azido group which results in highly efficient CuAAC reactions.35,37 Without chelation assistance, copper(I) has been shown to favor Nγ, the terminal nitrogen of an azido group, which is capable of accepting backbonding from copper(I).65
Figure 14.

Chelation between copper(II) and azides 1,27,37 5,37 and 635 found in the solid state structures. L represents a counter ion (Cl−, NO3−, SO42−,66 or BF4−66), a second ligand, or a water molecule.
The chelation-enforced coordination between Nα of the azido group and copper(II) center has been observed regardless the coordinating strength of the counter ion (BF4−,66 SO42−,66 NO3−,37 and Cl−27,35,37). Unexpectedly, when Cu(OAc)2, which is the most effective copper(II) salt among tested thus far that accelerates the CuAAC reaction in non-oxidizable solvents, forms complexes with 1 or 6 in CH3CN, (Figures 15A–B) chelation-assisted azido coordination to copper(II) is not observed. The common denominator of both structures is the dinuclear “paddle-wheel” core of Cu(OAc)2·H2O,67 on which the coordinated water molecules at the apical positions are replaced by the auxiliary pyridyl group in 1 or 6 monodentately.
In complex [Cu2(3)4]n (Figure 15C), acetate ion in [Cu2(OAc)4(H2O)2] is replaced by 3-azidopropionate (3) where the apical positions are substituted by the azido groups from the adjacent dinuclear [Cu2(3)4] units. Again, the dinuclear copper(II) core remains intact. The structure propagates into a two-dimensional network, which will be discussed in detail under a different context. Due to the stoichiometrical nature of the complex formation, all carboxylate moieties from 3 engage in the formation of the paddle wheel, leaving only the azido group of 3 to bind at the apical positions. It is conceivable that when a catalytic amount of [Cu2(OAc)4(H2O)2] is used, after exchange of acetate by 3-azidopropionate 3, the rest of compound 3 likely uses the carboxylate moiety, in addition to the azido group, to bind at the apical positions68,69 to result in a structure similar to [Cu2(1)2(OAc)4].
In all three structures in Figure 15, the apical N-Cu bonds are rather long (2.22–2.24 Å), indicating relatively weak and dynamic association that facilitates turnover. The Cu-Cu distances (2.59–2.64 Å) are within the range of a large collection of known apically-substituted copper(II) acetate structures.70 Both copper(II) centers in a paddle wheel are coordinatively saturated with 6 ligands each (counting the other copper(II) center) in an octahedral geometry, leaving no room for azido chelation. In the following subsections, our explanation on the unique role of Cu(OAc)2 in the chelation-assisted, copper(II)-accelerated CuAAC reaction will be presented based on the kinetic and structural data collected by us and others.
10-2. Dinuclear copper(I) catalytic center
There is a consensus on the second-order dependence on copper(I) of ligand-free CuAAC reactions,13–15,26,36,71 which implicates the involvement of a highly active dinuclear copper catalyst. It should be noted that the active dinuclear species is likely in equilibrium with monomeric copper species,72–74 which is the premise for the reaction to show a second-order dependence in copper. If a stable dinuclear copper catalyst which does not dissociate into mononuclear species is involved, a first order should result as reported in the dicopper(II) silicotungstate system by Mizuno, et al.26 Two structural models (A and B in Figure 16) have been proposed in a number of articles to account for the second-order dependence on copper in CuAAC reactions.13,16,17,28,41,75 Alkyne and azide are bonded to different copper(I) centers in A (Figure 16), whereas a single copper(I) center activates both alkyne and azide components in B. The two structural models may interconvert in an equilibrium where the copper(I) acetylide bond alternates from σ (solid bond) to π (dashed bond). Depending on which copper(I) is oxidized in the C2–N3 ligation step, four intermediate (or transition state) structures (I–IV) may result. Structure IV which contains a highly strained endocyclic copper(III)-alkenylidene moiety is deemed an unlikely high energy species.14 Structures I and III are consistent with the computed dicopper(I,III) μ-alkenylidene intermediate,14,15 whereas the larger ring size in structure II alleviates the ring strain of the endocyclic copper(III)-alkenylidene. One observation worth noting is that only one copper(I) center is needed for redox chemistry. The oxidation state of the other copper center is unaltered during the ligation step. In our subsequent discussions in the role of acetate under the copper(II)-accelerated CuAAC reactions, we will refer back to the structural models shown in Figure 16.
Figure 16.

Models of dinuclear pathways for azide/alkyne ligation. L: a bridging ligand or counter ion (e.g. acetate, acetylide, or iodide) that is not participating in the redox reaction, i.e., no electron exchange between L and copper. The formal charges on individual atoms are noted. All bonds surrounding L are considered to be coordinative rather than covalent for the ease in formal charge calculations. Orange: copper(I); purple: copper(III). The carbon and nitrogen atoms that engage in bonding are numbered 2 and 3, respectively.
In order for the cooperative dinuclear catalysis depicted in Figure 16 to work, an important parameter is the Cu-Cu distance that is appropriate for the formation of the (µ, η1,2)-C≡C→Cu2 in structures A and B (Figure 16). A Cu-Cu distance in the range of 2.54–2.64 Å was computed by Ahlquist and Fokin15 in a dinuclear transition state en route to the matallacycle intermediate similar to structure III (Figure 16). The dinuclear copper catalysts that have shown exceptional activities under conventional solution phase CuAAC reactions are included in Table 5. All four dinuclear copper reagents have Cu-Cu distances within or close to the computed range. The two copper(II) reagents (columns 1 and 2) experience induction periods in non-oxidizable solvents to generate copper(I) catalytic species via oxidative homocoupling (OHC) of the alkyne component. In comparison, the dismally low activity of other copper(II) salts in CuAAC reactions shown in Table 3 suggest that a dinuclear copper(II) core of an appropriate Cu-Cu distance be favorable for the OHC reactions to afford the much needed copper(I).78 In the following text, we will describe the mechanistic synergy between the OHC and the CuAAC reactions as well as the important role that a chelating azide such as 2-picolylazide 1 plays in this Cu(OAc)2-mediated OHC/AAC sequence.
Table 5.
The Cu-Cu distances in various copper salts or complexes evaluated in CuAAC reactions.
10-3. The mechanistic picture involving Cu(OAc)2 and the chelating 2-picolylazide
The induction process to generate copper(I) in a non-oxidizable solvent such as CH3CN is the OHC reaction. The structures of [Cu2(1)2(OAc)4] and [Cu2(6)2(OAc)4] (Figure 15) are isomorphic to that of the 1:1 complex of Cu(OAc)2 and pyridine (structure II in Figure 17. Five single crystal structures in the CCDC database, PYCUAC01–05), which is the active ingredient in an OHC reaction in the method of Eglinton.79,80 The functions of pyridine in the Eglinton coupling include deprotonation of alkyne to acetylide,81 which displaces two acetate to afford µ2,η1,2-C≡C→Cu2-containing III (Figure 17, step B). A formal metathesis process follows to release the diyne product56,82 and the dinuclear copper(I) acetate (step C).63,83,84 Based on our observations on the OHC in the induction periods of the CuAAC reaction, the deprotonation step B is rate-determining.
Figure 17.

Postulated Glaser/Eglinton coupling sequence enabled by Cu(OAc)2/pyridine complex. A delocalized negative charge is implied in the drawing of the acetate.
The mechanistic model for Cu(OAc)2-accelerated CuAAC reaction involving 2-picolylazide 1 is depicted in Figure 18. In the presence of a terminal alkyne, [Cu2(1)2(OAc)4] (Figure 15A) may also rapidly transform alkyne to diyne via the sequence shown in Figure 17 to afford [Cu2(1)2(OAc)2] (Structure I in Figure 18). With two acetate counter ions removed, [Cu2(1)2(OAc)2] has open coordination positions for the azido group to associate with copper(I), while an alkyne molecule upon deprotonation by 1 also binds a copper(I) center to afford structures II or II’ (Figure 18). The azide and alkyne components associate with different copper(I) centers in structure II. Intramolecular oxidative metallacycle formation affords a 7-membered ring containing a copper(III)-alkenylidene. Subsequently the rapid reductive ring contraction affords copper(I) triazolide which upon protonation results in the triazole product.
Figure 18.

Mechanistic model of the 2-picolylazide (1) involved, Cu(OAc)2-accelerated CuAAC reaction. Structure I could be generated via the OHC sequence depicted in Figure 17 in an induction period. Orange: +1 oxidation state; purple: +3 oxidation state. Formal charges on individual atoms are noted. CuA – oxidation state is undefined. See text.
If both azide and alkyne components bind the same copper(I) center, structure II’ results (Figure 18). The second copper(I) center participates in the reaction by forming a π-complex with the acetylide. A 6-membered metallacycle containing a µ-alkenylidene with minimal strain is afforded in the next step. Subsequent ring contraction followed by protonation completes the triazole formation. Based on the kinetic data, the formation of structures II or II’ (Figure 18), which involves deprotonation of alkyne, is rate-determining. The feasibility of either pathway may be interrogated computationally, which will be addressed in a future study.
The two copper centers in the model presented in Figure 18 carry different functions. CuB is oxidized to copper(III) upon the formation of the metallacycle intermediate. This redox reaction accounts for the regiospecificity of the CuAAC reaction where only 1,4-substituted 1,2,3-triazole is produced.1 CuA holds the azide component in position and as a Lewis acid increases the electrophilicity of the azido group. The oxidation state of CuA does not change over the course of the reaction. One may surmise that a stronger Lewis acid than copper(I) in place of CuA may increase the reactivity of the azido group in a CuAAC reaction. A copper(II) center which is more Lewis acidic than copper(I) is the most convenient choice.
10-4. Possible involvement of a highly active mixed valency copper(II)/copper(I) dinuclear catalyst?
A mixed-valency copper(II)/copper(I) acetate may fit the bill to activate alkyne and azide at different copper centers (as shown in II and II’ in Figure 18, when CuA has +2 oxidation state). copper(I) is necessary for the redox step in the mechanism, whereas copper(II) activates the azido group as a strong Lewis acid. Carboxylate-bridged mixed-valency copper(II)/copper(I) complexes are known.85 Its general structure is depicted in Figure 19 along with copper(II) and copper(I) acetates based on reported single crystal structures.86–92 In most cases, both copper centers adopt square planar geometry capped by a carboxylate bridge. The apical carboxylate may be displaced by other ligands,91 for example, by azide and alkyne, to afford structure II in Figure 18. The charges on most carboxylate-bridged [Cu2]3+ cores are fully delocalized where each copper takes a formal +1.5 oxidation number. However, when the two copper centers are asymmetrically substituted as in structure II (Figure 18), the charge density may redistribute to best accommodate the two electronically different apical ligands. Therefore, it is plausible that such an asymmetric [Cu2]3+ core may possess distinct reactivities of both copper(II) (a Lewis acid) and copper(I) (a soft, backbonding cation).
Figure 19.

Known structural modes for carboxylate-bridged dinuclear copper complexes. Left: copper(II) acetate, middle: mixed-valency copper(II)/copper(I) acetate; right: copper(I) acetate.
We have not captured the mixed-valency dinuclear copper catalyst in either free or substrate-bound form (II and II’ in Figure 18). However, the indirect evidence that supports the proposition that copper(II) is not merely a precursor to the copper(I) catalyst but actively engages in catalyzing the reaction as a Lewis acid is shown in Figure 20. The reaction between alkyne 7 and 2-picolylazide 1 aided by Cu(OAc)2 endures an induction period of ~ 40 min (case #1). With the addition of 4 molar equivalents of sodium ascorbate to copper(II), the reaction proceeds immediately after mixing without an induction period (case #2). However, the rate of the reaction (the slope of the curve) is consistently lower than the rate of the reaction phase of case #1. When sub-stoichiometric amount of sodium ascorbate is added into the reaction mixture (case #3), not only the induction period disappears, the rate of the reaction is enhanced over both copper(II) and copper(I)-accelerated cases. These data suggest that the synergistic effect of copper(I) and copper(II) lead to the high activity of the catalyst present in case #3. Presumably the highly catalytic species has a mixed-valency dinuclear copper core.
Figure 20.

The dependence of reaction time course on sodium ascorbate (NaAsc). Conditions: [1] = 10 mM, [7] = 10 mM, [Cu(OAc)2·H2O] = 1 mM, [NaAsc] = 0 (lime triangles); [NaAsc] = 4 mM (garnet squares); [NaAsc] = 0.25 mM (cornflower diamonds).
11. Evolution of the structure of the copper-containing catalyst
In the above and other mechanistic analyses of CuAAC reactions, conclusions are drawn based on data such as kinetic orders determined over the initial phase of the reaction, or the Cu-Cu distance of a dinuclear catalyst (or a precatalyst) that is introduced at the onset of the reaction. However, kinetic order, Cu-Cu distance, ratio of reactants to catalyst, and other mechanistically relevant parameters do likely change over the course of the reaction.16 Discretion must be used when the mechanistic conclusions drawn based on the data from the initial phase are extended to the full duration of the reaction.
The versatility of copper coordination chemistry renders the CuAAC reaction an excellent case study where the structure of the (pre)catalyst, in this case Cu(OAc)2, may evolve as the reaction proceeds, thus altering the mechanism.93 For example, during an attempt to prepare the Cu(OAc)2 complex with 2-picolylazide 1 in CH3OH, [Cu4(OAc)4(OCH3)4] (Figure 21), a known tetranuclear copper(II) cluster structure,94,95 is isolated in single crystal forms after 3–4 days. In the absence of an alkyne reaction partner, compound 1 deprotonates CH3OH over time to methoxide which displaces acetate to afford the Cu4 cluster structure. If the time scale of a CuAAC reaction is longer than that of this transformation, a change in reaction mechanism over the course of the reaction is expected.
Figure 21.

A tetranuclear copper(II) cluster structure [Cu4(OAc)4(OCH3)4] (50% probability ellipsoids) isolated after mixing Cu(OAc)2·H2O and azide 1 in CH3OH. Selected distances (Å): Cu1-O1 1.973, Cu1-O3 1.938, Cu1-O5 1.935, Cu1-O5i 1.924, Cu1-Cu2 2.958, Cu1-Cu1i 2.987, Cu1-O2 (opposite to Cu2 on the axial position; not shown) 2.586.
The second example highlights how copper/alkyne interaction transforms the structure of Cu(OAc)2. In the absence of an azide component, 3,3-dimethylbutyne slowly reacts with Cu(OAc)2 in CH3OH to result in a copper(I)14 cluster and the homocoupled diyne product (Figure 22). Evidently, the strong affinity between copper(I) and acetylide leads to the disintegration of the dinuclear Cu(OAc)2 structure. Phenylacetylene was initially used in this experiment which afforded an intractable yellow precipitate.77 3,3-Dimethylbutyne is known to afford discrete alkynyl/copper(I) cluster structures because the steric effect imposed by the t-butyl group prevents the aggregation of copper(I)-acetylide.77,96,97 This copper(I)14 cluster structure is interesting in its own right.97 The relevance of this structure to the current work is that in the absence of an azide, or in the presence of an azide substrate with low reactivity, copper centers preferentially interact with the alkyne component to transform to cluster type structures often with impaired catalytic reactivity.
Figure 22.

(A) A copper(I)14 cluster structure isolated via mixing Cu(OAc)2·H2O with 3,3-dimethylbutyne in CH3OH. (B) The co-crystallized 2,2,7,7-tetramethylocta-3,5-diyne.
Conclusions
A mechanism is proposed for the chelation-assisted, Cu(OAc)2-accelerated azide-alkyne cycloaddition reaction based on kinetic and structural investigations. The high reactivity of chelating azides, such as 2-picolylazide, is attributed to the rapid copper-azido interaction that occurs prior to copper(I) acetylide formation. Under this circumstance, the deprotonation of alkyne becomes rate-determining, as shown in deuterium KIE and substituent effect experiments. The specificity of Cu(OAc)2 in the reported reaction is attributed to its optimal Cu-Cu distance in the dinuclear copper(II) core in mediating both the alkyne oxidative homocoupling and CuAAC reactions. The two copper centers in the proposed dinuclear catalyst are carrying different functions – one is a Lewis acid and the other is a redox active copper(I) center. This observation leads us to speculate that a copper(II)/copper(I) mixed valency dinuclear species would be highly catalytic in CuAAC reactions, which is supported by the extraordinary CuAAC reactivity of a partially reduced Cu(OAc)2 system involving the chelating 2-picolylazide. In addition to continuing the hunt for intermediates in the chelation-assisted, Cu(OAc)2-accelerated variant of the CuAAC reaction, computational interrogation of the proposed mechanism will be conducted in the near future. This work may lead to the examination of the possible mechanistic significance of mixed valency dinuclear copper catalysts in other copper-mediated reactions.
Supplementary Material
Scheme 2.

Conditions: (a) 5 mol% Cu(OAc)2·H2O, CD3CN.
ACKNOWLEDGMENT
This work was supported in part by NIGMS (R01GM081382) and NSF (CHE0809201). We thank Professor Ronald Breslow (Columbia Univ.) for suggesting the deuterium kinetic isotope effect experiment.
Footnotes
Supporting Information Available. Experimental procedures, characterization of new compounds, and additional figures. This material is available free of charge via internet at http://pubs.acs.org.
REFERENCES
- 1.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 2.Tornøe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 3.Wu P, Fokin VV. Aldrichimica Acta. 2007;40:7–17. [Google Scholar]
- 4.Moses JE, Moorhouse AD. Chem. Soc. Rev. 2007;36:1249–1262. doi: 10.1039/b613014n. [DOI] [PubMed] [Google Scholar]
- 5.Meldal M, Tornøe CW. Chem. Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 6.Finn MG, Fokin VV. Chem. Soc. Rev. 2010;39:1231–1232. doi: 10.1039/c003740k. [DOI] [PubMed] [Google Scholar]
- 7.Kolb HC, Sharpless KB. Drug Discovery Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 8.Decréau RA, Collman JP, Hosseini A. Chem. Soc. Rev. 2010;39:1291–1301. doi: 10.1039/b901972n. [DOI] [PubMed] [Google Scholar]
- 9.Lutz J-F. Angew. Chem. Int. Ed. 2007;46:1018–1025. doi: 10.1002/anie.200604050. [DOI] [PubMed] [Google Scholar]
- 10.van Kasteren SI, Kramer HB, Gamblin DP, Davis BG. Nat. Protocols. 2007;2:3185–3194. doi: 10.1038/nprot.2007.430. [DOI] [PubMed] [Google Scholar]
- 11.Hong V, Presolski SI, Ma C, Finn MG. Angew. Chem. Int. Ed. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Himo F, Lovell T, Hilgraf R, Rostovtsev VV, Noodleman L, Sharpless KB, Fokin VV. J. Am. Chem. Soc. 2005;127:210–216. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- 13.Rodionov VO, Fokin VV, Finn MG. Angew. Chem. Int. Ed. 2005;44:2210–2215. doi: 10.1002/anie.200461496. [DOI] [PubMed] [Google Scholar]
- 14.Straub BF. Chem. Commun. 2007:3868–3870. doi: 10.1039/b706926j. [DOI] [PubMed] [Google Scholar]
- 15.Ahlquist M, Fokin VV. Organometallics. 2007;26:4389–4391. [Google Scholar]
- 16.Hein JE, Fokin VV. Chem. Soc. Rev. 2010;39:1302–1315. doi: 10.1039/b904091a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.“… pre-equilibrium coordination of organic azide to Cu may be important, …” was noted in the following reference: Rodionov VO, Presolski SI, Díaz DD, Fokin VV, Finn MG. J. Am. Chem. Soc. 2007;129:12705–12712. doi: 10.1021/ja072679d..
- 18.Reddy KR, Rajgopal K, Kantam ML. Synlett. 2006:957–959. [Google Scholar]
- 19.Bonnamour J, Legros J, Crousse B, Bonnet-Delpon D. Tetrahedron Lett. 2007;48:8360–8362. [Google Scholar]
- 20.Fukuzawa S-I, Shimizu E, Kikuchi S. Synlett. 2007:2436–2438. [Google Scholar]
- 21.Reddy KR, Rajgopal K, Kantam ML. Catal. Lett. 2007;114:36–40. [Google Scholar]
- 22.Song Y-J, Yoo C, Hong J-T, Kim S-J, Son SU, Jang H-Y. Bull. Korean Chem. Soc. 2008;29:1561–1564. [Google Scholar]
- 23.Namitharan K, Kumaaraja M, Pitchumani K. Chem. Eur. J. 2009;15:2755–2758. doi: 10.1002/chem.200802384. [DOI] [PubMed] [Google Scholar]
- 24.Fiandanese V, Bottalico D, Marchese G, Punzi A, Capuzzolo F. Tetrahedron. 2009;65:10573–10580. [Google Scholar]
- 25.Copper(II)/copper(I) redox equilibrium is dependent on other redox couple(s) in the reaction mixture. Therefore, there is always a certain level of copper(I) presence under any circumstance. For example, in water, copper(I) may exist at ppb level per mole of copper(II) as estimated using the Nerst Equation. Extremely low level of copper(I) salt may indeed do wonders (Buchwald SL, Bolm C. Angew. Chem. Int. Ed. 2009;48:5586. doi: 10.1002/anie.200902237.). However, the comparable or even higher reactivity that we observed under the copper(II)-promoted conditions than those under the typical copper(I)-catalyzed conditions cannot be simply explained by the extremely high reactivity of a miniscule amount of copper(I) contaminant. Furthermore, we and others have presented evidence for the transformation of copper(II) to copper(I) over the course of a copper(II)-accelerated CuAAC reaction.
- 26.Kamata K, Nakagawa Y, Yamaguchi K, Mizuno N. J. Am. Chem. Soc. 2008;130:15304–15310. doi: 10.1021/ja806249n. [DOI] [PubMed] [Google Scholar]
- 27.Brotherton WS, Michaels HA, Simmons JT, Clark RJ, Dalal NS, Zhu L. Org. Lett. 2009;11:4954–4957. doi: 10.1021/ol9021113. [DOI] [PubMed] [Google Scholar]
- 28.Bock VD, Hiemstra H, van Maarseveen JH. Eur. J. Org. Chem. 2006:51–58. [Google Scholar]
- 29.Copper(I)/copper(III) redox pair has been recognized to involve in copper-mediated cross coupling reactions. See refs 29 and 30. Hartwig J. Organotransition Metal Chemistry - From Bonding to Catalysis. Sausalito, CA: University Science Books; 2010. .
- 30.Casitas A, King AE, Parella T, Costas M, Stahl SS, Ribas X. Chem. Sci. 2010;1:326–330. [Google Scholar]
- 31.Nolte C, Mayer P, Straub BF. Angew. Chem. Int. Ed. 2007;46:2101–2103. doi: 10.1002/anie.200604444. [DOI] [PubMed] [Google Scholar]
- 32.Diéz-González S, Nolan SP. Angew. Chem. Int. Ed. 2008;47:8881–8884. doi: 10.1002/anie.200803289. [DOI] [PubMed] [Google Scholar]
- 33.Buckley BR, Dann SE, Harris DP, Heaney H, Stubbs EC. Chem. Commun. 2010;46:2274–2276. doi: 10.1039/b924649e. [DOI] [PubMed] [Google Scholar]
- 34.Evidence suggests that under ligand-assisted conditions (see Díez-González S. Catal. Sci. Technol. 2011;1:166–178.) mononuclear pathways could be operative (see references 17 and 32).
- 35.Kuang G-C, Michaels HA, Simmons JT, Clark RJ, Zhu L. J. Org. Chem. 2010;75:6540–6548. doi: 10.1021/jo101305m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buckley BR, Dann SE, Heaney H. Chem. Eur. J. 2010;16:6278–6284. doi: 10.1002/chem.201000447. [DOI] [PubMed] [Google Scholar]
- 37.Brotherton WS, Guha PM, Hoa P, Clark RJ, Shatruk M, Zhu L. Dalton Trans. 2011;40:3655–3665. doi: 10.1039/c0dt01702g. [DOI] [PubMed] [Google Scholar]
- 38.The concentration of alkyne used in the fluorescence assay in CH3OH is 103-fold lower than that in the 1H NMR assay in CD3CN.
- 39.Blackmond DG, Rosner T, Pfaltz A. Org. Process Res. & Dev. 1999;3:275–280. [Google Scholar]
- 40.As a piece of circumstantial evidence to support this argument, addition of 5% (v/v) tBuOH in CH3CN (entry 2 in Table 1, T1) shortens the reaction duration by half.
- 41.Presolski SI, Hong V, Cho S-H, Finn MG. J. Am. Chem. Soc. 2010;132:14570–14576. doi: 10.1021/ja105743g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kratochvil B, Zatko DA, Markuszewski R. Anal. Chem. 1966;38:770–772. [Google Scholar]
- 43.Azide 1 shows similar counter ion dependence as previously reported in ref. 27.
- 44.A recent report detailed the use of infrared spectroscopy in following the kinetics of CuAAC reaction: Sun S, Wu P. J. Phys. Chem. A. 2010;114:8331–8336. doi: 10.1021/jp105034m..
- 45.Zhou Z, Fahrni CJ. J. Am. Chem. Soc. 2004;126:8862–8863. doi: 10.1021/ja049684r. [DOI] [PubMed] [Google Scholar]
- 46.Valeur B. Molecular Fluorescence. Principles and Applications. Wiley-VCH; 2002. [Google Scholar]
- 47.The need for high concentration of alkyne in CH3CN is due to the second-order dependence on alkyne in the oxidative homocoupling induction reaction as shown later.
-
48.A plausible base-dependent CH3OH oxidation sequence is the following:
- 49.It is challenging to monitor the diyne (9) formation in the reaction in CH3CN because the signal from the triazole product 8 always overwhelms the absorption of diyne 9 of a very small abundance.
- 50.In the absence of azide, the amount of 9 formed in the experiment described after 60 min is estimated at 0.4% conversion (based on the data in Figure 5B), which should be considered as the upper end of the amount of 9 that forms under the CuAAC conditions.
- 51.Anslyn EV, Dougherty DA. Sausalito, CA: University Science Books; 2006. Vol. Chapter 7; p. 389. [Google Scholar]
- 52.Wierzchowski J, Defeldecker WP, Holmquist B, Vallee BV. Anal. Biochem. 1989;178:57–62. doi: 10.1016/0003-2697(89)90356-4. [DOI] [PubMed] [Google Scholar]
- 53.See Figure S7 for the diagram of Vint vs. [1].
- 54.We also attempted to determine the reaction orders in CD3OD in a 1H NMR assay (Figure S8). However it was not satisfactory due to the difficulty in integrating the NMR data which was caused by the significant blurring of NMR signals via associations of azide 1 and triazole 8 with the paramagnetic copper(II) ion.
- 55.Blackmond DG. Angew. Chem. Int. Ed. 2009;48:386–390. doi: 10.1002/anie.200804565. [DOI] [PubMed] [Google Scholar]
- 56.Bohlmann F, Schönowsky H, Inhoffen E, Grau G. Chem. Ber. 1964;97:794–800. [Google Scholar]
- 57.Mykhalichko BM, Temkin ON, Mys'kiv MG. Russ. Chem. Rev. 2000;69:957–984. [Google Scholar]
- 58.In a separate scenario, the positive order of azide 1 acquired from being a base in step C may be offset by its ability to aid the dissociation of the active dinuclear copper(II) acetate ([Cu2(OAc)4L2], see Section 10) into unproductive mononuclear species. We are not able to distinguish these two scenarios at this point.
- 59.The impact of 2-picolylazide 1 on the dissociation of [Cu2(OAc)4L2] in CH3OH is attenuated comparing to that in CD3CN (see the previous note).
- 60.Zhu L, Lynch VM, Anslyn EV. Tetrahedron. 2004;60:7267–7275. [Google Scholar]
- 61.Gonda Z, Novák Z. Dalton Trans. 2010;39:726–729. doi: 10.1039/b920790m. [DOI] [PubMed] [Google Scholar]
- 62.Shao C, Cheng G, Su D, Xu J, Wang X, Hu Y. Adv. Synth. Catal. 2010;352:1587–1592. [Google Scholar]
- 63.Ogura T, Mounts RD, Fernando Q. J. Am. Chem. Soc. 1973;95:949–951. [Google Scholar]
- 64.Barz M, Herdtweck E, Thiel WR. Angew. Chem. Int. Ed. 1998;37:2262–2265. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2262::AID-ANIE2262>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 65.Dias HVR, Polach SA, Goh S-K, Archibong EF, Marynick DS. Inorg. Chem. 2000;39:3894–3901. doi: 10.1021/ic0004232. [DOI] [PubMed] [Google Scholar]
- 66.Unpublished results.
- 67.van Niekerk JN, Schoening FRL. Nature. 1953;171:36–37. [Google Scholar]
- 68.Rao VM, Sathyanarayana DN, Manohar H. J. Chem. Soc. Dalton Trans. 1983:2167–2173. [Google Scholar]
- 69.Vives G, Mason SA, Prince PD, Junk PC, Steed JW. Cryst. Growth Des. 2003;3:699–704. [Google Scholar]
- 70.184 hits in CCDC in Oct. 2010.
- 71.Dinuclear-copper catalysis is favorable, but not required. See Nolte C, Mayer P, Straub BF. Angew. Chem. Int. Ed. 2007;46:2101–2103. doi: 10.1002/anie.200604444..
- 72.Kondo M, Kubo M. J. Phys. Chem. 1958;62:468–469. [Google Scholar]
- 73.Kochi JK, Subramanian RV. Inorg. Chem. 1965;4:1527–1533. [Google Scholar]
- 74.Cheng ATA, Howald RA. Inorg. Chem. 1968;7:2100–2105. [Google Scholar]
- 75.Aucagne V, Berna J, Crowley JD, Goldup SM, Hanni KD, Leigh DA, Lusby PJ, Ronaldson VE, Slawin AMZ, Viterisi A, Walker DB. J. Am. Chem. Soc. 2007;129:11950–11963. doi: 10.1021/ja073513f. [DOI] [PubMed] [Google Scholar]
- 76.Yamaguchi K, Kamata K, Yamaguchi S, Kotani M, Mizuno N. J. Catal. 2008;258:121–130. [Google Scholar]
- 77.Chui SSY, Ng MFY, Che C-M. Chem. Eur. J. 2005;11:1739–1749. [Google Scholar]
- 78.The low reactivity of Cu(CF3CO2)2 shown in Table 3 can be explained by its propensity to decomposition into mononuclear species. Furthermore, in a dinuclear Cu(CF3CO2)2 complex where quinoline is the apical ligand, the Cu-Cu distance is 2.886 Å in the solid state structure. It is a bit over the computed optimal range for CuAAC reactions. In contrast, the Cu-Cu separation in the complex of Cu(OAc)2 and quinoline is 2.642 Å. See Moreland JA, Doedens RJ. J. Am. Chem. Soc. 1975;97:508–513..
- 79.Eglinton G, Galbraith AR. Chem. Ind. (London) 1956:737–738. [Google Scholar]
- 80.Toda F, Tokumaru Y. Chem. Lett. 1990:987–990. [Google Scholar]
- 81.Siemsen P, Livingston RC, Diederich F. Angew. Chem. Int. Ed. 2000;39:2632–2657. [PubMed] [Google Scholar]
- 82.Mizuno N, Kamata K, Nakagawa Y, Oishi T, Yamaguchi K. Catal. Today. 2010;157:359–363. [Google Scholar]
- 83.Drew MGB, Edwards DA, Richards R. J. Chem. Soc. Chem. Commun. 1973:124–125. [Google Scholar]
- 84.The details of this step remain unclear. Based on the suggestion by Stahl et al. that a copper(III) acetylide be involved, we envision that structure III may undergo disproportionation (or electron transfer) to afford transient copper(III) and copper(I) acetylides, which subsequently couple to afford a diyne and two copper(I) centers. See King AE, Huffman LM, Casitas A, Costas M, Ribas X, Stahl SS. J. Am. Chem. Soc. 2010;132:12068–12073. doi: 10.1021/ja1045378..
- 85.Sigwart C, Hemmerich P, Spence JT. Inorg. Chem. 1968;7:2545–2548. [Google Scholar]
- 86.LeCloux DD, Davydov R, Lippard SJ. J. Am. Chem. Soc. 1998;120:6810–6811. [Google Scholar]
- 87.LeCloux DD, Davydov R, Lippard SJ. Inorg. Chem. 1998;37:6814–6826. doi: 10.1021/ic980763b. [DOI] [PubMed] [Google Scholar]
- 88.Lo SM-F, Chui SS-Y, Shek L-Y, Lin Z, Zhang XX, Wen G-h, Williams ID. J. Am. Chem. Soc. 2000;122:6293–6294. [Google Scholar]
- 89.Zhang X-M, Tong M-L, Chen X-M. Angew. Chem. Int. Ed. 2002;41:1029–1031. doi: 10.1002/1521-3773(20020315)41:6<1029::aid-anie1029>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 90.Zhang X-M, Tong M-L, Gong M-L, Lee H-K, Luo L, Li K-F, Tong Y-X, Chen X-M. Chem. Eur. J. 2002;8:3187–3194. doi: 10.1002/1521-3765(20020715)8:14<3187::AID-CHEM3187>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 91.Hagadorn JR, Zahn TL, Que JL, Tolman WB. Dalton Trans. 2003:1790–1794. [Google Scholar]
- 92.Lu JY, Schauss AB, Julve M. Inorg. Chim. Acta. 2006;359:2565–2568. [Google Scholar]
- 93.See the discontinuous time courses of reactions between alkyne 7 and azide 2 in Figure S10.
- 94.Wu LP, Kuroda-Sowa T, Maekawa M, Suenaga Y, Munakata M. J. Chem. Soc., Dalton Trans. 1996:2179–2180. [Google Scholar]
- 95.Koval IA, Gamez P, Roubeau O, Driessen WL, Lutz M, Spek AL, Reedjik J. Inorg. Chem. 2003;42:868–872. doi: 10.1021/ic026034k. [DOI] [PubMed] [Google Scholar]
- 96.Olbrich F, Kopf J, Weiss E. Angew. Chem. Int. Ed. Engl. 1993;32:1077–1079. [Google Scholar]
- 97.Baxter CW, Higgs TC, Bailey PJ, Parsons S, McLachlan F, McPartlin M, Tasker PA. Chem. Eur. J. 2006;12:6166–6174. doi: 10.1002/chem.200501379. [DOI] [PubMed] [Google Scholar]

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.