

Abstract
Mimimized β hairpins have provided additional data on the geometric preferences of Trp interactions in TW-loop-WT motifs. This motif imparts significant fold stability to peptides as short as 8 residues. High-resolution NMR structures of a 16- (KKWTWNPATGKWTWQE, ΔGU298 ≥ +7 kJ/mol) and 12-residue (KTWNPATGKWTE, ΔGU298 = +5.05 kJ/mol) hairpin reveal a common turn geometry and edge-to-face (EtF) packing motif and a cation-π interaction between Lys1 and the Trp residue nearest the C-terminus. The magnitude of a CD exciton couplet (due to the two Trp residues) and the chemical shifts of a Trp Hε3 site (shifted upfield by 2.4 ppm due to the EtF stacking geometry) provided near-identical measures of folding. CD melts of representative peptides with the –TW-loop-WT- motif provided the thermodynamic parameters for folding, which reflect enthalpically driven folding at laboratory temperatures with a small ΔCp for unfolding (+420 JK−1/mol). In the case of Asx-Pro-Xaa-Thr-Gly-Xaa loops, mutations established that the two most important residues in this class of direction-reversing loops are Asx and Gly: mutation to alanine is destabilizing by about 6 and 2 kJ/mol, respectively. All indicators of structuring are retained in a minimized 8-residue construct (Ac-WNPATGKW-NH2) with the fold stability reduced to ΔGU278 = −0.7 kJ/mol. NMR and CD comparisons indicate that -TWXNGKWT- (X = S, I) sequences also forms the same hairpin-stabilizing W/W interaction.
Introduction
Peptide models of β sheets have been a major area of study over the last decade. There has been considerable success in preparing peptide sequences that form β hairpins1,2,3,4,5 and the extension of such systems to three- and four-stranded antiparallel-sheet models.6 These studies have provided data indicating that both the turn propensity within the reversing loops and cross-strand interactions contribute to hairpin stability. However with only a few exceptions,4,5,7,8,9,10,11 the studies have not included extensive series of hairpin analogs with the same strands and different turns or a common turn with systematic variation in the strand sites. As a result, a number of questions remain both regarding the specific contributions of cross-strand interactions and requirements for matching specific turn sequences with appropriate strands in designing stable hairpin models.
The C-terminal hairpin (GB1p), residues 41 – 56 of the B1 domain of protein G, was the first hairpin sequence12,13 to be examined extensively both with regard to folding kinetics14,15 and thermodynamics16. We recently prepared a series of more stable GB1 hairpin analogs (see Table 1) with melting temperatures in the 47 – 85 °C range.5 Extensive spectral comparisons revealed that GB1p is less than 30% folded at 25 °C. Cochran and co-workers4 prepared a series of “trpzip” hairpins that also display notable stability. Of these, trpzip4 is a direct analog of GB1p with the only change being the introduction of three Trp residues at non-H-bonded (S±even) sites. So long as the opposed WTW/WTW strand positions are retained, trpzip analogs display substantial hairpin stability for a variety of loop sequences and lengths. This motif introduces direct cross-strand W/W interactions; stabilizing diagonal Trp/Trp interactions have also been exploited in hairpin design.10 Throughout, we will refer to strand and turn positions by the nomenclature in Figure 1. The placement of the Trp residues allows their aromatic sidechains to engage in both direct cross-strand edge-to-face17 and diagonal (S-4/S+2) stacking interactions.
Table 1.
Representative sequences and stabilities of trpzip hairpins and GB1 analogs.
| name | sequence | Tm (°C) |
|---|---|---|
| trpzip2 | SWTW- ENGK -WTWK-NH2 | 72° (a) |
| (T3V)-HP6 | KYVW- SNGK –WTVE | 58° |
| Espinosa-GB1 (b) | RWQY- VNGK –FTVQ | 33% folded at 5° (b) |
| GB1p | GEWTY-DDATKT-FTVTE | 1–11° (c) |
| GB1m2 | GEWTY-NPATGK-FTVTE | 47° |
| GB1m3 | KKWTY-NPATGK-FTVQE | 59 – 62° (c) |
| HP5W4 | KKWTW-NPATGK-WTWQE | 85° |
| trpzip4 | GEWTW-DDATKT-WTWTE-NH2 | 70° (a) |
| trpzip6 | GEWTW-DDATKT-WTVTE-NH2 | 44.5° (a) |
| HP5W | KKYTW-NPATGK-WTVQE | 66° |
| HP5A | KKYTW-NPATGK-ATVQE | <30% folded at 7° |
| HP7 | KTW-NPATGK-WTE | 66° |
| Chignolin | GY-DPETGT-WG | 39° (d) |
Figure 1.

The nomenclature for positions in [2:2]/[2,4], [3:5], and [4:4]/[4:6] (Thornton18 classification) β hairpins; T indicates turn or loop positions, S indicates strand positions: [2:2]- and [4:4]-hairpins have an H-bond between the S+1 NH and the S−1 carbonyl. The S±even, non-hydrogen bonded (NHB), positions have their Hα atoms directed inward and display short Hα/Hα distances. The S±odd positions have their Hα’s outwardly directed with the sidechains displayed on the top surface of the hairpin as depicted here. The odd-numbered strand positions are also designated as “H-bonded pairs”.
GB1 hairpin analogs (and trpzip sequences) in which the 6-residue loop has been replaced by 4-residue sequences favoring type I’ or II’ (p = D-Pro) β turns have also been reported (Table 1). The Espinosa & Gellman analog19 retains the wild-type GB1 hydrophobic cluster. Our efforts in this area are illustrated by the (T3V)-HP6 sequence, which bears only one pair of cross-strand tryptophans but is otherwise analogous to trpzip2. The HP5 and HP7 series of peptides are built around the same optimized loop sequence, NPATGK. Chignolin20, a truncated hairpin with an analogous loop sequence (DPETGT), is also included in the tabulation. Melting temperatures (Tm), or an alternative measure of the thermodynamic stability, of each hairpin state are also included in Table 1.
Cochran and coworkers reported4 that returning W5 and W12, or W14 of trpzip4 to the ‘native’ amino acid, as found in GB1p, resulted in at least a 5 kJ/mol destabilization. Whether this represents specific cross-strand interactions or only the previously noted stabilizing effect of Trp at NHB positions is not clear. The modest stability of trpzip6 is intriguing since the sequence includes both a cross-strand W5/W12 pair and a diagonal W5/W14 pair. Even so, the construct is less stable than HP5W, despite having only two Trp’s at NHB strand positions. The enhanced fold stability of HP5W and HP5W4 likely reflects of the DDATKT NPATGK loop replacement with an additional contribution associated with the attractive Coulombic interactions at the chain termini.5,15
A comparison of the NMR data for trpzip4 and HP5W4 suggested the need for additional studies to determine the structural basis of hairpin stabilization associated with cross-strand Trp residues at NHB strand sites. Despite the obvious analogy between trpzip4 and HP5W4, the chemical shifts observed for the indole aryl-H resonances were very different. The indole ring chemical shifts reported for trpzip4 include two Hε3 sites, W5 (4.97) and W14 (5.21 ppm), with extreme upfield shifts; the other two Hε3 resonances appear at essentially random-coil values (7.59 ± 0.01 ppm). In contrast, the Hε3 shifts for HP5W4 are: 5.53 (W5), 6.74 (W14), 6.31 (W3), and 6.90 ppm (W12) – only W5 displays an extreme upfield Hε3. These discrepancies, and the current interest in edge-to-face (EtF) versus parallel-displaced (PD) aryl/aryl interaction energies17,21, prompted us to prepare additional peptides for determining the packing motifs of such cross-strand interactions. Herein we report two high-definition NMR structures and the thermodynamic stabilities observed for peptide folds with TW-loop-WT sequences. The fold stability associated with these sequences allowed us to examine loop substitutions to determine the essential features that allow or facilitate hairpin formation about Asx-Pro-Xaa-Thr-Gly-Xaa loops.
Results
Chemical shift deviations (CSDs) are established measures of peptide β-hairpin formation and provide multiple probes and thus address the folding cooperativity issue.5,11,15 The method does have some limitations: 1) a two-state folding model must be assumed to obtain meaningful ΔGU values, 2) 100%-folded CSD values are rarely available, and 3) only a limited temperature range can be probed. While circular dichroism shares the 100%-folded baseline concern, the data can be collected over a wider temperature range and at much lower concentrations (typically 10 – 30 μM). Agreement between NMR- and CD-derived equilibrium constants (measured at mM and μM concentrations) provides additional evidence that aggregation is not influencing the measurement of folding equilibria.
Melting curves for the trpzip peptides, from which the folding thermodynamics were derived, were based on CD data. The trpzip hairpins display a large amplitude exciton couplet with a maximum at 228 nm and a minimum at 213 nm. The intensity at the maximum, a wavelength where there is very little background ellipticity due to the peptide backbone, was used in those studies.4 Further, the reduced [θ]228-values for other systems have been attributed to incomplete population of the hairpin fold. 9 As a result, we set out to obtain fold-population estimates using both spectroscopic methods and to determine the structural basis for the exciton couplet observed for hairpins with cross-strand W/W interactions.
A Detailed Structural Comparison: HP5W4 versus Trpzip4
As previously noted, HP5W4, and trpzip4 share many structural features. The CD exciton couplets are qualitatively identical; see Figure 2 for representative spectra of HP5W4. The HP5W4 melting curve based on [θ]228 indicates a Tm on the order of 85 °C and a small degree of cold-denaturation. Based on data in the original trpzip report4, molar [θ]229-values approaching +1,000,000° (+88,000° on a residue-molar basis for a 12-residue peptide) are indicative of 100% folding for trpzip1. However, the maximum amplitude, [θ]228 = +608,000° (on a molar basis), attained by HP5W4 is significantly smaller. The ring current shift observed for Trp residue sites (the β-methylene and aryl-H resonances) in HP5W4 and trpzip4 are also remarkably different. A priori, the most comparable data should be for the Trp immediately preceding the loop with upfield CSDs of 1.55, 0.63 and 2.63 ppm for HβS, HβR, and Hε3, respectively, in trpzip4. The corresponding values for HP5W4 were 2.02, 0.83, and 2.07 ppm. The shift comparison does not support the view that HP5W4 is notably less well folded. Thus, the CD difference cannot be attributed to a lower fold population.
Figure 2.
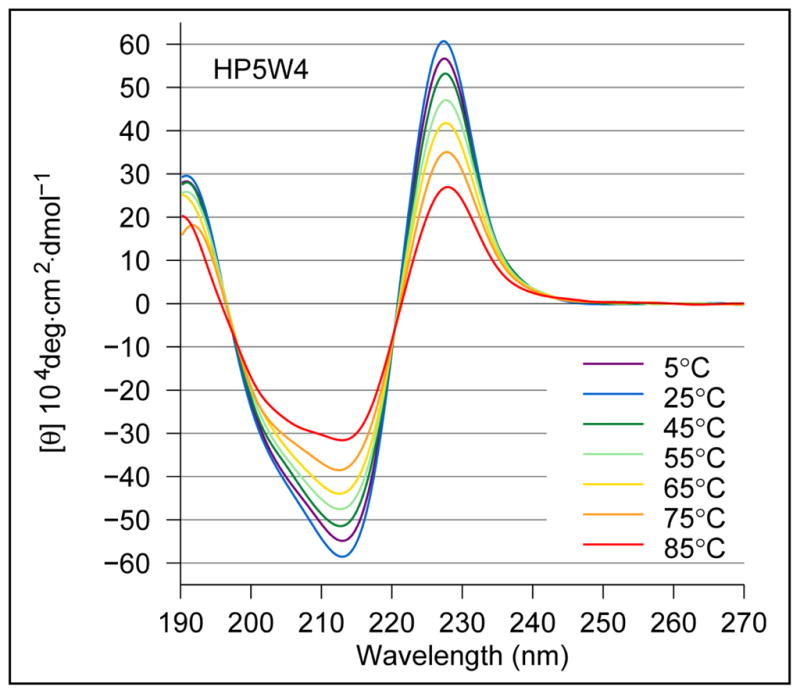
CD spectra recorded for peptide HP5W4.
With the aim of providing a structural rationale for these differences, we calculated an NMR structure ensemble for HP5W4 (Figure 3) for comparisons with trpzip4 and the native fold seen in the intact B1 domain of protein G.22 The structure was well converged for residues 3–14: key comparisons appear in Table 2, extended comparisons appear in Table 5. The backbone of HP5W4 and trpzip4 were nearly identical: the RMSD, excluding two terminal residues in each strand, is greater by just over 0.2Å than the RMSD observed over the same span within each ensemble. Despite four mutations in the loop, the conformational match is even closer over this region. Similar backbone comparisons between HP5W4 and chignolin (PDB #1UAO), over the 4-residue turn (0.27 ± 0.10 Å) and from aryl to aryl site (residues 5–12 of HP5W4, 0.58 ± 0.19 Å), indicate that all of these constructs belong to the same tightly defined hairpin class.
Figure 3.

An overlay (least squares fitted over the residue 3 – 14 backbones) of representative structures from the NMR ensembles of trpzip4 and peptide HP5W4.
Table 2.
| region | trpzip4 vs HP5W4 | trpzip4 vs GB1 | HP5W4 vs GB1 |
|---|---|---|---|
| backbone (3 – 14) | 0.57 ± 0.09 | 1.25 ± 0.11 | 1.22 ± 0.15 |
| turn backbone (T1 – T4) | 0.23 ± 0.06 | 0.22 ± 0.05 | 0.14 ± 0.02 |
| Cβ,Cγ (3,5,12,14) c | 1.09 ± 0.14 | 1.33 ± 0.11 | 1.42 ± 0.11 |
| indole rings (5,12) d | 0.27 ± 0.07 | n. a. | n. a. |
| indole rings (3,5,12,14) d | 1.58 ± 0.42 | n. a. | n. a. |
RMSDs are pairwise over the complete ensembles and were calculated using MolMol.23
PDB# 1LE3.
For GB1, 14Cγ is excluded since it is ambiguous when this residue is a valine.
The 9 heavy atoms of the indole rings.
Table 5.
NMR Structure Ensemble Statistics
| A. Structure Generation Summary
| ||
|---|---|---|
| HP5W4 | HP7 | |
| Distance Constraints | ||
| total | 137 | 82 |
| intraresidue | 15 | 5 |
| sequential | 48 | 31 |
| i/i+n (n = 2–4) | 18 | 16 |
| i/i+n (n ≥ 5) | 56 | 30 |
|
| ||
| Ensemble Statistics1 (with standard deviation) | ||
| accepted/random starts | 27 / 50 | 43 / 50 |
| ELJ (kcal/mole) | −93.75±4.55 | −62.53±2.82 |
| ENOE (kcal/mole) | 1.15±0.37 | 0.43±0.12 |
| Bond RMSD Å | 0.002±0.000 | 0.002±0.000 |
| Angles RMSD (deg) | 0.415±0.005 | 0.460±0.004 |
| Improper RMSD (deg) | 0.153±0.012 | 0.130±0.014 |
| B. Ensemble RMSD2 Values (Å)
| |||
|---|---|---|---|
| trpzip43 | HP5W4 | HP7 | |
| Backbone4 | 0.36±0.08 | 0.32±0.12 | 0.19±0.06 |
| All heavy atoms4 | 0.97±0.13 | 0.72±0.20 | 0.81±0.29 |
| Turn backbone5 | 0.20±0.07 | 0.08±0.03 | 0.09±0.05 |
| Turn heavy atoms5 | 1.21±0.256 | 0.14±0.06 | 0.18±0.08 |
| Indole rings7 | 0.43±0.18 | 0.36±0.12 | 0.12±0.06 |
| C. RMSD Comparison of HP5W4 and HP7 Ensembles (Å) | |
|---|---|
| 4–13 Backbone | 0.46±0.08 |
| 4–13 Heavy | 1.00±0.22 |
| 7–10 Backbone | 0.21±0.06 |
| 7–10 Heavy | 0.33±0.07 |
| Indole Rings 5,12 | 0.24±0.06 |
Calculated with CNS prior to final minimization using the AMBER forcefield.
Calculated (pair-wise, over the complete ensembles) using MolMol23; the mean and standard deviation are given for both intra- and inter-ensemble comparisons.
Calculated using PDB# 1LE3.
Residues 3–14 for trpzip4 and HP5W4, 4–13 for HP7
Residues 7–10 are the 4 turn residues: PATG for HP5W4/HP7 and DATK for trpzip4.
The large difference in RMSD for trpzip4 versus the two new constructs may reflect the amount of disorder possible in the sidechains of the DATK turn sequence as opposed to PATG.
This measure includes only the 9 heavy atoms of the indole rings.
The overlay of the trpzip4 and HP5W4 structures (Figure 3) and the indole ring RMSDs provide a direct comparison of the indole ring interaction geometries. While residues 5 and 12 structure similarly, as indicated by their 0.27Å RMSD, the tryptophans located closer to the termini exhibit different packing motifs in the two structures. In trpzip4, both cross-strand Trp pairs display an edge-to-face (EtF) interaction. In HP5W4, only residues 5 and 12 displays the EtF interaction geometry; the other indole/indole interactions are closer to PD stacking.
The greater CD signal intensity for trpzip peptides (which have two chiral EtF interactions), can be rationalized by assuming that only the EtF interaction geometry results in exciton coupling in the CD. This is also the interaction geometry that produces extreme upfield shifts for Hε3 of an indole ring (the “E” ring in the EtF interaction). CSDs at strand backbone positions would normally provide an orthogonal measure of folding, however variations in the apparent orientation of the indole rings, and the resulting ring-current effects on chemical shifts, make a direct comparison difficult. In order to validate both CD and multiple NMR measures of folding, we turned to HP5W mutants bearing a close resemblance to tripzip2 but lacking the W/W NHB pair more remote from the turn. Validated CSD measures11 of structuring at XNGK turns were viewed as an additional cross-check for spectroscopic measures of both β-strand association and those associated with the W/W interaction geometry.
Quantitating Folding Associated W/W interactions by CD and NMR
In our first test system (HP6), the loop sequence of HP5W was replaced by SNGK and the 2nd and penultimate residues were eliminated. HP6 was also examined in its N-acetylated form (Ac-HP6) and with a T3V mutation. In analogy to a fold-stabilizing modification reported from the Cochran laboratory8, (T3V)-HP6 was significantly more stable. We prepared AWSNGKAT to serve as a non-folded control for the CD spectrum and chemical shifts for an unstructured Trp.
Our initial estimates of the extent of folding of these constructs were based on CSD comparisons with trpzip2 and HP5W (Figure 1S). The latter comparison was, of necessity, limited to the common strand positions which included four sites with the expected downfield shifts.11 Two were shifted downfield due to cross-strand H-bonding (T3HN, T10HN) and the others due to cross-strand diamagnetic anisotropy effects (Y2Hα, W9Hα). In addition there were large, diagnostic CSDs at T3Hα, T10Hα, W4HN, and K8HN. The 300K fraction-folded estimates, fF, from these comparisons were: 0.82 ± 0.08 ((T3V)-HP6), 0.61 ± 0.07 (HP6), and 0.52 ± 0.07 (Ac-HP6). Fraction-folded (χF) estimates could also be derived from shift data for the sites in the turn. For XNGK turns, two diagnostics are available: the downfield shift of Asn HN and the diastereotopicity at the Gly-CH2 (Gly-ΔδHα). The latter has a long history as a quantitation of hairpin folding.2,24,25,26,27 For trpzip2 (at χF = 0.90), these two turn shift measures are 0.79 and 0.59 ppm. The corresponding values for (T3V)-HP6 are 0.55 and 0.56 ppm; χF ≈ 0.63 and 0.85, respectively at 300K.
As with trpzip2, the largest structuring shift observed for each of the HP6 peptides was Hε3 of Trp4 and these shifts correlated with the amplitude of the CD exciton couplet (Figure 4). The CD data and the ring current shift at W4Hε3 (as large as –2.07 ppm) provided the same ranking of the stability of (T3V)-HP6, HP6 and Ac-HP6. If we assume that an Hε3 structuring shift of –2.25 ppm [CSD = −2.03 for trpzip2 at χF = 0.90] corresponds to 100% folding, we obtain χF estimates concordant with those from backbone sites and the following melting temperatures (°C) for the peptides: 36° (Ac-HP6), 45° (HP6) and 58° ((T3V)-HP6). The same analysis of the CD melt suggests that the Δ[θ]228-value for 100% folded should be 490,000°, one-half of the value observed for trpzip peptides with two, rather than one, EtF W/W interactions. The CD melting temperatures (see Methods) for Ac-HP6, HP6 and (T3V)-HP6 were 25, 45 and 58 °C, respectively.
Figure 4.

NMR and CD melts for the HP6 series of peptides.
An alternative, second derivative, analysis of the CD melts (which makes no assumptions regarding the limiting values or their temperature dependence) is also available. Tm can be defined as the point where ∂[θ]/∂T is maximal. This places the Tm’s for Ac-HP6 and HP6 in the 30 – 45° and 40 – 50° ranges, respectively, with the distinctly sigmoidal (T3V)-HP6 melt clearly indicating Tm = 59 ± 4 °C. The agreement between multiple NMR probes (measured at 1.5 mM) and the CD melts (30 μM) is indicative of two-state folding transitions for monomeric peptides. It is also apparent that (T3V)-HP6 is nearly as stable as the 12-residue trpzip peptide with an analogous Asn-Gly turn locus. The chemical shifts observed in the turn region of control peptide AWSNGKAT imply little turn formation (≈14 % at 298K, < 19 % at 280K) based on the Gly ΔδHα calibration.
Hairpin Strand Truncation
Since we anticipated that the SNGK turn of HP6 might not have sufficient turn-forming propensity to support hairpin formation when the β strands are only of three residue length, we returned to the NPATGK loop for this study: HP7 = KTW-NPATGK-WTE. This additional strand deletion returns the terminal Lys/Glu pair to the non-H-bonded positions that they occupied in HP5 peptides and GB1m3. The cross-strand W/W placement and β-strand favoring threonines were retained. Peptide HP7 formed a stable hairpin, displaying the diagnostic upfield shift of Hε3 (5.33 ppm) and the CD exciton couplet (molar [θ]228 = +406,000) at 280K. A comparison of the five backbone CSDs used5 to determine the extent of hairpin formation in HP5W with those observed for HP7 (Figure 2S, Supporting Information) suggested the latter was 93% folded at 280K. For HP7 mutants, large upfield shifts at the T7 and K9 HN sites provide turn-based measures of folding that are in excellent agreement with the strand probes.
The upfield shift of W3-Hε3 and CD (the [θ]228-value), which could be readily monitored over a large temperature range, provide melting temperatures for HP7 and mutants of the sequence. In the case of the NMR probe, we assumed that both the random-coil value (7.62 ppm) and the 100% folded value (5.22 ppm) were temperature invariant. This NMR probe (panel A of Figure 6) provided a Tm = 69 °C for HP7. The second derivative analysis of the CD melt afforded Tm = 63 °C. A full thermodynamic analysis (Table 4, vide infra) of the CD melt also provided a Tm of 63 °C. By all analyses, peptide HP7 forms a stable, monomeric hairpin (ΔGU298 = +5.05 kJ/mol) displaying reversible melting by both NMR and CD.
Figure 6.

Representative melting studies of a series of HP7 analogs. A. The temperature dependence of the most upfield aryl-H signal (Hε3 of W3), B. CD melts of the same series; the observed “adjusted-[θ]227” (see Methods) values for (K1R)-HP7 and (N4D)-HP7 are multiplied by factors of 1.14 and 1.07, respectively, to raise them just above the values recorded for HP7 since all other data indicates that these analogs are at least as well-folded as the parent system. The fitted curves in panel B are fourth order polynomials, rather than based on a folding model; presumably they capture the curvature of the experimental data and can be used to obtain Tm values by the second derivative method.
Table 4.
Thermodynamic Parameters for Hairpin Unfolding.
| Tm a | ΔHma | ΔH298 | ΔS298 | ΔCp | |
|---|---|---|---|---|---|
| Peptide b | ( °C) | ( kJ/mol ) | ( JK−1/mol ) | ||
| KYVW- SNGK –WTVE | 59 | 43.5 | 29.2 ± 0.8 | 84.5 ± 2.5 | 420 ±40 c |
| KTW-NPATGK-WTE | 63 | 46.7 | 31.2 ±1.4 | 88.7 ±4.2 | 410 ±60 c |
| KTW-NAATGK-WTE | 59 | 47.3 | 30.8 ±1.3 | 89.1 ±3.8 | 480 ±60 c |
| KTW-NPAAGK-WTE | 53 | 35.7 | 25.2 ±1.0 | 74.5 ±2.9 | 370 ±40 |
| KTW-NPATAK-WTE | 41 | 35.7 | 28.1 ±0.8 | 87.0 ±2.5 | 470 ±40 |
| Ac-TW-NPATGK-WT-NH2 | 21 | 32.2 | 33.7 ±1.5 | 114 ± 4 | 380 ±50 |
| Ac-W-NPATGK-W-NH2 | 3 | 21.2 | 25.4 ±0.8 | 92.2 ± 2.6 | 190 ±80 |
| KTW-APATGK-WTE | −2 | 5.2 | 18.0 ±1.7 | 81.1 ±5.4 | −80 ±80 |
| Prior Literature Examples
| |||||
| Ac-RFVO VNGK EIFQ-NH2 d | 18 | 64 | 360 | ||
| SWVW ENGK WTWK-NH2 e | 69 | 70.3 | 14.9 | 31 | 1180 |
| GEWVWDDATKTWTVTE-NH2 e | 37 | 43.1 | 23.6 | 72 | 990 |
| Ac-RWVE VNGK KMIQ-NH2 d | −3 | −5 | 850 | ||
| Ac-KKYTVEINGKKITVEI f | −11.9 | −38 | 465 | ||
| Ac-KKYTVSINGKKITVSI g, h | −5 ±1 | −10 ±3 | 700 ±75 h | ||
Tm and the enthalpy of unfolding at that temperature are calculated from the best fit ΔH298, ΔS298 and ΔCp values.
From the present study.
The ΔCp values increase by 125 JK−1/mol, or less, if we assume that the 100% folded value is under-estimated by 8 %; for analogs that are less well folded the increase associated with potential errors in χF estimates are not significant.
Data from Cochran et al.4
Data from the Searle laboratory.34
Tabulated data from Andersen et al.29
With different assumption regarding the CD and NMR parameters for the folded state, Griffiths-Jones et al.2 report ΔH298 = −3.7 kJ/mol, ΔS298 = −10±2 JK−1/mol, and ΔCp = +1230±175 JK−1/mol; while the differences are large, both analyses conclude that folding is entropically driven at 298 K.
An NMR structure ensemble (Figure 5) was calculated for HP7 using the same protocol as for HP5W4. The residue 2 – 11 backbone conformation is well converged, with a low pairwise-RMSD over the ensemble (0.19 Å), and essentially the same as that observed for the same sequence in HP5W4 (0.46±0.08 Å inter-ensemble RMSD, see Table 5). The interaction geometries for the Trp/Trp pair are also identical (0.24±0.04 Å RMSD).
Figure 5.
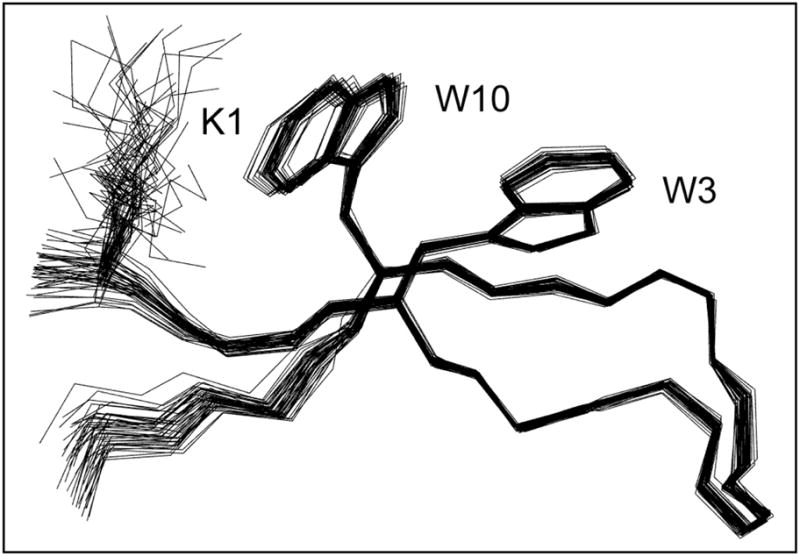
Overlaid structures in the HP7 NMR ensemble, the N-terminal strand is in front of the C-terminal strand in this view.
The HP7 NMR ensemble also shows a close association of the K1 and W10 sidechains suggestive of a cation-π interaction. This feature of the ensemble predominantly reflects residue-specific cross-strand 1H nOe’s: 1β/10ε3, 1β/10β, 1δ/10ε3, 1δ/10β, and 1α/10ε3. The Lys/Trp interaction was confirmed by ring-current shifts at K1, position (upfield shift in ppm): Hβ (0.22 and 0.16), Hδ (0.30 and 0.49), Hε (0.62 and 0.74). The nOe-derived ensemble places this sidechain within the shielding cone of the W10 indole ring. Unambiguous K1/W14 distance assignments were sparser in HP5W4 due to signal overlap. Even so, ring current shifts (e.g. 0.32 at Hδ and 0.52 ppm at Hε) comparable to those in HP7 and other β-hairpins10,28 indicate an analogous cross-strand Lys/Trp cation-π interaction.
A more complete examination of the extent to which the distinctive NMR structure ensembles of trpzip4, HP5W4, and HP7 predict experimental chemical shifts, with particular attention to the ring-current shifted, indole-ring sites appears in the Supporting Information. The differences observed in the NMR structure ensembles are largely corroborated by the chemical shifts. All three structures (trpzip4, HP5W4 and HP7) share the same turn conformation and turn-flanking Trp-sidechain geometries; the latter indicated by identical Jαβ2/Jαβ3 coupling constants reflecting a highly populated anti rotamer at Trp3 and (−)-gauche rotamer at Trp10 (Jαβ2 = 12.0, Jαβ3 = 2.5 Hz) in HP7. However the packing of the terminal Trp sidechains differs in the trpzip4 and HP5W4 ensembles and the latter does not predict the shifts observed for the aryl-H signals of W3 and W14. The NMR ensemble fails to include alternate sidechain conformers for this Trp/Trp pair; these are reflected in averaged Jαβ2 /Jαβ3 coupling constants. The residual upfield shift at Hε3 of W14, which is not expected based on the structures in the NMR ensemble, likely implies that the EtF orientation of W14 and W3 seen in trpzip4 is partially populated for HP5W4.
Truncation and Mutation of the HP7 Sequence
Representative NMR and CD melting curves for HP7 and a series of mutants appear in Figure 6. Even a cursory inspection of Figure 6 reveals that (N4A)-HP7 and HP7(-2) are significantly less folded than all of the other species at the low temperature limit. Of the remaining analogs, the G8A mutant is the least stable. This data provides experimental evidence for hairpin stabilization associated with the S-1 Asx and T4 Gly residues. Panel A of Figure 6 provides qualitative, but not quantitative, support for our assumption that 5.2 ppm is the 100% fold expectation-value for W3 Hε3: the observed shifts plateau near this value at the lowest temperatures studied. Tm’s were derived from CD melts using the two methods previously described; the results from both appear in Table 3. Unlike many designed hairpins11,25,26,27,29, none of these peptides displayed cold denaturation over the 275 – 330 °C temperature range in either water or upon addition of hexafluoro-isopropanol (HFIP).
Table 3.
Hairpin stability effects of mutations and truncations in the HP7 hairpin series. Measurements are at pH 6 unless otherwise noted.
| Name | Sequence | Tm ( | °C) | %-folded | at 298K |
|---|---|---|---|---|---|
| CDa | NMRb | strandc | turnd | ||
| HP7 | KTW-NPATGK-WTE | 66 (63) | 69 | 88 | 89 ± 1 |
| + 8% HFIP, pH ≈5.5 | 59 (54) | 57 | 85 | 82 ± 1 | |
| at pH 3 | 47 | 77 | 79 ± 2 | ||
| Strand Mutations and Truncations
| |||||
| K1R | RTW-NPATGK-WTE | 71 (68) | 71 | 90 | <90> |
| K1A/E12A | ATW-NPATGK-WTA | 43 (43) | 48 | 74 | 82 ± 2 |
| HP7(-2) | Ac-TW-NPATGK-WT-NH2 | 22 (22) | 17 | (41b) | 55 ± 6 |
| HP7(-4) | Ac-W-NPATGK-W-NH2 | ~1e | < 7e | (24b) | 41 ± 9 |
| Des-W10 | KTW-NPATGK-TE | n. a. | n. a. | n. a. | 20 ± 10 |
| loop | Ac-NPATGK-NH2 | n. a. | n. a. | n. a. | 6 ± 7 |
|
| |||||
| Loop Mutations
| |||||
| N4D | KTW-DPATGK-WTE | 66 (62) | 73 | 87 | 89 ± 1 |
| N4A | KTW-APATGK-WTE | −1 (≤15) | 13 f | 41 | 43 ± 2 |
| P5A | KTW-NAATGK-WTE | 61 (59) | 56 | 88 | 81 ± 7 |
| T7A | KTW-NPAAGK-WTE | 57 (55) | 65 | 83 | 80 ± 15 |
| G8A | KTW-NPATAK-WTE | 45 (44) | 56 | 81 | n. a. |
| Substitution with Shorter Loops
| |||||
| INGK | KTW- INGK -WTE | 54 (49) | 42 | 63(67b) | (63 – 70) |
| NPDGT | KTW- NPDGT -WTE | 32g(50) | < 7g | 39(29b) | (46 / 50) |
| EPDGK | KTW- EPDGK -WTE | ≪ 5 | ≪ 7 | 23(15b) | (23 / 31 ) |
The CD Tm is derived assuming a warming-induced decrease (0.36%/ °C, see Methods) in the folding-induced change in [θ]228 with Δ[θ]228 = +434,000° at 0 °C for all systems. The model-free Tm’s based on the second derivative of the fitted melting curve are shown in parentheses; italicized values are viewed as inaccurate due to the near linearity of the CD melt.
Based on the W3-Hε3 shift.
Based on T2 and W10 Hα and T2, W3, and T11 HN CSDs.
For the 6-residue loop species, the average and standard deviation of %-folded as determined from the T7 and K9 HN CSDs and G8-ΔδHα, assuming that (K1R)-HP7 is 90% folded at 298K. See text for the probes employed for the 5-residue loops and the INGK turn.
38% folded at 7 °C, 24% at 25 °C based on the Hε3 shift probe versus 48% folded at 5 °C based on CD.
Two additional NMR melting points are available: 12 °C (strand-CSDs) and 17 °C (turn CSDs).
34% folded at 280K based on the W3-Hε3 shift; 61% by CD assuming the expectation values for HP7 applies to this loop.
Table 3 also records the %-folded estimate at 298K derived from the Hα and HN sites in the strands displaying large CSDs. Although there are larger CSDs (HN at T7 and K9) within the turn region, and these yield comparable results, the strand residue probes have the potential advantage of being applicable to peptides with the same strands but modified loop and turn sequences. We replaced the 6-residue NPATGK loop with both 5-residue (NPDGT3 and EPDGK9) and 4-residue (INGK) loops, forming [3:5] Gly-bulge and type-I’ turns, respectively.
Since the Asx-PATGK sequence appears to be so well suited for chain reversal with β-hairpin formation, we also examined two extreme variations: complete truncation of the β strands (loop-only = Ac-NPATGK-NH2 ) and the deletion of one of the Trp residues involved in the stabilizing W/W interaction (des-W10, see Table 3). The aim was to determine the extent of structuring by the turn sequence in the absence of the stabilizing W/W interaction. Both changes produced constructs that had very poorly formed turns (< 25% turn population); for the loop-only sequence, all available folding measures yield %folded values of 14% or less.
Although the different probes do not yield exactly the same folding estimates, the stability ranking that follows from the average CD melting values is in agreement with the ranking based on the magnitude of the strand CSDs at 298K. The most likely source of error in the Tm values based on the Hε3 shift and CD is that the value for 100% folded varies to some extent over this series of analogs due to subtle changes in the relative orientations of the W3 and W10 indole rings. Such changes should not, however, complicate the second derivative analysis of the CD melts which makes no assumptions concerning the 100% folded value. Given the broadness of the melting transitions, these Tm estimates are probably accurate to ±5 °C. With the exception of the 5-residue loop analogs, the Tm determined by the second-derivative method agrees (ΔTm < 5°C) with the Tm derived based on a fixed [θ]228-value for 100% folding at the low temperature limit. Apparently, the amplitude of the exciton couplet is comparable in the folded state of all the [4:4]- and [2:2]-hairpins. Defining the extent of folding of the [3:5]-hairpin systems is more difficult. However, all possible analyses based on CD or NMR data imply that the [3:5]-hairpins are less stable than the optimized [4:4]- and [2:2]-hairpins.
For severely truncated species and shorter loops, we had to rely increasingly on chemical shift probes in the turn region. In the case of XNGK turns, we employed Asn-HN, Gly-ΔδHα, and Lys-HN as the turn folding probes with the same calibration procedure employed for HP6. For the 5-residue XPDGX loop sequences, analogies to well-folded [3:5]-hairpins11,30 would predict that Gly-ΔδHα and the Asp-HN CSD should approach +0.6 and −1.0 ppm, respectively. The CSD diagnostics for turn formation in the 6-residue loops were HN of the Thr and Lys and Gly-Hα, the latter quantitated as Gly-ΔδHα. The G8-ΔδHα value for our most stable systems, HP7 and (K1R)-HP7, was 0.520±0.002 ppm throughout the 7 – 25°C range; this was equated with 90% folding for Table 3. For (N4A)-HP7, the least stable analog retaining the ATGKW sequence, the Gly-ΔδHα values at 300K dropped to 0.236 (41%) and 0.069 ppm (12%) at pH 6 and 3, respectively. The decrease at pH 3 reflects the fold-destabilizing effect of protonation of the C-terminal carboxylate sites. These folded measures, and ones derived from the HN CSDs of T7 and K9, are in excellent agreement with the CD and strand Hα CSD values.
DISCUSSION
A large set of sequences with a pair of tryptophans immediately flanking 4 – 6 residue loops favoring chain reversal and β-hairpin formation have been examined. The common features indicating folding are a strong CD exciton couplet and an extreme upfield shift for the Hε3 indole ring proton of the Trp that precedes the turn. These became our primary probes for melting studies. Backbone CSD measures of folding were generally in excellent agreement with the probes that report on the turn-flanking W/W interaction. The derivation of similar fraction-folded estimates and melting points using multiple probes provides evidence for folding cooperativity and the observation of the same melting point by CD (15 – 30 μM) and NMR (1 – 2 mM peptide concentrations) implies that these structuring parameters are not concentration dependent and thus attributable to a folded monomer.
Both 16- and 12-residue constructs with a TWNPATGKWT core proved to be folded in water (ΔGU290 ≥ +6 kJ/mol). This level of fold stability allowed for high-resolution NMR structure determination. Although the loop sequences of HP5W4 and trpzip4 are different, we found no significant changes in the loop backbone conformation or in the β strands near the loop; both models displayed an excellent fit to the protein-context GB1p structure. The sidechain conformations of the two common tryptophan rings in these hairpins was identical to that of the same sites4 in trpzip peptides. However, a comparison of the chemical shifts and structure ensembles of HP5W4 and trpzip4 revealed differences (Figure 3) in the orientation of the terminal Trp pair. Recent MD simulations17 suggest that EtF W/W interactions in trpzip hairpins are 24 kJ/mol more stable than parallel-displaced (PD) interactions and that the preference is associated with Coulombic effects (due to sidechain multipole, MP, moments) rather than a strictly hydrophobic effect. When the MP terms are not included, the trpzip2 structure simulation deviates from the NMR structure: W2 and W4 maintain the same χ1/χ2 angles but changes at W9 and W11 place W4/W9 in a ‘looser’ EtF interaction with W9/W2/W11 in an extended PD stack that resembles that present for W12/W3/W14 in the NMR ensemble of HP5W4. The absence of folding observed for a W7A mutation in a truncated HP6 peptide (AWSNGKAT) suggests that the Trp sites in W-loop-W sequences provide at least 5 kJ/mol of hairpin stabilization.
Seven of the HP7 peptides and (T3V)-HP6 displayed excellent agreement between NMR (multiple probes) and CD melts, with the latter displaying sufficient curvature to allow us to derive the thermodynamic parameters of the folding transitions. A caveat must be raised, however, as defining the 100% value can be problematic. Even when chemical shifts appear to be leveling out at low temperatures for the most stable member of a series of analogs, one cannot eliminate the possibility that this represents a broad inflection point, where the hot and cold denaturation transitions meet; such a plateau cannot be equated with >90% folded.15 Additional studies using aqueous HFIP (data not shown) did not provide any instances in which spectroscopic measures of folding were enhanced.31
The folding thermodynamics derived for these systems are compared to those of other hairpins in Table 4. They were derived assuming a temperature invariant ΔCp would apply. We first determined the range of ΔCp values consistent with the observed curvature of the ln KU versus T plot (see Methods); this provided the allowed range of ΔH and ΔS values. With the exception of the species with the highly destabilized turn, mutations within the turn region were accommodated with no significant change in the ΔCp value. A common ΔCp is a reasonable expectation for a series of peptides that share the same hydrophobic side-chain interactions in the folded state.
The comparisons included in Table 4 provide some additional insights into hairpin folding. The Waters hairpin with a cross-strand F/F cluster has a ΔCp similar to that observed in our systems. The trpzip peptides have larger ΔCp-values but folding is still favored by enthalpy rather than entropy throughout the normal temperature range. The larger ΔCp likely reflects the greater extent of apolar surface burial that results due to the PD stacking in the additional (diagonal) W/W interactions. The three hairpin sequences included at the bottom of Table 4 lack cross-strand aryl/aryl interactions and display large ΔCp-values; the ΔSU is negative in the vicinity of the melting temperature, another diagnostic of hydrophobically driven folding.32 The last three peptides listed also display dramatic cold-denaturation. In the case of a similar sequence, KKYTVSINGKKITVSA, cold-denaturation becomes more substantial with a Y3L mutation, which replaces the only aryl sidechain with a aliphatic one, and upon addition of 8% HFIP.11,27,29 The thermodynamic parameters for the present series of peptides predict temperatures of maximal structuring that are below 250 K.
The Trp-packing differences between HP5W4 and trpzip4 are attributed to changes at the chain termini: KKW----WQE versus GEW----WTE-NH2. The trpzip4 termini contain a potentially repulsive E/E sidechain interaction, while the HP5W4 sequence has only attractive interactions between the termini and provides the possibility of a Lys/Trp cation-π interaction (K1/W14). The NMR data for both HP5W4 and the 12-residue HP7 revealed ring-current shifts at K1 that are best rationalized by a cation-π interaction. A cation-π interaction requires a W14 indole ring orientation that is incompatible with a W14/W3 EtF interaction while the W3/W10 EtF geometry in HP7 allows a K1/W10 interaction. In the trpzip peptides, both cross-strand indole rings pairs adopt the EtF interaction geometry. The partial population of the EtF W3/W14 interaction in peptide HP5W4 would be consistent with the observed [θ]228 value which was greater than 50% of the value observed for well-folded trpzip systems containing two EtF interactions. We posit that HP5W4 samples both a K1/W14 cation-π interaction and a W3/W14 stacking interaction and that the more remote Trp pair provides less hairpin stabilization than the W5/W12 pair. Strand mutations that eliminate the possibility of a cation-π interaction decrease the fold population. Fold population decreases are also observed upon acidification. As a result, we cannot quantitate the hairpin stabilization associated with the terminal Lys/Trp interaction: mutations at K1 also change other hairpin-stabilizing Coulombic interactions2,5,15,35 between the strand termini. We attribute the enhanced folding associated with the K1R mutation to a more favorable cation-π interaction.
The present study also included W-loop-W hairpins with both 4- and 5-residue loops. The 4-residue loops were XNGK (X = S, I); these favor type-I’ [2:2]-hairpin formation. The INGK mutant of HP7, although folded, was distinctly less stable (ΔTm = −18 °C, ΔΔGF ≥ 2 kJ/mol) than the original construct. The [3:5]-hairpins prepared for this study, however, presented a number of problems. The CD exciton couplet and ring-current shifts confirm chain reversal and formation of a W/W interaction similar to that seen in the other series. But the folding probes that gave consistent estimates of stability for the [4:4]- and [2:2]-hairpins now diverge. Comparisons with stable [3:5]-hairpins with XPDGX loop sequences11,30 provided fold estimates ranging from χF = 0.26–0.71 for the NPDTG loop analog of HP7.36 We conclude that a favorable the W/W interaction geometry can be achieved in [3:5]-hairpins, but the packing motif may not be identical to that observed for the 4- and 6-residue loops.
In our optimization of the GB1 hairpin, we observed significant stabilization for a DDATKT → NPATGK loop substitution.5 A statistical analysis5 of 285 [4:4]- and [4:6]-hairpins in proteins revealed strong preferences for specific residues at the S-1, T1, T3 and T4 positions which can be summarized as a preferred Asx-Pro-Xaa-Thr-Gly-Xaa loop motif. Fully 65% of the hairpins had Asx at S-1; 44% Thr(or Ser) at T3; 28% Gly at T4. The preference for Gly at T4 can be attributed to positive phi/psi values required at this site for chain reversal. The contributions of individual sites were examined in the HP7 peptide series; ΔΔGU estimates were obtained from changes in Tm. The N4D mutation, which results in a non-significant +1 °C ΔTm (using the average of the three Tm determinations in Table 3), revealed that Asx is the correct designation for the first loop position. In contrast, the N4A mutation results in ΔTm = −52 °C (based on three NMR measures of folding) and reduces the %-fold at 298K from 88.5 ± 1 to 42% (ΔΔGU = −5.9 kJ/mol). This provided the calibration of the relationship between ΔTm and ΔΔGU. The typical spread in Tm-values corresponds to ±0.4 kJ/mol precision in ΔΔGU-values. The data in Table 3 provides the following mutational destabilization measures: P5A and T7A (ΔΔGU = −0.9 kJ/mol), G8A (−2.1). Although earlier studies of GB1p mutants indicated that the Asp at the S-1 site is important for folding,13 the magnitude of the effect was surprising.
The NMR ensembles for HP5W4 and HP7 provide a potential rationale for the fold-stabilizing effect of the S-1 Asn and Thr at T3/S+3: an extensive series of H-bonding interactions: Asn-Oδ/T2-HN, Asn-Oδ/T3-HN, Asn-Hδ21/Thr-Oγ (at S+3), and Thr9-Oγ/Lys11-HN (T3/S+1) which could represent stabilizing interactions within this particular loop geometry. Some, but not all of these interactions, appear over the DXATXXXT sequence in GB1 crystal structures (PDB entries 1PGb and 1PGBA). The chemical shifts of the Asn sidechain provide additional evidence for a structuring effect. The δ21 and δ22 resonances appear at 7.18 and 7.24 ppm rather than at the solvated, random-coil values of 7.60 and 6.92 ppm, respectively. In thermal melts, the sidechain CONH2 CSDs melt out together with the cross-strand Hα and HN CSDs.37 Backbone torsion angle preferences do not appear to play any role in the propensity for Asx at the S-1 site; this residue displays phi/psi values (−110°/+94°) that are unremarkable (close to the minimum energy point in the extended/poly-ProII region of the Ramachandran plot).
With the possible exception of ‘unnatural’ Aib-Gly and D-Pro-Gly sequences11,39, no known loop sequences yield stable turns in the absence of cross-strand hydrophobic or aromatic stacking interactions. This also appears to be case for Asx-Pro-Xaa-Thr-Gly-Xaa loops. Further truncation of the β strands linked by the NPATGK loop reduced fold stabilities. The basis for these changes remains speculative. In the HP7(-2) → HP7(-4) truncation, Thr residues flanking the Trp residues are removed. Threonine is known to have a greater propensity for extended, β-chain configurations. The removal of this conformational restriction could increase the tendency for disorder in the Trp sidechains and thus reduce the contribution of a fold-favoring W/W interaction. The truncation also removes amide NH and C=O functions that could form an additional cross-strand H-bond. The CD exciton couplet and ring-current shifts observed for an 8-residue peptide (Ac-WNPATGKW-NH2) indicate that the cross-strand EtF W/W interaction is retained. This does not reflect a loop sequence that forms a turn in the absence of cross-strand interactions. At 280K, the turn population of Ac-NPATGK-NH2 was 0.09 ± 0.05 while that of Ac-WNPATGKW-NH2 was 0.41 ± 0.09. While we cannot attribute the fold stabilization exclusively to an EtF W/W interaction, the ΔΔGF (circa −5 kJ/mol) and geometry preference is preserved throughout the series, from 16-mer hairpins to this 8-mer that is nothing more than a stabilized loop. Subsequent studies (to be reported elsewhere) have uncovered additional W-loop-W folds all of which have the EtF geometry even though the “edge-on” Trp can be either at the N- or C-terminal site of the W-loop-W sequence.
Methods and Materials
Materials and Peptide Synthesis
Peptides HP5W4, HP5W and HP5A were available from a previous study.5 All of the other peptides appearing in Tables 1 and 3 were constructed using fast FMOC chemistry on an ABI 433A peptide synthesizer and purified by reverse-phase HPLC (C18 column) with a water (0.1% TFA)/acetonitrile (0.085% TFA) gradient. All peptides displayed the expected (M+2H)+2 and/or (M+3H)+3 mass spectral ions on a Bruker Esquire ion-trap mass spectrometer. In some cases, an additional HPLC purification using a C8 column was required to obtain peptide samples of > 95% purity by NMR. Two additional control peptides -- Ac-RKAWAGK, and AWSNGKAT -- were also prepared. All of the sequences were confirmed by 2D NMR connectivities observed in NOESY or ROESY spectra.
CD Spectroscopy
CD stock solutions were prepared by dissolving weighed amounts of peptides in 20 mM aqueous pH 6 potassium phosphate buffer to make a solution of about 500 μM. The concentrations of stock solutions were measured by the combined expectation UV absorption of Trp (ε = 5580 M−1 cm−1 per Trp) and Tyr (ε = 1280 M−1 cm−1 per Tyr) at 280 nm. CD stocks were diluted to obtain 15 – 30μM peptide solutions. Spectra were recorded on a Jasco J715 spectropolarimeter using 0.10 cm pathlength cells. The calibration method, for the wavelength and degree-ellipticity scales, and spectral-accumulation parameters have been described previously.40 The procedure for CD melting studies is modeled on previous studies.5 Although peptide CD data is typically reported in residue-molar ellipticity units (deg cm2 residue–dmol−1), we employ molar units (deg cm2 dmol−1): the dominant feature in most of the CD spectra is due to a single chromophore in the peptide rather than the backbone amide units. This dominant feature is an exciton couplet appearing as a maximum at 227–229 nm and minimum at ~213 nm in all folded systems; it is illustrated by the temperature dependence of the CD spectrum of HP5W4 (Figure 2). The control peptides (listed above) and (des-W10)-HP7 have, on a molar basis, [θ]228-values of ~ –11000°, and display little temperature dependence. As a result, we add 11000°/Trp to the observed molar [θ]228-values to afford a measure of folding that should approach zero as the fold population of HP7 analogs goes to zero. For the HP6 peptide series, AWSNGKAT provides a specific control and the small temperature dependence of its CD was included in the analysis.
NMR Spectroscopy
NMR spectra were recorded on 500 and 750MHz NMR instruments with Bruker DRX and DMX consoles, respectively. All 2D spectral experiments employed a WATERGATE41 pulse train for solvent suppression. TOCSY spectra employed a 60 ms MLEV-17 spinlock42 and NOESY spectra had a 150 ms mixing time. Samples contained 1 – 2 mM peptide in buffered H2O with 10% D2O. Sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) was used as an internal chemical shift reference and was set to 0 ppm for all temperature and pH conditions. The buffers employed were as follows: pH 6 (20 mM sodium phosphate for the HP5W4 and HP7 structure ensemble calculation), otherwise pH 6 (50 mM sodium phosphate) and pH 3 (20mM formate/formic acid). Complete assignments for eight representative peptides, including the most- and least-folded representative of each series, appear in the Supporting Information (Tables 8 – 15).
With the exception of resonances that display CSDs in excess of 1 ppm, all 1H-C resonances were sharp singlets or multiplets implying linewidths (Δ0 < 2.4 Hz) consistent with the rapid tumbling times expected for monomeric species in the 1000 – 2100 amu molecular weight range. Sites with large folded-state CSDs display differential exchange broadening that scales as (CSD)2 consistent with folded/unfolded state relaxation phenomena on the 1 – 10 μsec timescale.15
Extracting fraction-folded values and melting behavior
Fraction-folded measures based on backbone Hα and HN shifts, employ CSD comparisons. An updated version (http://andersenlab.chem.washington.edu/CSDb)5 of our previously published43 method for determining random-coil values and CSDs was used. For HN’s, alternative temperature gradients are used depending on the degree of solvent sequestration present in the folded state and the degree of unfolding observed over the temperature range examined.11 The probes employed are given in the text or in Table 3. For this analysis, the largest CSD value observed, at each probe site, for any HP7 series peptide at 280K was equated with 94% folding. The CD melt analysis indicates χF = 0.935 for HP7 at 278K.
%-Fold measures were also derived from the diagnostic measures of the W/W interaction: [θ]228 and the upfield shift of Trp Hε3 nearest the N-terminus of the turn. In the case of the NMR probe, we assumed that both the random-coil value (7.62 ppm) and a 100%-folded value (5.22 ppm for the HP7 series, 5.37 ppm for the HP6 series) were temperature invariant. Turning to the CD analysis, we obtained better agreement between CD and NMR Tm values by assuming a small linear, warming-induced decrease in the Δ[θ]228 associated with folding. For example, when we assume a temperature-invariant Δ[θ]228 = +434,000° for the folding transition, a lower melting point (58 °C) results for HP7 (versus 69 °C by NMR and 63 °C by applying the second derivative method or a full melting curve fit to the CD data, below). A 0.36%/°C Δ[θ]228 correction brought Tm measures within 4 °C agreement for all HP7 analogs.
Folding thermodynamics were derived from the temperature dependence of the CD measures of folding. We first determined the range of ΔCp values that produced a linear relationship for Eqn. 1, as measured by R2. In every case, linearity was lost if the χF values were decreased by more than 8 %, which serves as further support for our 100%-folded assumption. Even a small increase in the χF values for (T3V)-HP6 and the better-folded HP7 analogs, also had a negative effect on R2. With the exception of HP7(-2) (Ac-TWNPATAKWT-NH2 , R2 > 0.98), Table 4 reports the best (and range) of ΔS and ΔH values allowed (R2 > 0.996) and the corresponding range of ΔCp values.
| Eqn 1 |
We report the thermodynamic parameters for T* = 298K. The thermodynamic parameters so derived were also used to calculate both a Tm value and the temperature below which cold denaturation would be expected.
NMR Structure Ensemble Generation
Distance constraints were derived from the intensity of nOe crosspeaks in 750 MHz NOESY spectra with 160ms and 180ms mixing times for HP5W4 (290K) and HP7 (280K), respectively. Peak intensities were converted to distance ranges, (d – d−) – (d + d+), by an in-house program containing corrections for sharp aromatic peaks and contributions from multiple chemically equivalent protons (e.g., methyls). The initial constraint set was relatively tight, which aided in structure refinement. For generation of the final ensemble, the constraints were made more conservative: d+ and d− values were increased by 0.1 Å, with d-values adjusted so that no constraint range included a value less than 1.9 Å.
NMR structure ensembles were generated using v1.1 of CNS44. The standard simulated-annealing script was modified to incorporate a Lennard-Jones potential, rather than a ‘repel’ function, to calculate van der Waals interaction energies during the final minimization step. A complete description of the protocol and the acceptance criteria, which allows no nOe constraint violations greater than 0.12Å for individual structures, have been published previously45; we have since reduced the nOe scaling factor in the final minimization to 40, as opposed to the previously reported 75 kcal/(mol Å2). The constraints employed for the ensembles are summarized in Table 5 and appear in their entirety as Table 6 and 7 (Supporting Information). RMSD values for the ensembles appear in part B of Table 5. The corresponding data for trpzip4, PDB #1LE34, is included for comparison.
Since some bond lengths and angles are parameterized differently by CNS and the structure analysis algorithm of the PDB, the structures presented here (and the HP7 ensemble submitted to the PBD, accession #2EVQ), are the CNS-generated structures modified by 500 steps of steepest-descent minimization using the SANDER application of AMBER6. This minimization has no significant effect on the nOe–constraint deviations. All molecular structures appearing in this report were visualized using MolMol23. Chemical shift predictions based on structure coordinates employ the Shifts program38, version 4.1.
Supplementary Material
Acknowledgments
This work was supported by a grant from the NSF (CHE0315361); the synthesis and structure determination of peptide HP5W4 were accomplished under NIH support (GM059658).
Footnotes
Supporting Information Available: Supporting information contains additional structural comparisons and full chemical shift assignments. The material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Ramírez-Alvarado M, Blanco FJ, Serrano L. Nat Struct Biol. 1996;3:604–612. doi: 10.1038/nsb0796-604. [DOI] [PubMed] [Google Scholar]; Maynard AJ, Sharman GJ, Searle MS. J Am Chem Soc. 1998;120:1996–2007. [Google Scholar]; Stanger HE, Gellman SH. J Am Chem Soc. 1998;120:4236–4237. [Google Scholar]; Syud FA, Espinosa JF, Gellman SH. J Am Chem Soc. 1999;121:11577–11578. [Google Scholar]
- 2.Griffiths-Jones SR, Maynard AJ, Searle MS. J Mol Biol. 1999;292:1051–1069. doi: 10.1006/jmbi.1999.3119. [DOI] [PubMed] [Google Scholar]
- 3.de Alba E, Jiménez MA, Rico M. J Am Chem Soc. 1997;119:175–183. [Google Scholar]
- 4.Cochran AG, Skelton NJ, Starovasnik MA. Proc Natl Acad Sci U S A. 2001;98:5578–5583. doi: 10.1073/pnas.091100898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fesinmeyer RM, Hudson FM, Andersen NH. J Am Chem Soc. 2004;126:7238–7243. doi: 10.1021/ja0379520. [DOI] [PubMed] [Google Scholar]
- 6.Kortemme T, Ramírez-Alvarado M, Serrano L. Science. 1998;281:253–256. doi: 10.1126/science.281.5374.253. [DOI] [PubMed] [Google Scholar]; Schenck H, Gellman S. J Am Chem Soc. 1998;120:4869–4870. [Google Scholar]; Sharman GJ, Searle MS. J Am Chem Soc. 1998;120:5291–5300. [Google Scholar]; de Alba E, Santoro J, Rico M, Jiménez MA. Protein Sci. 1999;8:854–865. doi: 10.1110/ps.8.4.854. [DOI] [PMC free article] [PubMed] [Google Scholar]; Griffiths-Jones SR, Searle MS. J Am Chem Soc. 2000;122:8350–8356. [Google Scholar]; López de la Paz M, Lacroix E, Ramírez-Alvarado M, Serrano L. J Mol Biol. 2001;312:229–246. doi: 10.1006/jmbi.2001.4918. [DOI] [PubMed] [Google Scholar]; Carulla N, Woodward C, Barany G. Protein Sci. 2002;11:1539–1551. doi: 10.1110/ps.4440102. [DOI] [PMC free article] [PubMed] [Google Scholar]; Syud FA, Stanger HE, Mortell HS, Espinosa JF, Fisk JD, Fry CG, Gellman SH. J Mol Biol. 2003;326:553–568. doi: 10.1016/s0022-2836(02)01304-9. [DOI] [PubMed] [Google Scholar]
- 7.Cochran AG, Tong RT, Starovasnik MA, Park EJ, McDowell RS, Theaker JE, Skelton NJ. J Am Chem Soc. 2001;123:625–632. doi: 10.1021/ja003369x. [DOI] [PubMed] [Google Scholar]; Phillips ST, Piersanti G, Bartlett PA. Proc Natl Acad Sci U S A. 2005;102:13737–13742. doi: 10.1073/pnas.0506646102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell SJ, Blandl T, Skelton NJ, Cochran AG. J Am Chem Soc. 2003;125:388–395. doi: 10.1021/ja028075l. [DOI] [PubMed] [Google Scholar]
- 9.Blandl T, Cochran AG, Skelton NJ. Protein Sci. 2003;12:237–247. doi: 10.1110/ps.0228603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butterfield SM, Sweeney MM, Waters MJ. J Org Chem. 2005;70:1105–1114. doi: 10.1021/jo0491105. [DOI] [PubMed] [Google Scholar]
- 11.Fesinmeyer RM, Hudson FM, White GWN, Olsen KA, Euser A, Andersen NH. J Biomol NMR. 2005;33:213–231. doi: 10.1007/s10858-005-3731-7. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi N, Endo S, Munekata E. Peptide Chem. 1993:278–280. [Google Scholar]; Kobayashi N, Honda S, Yoshii H, Uedaira H, Munekata E. FEBS Lett. 1995;366:99–103. doi: 10.1016/0014-5793(95)00503-2. [DOI] [PubMed] [Google Scholar]; Blanco FJ, Rivas G, Serrano L. Nat Struct Biol. 1994;1:584–590. doi: 10.1038/nsb0994-584. [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi N, Honda S, Yoshii H, Munekata E. Biochemistry. 2000;39:6564–6571. doi: 10.1021/bi000013p. [DOI] [PubMed] [Google Scholar]
- 14.Muñoz V, Thompson PA, Hofrichter J, Eaton WA. Nature. 1997;390:196–199. doi: 10.1038/36626. [DOI] [PubMed] [Google Scholar]
- 15.Olsen KA, Fesinmeyer RM, Stewart JM, Andersen NH. Proc Natl Acad Sci U S A. 2005;102:15483–15487. doi: 10.1073/pnas.0504392102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Honda S, Kobayashi N, Munekata E. J Mol Biol. 2000;295:269–278. doi: 10.1006/jmbi.1999.3346. [DOI] [PubMed] [Google Scholar]
- 17.Guvench O, Brooks CL., III J Am Chem Soc. 2005;127:4668–4674. doi: 10.1021/ja043492e. [DOI] [PubMed] [Google Scholar]
- 18.Thornton K, Gorenstein DG. Biochemistry. 1994;33:3532–3539. doi: 10.1021/bi00178a009. [DOI] [PubMed] [Google Scholar]
- 19.Espinosa JF, Gellman SH. Angew Chem Int Ed. 2000;39:2330–2333. doi: 10.1002/1521-3773(20000703)39:13<2330::aid-anie2330>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 20.Honda S, Yamasaki K, Sawada Y, Morii H. Structure. 2004;12:1507–1518. doi: 10.1016/j.str.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 21.Chelli R, Gervasio FL, Procacci P, Schettino V. J Am Chem Soc. 2002;124:6133–6143. doi: 10.1021/ja0121639. [DOI] [PubMed] [Google Scholar]
- 22.Kuszewski J, Gronenborn AM, Clore GM. J Am Chem Soc. 1999;121:2337–2338. [Google Scholar]
- 23.Koradi R, Billeter M, Wüthrich K. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 24.Searle MS, Griffiths-Jones SR, Skinner-Smith H. J Am Chem Soc. 1999;121:11615–11620. [Google Scholar]
- 25.Kiehna SE, Waters ML. Protein Sci. 2003;12:2657–2667. doi: 10.1110/ps.03215403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tatko CD, Waters ML. Protein Sci. 2004;13:2515–2522. doi: 10.1110/ps.04820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dyer RB, Maness SJ, Franzen S, Fesinmeyer RM, Olsen KA, Andersen NH. Biochemistry. 2005;44:10406–10415. doi: 10.1021/bi050698z. [DOI] [PubMed] [Google Scholar]
- 28.Hughes RM, Waters ML. J Am Chem Soc. 2005;127:6518–6519. doi: 10.1021/ja0507259. [DOI] [PubMed] [Google Scholar]
- 29.Andersen NH, Dyer RB, Fesinmeyer RM, Gai F, Liu Z, Neidigh JW, Tong H. J Am Chem Soc. 1999;121:9879–9880. [Google Scholar]
- 30.de Alba E, Rico M, Jiménez MA. Protein Sci. 1999;8:2234–2244. doi: 10.1110/ps.8.11.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.We originally included CD and NMR studies of the HP7 peptides in HFIP-water mixtures to provide a confirmation the 100%-folded values. Numerous hairpins that fold incompletely in water, including at least one11 with strands shorter than those present in this series, display a dramatic increase in fold population upon addition of 8 – 10 vol-% HFIP. In distinct contrast, HP7 peptides are, with the possible exception of the poorly folded EPDGK turn mutant, less stable in aqueous HFIP. This in itself suggests that hairpin folding is not strictly a hydrophobic effect2,29 in the HP7 peptide series.
- 32.A reviewer suggested that “it is equally plausible that the thermodynamic parameters for the last three peptides in Table 4 have something to do with their relatively higher content of charged residues”, rather than an increased ΔCp due to greater burial of apolar surface area in the folded state. We do not see a correlation between ΔCp and the total number of charged sites for the peptides of Table 4.
- 33.Tatko CD, Waters ML. J Am Chem Soc. 2002;124:9372–9373. doi: 10.1021/ja0262481. [DOI] [PubMed] [Google Scholar]
- 34.Ciani B, Jourdan M, Searle MS. J Am Chem Soc. 2003;125:9038–9047. doi: 10.1021/ja030074l. [DOI] [PubMed] [Google Scholar]
- 35.Searle MS. J Chem Soc, Perkin Trans. 2001;2:1011–1020. [Google Scholar]; de Alba E, Blanco FJ, Jiménez MA, Rico M, Nieto JL. Eur J Biochem. 1995;233:283–292. doi: 10.1111/j.1432-1033.1995.283_1.x. [DOI] [PubMed] [Google Scholar]; Ramírez-Alvarado M, Blanco FJ, Serrano L. Protein Sci. 2001;10:1381–1392. doi: 10.1110/ps.52901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.In the stable [3:5]-hairpins, the Gly-Δδα, Hα CSDs at S-3 and S+2, and HN CSDs at S-3, T2 and S+2 approach 0.6, +0.45, +0.90, +1.2, −1.0, and +0.8 ppm, respectively. The corresponding values for (NPDGT)-HP7 at 7 °C were 0.42, +0.31, +0.45, +0.31, −0.49, and +0.57 ppm. The EPDGK mutant also displayed the correct signs for each of these CSDs but was judged to be significantly less stable than (NPDGT)-HP7.
- 37.The Asn Hδ21 and δ22 CSDs are attributed to H-bonding interactions, they cannot be rationalized as ring-current shifts. Chemical shifts of 7.51 and 6.81 ppm, respectively, are calculated when the NMR structure ensemble is evaluated using Case’s Shifts 4.138 program.
- 38.Xu XP, Case DA. J Biomol NMR. 2001;21:321–333. doi: 10.1023/a:1013324104681. [DOI] [PubMed] [Google Scholar]
- 39.Setnicka V, Huang R, Thomas CL, Etienne MA, Kubelka J, Hammer RP, Keiderling TA. J Am Chem Soc. 2005;127:4992–4993. doi: 10.1021/ja043007f. [DOI] [PubMed] [Google Scholar]
- 40.Andersen NH, Cort JR, Liu Z, Sjoberg SJ, Tong H. J Am Chem Soc. 1996;118:10309–10310. [Google Scholar]; Andersen NH, Brodsky Y, Neidigh JW, Prickett KS. Bioorg Med Chem. 2002;10:79–85. doi: 10.1016/s0968-0896(01)00263-2. [DOI] [PubMed] [Google Scholar]
- 41.Piotto M, Saudek V, Sklenar V. J Biomol NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 42.Bax A, Davis DG. J Magn Reson. 1985;65:355–360. [Google Scholar]
- 43.Andersen NH, Neidigh JW, Harris SM, Lee GM, Liu Z, Tong H. J Am Chem Soc. 1997;119:8547–8561. [Google Scholar]
- 44.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr D Biol Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 45.Neidigh JW, Fesinmeyer RM, Prickett KS, Andersen NH. Biochemistry. 2001;40:13188–13200. doi: 10.1021/bi010902s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.