

Abstract
The polycystic kidney disease-1 (Pkd1) gene encodes a large transmembrane protein (polycystin-1, or PC-1) that is reported to function as a fluid flow sensor in the kidney. As a member of the transient receptor potential family, PC-1 has also been hypothesized to play a role in the elusive mechanoelectrical transduction (MET) channel in inner ear hair cells. Here, we analyze two independent mouse models of PC-1, a knock-in (KI) mutant line and a hair cell-specific inducible Cre-mediated knock-out line. Both models exhibit normal MET channel function at neonatal ages despite hearing loss and ultrastructural abnormalities of sterecilia that remain properly polarized at adult ages. These findings demonstrate that PC-1 plays an essential role in stereocilia structure and maintenance but not directly in MET channel function or planar cell polarity. We also demonstrate that PC-1 is colocalized with F-actin in hair cell stereocilia in vivo, using a hemagglutinin-tagged PC-1 KI mouse model, and in renal epithelial cell microvilli in vitro. These results not only demonstrate a novel role for PC-1 in the cochlea, but also suggest insight into the development of polycystic kidney disease.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) affects >12 million people worldwide with ∼85% of cases caused by polycystic kidney disease-1 (Pkd1) gene mutations (Wilson, 2008). ADPKD is classically characterized by cysts in the kidneys, pancreas, and liver (Gabow, 1993).
Pkd1 encodes polycystin-1 (PC-1), a protein with 9–11 transmembrane domains and a large N-terminal extracellular tail with multiple domains. The intracellular C-terminal contains a coil–coil domain that affects actin cytoskeleton (Boca et al., 2007). PC-1 is a member of the transient receptor potential (TRP) family of proteins, and the polycystin subfamily (TRPP) (Delmas, 2005). PC-1 is expressed in fetal and adult tissues but has been studied primarily in the kidney (Yoder et al., 2002), where it is localized to the basolateral membrane of tubular epithelial cells (Palsson et al., 1996; Ward et al., 1996; Ibraghimov-Beskrovnaya et al., 1997; Wilson et al., 1999), and adherens junctions, which are linked to epithelial cells' actin cytoskeleton (Markoff et al., 2007).
While both function and subcellular localization of PC-1 are unclear, PC-1 has been proposed to play a role in planar cell polarity (PCP) (Grantham, 1996; Carone et al., 1998; Ong et al., 1999) and flow sensing in renal epithelial cells. Cells from mouse models lacking PC-1 or with PC-1 mutations (Nauli et al., 2003) or in an ADPKD patient-derived cell line (Xu et al., 2007) show limited Ca2+ uptake. This mechanosensitive role, together with the proposed topology of PC-1, is similar to other TRP members implicated in mechanically gated ion channels of the inner ear (Christensen and Corey, 2007; Fettiplace, 2009).
In the inner ear, hair cells (HCs) are specialized epithelial cells crowned with F-actin-based stereocilia. In mice, at approximately embryonic day 15 (E15), cochlear HC stereocilia appear together with a tubulin-based kinocilium (Anniko, 1983; Richardson, 2006). The bundle development makes the stereocilia of inner ear HCs a standard model for the study of PCP defects. The cochlear mechanoelectrical transduction (MET) channel is a nonselective cation channel permeable to Ca2+ and K+ (Corey and Hudspeth, 1979), and the influx of ions causes HC depolarization as early as postnatal day 0 (P0) in the basal turn of the cochlea (Lelli et al., 2009). The MET channel is thought to open via a tip link connector, which is composed of cadherin-23 and protocadherin-15 (Siemens et al., 2004; Kazmierczak et al., 2007; Lelli et al., 2010). Although the MET channel has been studied extensively, including a recent candidate protein (Coste et al., 2010), its molecular identity remains elusive due to the small number of MET channels per animal.
To investigate whether PC-1 plays a role in MET channels and stereocilia in inner ear HCs, we analyzed three independent mouse models of PC-1: (1) a PC-1-hemagglutinin (HA) knock-in (KI) mutant, in which PC-1 is tagged at its C terminus with HA (Wodarczyk et al., 2009); (2) a PC-1 KI mutant, in which PC-1 is mutated at residue T3041V (Yu et al., 2007); and (3) a PC-1 conditional knock-out (CKO) mutant, in which PC-1 is deleted specifically in HCs (Piontek et al., 2004; Chow et al., 2006). Here, we found that PC-1 is localized throughout F-actin HC stereocilia using the PC-1-HA KI mouse model. The other two independent mutations in Pkd1 demonstrate hearing defects and an ultrastructural abnormality of stereocilia, which do not involve PCP defects, in adult mice; however, the MET channel is functional at neonatal ages. Our findings demonstrate that PC-1 does not directly play a role in the MET channel; rather, it plays a structural role, likely with F-actin, in HC stereocilia, which may provide a novel mechanism for polycystic kidney disease.
Materials and Methods
Mouse models.
All animal experiments were approved by the Institutional Animal Care and Use Committee at St. Jude Children's Research Hospital and were performed according to National Institutes of Health guidelines. Experimental animals of both genders were used. To obtain a homozygous PC-1-HA KI mouse model, heterozygous PC-1-HA KI mice were bred together (Wodarczyk et al., 2009). PC-1 KI heterozygous mice were crossed to obtain PC-1 KI homozygous (experimental) and wild-type (WT) control littermates (Yu et al., 2007). The Pkd1-floxed allele was described previously (Piontek et al., 2004), as was the transgenic Atoh1-CreER mouse model (Chow et al., 2006, Weber et al., 2008). To obtain the PC-1 CKO model, Atoh1-CreER-positive; Pkd1loxP/+ mice were bred together to obtain Atoh1-CreER-positive; Pkd1loxP/loxP (experimental), Atoh1-CreER-positive; Pkd1loxP/+ (control), and Atoh1-CreER-positive; Pkd1+/+ (control) mice with tamoxifen injections given as described previously (Weber et al., 2008). Whereas only auditory brainstem response (ABR) was performed in PC-1 KI mutants and their paired WT littermates, both ABR and distortion product otoacoustic emissions (DPOAEs) were performed in PC-1 CKO mutants and their paired WT littermates with tamoxifen injection. We determined that tamoxifen alone with the amount we injected had no effect in WT littermates in ABR and DPOAE tests.
Quantitative real-time PCR.
Animal samples were collected and quickly snap frozen with liquid nitrogen. mRNA was isolated using the Perfect Pure mRNA isolation kit (5 Prime). mRNA yield and purity were determined with a NanoDrop spectrophotometer as well as gel electrophoresis. cDNA was produced using a high-capacity cDNA reverse transcription kit (Applied Biosystems). Reactions were completed with primer/probe sets designed using Primer Express software (Applied Biosystems) and TaqMan universal master mix (Applied Biosystems). Probes were designed with a 5′ FAM and 3′ BHQ. Each age group was n ≥ 4, where each sample was taken from a different mouse. Each reaction was performed in duplicate and run with GAPDH controls as well as nontemplate reaction controls. Experiments were performed using the ABI 7900 HT Sequence Detection System (Applied Biosystems). Results were fit to a standard curve and then normalized to GAPDH controls.
Immunostaining.
Inner ears were removed from perfused animals and postfixed for 30 min at room temperature in 4% paraformaldehyde. Adult-age inner ears were decalcified in 120 mm EDTA, whereas younger samples did not require this decalcification step. For sections, ears were placed into 30% sucrose overnight and then sectioned using a Leica CM3050 S at 12 μm. For whole-mount immunostaining, cochleae were dissected into basal, midbasal, and apical sections. Sections or whole mounts were washed with 10 mm PBS and blocked (1% bovine serum albumin, 10% goat serum, and 1% Triton X-100) for 1 h at room temperature. Primary antibodies were then applied in 1% bovine serum albumin, 5% goat serum, and 0.1% Triton X-100 and incubated overnight at 4°C. Primary antibodies included rat anti-HA (1:100; Roche), rabbit anti-α acetylated tubulin (1:200; Sigma), rabbit anti-cadherin-23 (1:500; a gift from Dr. Ulrich Mueller, Scripps Research Institute, La Jolla, CA), and rabbit anti-espin (1:5000; a gift from Dr. Stefan Heller, Stanford, Stanford, CA). Samples were washed with PBS and incubated at room temperature for 2 h in secondary antibodies (goat anti-rabbit Alexa-488 or Alexa-568, 1:1000, or goat anti-rat Alexa-488 or Alexa-568, 1:1000; Invitrogen) diluted in the same solution as primary antibodies. Samples were again washed with PBS, and rhodamine phalloidin (1:40; Invitrogen) and nuclear stain (Hoechst 33342; 1:2000; Invitrogen) were applied to the samples for 20 min at room temperature, followed by a final PBS wash. Cochleae were mounted with Prolong Gold anti-fade reagent (Invitrogen) and imaged with a Zeiss LSM 510 NLO Meta or LSM 700 confocal microscope.
Intensity analysis.
Pixel intensity was measured using Zeiss Zen 2009 software. Pixel intensity was recorded for each fluorophore and then plotted as intensity of signal versus distance on the line. Pearson's correlation coefficient and the overlapping Manders' coefficient were determined for the organ of Corti stereocilia region only, and, for the case of the CL1 cells, single cells were analyzed.
Cell culture.
Renal epithelial kidney cells (CL1) were purchased from the American Type Culture Collection (catalog #Cl-101) and were grown in Minimum Essential Medium (Invitrogen) supplemented with 10% fetal calf serum (Invitrogen). Cells were transfected with Halo-mPkd1 using Lipofectamine 2000 (Invitrogen). The construct was prepared by cloning a full-length mPkd1 construct into the pcDNA5 vector (Invitrogen) that contains HaloTag (Promega). A flexible linker with the sequence GGGGS (Halo-GGGGS) was used at the N terminus immediately following the signal peptide sequence, and a FLAG tag (DYKDDDDK) was used at the C terminus. The Halo-GGGGS cloning was performed in three steps. First, the Halo-GGGGS cassette was generated by annealing and PCR extension with the two primers (G4S-BamHI-F, 5′-AAAAGGATCCTCAGGTGGTGGCGGTTCAGGCGGAGGTGG-3′; G4S-BglII-R, 5′-TTTTAGATCTCGATCCGCCACCGCCAGAGCCACCTCCGCCTG-3′). The 76 bp PCR fragment was cloned at the 3′ end of Halo in pHT2 vector (Promega) using BamHI and BglII sites. Next, the mPkd1 construct was subjected to site-specific mutagenesis to generate an AgeI site on the immediate C-terminal end of the signal peptide sequence, whereby CCTGGG (base pairs 391–396; Accession no. U70209) was changed to ACCGGT (AgeI site). Finally, the Halo-GGGGS cassette was amplified by PCR and inserted at the AgeI site generated in the second step. The final construct was confirmed by DNA sequencing for the correct and in-frame insertion of the Halo-GGGGS cassette. After transfection, the cells were stained with a rabbit anti-Halo antibody (1:200; Promega) and rhodamine phalloidin (1:40; Invitrogen).
Scanning electron microscopy.
Mice were anesthetized using Avertin (0.5 mg/g) and perfused with Super reagent perfusion wash and Super reagent perfusion fixation (Electron Microscopy Science). Cochleae were removed and decalcified with a Pelco Biowave microwave tissue processor (Ted Pella). The tissue was dissected and fixed with 2.5% glutaraldehyde in 0.1 m sodium cacodylate buffer, pH 7.35 (Tousimis Research), and 2% osmium tetroxide, pH 7.35, in 0.1 m sodium cacodylate buffer (Electron Microscopy Sciences) before dehydration in an ethanol series and critically point dried (Tousimis Sandai 790). Samples were mounted, coated with gold/palladium, and imaged using a JEOL 7000 field emission gun scanning electron microscope. Stereocilia counts were performed on the middle row of outer HCs in the middle basal turns of the cochlea. Individual stereocilia were counted in each bundle and averaged (n ≥ 4 animals per genotype, with four HCs from each animal used for the stereocilia measurements and counts). Comparison with controls was determined using a one-way ANOVA followed by a Student's t test with a Bonferroni correction. Stereocilia height was measured from the tallest row of the stereocilia bundles directly adjacent to the location were the kinocilium was located, again from the middle row of outer HCs in the middle and basal turns of the cochlea. The heights were averaged and compared with controls using a one-way ANOVA followed by a Student's t test with a Bonferroni correction with GraphPad Prism 5.0 software.
ABR.
Mice were anesthetized with Avertin (0.5 mg/g, i.p.). Three electrodes were placed subcutaneously in the anesthetized animals: one in the vertex, one underneath the external ear, and one on the back close to the tail. Tone pips of 4, 6, 12, 16, 22, 32, and 44 kHz were generated using a Tucker Davis Technologies (TDT) workstation (System III) running SigGen32 software (TDT) and delivered to the animal via the external auditory meatus by a 2 mm plastic tube connected to a high-frequency transducer in a controlled acoustic chamber (Industrial Acoustics). The output was fed to an amplifier (HS4 Bioamp headstage; TDT), viewed on an oscilloscope, and recorded. Maximum stimuli were at 75 dB sound pressure level in tone pips. The sampling rates were 50 kHz, and durations were 10 ms. The signals were bandpass filtered (300 Hz to 3 kHz) and amplified 100,000 times. The number of acquisition trials was set at 500 averages with 250 rarefaction and 250 condensation stimuli. Auditory thresholds were determined by decreasing the sound intensities, from 75 to 5 dB, until reaching the lowest sound intensity at which reproducible waves could be recognized. Comparison with the corresponding frequency in controls was performed using a two-way ANOVA followed by a Student's t test with a Bonferroni correction with GraphPad Prism 5.0 software, with n ≥ 4 animals per genotype.
DPOAEs.
Mice were anesthetized with Avertin (0.5 mg/g), and DPOAE 2f1 − f2 responses were recorded at a range of frequencies (6–22 kHz) using two EC1 speakers coupled via an ER10B microphone probe (Etymotic Research) within the acoustic probe and the TDT BioSig II system. The signal duration was 83.88 ms, and the repetition rate was 11.92 per second. Emissions f1 and f2 were each passed through an RX6 multifunction processor (TDT) for digital/analog conversion to PA5 programmable attenuators. Ear canal sound pressure was recorded with an ER10B+ low-noise microphone and probe (Etymotic Research) housed in the same coupler as the f1 and f2 speakers. The output of the ER10B+ amplifier directly went to an RX6 multifunction processor (TDT) for analog/digital conversion for sampling at 200 kHz. Fast-Fourier transforms were performed from averaged responses using TDT BioSig software on the resultant waveform. The noise floor was defined as the average of the sound levels of 10 frequency bins above and below the 2f1 − f2 frequency bin. Thresholds were determined if the 2f1 − f2 measurement was higher than two standard deviations from the noise floor. Individual mouse thresholds were averaged for each frequency tested and compared with corresponding frequencies in controls using a two-way ANOVA followed by a Student's t test with a Bonferroni correction with GraphPad Prism 5.0 software, with n ≥ 4 animals per genotype.
Electrophysiology measurements.
Cochlear HC measurements were recorded from P4/P5 explant cultures as described previously (Lelli et al., 2009), with n = 3 animals per genotype and ≥4 cells tested per genotype. All data sets were nonsignificant, with p > 0.05 as determined by a Student's t test when comparing mutants to controls.
Results
Polycystin-1 is associated with HC stereocilia
For a gene to be part of the MET channel in the inner ear, it must be expressed both temporally and spatially in stereocilia bundles. To determine the temporal expression of Pkd1, we first performed quantitative real-time PCR with RNA isolated from the whole cochlea of WT mice and found that Pkd1is expressed at perinatal as well as postnatal ages (Fig. 1), with Pkd1 mRNA peaking around E17.5 and leveling off in postnatal ages. This temporal pattern is also comparable to genes associated with the MET channel, specifically tip link proteins (Corey et al., 2004; Cuajungco et al., 2007; Lelli et al., 2009).
Figure 1.

Developmental profile of Pkd1 mRNA expression levels in the murine cochlea. Quantitative real-time PCR from mRNA extracted from the whole cochlea is shown. Experiments were performed in duplicate with n ≥ 4 animals at each time point. All samples are normalized to the housekeeping gene GAPDH and expressed as mean ± SEM.
To confirm mRNA expression and determine the subcellular localization of PC-1 in inner ear HCs, we examined the protein expression pattern of PC-1 in the cochlea, specifically in the stereocilia. Since most PC-1 antibodies do not work well for immunohistochemistry, and the expression level of PC-1 is generally low, we took advantage of a recently developed KI mouse model in which PC-1 is tagged with three HA sequences on the C terminus before the stop codon (PC-1-HA KI). The addition of these tags has been shown to not interfere with subcellular localization and function of PC-1 in kidney epithelial cells (Wodarczyk et al., 2009). To further confirm that PC-1-HA is normal in the cochlea, we performed ABR analysis of homozygous PC-1-HA KI mice at 5 weeks of age and found normal ABR responses at all frequencies tested except 32 kHz (Fig. 2). We did observe a significant threshold increase compared to WT at 32 kHz, but attributed the slight threshold elevation to variable age-related hearing loss known in the C57BL/6 stain of mice.
Figure 2.

ABR measurements of PC-1-HA KI mice. PC-1-HA KI mice (gray) show no differences from WT (black) littermates from 4 to 22 kHz, with a significant difference detected at 32 kHz, likely due to age related hearing loss common in C57BL/6 mice. Data are expressed as mean ± SEM. *p < 0.05 compared with WT threshold at the corresponding frequency as determined by a one-way ANOVA followed by a Student's t test with a Bonferroni correction, with n ≥ 5 animals for each genotype. Black bars, WT; gray bars, PC-1-HA KI.
After confirming that PC-1-HA does not affect normal PC-1 function in HCs, we went on to examine the distribution of PC-1-HA in inner ears at P0. We found that PC-1-HA was expressed throughout the actin-based stereocilia of HCs (Fig. 3A–C′) but not in the kinocilium of HCs or colocalized with α-tubulin, which labels the kinocilium (Fig. 3D–H). We examined the staining intensity patterns of F-actin and HA in both WT and PC-1-HA KI mice at levels of both the stereocilia (Fig. 3A–B′) and the lateral wall of the HC body (Fig. 3C,C′). WT HCs had similar line intensity plots for HA staining in both stereocilia (Fig. 3A,A′) and the cuticular plate (data not shown), whereas the line intensity plots of HA staining in the PC-1-HA KI model were increased at the stereocilia level (Fig. 3B,B′) compared with the lateral wall (Fig. 3C,C′). This can be further seen through the line intensity analysis of the HA signal between the WT (Fig. 3A′), PC-1-HA KI stereocilia (Fig. 3B′), and the PC-1-HA KI cell body (Fig. 3C′). When analyzed for colocalization of HA with F-actin in the stereocilia, PC-1-HA KI HCs had a correlative overlapping Manders' coefficient of 0.86 and a Pearson's correlation coefficient of 0.68 (Fig. 3B′). The intensity data convincingly demonstrate that PC-1-HA is localized to the F-actin-based stereocilia bundles. Additionally, we found HA staining throughout all turns of the cochlea and throughout the length of each stereocilium (data not shown), indicating that PC-1-HA and F-actin are distributed in similar patterns in HC stereocilia. Similar to outer hair cells (OHCs), PC-1 is also localized to stereocilia of inner hair cells (IHCs) (data not shown).
Figure 3.
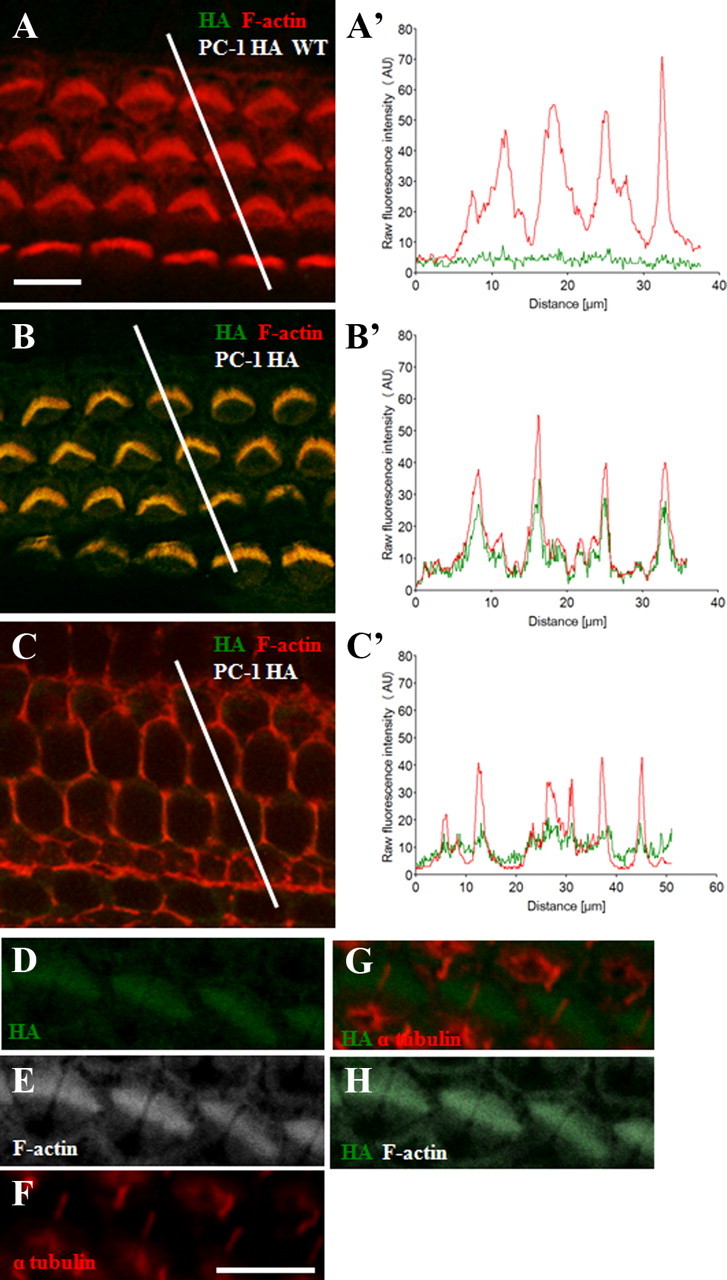
Colocalization of PC-1-HA with F-actin in HC stereocilia in PC-1-HA KI mice. A–C, Cochlear whole mounts of WT littermates (A) and PC-1-HA KI mice (B, C) imaged at the HC stereocilia layer (A, B) and in the lateral wall of outer HCs (C) in P4 animals. Samples are stained with HA (green) and F-actin (red). A′–C′, Raw fluorescence intensity data for the white lines drawn in A–C, respectively. Differences between A′ (WT) and B′ (PC-1-HA KI) indicate the specificity of the HA antibody. PC-1-HA KI animals analyzed for HA colocalization with F-actin had a correlative overlapping Manders' coefficient of 0.86 and a Pearson's correlation coefficient of 0.68. D–F, Cochlear whole mounts from P0 PC-1-HA KI mice stained for HA (green, D), F-actin (white, E), and α-tubulin (red, F). G, H, Merged images showing HA localized to the stereocilia but not to the kinocilium of the developing bundle. Scale bars: A (for A–C), 10 μm; F (for D–H), 5 μm.
By examining ages after the kinocilium regressed, we found that PC-1-HA continues to colocalize with F-actin in the stereocilia in PC-1-HA KI animals (data not shown). Consistent with the age-dependent decrease in Pkd1 mRNA, we also saw a decrease in PC-1-HA-tagged protein expression with age. Our colocalization of PC-1-HA with F-actin is consistent with previously described in vitro findings indicating PC-1 plays a role in F-actin cytoskeleton rearrangement (Boca et al., 2007).
Colocalization of polycystin-1 with F-actin in transiently transfected CL1 and HeLa cells
To further examine PC-1's colocalization with F-actin, we used the CL1 renal epithelial kidney cell line, the parental strain of CL4, both of which have been widely used for localization studies of other HC stereocilia proteins (Loomis et al., 2003; Sekerkova et al., 2004; Zheng et al., 2010). We transiently transfected CL1 cells with a mouse Halo-Pkd1 fusion construct (Halo-mPkd1; see Materials and Methods). HaloTag is a new protein tagging system that allows for multiple analytical processes to be monitored with one tag. In addition to localization through the use of antibodies, HaloTag can also be localized through ligand binding (Los et al., 2008). Using this technology, we localized Halo-mPkd1 to the actin-based microvilli (similar to actin-based stereocilia of the cochlea) of transfected CL1 cells (Fig. 4A–D, dashed lines) compared with control nontransfected cells (Fig. 4A–D, asterisks). We also confirmed colocalization of F-actin and Halo-mPkd1 protein with measures of fluorescent intensities of Halo (green line) and F-actin (red line) staining (Fig. 4A′–D′). Line intensity plots indicate that there was colocalization of F-actin and Halo-mPkd1 in the microvilli of these cells (Fig. 4C′,D′) but little to no colocalization of Halo-mPkd1with F-actin in the cytosol or nucleus (Fig. 4A′,B′). As further confirmation of the localization of the Halo-mPkd1 protein in CL1 cells, we transfected a mutated version of Halo-mPkd1, which contains a mutation in the GPS domain of the PC-1 protein and is known to render the protein less functional in the kidney, into CL1 cells (Yu et al., 2007). The mutant protein, termed Halo-PC-1T3041V, does not appear to localize to the microvilli (Fig. 4E, microvilli layer), but rather to the cytoplasm (likely endoplasmic reticulum) of CL1 cells (Fig. 4F, cell body layer). In addition, we also transfected HeLa cells with the Halo-mPkd1 construct and confirmed the localization of Halo-mPkd1 to the actin-based filopodia (Fig. 4G). Halo-mPkd1 was also present in other areas of CL1 and HeLa cells, likely due to the overexpression of the mPkd1-Halo construct. Together, these observations, along with the PC-1-HA KI staining pattern in HCs, are consistent with PC-1 colocalizing with F-actin in the stereocilia of inner ear HCs. Since CL1 cells are a renal epithelial cell line, and we found localization of Halo-mPkd1 with F-actin in vitro, it is also likely that in vivo PC-1 localizes with F-actin in the kidney. However, in our experiments we did not see a change in F-actin organization as shown by Boca et al. (2007), but attribute this difference to cell type specificity as well as differences in constructs.
Figure 4.
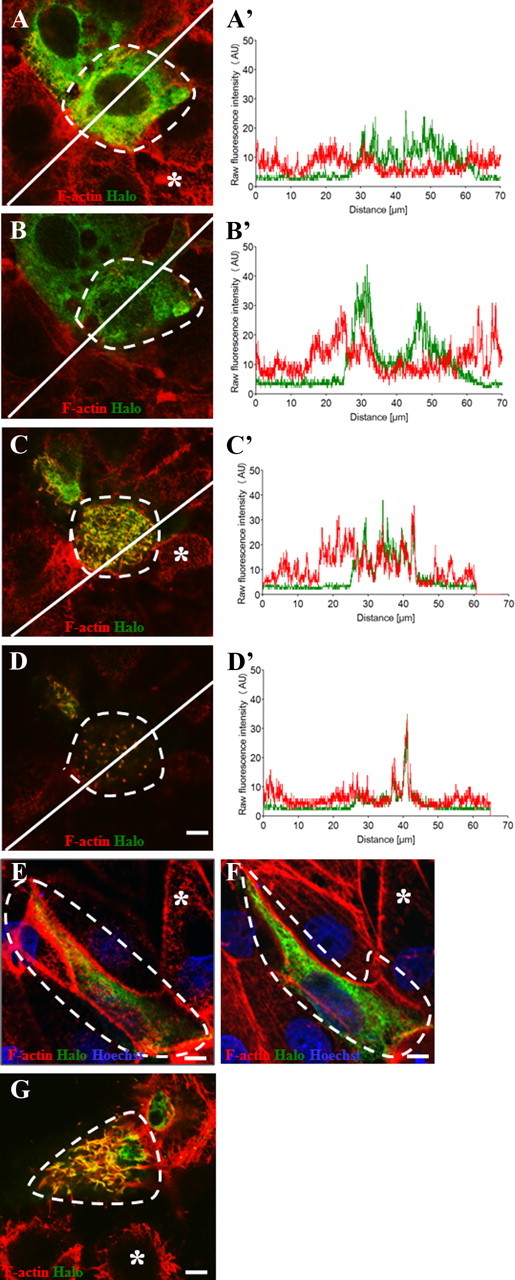
CL1 and HeLa cells transiently transfected with a Halo-mPkd-1 construct. A–D, Confocal image slices of a transfected CL1 cell at the base of the cell (A), the middle of the cell (B), and the top of epithelium including microvilli (C), and microvilli alone (D) stained with Halo (green) and F-actin (red). The transfected cell of interest is surrounded by a dashed white line, with nontransfected control cells indicated by asterisks. A′–D′, Corresponding line intensity measurements for each protein indicate strong colocalization of Halo (green) and F-actin (red) in microvilli. E, F, A mutated version of mPkd-1, termed Halo-PC-1T3041V, shows no localization of Halo to the microvilli (E), but rather localization to the cytosol, likely the endoplasmic reticulum (F). G, Halo (green) and F-actin (red) are also colocalized in the filopodia of HeLa cells transfected with Halo-mPkd1. Transfected cells are also surrounded by a dashed white line, with nontransfected control cells indicated by an asterisks. Scale bars: D (for A–D), E–G, 5 μm.
Pkd1 mutations lead to hearing defects in two independent mouse models
After localization of PC-1 in HCs, we examined the in vivo effect of Pkd1 mutations in the inner ear using two independent Pkd1 mutant mouse models. The first mouse model examined was a KI mouse model (PC-1 KI) in which there was an amino acid substitution (T3041V) in the G-protein-coupled proteolytic site (GPS) domain of PC-1's extracellular N terminus. This mutation is the same used as a negative control in the CL1 transfection studies (Fig. 4E,F). Substitution of a valine at this site prevents the post-translational cleavage of the extracellular N terminus of the protein at the juxtamembrane GPS; however, the CL1 study revealed mislocalization of the PC-1 protein, which could cause a lack of protein function. PC-1 KI mice are visibly smaller, develop polycystic kidney disease characterized by enlarged abdomens and cyst filled kidneys, and die at ∼P25 (Yu et al., 2007). The body defects as well as the premature death in the PC-1 KI imposed some limitations in our studies; thus, we used a second Pkd1 mouse model. The second model is an HC-specific conditional knock-out mouse model of Pkd1 (PC-1 CKO), in which the Pkd1 gene is deleted specifically in inner ear HCs. We developed the PC-1 CKO model by crossing a transgenic Atoh1-CreER mouse model (Chow et al., 2006; Weber et al., 2008) with a PC-1 floxed mouse model (Piontek et al., 2004) and induced Pkd1 gene deletion with tamoxifen at P0 and P1. In the PC-1 CKO model, the deletion of the PC-1-targeted exons is detected as early as 48 h after the second tamoxifen injection when examined through genomic DNA PCR (data not shown), which is similar to previous reports (Weber et al., 2008). This PC-1 CKO mouse is viable without obvious abnormalities in other systems examined.
Both mutant PC-1 models underwent ABR evaluations upon hearing maturation (P25 for PC-1 KI and P35 for PC-1 CKO) and displayed significant hearing threshold elevations at all frequencies tested (4 to 32 kHz) (Fig. 5A,B). While the mice were not completely deaf, mutation or deletion of Pkd1 resulted in a 20 to 30 dB threshold elevation in hearing response from that of their WT control littermates. Because of the possibility of incomplete tamoxifen-mediated deletion of PC-1 in the PC-1 CKO mice (Chow et al., 2006; Weber et al., 2008) or residual PC-1 activity in the PC-1 KI mice (Yu et al., 2007), the measured ABR threshold shifts may underestimate threshold shifts in the complete deletion of PC-1. Nevertheless, these findings show that PC-1 plays an important role in cochlear HCs and the hearing cascade. Similar phenotypes between the two Pkd1 mutant strains also demonstrate that hearing defects in the PC-1 KI mice are due primarily to defects in HCs, further confirming the functional role of PC-1 specifically in HCs.
Figure 5.

Hearing analysis of PC-1 KI (T3041V) and PC-1 CKO mutant mouse models. A, B, ABR measurements of PC-1 CKO mice at P35 (A) and PC-1 KI mice at P25 (B), where squares indicate WT littermates and circles indicate mutant animals. C, DPOAE measurements of PC-1 CKO mice (squares) and WT (circles) at P35. D, DPOAE measurements plotted as an input/output function for 20 kHz. The noise floor for the input/output function is indicated by the gray shaded area in D. *p < 0.05; **p < 0.01; ***p < 0.001 compared with WT threshold at the corresponding frequency as determined by a one-way ANOVA followed by a Student's t-test with a Bonferroni correction; n ≥ 4 animals for each experiment and genotype.
While ABR measurements showed a deficit in hearing ability for both Pkd1 mutant mouse models, this technique is a measurement of the entire hearing cascade. We conducted another in vivo hearing assessment, DPOAE, which specifically measures the function of outer HCs. Due to the extended time period of the test and the disease phenotype seen in the PC-1 KI model (Yu et al., 2007), only PC-1 CKO mice were tested. PC-1 CKO mice had a DPOAE threshold elevation of 15–30 dB at frequencies between 16 and 22 kHz (Fig. 5C), similar to results seen with ABR (Fig. 5A). Upon examination of DPOAEs as an input/output function, we observed a rightward shift in PC-1 CKO mice, indicating that outer HCs are defective in responding to sounds at various intensities compared with controls (Fig. 5D). Both DPOAE analyses are consistent with a defect in the outer HCs of our PC-1 CKO model.
HC mechanoelectrical transduction is not affected by deletion of PC-1
To elucidate whether the loss of PC-1 affects the MET channel, we examined the function of the channel in both mutant mouse models, PC-1 KI and PC-1 CKO. PC-1 CKO mice were killed at P2, and cochleae were dissected and maintained in culture for 3 d (equivalent to P5). PC-1 KI mouse models were killed at P3, and cochleae were dissected and maintained in culture for 1 d (equivalent to P4). Transduction currents as well as the open probability of the channel, operating range, and slow/fast adaptation time constants were recorded as described previously (Lelli et al., 2009). Both PC-1 KI and PC-1 CKO produced normal OHC MET channel currents compared with WT littermates (Fig. 6A). The maximal transduction current in both models appeared normal and showed no significant differences from WT (Fig. 6B). Furthermore, there were no significant differences between either mutant mouse model and their WT littermates with regard to the open probability and operating range of the MET channel (Fig. 6C). We also saw no change in the channel's slow or fast adaptation time constants (Fig. 6D). These data demonstrate that mutations in PC-1 do not affect MET channel activity in cochlear OHCs at P4–P5, during the time when the MET channel is normally mature.
Figure 6.
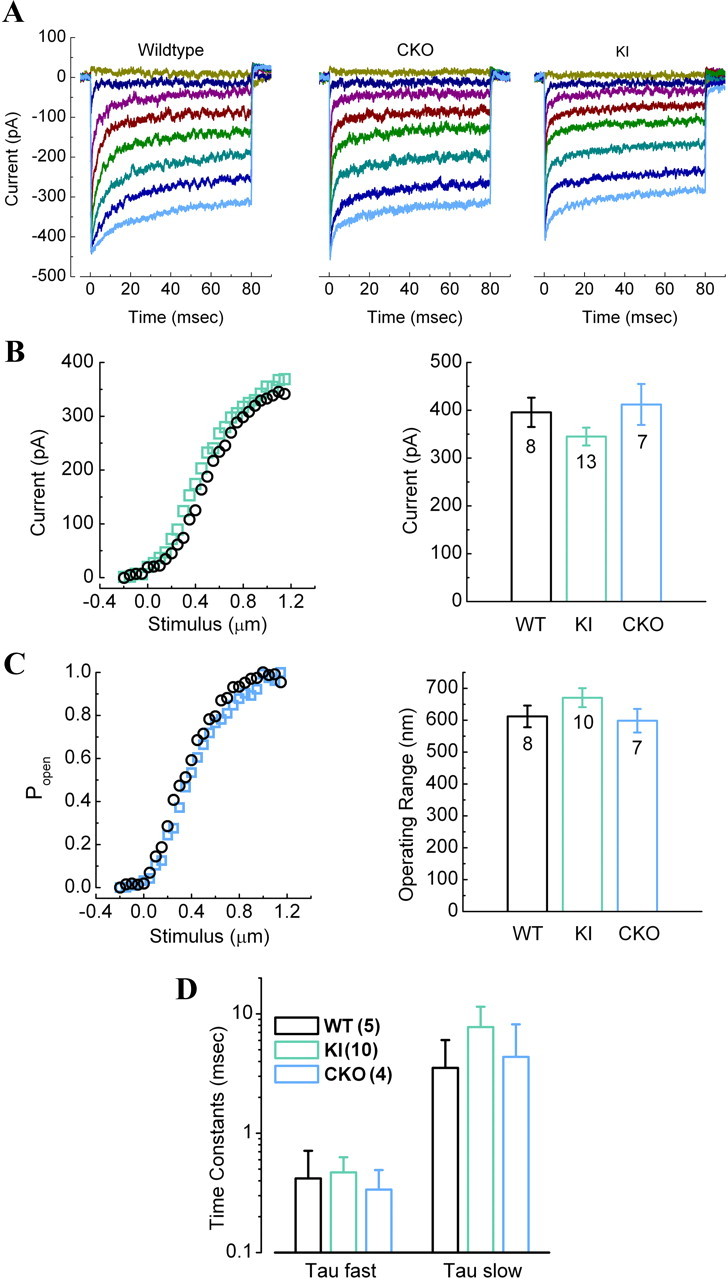
MET channel electrophysiology measurements in PC-1 KI (T3041V) and PC-1 CKO mice. A, Representative families of transduction currents recorded from WT, PC-1 CKO, and PC-1 KI OHCs. B, Left, Stimulus response relationships from WT (black) and PC-1 KI mice (green) at P4–P5. The bar graph shows the mean maximal transduction current amplitudes (±SEM) for the three genotypes. The number of cells recorded from each genotype (3 animals per genotype) is indicated on the graph. No significant differences were noted between genotypes. C, Left, The open probability of the MET channel in WT (black) and PC-1 CKO (blue) mice. Right, The mean 10–90% operating range measured in nanometers. The number of cells is indicated. Again, no significant differences were noted between genotypes. D, Adaptation rate of the MET channel in both mutant models (PC-1 CKO and PC-1 KI) were measured. We found no significant differences in the tau fast and tau slow values of adaptation between the mutants and wild-type littermate controls. Data are expressed as mean ± SEM.
Pkd1 mutations result in stereocilia ultrastructural abnormalities
To uncover the underlying reason for the hearing phenotype in the PC-1 mutant mice, we examined both the gross and ultrastructural morphology of inner ear HCs. Cochlear whole mounts from both PC-1 KI and PC-1 CKO animals were stained with phalloidin and analyzed for HC bundle abnormalities. In outer, inner, and vestibular HCs at P0, P4–P5, P14, and adult ages (P25 for PC-1 KI and P35 for PC-1 CKO), we found no obvious phenotypic differences among genotypes (data not shown). The stereocilia displayed normal bundle structure and polarity throughout all ages of the inner ear. Examination at the ultrastructural level using scanning electron microscopy also revealed normal stereocilia bundles with respect to bundle polarity and structure at P0, P4–P5, and P14 compared with WT littermates (Fig. 7A–C).
Figure 7.

Scanning electron microscopy analysis of HC bundles in PC-1 KI (T3041V) andPC-1 CKO mice. A–F, At age P14 there were no differences between the WT (A), PC-1 KI (B), and PC-1 CKO (C) whole mounts. At adult ages, WT (D), PC-1 KI (E), and PC-1 CKO (F) whole mounts (5000× magnification) show bundle malformations in all three rows of the OHCs. G–I, At 20,000× magnification, individual HC bundles can be seen, with WT (G) bundles having a unique stair-step, three-row pattern; however, the PC-1 KI (H) and PC-1 CKO (I) bundles have an obvious defect in bundle organization. Stereocilia in both mutants appear different in length (arrow), rigidity (arrowheads), and organization from the WT control cell. J, K, Quantification of the adult bundle phenotype indicates that both the stereocilia number per bundle (J) and the height of individual stereocilia (K) were significantly different in the mutants compared with controls. Four mice were analyzed from each genotype, with four cells analyzed from each mouse. PC-1 mutant mice had a decrease in the stereocilia number and an increase in the stereocilia height. Scale bar: I (for A–I), 1 μm. **p < 0.01; ***p < 0.001 as determined by a one-way ANOVA followed by a Student's t-test with a Bonferroni correction.
However, when we examined the bundles at adult ages (P25 for PC-1 KI and P35 for PC-1 CKO), we found significant structural differences between the mutants and WT cochleae (Fig. 7D–I). OHC bundles no longer maintained their typical stair-step pattern of stereocilia rows increasing in height; instead, individual stereocilia appeared farther apart from their neighbors and lacking in rigidity. The numbers of stereocilia per bundle in both mutants were significantly reduced compared with their WT littermates (Fig. 7J). Interestingly, the height of mutant OHC stereocilia was significantly increased compared with WT, based upon measurements of the last row of stereocilia in the bundles (Fig. 7K). IHC stereocilia were affected less than OHCs, consistent with the less number of IHCs that were Cre positive (Chow et al., 2006; Weber et al., 2008). Ultrastructural examination of the bundles at P14 revealed no visual differences between mutant and WT stereocilia (Fig. 7A–C), and, as further confirmation, there were no quantitative differences in stereocilia bundle height or stereocilia number per bundle between the two mutant models and their WT littermates (data not shown). Comparison of control littermates from each mutant model exhibited similar stereocilia number and height, confirming the validity of our measurements (Fig. 7J,K). Together, the data indicate that the ultrastructural defects occur between P14 and adult ages in both mutants and are consistent with the hearing defects detected at adult ages (Fig. 5). Since many stereocilia bundle mutants show a regression in stereocilia over time (Nayak et al., 2007), we examined PC-1 CKO mice at 1 year of age, and there was no HC loss revealed through whole mount immunostaining (data not shown).
Stereocilia proteins remain intact and present in PC-1 mutants
To further analyze the potential role of PC-1 in the stereocilia, we examined known adult stereocilia markers, including cadherin-23, a component of the tip link, and espin, an actin treadmilling protein. We found no obvious differences in stereocilia localization of cadherin-23 and espin between the WT and PC-1 CKO mutants at P35 (Fig. 8).
Figure 8.
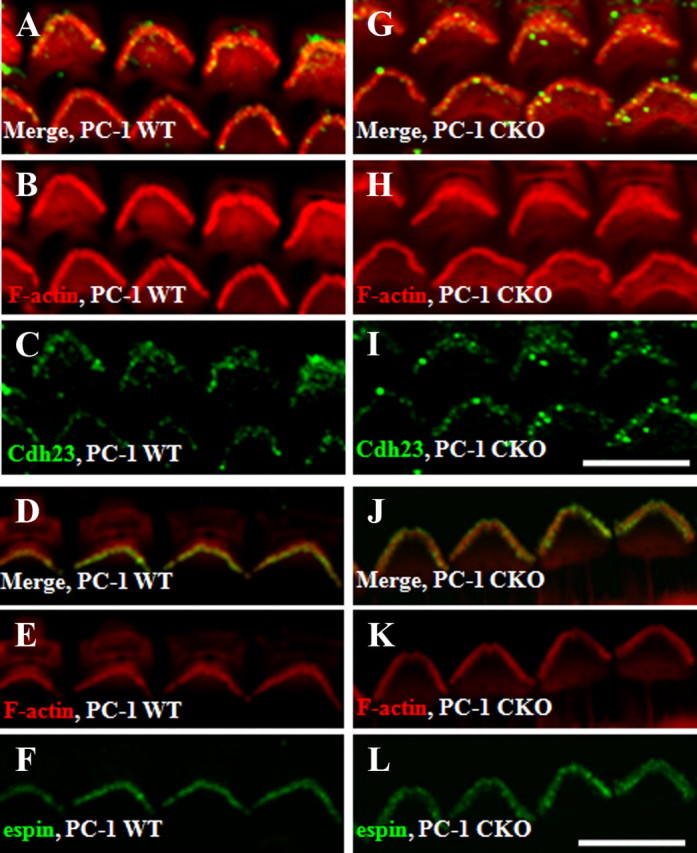
Known stereocilia components are not mislocalized in PC-1 CKO mice. A–M, WT littermates show localization of cadherin-23 (green) (merge, A; F-actin, red, B; cadherin-23, C) and espin (green) (merge, D; actin red, E; espin, F), with similar staining patterns seen in the PC-1 CKO mice with cadherin-23 (G, merge; F-actin, H; cadherin-23, I) and espin (merge, J; F-actin, K; espin, L). Scale bars: I (for A–C, G–I), L (for D–F, J–L), 10 μm. Three or more mice were examined for each genotype and protein.
Discussion
Since many members of the TRP family have been implicated as part of the MET channel (Colbert et al., 1997; Liedtke et al., 2000; Walker et al., 2000; Mutai and Heller, 2003; Corey et al., 2004), we investigated PC-1 through analysis of three independent mutant mouse models. We found in PC-1-HA KI mice that PC-1 is located throughout HC stereocilia, colocalized with F-actin. Two independent PC-1 mutations produced no MET channel abnormalities, similar ABR and DPOAE hearing threshold elevations, and abnormal HC bundle phenotypes. OHC bundles showed a significant increase in height of stereocilia and a decrease in the number of stereocilia per bundle, without defects in PCP. Therefore, the data suggest that PC-1 is not necessary for establishment of MET channel function but instead may be required to maintain normal stereocilia structure, possibly via localization with F-actin.
Due to previous reports of TRP family members' roles in other mechanotransducing systems, it is thought that they likely play a role in the HC MET channel (Colbert et al., 1997; Walker et al., 2000; Mutai and Heller, 2003). PC-1 can sense fluid flow and help in the uptake of Ca2+ in the kidney (Nauli et al., 2003). Additionally, TRPP family members have conductance and Ca2+ permeability similar to the MET channel (Owsianik et al., 2006). This suggested that PC-1 was an excellent candidate for the MET channel in HCs. We measured the MET channel transduction current, the operating range, and adaptation ability of the channel in PC-1 mutant mouse models at P4/P5, when the MET channel is fully functional. In all areas examined, there were no significant differences between the mutant models and the controls. Also, HC bundle morphology was normal at this age in both mutants. One explanation for the lack of a MET channel phenotype is that the half-life of PC-1 in HCs may be long, and thus it has not been effectively depleted in PC-1 CKO mice at P5 with induction at P0/P1. In renal cells of the germline PC-1 KI mice, PC-1 is expressed as a full-length, uncleaved protein due to the lack of cleavage in the membrane (Yu et al., 2007). If the cochlea mimics either the renal epithelial cells or CL1 transfected cells, the mutant PC-1T3041V protein likely does not target the stereocilia of HCs, eliminating the function of PC-1 associated with stereocilia. Even though it remains possible that a small amount residual normal PC-1 remains in the stereocilia, the PC-1 KI mouse should show significant defects in MET channel function at P4/P5 if PC-1 were an essential component of the MET channel. Further support comes from previous studies that no MET channel defects have been observed in other stereocilia structural mutants at this age, such as the TRIOBP mutants (Kitajiri et al., 2010). Our data cannot rule out the possibility that PC-1 acts as a regulatory or accessory protein that is associated with the MET channel, but it does show that it is not required for MET channel maturation and function at neonatal ages. Unfortunately, due to limitations in the ability to measure the MET channel function at adult ages, we cannot exclude that PC-1 plays a role in the MET channel at adult ages. However, our data do indicate that the phenotype we are observing is due to the lack of PC-1 in the HCs and not other cell types in the cochlea. Since PC-1 is thought to be ubiquitously expressed, it is possible that a defect in PC-1 in the stria vascularis could cause a distinct phenotype; however, the HC-specific PC-1 CKO mouse model had the same phenotype as the PC-1 KI model, which excludes the contribution made by dysfunction in the stria vascularis. Additionally, PC-1 is also localized to the IHC stereocilia, and we found the defects in IHC stereocilia in both mutants (PC-1 KI and PC-1 CKO; data not shown). Although it remains possible that only OHCs are defective, our data are consistent with the notion that ABR/DPOAE defects in either mutant are likely caused directly by the combined defects in IHCs and OHCs. Even though PC-1 does not play a direct role in the MET channel in either PC-1 mutant model, as we and others originally expected (Fettiplace, 2009), PC-1 still plays a distinct role in the stereocilia, based on abnormal stereocilia phenotypes and hearing deficits we observed in both mutants at adult ages. One of the most striking findings in our study was the colocalization of PC-1 with F-actin throughout the length of HC stereocilia. In support, we also saw colocalization of PC-1 with F-actin-based microvilli in a renal epithelium cell line. Previous studies have shown PC-1 plays a structural role in other systems through its ability to regulate the actin cytoskeleton (Boca et al., 2007; Markoff et al., 2007). In support, PC-1's coil–coil domain located in its C-terminal tail (Qian et al., 1997) often colocalizes with F-actin (Boeda et al., 2002). In our PC-1 mutant mouse models, we found disorganization of actin stereocilia bundles, providing strong evidence that PC-1 is indeed involved in stereocilia F-actin structure. Mutations in the known HC stereocilia components myosin XVa, whirlin, and espin result in a decrease in the height of bundles (Nayak et al., 2007). In the case of PC-1 deficiency in our models, we believe that the balance or maintenance of F-actin assembly is disrupted, resulting in the increase in stereocilia length and decrease in stereocilia number per bundle, which ultimately causes hearing loss. Further evidence that PC-1 is working together with the F-actin core of the bundles is shown through the normal localization of other bundle proteins such as espin and cadherin-23.
The stereocilia phenotype we observed, while not as extreme as some other bundle mutants, does indicate a role for PC-1 in stereocilia structural maintenance (Nayak et al., 2007). However, PC-1 has multiple family members with similar structural motifs (Hughes et al., 1999; Veldhuisen et al., 1999; Yuasa et al., 2002), so it is possible that other family members or other proteins compensate for the loss of PC-1 in our mutant mouse models. It should be noted that polycystin-2 (PC-2) is commonly thought to form a cation channel with PC-1 (Qian et al., 1997; Hanaoka et al., 2000; AbouAlaiwi et al., 2009). This leads to an interesting question of what would happen if both molecules were deleted in inner ear HCs and whether this double mutant would produce a MET channel phenotype. Since germline KO mice of either PC-1 or PC-2 are embryonic lethal (Boulter et al., 2001; Pennekamp et al., 2002), a conditional or inducible system is needed to delete both genes; however, the expression pattern of PC-2 in the inner ear is still unknown (it is entirely possible that PC-2 may not be even localized in stereocilia). It should also be noted that PC-2 can interact with actin cross-linkers as well as α actinins, actin binding and bundling partners (Schumann et al., 2009).
Due to the lack of hearing phenotypes reported in the polycystic kidney disease patient population, it might seem surprising to study PC-1 in the cochlea; however, it is now understood that polycystic kidney disease is a result of a second hit or loss of heterozygosity in the kidney, while the normal allele in the cochlea remains untouched (Qian et al., 1996; Koptides et al., 1998; Badenas et al., 2000; Perez-Oller et al., 2000). We know from examining the heterozygous PC-1 mutants (both mouse models) that the loss of one allele is not significant to cause hearing loss (data not shown). Hence, we believe this is the most likely explanation for normal hearing in polycystic kidney disease patients. In addition, many have thought that PC-1 is an important regulator of PCP in the kidney (Grantham, 1996; Carone et al., 1998; Ong et al., 1999). Upon examination of PC-1 in the inner ear, an organ known for PCP (Dabdoub and Kelley, 2005; Jones and Chen, 2007; Kelly and Chen, 2007), we did not detect any change in the orientation of stereocilia bundles, and thus no evidence that the PCP pathway is defective. The lack of phenotype can easily be explained in the PC-1 CKO model, since inactivation of the Pkd1 gene occurs at P0/P1 after stereocilia patterning is completed; however, there is also no PCP phenotype seen in the PC-1 KI model. Our results demonstrate that either PCP in the cochlea does not require PC-1 or the residual PC-1T3041V that is uncleaved in the membrane is sufficient to regulate PCP.
Both subcellular localization and binding partners of PC-1 are still unclear, even in the kidney; however, an effective avenue to precisely localize proteins such as PC-1 is to use biochemically tagged KI mouse models. These tags are small and easily traceable using well-characterized antibodies and do not affect the subcellular localization of endogenous proteins. We have successfully used this approach here and revealed the previously unknown PC-1 subcellular localization in the cochlea. Given our findings that PC-1 colocalizes with F-actin in HC stereocilia and in CL1 renal epithelial cells, we believe that PC-1 and F-actin cooperate with each other to help maintain the normal structure of microvilli in the kidney, and defects in this relationship may eventually lead to polycystic kidney disease. More experiments are necessary to elucidate the relationship between F-actin and PC-1 in the stereocilia of the inner ear and microvilli of the kidney.
Footnotes
This work was supported by NIH Grants 1F31DC009393 (K.S.), DC06471 (J.Z.), DC08800 (J.Z.), CA21765 (J.Z.), 5R25CA023944 (S.L.), DC006183 (G.S.G.G.), DC05439 (J.R.H.), DK062199 (Q.F.), DK090868 (Q.F.), and DK48006 (G.G.), as well as the American Lebanese Syrian Associated Charities, The Hartwell Foundation (J.Z.), the Intramural Research Program of the NIDDK (G.G.), Johns Hopkins Polycystic Kidney Disease Research, and Office of Naval Research Grant N000140911014 (J.Z.). We thank Sharon Frase (St. Jude Children's Research Hospital, Memphis, TN), Rich Martens (University of Alabama, Tuscaloosa, AL), and Lou Boykins (University of Memphis, Memphis, TN) for their help and guidance with scanning electron microscopy images; Stefan Heller (Stanford University, Stanford, CA), Ulrich Mueller (Scripps Research Institute, La Jolla, CA), James Bartles (Northwestern, Evanston, IL), and Peter Gillespie (Oregon Hearing Research Center, Portland, OR) for sharing valuable reagents (antibodies and constructs); Stanislav S. Zakharenko (St. Jude Children's Research Hospital) for his discussion on protein colocalization; and the Zuo lab, especially Dr. Brandon C. Cox, for their helpful comments and suggestions.
The authors declare no competing financial interests.
References
- AbouAlaiwi WA, Takahashi M, Mell BR, Jones TJ, Ratnam S, Kolb RJ, Nauli SM. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ Res. 2009;104:860–869. doi: 10.1161/CIRCRESAHA.108.192765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anniko M. Cytodifferentiation of cochlear hair cells. Am J Otolaryngol. 1983;4:375–388. doi: 10.1016/s0196-0709(83)80043-x. [DOI] [PubMed] [Google Scholar]
- Badenas C, Torra R, Perez-Oller L, Mallolas J, Talbot-Wright R, Torregrosa V, Darnell A. Loss of heterozygosity in renal and hepatic epithelial cystic cells from ADPKD1 patients. Eur J Hum Genet. 2000;8:487–492. doi: 10.1038/sj.ejhg.5200484. [DOI] [PubMed] [Google Scholar]
- Boca M, D'Amato L, Distefano G, Polishchuk RS, Germino GG, Boletta A. Polycystin-1 induces cell migration by regulating phosphatidylinositol 3-kinase-dependent cytoskeletal rearrangements and GSK3beta-dependent cell cell mechanical adhesion. Mol Biol Cell. 2007;18:4050–4061. doi: 10.1091/mbc.E07-02-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeda B, El-Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, Perfettini I, Fath KR, Shorte S, Reiners J, Houdusse A, Legrain P, Wolfrum U, Richardson G, Petit C. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 2002;21:6689–6699. doi: 10.1093/emboj/cdf689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter C, Mulroy S, Webb S, Fleming S, Brindle K, Sandford R. Cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proc Natl Acad Sci U S A. 2001;98:12174–12179. doi: 10.1073/pnas.211191098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carone FA, Bacallao R, Kanwar Y. Role of the matrix in autosomal dominant polycystic kidney disease. Ren Fail. 1998;20:181–189. doi: 10.3109/08860229809045101. [DOI] [PubMed] [Google Scholar]
- Chow LM, Tian Y, Weber T, Corbett M, Zuo J, Baker SJ. Inducible Cre recombinase activity in mouse cerebellar granule cell precursors and inner ear hair cells. Dev Dyn. 2006;235:2991–2998. doi: 10.1002/dvdy.20948. [DOI] [PubMed] [Google Scholar]
- Christensen AP, Corey DP. TRP channels in mechanosensation: direct or indirect activation? Nat Rev Neurosci. 2007;8:510–521. doi: 10.1038/nrn2149. [DOI] [PubMed] [Google Scholar]
- Colbert HA, Smith TL, Bargmann CI. OSM-9, a novel protein with structural similarity to channels, is required for olfaction, mechanosensation, and olfactory adaptation in Caenorhabditis elegans. J Neurosci. 1997;17:8259–8269. doi: 10.1523/JNEUROSCI.17-21-08259.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey DP, Hudspeth AJ. Ionic basis of the receptor potential in a vertebrate hair cell. Nature. 1979;281:675–677. doi: 10.1038/281675a0. [DOI] [PubMed] [Google Scholar]
- Corey DP, Garcia-Anoveros J, Holt JR, Kwan KY, Lin SY, Vollrath MA, Amalfitano A, Cheung EL, Derfler BH, Duggan A, Geleoc GS, Gray PA, Hoffman MP, Rehm HL, Tamasauskas D, Zhang DS. TRPA1 is a candidate for the mechanosensitive transduction channel of vertebrate hair cells. Nature. 2004;432:723–730. doi: 10.1038/nature03066. [DOI] [PubMed] [Google Scholar]
- Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, Dubin AE, Patapoutian A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330:55–60. doi: 10.1126/science.1193270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuajungco MP, Grimm C, Heller S. TRP channels as candidates for hearing and balance abnormalities in vertebrates. Biochim Biophys Acta. 2007;1772:1022–1027. doi: 10.1016/j.bbadis.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabdoub A, Kelley MW. Planar cell polarity and a potential role for a Wnt morphogen gradient in stereociliary bundle orientation in the mammalian inner ear. J Neurobiol. 2005;64:446–457. doi: 10.1002/neu.20171. [DOI] [PubMed] [Google Scholar]
- Delmas P. Polycystins: polymodal receptor/ion-channel cellular sensors. Pflugers Arch. 2005;451:264–276. doi: 10.1007/s00424-005-1431-5. [DOI] [PubMed] [Google Scholar]
- Fettiplace R. Defining features of the hair cell mechanoelectrical transducer channel. Pflugers Arch. 2009;458:1115–1123. doi: 10.1007/s00424-009-0683-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med. 1993;329:332–342. doi: 10.1056/NEJM199307293290508. [DOI] [PubMed] [Google Scholar]
- Grantham JJ. The etiology, pathogenesis, and treatment of autosomal dominant polycystic kidney disease: recent advances. Am J Kidney Dis. 1996;28:788–803. doi: 10.1016/s0272-6386(96)90378-9. [DOI] [PubMed] [Google Scholar]
- Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature. 2000;408:990–994. doi: 10.1038/35050128. [DOI] [PubMed] [Google Scholar]
- Hughes J, Ward CJ, Aspinwall R, Butler R, Harris PC. Identification of a human homologue of the sea urchin receptor for egg jelly: a polycystic kidney disease-like protein. Hum Mol Genet. 1999;8:543–549. doi: 10.1093/hmg/8.3.543. [DOI] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, Dackowski WR, Foggensteiner L, Coleman N, Thiru S, Petry LR, Burn TC, Connors TD, Van Raay T, Bradley J, Qian F, Onuchic LF, Watnick TJ, Piontek K, Hakim RM, Landes GM, Germino GG, Sandford R, Klinger KW. Polycystin: in vitro synthesis, in vivo tissue expression, and subcellular localization identifies a large membrane-associated protein. Proc Natl Acad Sci U S A. 1997;94:6397–6402. doi: 10.1073/pnas.94.12.6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C, Chen P. Planar cell polarity signaling in vertebrates. Bioessays. 2007;29:120–132. doi: 10.1002/bies.20526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak P, Sakaguchi H, Tokita J, Wilson-Kubalek EM, Milligan RA, Muller U, Kachar B. Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature. 2007;449:87–91. doi: 10.1038/nature06091. [DOI] [PubMed] [Google Scholar]
- Kelly M, Chen P. Shaping the mammalian auditory sensory organ by the planar cell polarity pathway. Int J Dev Biol. 2007;51:535–547. doi: 10.1387/ijdb.072344mk. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajiri S, Sakamoto T, Belyantseva IA, Goodyear RJ, Stepanyan R, Fujiwara I, Bird JE, Riazuddin S, Ahmed ZM, Hinshaw JE, Sellers J, Bartles JR, Hammer JA, III, Richardson GP, Griffith AJ, Frolenkov GI, Friedman TB. Actin-bundling protein TRIOBP forms resilient rootlets of hair cell stereocilia essential for hearing. Cell. 2010;141:786–798. doi: 10.1016/j.cell.2010.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koptides M, Constantinides R, Kyriakides G, Hadjigavriel M, Patsalis PC, Pierides A, Deltas CC. Loss of heterozygosity in polycystic kidney disease with a missense mutation in the repeated region of PKD1. Hum Genet. 1998;103:709–717. doi: 10.1007/s004390050896. [DOI] [PubMed] [Google Scholar]
- Lelli A, Asai Y, Forge A, Holt JR, Geleoc GS. Tonotopic gradient in the developmental acquisition of sensory transduction in outer hair cells of the mouse cochlea. J Neurophysiol. 2009;101:2961–2973. doi: 10.1152/jn.00136.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelli A, Kazmierczak P, Kawashima Y, Muller U, Holt JR. Development and regeneration of sensory transduction in auditory hair cells requires functional interaction between cadherin-23 and protocadherin-15. J Neurosci. 2010;30:11259–11269. doi: 10.1523/JNEUROSCI.1949-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Choe Y, Marti-Renom MA, Bell AM, Denis CS, Sali A, Hudspeth AJ, Friedman JM, Heller S. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell. 2000;103:525–535. doi: 10.1016/s0092-8674(00)00143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis PA, Zheng L, Sekerkova G, Changyaleket B, Mugnaini E, Bartles JR. Espin cross-links cause the elongation of microvillus-type parallel actin bundles in vivo. J Cell Biol. 2003;163:1045–1055. doi: 10.1083/jcb.200309093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- Markoff A, Bogdanova N, Knop M, Ruffer C, Kenis H, Lux P, Reutelingsperger C, Todorov V, Dworniczak B, Horst J, Gerke V. Annexin A5 interacts with polycystin-1 and interferes with the polycystin-1 stimulated recruitment of E-cadherin into adherens junctions. J Mol Biol. 2007;369:954–966. doi: 10.1016/j.jmb.2007.03.070. [DOI] [PubMed] [Google Scholar]
- Mutai H, Heller S. Vertebrate and invertebrate TRPV-like mechanoreceptors. Cell Calcium. 2003;33:471–478. doi: 10.1016/s0143-4160(03)00062-9. [DOI] [PubMed] [Google Scholar]
- Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- Nayak GD, Ratnayaka HS, Goodyear RJ, Richardson GP. Development of the hair bundle and mechanotransduction. Int J Dev Biol. 2007;51:597–608. doi: 10.1387/ijdb.072392gn. [DOI] [PubMed] [Google Scholar]
- Ong AC, Harris PC, Biddolph S, Bowker C, Ward CJ. Characterisation and expression of the PKD-1 protein, polycystin, in renal and extrarenal tissues. Kidney Int. 1999;55:2091–2116. doi: 10.1046/j.1523-1755.1999.00404.x. [DOI] [PubMed] [Google Scholar]
- Owsianik G, Talavera K, Voets T, Nilius B. Permeation and selectivity of TRP channels. Annu Rev Physiol. 2006;68:685–717. doi: 10.1146/annurev.physiol.68.040204.101406. [DOI] [PubMed] [Google Scholar]
- Palsson R, Sharma CP, Kim K, McLaughlin M, Brown D, Arnaout MA. Characterization and cell distribution of polycystin, the product of autosomal dominant polycystic kidney disease gene 1. Mol Med. 1996;2:702–711. [PMC free article] [PubMed] [Google Scholar]
- Pennekamp P, Karcher C, Fischer A, Schweickert A, Skryabin B, Horst J, Blum M, Dworniczak B. The ion channel polycystin-2 is required for left-right axis determination in mice. Curr Biol. 2002;12:938–943. doi: 10.1016/s0960-9822(02)00869-2. [DOI] [PubMed] [Google Scholar]
- Pérez-Oller L, Torra R, Badenas C, San Millan JL, Darnell A. [Heterozygosity loss and somatic mutations in type I and II dominant autosomal renal polycystic kidney disease: evidence of a recessive mechanism at a cell level in cystogenesis] Nefrologia. 2000;20:130–138. [PubMed] [Google Scholar]
- Piontek KB, Huso DL, Grinberg A, Liu L, Bedja D, Zhao H, Gabrielson K, Qian F, Mei C, Westphal H, Germino GG. A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J Am Soc Nephrol. 2004;15:3035–3043. doi: 10.1097/01.ASN.0000144204.01352.86. [DOI] [PubMed] [Google Scholar]
- Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87:979–987. doi: 10.1016/s0092-8674(00)81793-6. [DOI] [PubMed] [Google Scholar]
- Qian F, Germino FJ, Cai Y, Zhang X, Somlo S, Germino GG. PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet. 1997;16:179–183. doi: 10.1038/ng0697-179. [DOI] [PubMed] [Google Scholar]
- Richardson GP. The development of hair cells in the inner ear. In: Fay RR, Popper AN, editors. Vertebrate hair cells. New York: Springer; 2006. p. 454. [Google Scholar]
- Schumann FH, Hoffmeister H, Schmidt M, Bader R, Besl E, Witzgall R, Kalbitzer HR. NMR-assignments of a cytosolic domain of the C-terminus of polycystin-2. Biomol NMR Assign. 2009;3:141–144. doi: 10.1007/s12104-009-9160-x. [DOI] [PubMed] [Google Scholar]
- Sekerkova G, Zheng L, Loomis PA, Changyaleket B, Whitlon DS, Mugnaini E, Bartles JR. Espins are multifunctional actin cytoskeletal regulatory proteins in the microvilli of chemosensory and mechanosensory cells. J Neurosci. 2004;24:5445–5456. doi: 10.1523/JNEUROSCI.1279-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemens J, Lillo C, Dumont RA, Reynolds A, Williams DS, Gillespie PG, Muller U. Cadherin 23 is a component of the tip link in hair-cell stereocilia. Nature. 2004;428:950–955. doi: 10.1038/nature02483. [DOI] [PubMed] [Google Scholar]
- Veldhuisen B, Spruit L, Dauwerse HG, Breuning MH, Peters DJ. Genes homologous to the autosomal dominant polycystic kidney disease genes (PKD1 and PKD2) Eur J Hum Genet. 1999;7:860–872. doi: 10.1038/sj.ejhg.5200383. [DOI] [PubMed] [Google Scholar]
- Walker RG, Willingham AT, Zuker CS. A Drosophila mechanosensory transduction channel. Science. 2000;287:2229–2234. doi: 10.1126/science.287.5461.2229. [DOI] [PubMed] [Google Scholar]
- Ward CJ, Turley H, Ong AC, Comley M, Biddolph S, Chetty R, Ratcliffe PJ, Gattner K, Harris PC. Polycystin, the polycystic kidney disease 1 protein, is expressed by epithelial cells in fetal, adult, and polycystic kidney. Proc Natl Acad Sci U S A. 1996;93:1524–1528. doi: 10.1073/pnas.93.4.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Corbett MK, Chow LM, Valentine MB, Baker SJ, Zuo J. Rapid cell-cycle reentry and cell death after acute inactivation of the retinoblastoma gene product in postnatal cochlear hair cells. Proc Natl Acad Sci U S A. 2008;105:781–785. doi: 10.1073/pnas.0708061105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson PD. Mouse models of polycystic kidney disease. Curr Top Dev Biol. 2008;84:311–350. doi: 10.1016/S0070-2153(08)00606-6. [DOI] [PubMed] [Google Scholar]
- Wilson PD, Geng L, Li X, Burrow CR. The PKD1 gene product, “polycystin-1,” is a tyrosine-phosphorylated protein that colocalizes with alpha2beta1-integrin in focal clusters in adherent renal epithelia. Lab Invest. 1999;79:1311–1323. [PubMed] [Google Scholar]
- Wodarczyk C, Rowe I, Chiaravalli M, Pema M, Qian F, Boletta A. A novel mouse model reveals that polycystin-1 deficiency in ependyma and choroid plexus results in dysfunctional cilia and hydrocephalus. PLoS One. 2009;4:e7137. doi: 10.1371/journal.pone.0007137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Rossetti S, Jiang L, Harris PC, Brown-Glaberman U, Wandinger-Ness A, Bacallao R, Alper SL. Human ADPKD primary cyst epithelial cells with a novel, single codon deletion in the PKD1 gene exhibit defective ciliary polycystin localization and loss of flow-induced Ca2+ signaling. Am J Physiol Renal Physiol. 2007;292:F930–F945. doi: 10.1152/ajprenal.00285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13:2508–2516. doi: 10.1097/01.asn.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- Yu S, Hackmann K, Gao J, He X, Piontek K, Garcia-Gonzalez MA, Menezes LF, Xu H, Germino GG, Zuo J, Qian F. Essential role of cleavage of Polycystin-1 at G protein-coupled receptor proteolytic site for kidney tubular structure. Proc Natl Acad Sci U S A. 2007;104:18688–18693. doi: 10.1073/pnas.0708217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa T, Venugopal B, Weremowicz S, Morton CC, Guo L, Zhou J. The sequence, expression, and chromosomal localization of a novel polycystic kidney disease 1-like gene, PKD1L1, in human. Genomics. 2002;79:376–386. doi: 10.1006/geno.2002.6719. [DOI] [PubMed] [Google Scholar]
- Zheng L, Zheng J, Whitlon DS, Garcia-Anoveros J, Bartles JR. Targeting of the hair cell proteins cadherin 23, harmonin, myosin XVa, espin, and prestin in an epithelial cell model. J Neurosci. 2010;30:7187–7201. doi: 10.1523/JNEUROSCI.0852-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]