

Abstract
In Drosophila, the correct formation of the segmental commissures depends on neuron–glial interactions at the midline. The VUM midline neurons extend axons along which glial cells migrate in between anterior and posterior commissures. Here, we show that the gene kette is required for the normal projection of the VUM axons and subsequently disrupts glial migration. Axonal projection defects are also found for many other moto- and interneurons. In addition, kette affects the cell morphology of mesodermal and epidermal derivatives, which show an abnormal actin cytoskeleton. The KETTE protein is homologous to the transmembrane protein HEM-2/NAP1 evolutionary conserved from worms to vertebrates. In vitro analysis has shown a specific interaction of the vertebrate HEM-2/NAP1 with the SH2–SH3 adapter protein NCK and the small GTPase RAC1, which both have been implicated in regulating cytoskeleton organization and axonal growth. Hypomorphic kette mutations lead to axonal defects similar to mutations in the Drosophila NCK homolog dreadlocks. Furthermore, we show that kette and dock mutants genetically interact. NCK is thought to interact with the small G proteins RAC1 and CDC42, which play a role in axonal growth. In line with these observations, a kette phenocopy can be obtained following directed expression of mutant DCDC42 or DRAC1 in the CNS midline. In addition, the kette mutant phenotype can be partially rescued by expression of an activated DRAC1 transgene. Our data suggest an important role of the HEM-2 protein in cytoskeletal organization during axonal pathfinding.
Keywords: Drosophila, kette, PAK, axon guidance, HEM-2/NAP1
Development of a multicellular organism requires extensive cell movements. In the developing nervous system, many neuronal and glial cells migrate away from their place of birth to their final destination. Subsequently, neuronal growth cones have to navigate often over long distances through the body. To date, a number of signaling molecules and their receptors function in the guidance of cells and growth cones (Tessier-Lavigne and Goodman 1996). Eventually these guidance signals are transmitted to the cellular cytoskeleton to direct cell growth and movement (Tanaka and Sabry 1995; Hu and Reichardt 1999).
How is the signal perception at the membrane connected to the cytoskeleton? The RHO family of small GTPases is implicated in a range of cellular functions including cell migration and neurite outgrowth (Hall 1998). Notably, the expression of the CDC42 GTPase in fibroblasts leads to the formation of filopodia, whereas expression of RAC1 induces the formation of lamellipodia (Nobes and Hall 1995; Tapon and Hall 1997). Function of RAC1 during axonal outgrowth is conserved between flies and mammals (Luo et al. 1996). A connection between these small G proteins and membrane-bound receptors is beginning to emerge. A central role has the SH2–SH3 adapter protein NCK, which is involved in multiple cellular signaling events (McCarty 1998). NCK can bind via the p21-activated kinase (PAK) to both CDC42 and RAC1. Both PAK and small G proteins are involved in actin organization (Sells et al. 1997). Additional effector proteins have been reported to bind to the different SH3 domains of the NCK adapter protein (Hu et al. 1995; Rivero-Lezcano et al. 1995; Quilliam et al. 1996; Symons et al. 1996; Tang et al. 1997; McCarty 1998; Rohatgi et al. 1999). The first SH3 domain of NCK interacts with the transmembrane protein HEM-2/NAP1 (Kitamura et al. 1996).
The SH2 domain of NCK is capable to interact with a variety of receptor tyrosine kinases (RTK) coupling activation of RTK by extracellular signals to intracellular signaling (Li et al. 1992; Stein et al. 1998). Expression of mutagenized NCK protein variants in Xenopus embryos affected dorsoventral patterning and the migratory properties of mesodermal cells (Tanaka et al. 1997; Gupta and Mayer 1998). A Drosophila NCK homolog is encoded by the gene dreadlocks (dock). dock mutations disrupt axon guidance and targeting in the developing nervous system (Garrity et al. 1996; Desai et al. 1999). Similar axonal phenotypes were observed following disruption of a PAK homolog (Hing et al. 1999).
During Drosophila embryogenesis, a ladder-like axon pattern is established in the ventral nerve cord (Goodman and Doe 1993). The individual neuromeres are connected by two lateral longitudinal connectives, whereas the individual neuromeres are interconnected by two segmental commissures, the anterior and the posterior commissure. Over the last few years, a number of genetic studies have identified key players during the formation of this axonal network (Klämbt et al. 1991; Seeger et al. 1993; Kolodziej et al. 1995; Harris et al. 1996; Mitchell et al. 1996; Hummel et al. 1999a,b).
Basically, axonal pattern formation occurs in a series of sequential steps. First, commissural growth cones are guided toward and across the midline (Tessier-Lavigne and Goodman 1996). Once the commissural axon tracts are established, the commissures need to be arranged into their final ladder-like appearance. This developmental process requires the migration of the midline glial cells along cell processes of the VUM-midline neurons (Klämbt et al. 1991).
In a large EMS mutagenesis, we screened for mutations affecting the organization of the CNS axon pattern. We identified >20 genes required for the correct migration of the midline glial cells (Hummel et al. 1999a). These mutations fall into two different classes, the pointed group and the tramtrack group. One member of the pointed group is kette. The newly identified genes could control the differentiation or migration of the midline glial cells or the formation and correct projection of the midline axons along which the midline glial cells migrate. Double-mutant analyses provided the first hints that most of the genes affected by these fused commissure mutations function in the midline glial cells, whereas the gene kette is likely to act in CNS midline neurons (Hummel et al. 1999b).
Here we describe the molecular identification and detailed phenotypic analysis of kette. Mutations in kette disrupt the function of the Drosophila homolog of the HEM-2/NAP1 protein, which has been first identified by Baumgartner et al. (1995). kette mRNA is widely expressed in the early embryo, but expression becomes restricted to the CNS by the end of embryogenesis. Loss of kette function leads to strong defects in axonal pattern formation. In the CNS midline, the VUM neurons show distinct projection defects, which correlate to the migratory defects of the midline glial cells. Overexpression studies demonstrate an important role of the KETTE protein level during axonogenesis. Further analyses of the non-neuronal kette phenotype imply a more general function during the organization of the actin cytoskeleton. Phenotypic and genetic studies suggest functional interactions of KETTE with the SH2–SH3 domain protein DOCK and with small GTPases such as DRAC1.
Results
Isolation of kette alleles
Five different kette mutations were recovered on the basis of a characteristic CNS axon pattern phenotype consisting of fused commissures and reduced longitudinal connectives (Fig. 1). In an independent screen, a large number of P-element-induced lethal mutations were screened for defects in axonal pattern formation (T. Hummel, unpubl.). The P-element insertion l(3)03335 (Karpen and Spradling 1992) leads to strong fused commissure phenotype and failed to complement all EMS-induced kette alleles. The P element mapped at the cytological position 79E, which corresponds well to the meiotic position 3–46.8 determined for the EMS-induced kette mutations (Hummel et al. 1999a). Both the kette phenotype and the lethality associated with the P-element insertion could be reverted by mobilizing the P element. From 160 independent excision lines, 53 remained associated with a lethal mutation or showed reduced viability. In 36 lines, the mutations lead to embryonic lethality as the original l(3)03335 P-element insertion and 13 excision mutations lead to larval or pupal lethality. In four lines <10% of the expected numbers of homozygous adult flies eclosed. The largest deficiency isolated, ketteΔ14–2 (see Fig. 4, below), removes the entire kette gene and shows the same axonal phenotype in homozygous condition and in transheterozygosity to ketteC3–20 or ketteJ4–48 (Fig. 1), indicating that ketteC3–20 and ketteJ4–48 are amorphic mutations.
Figure 1.
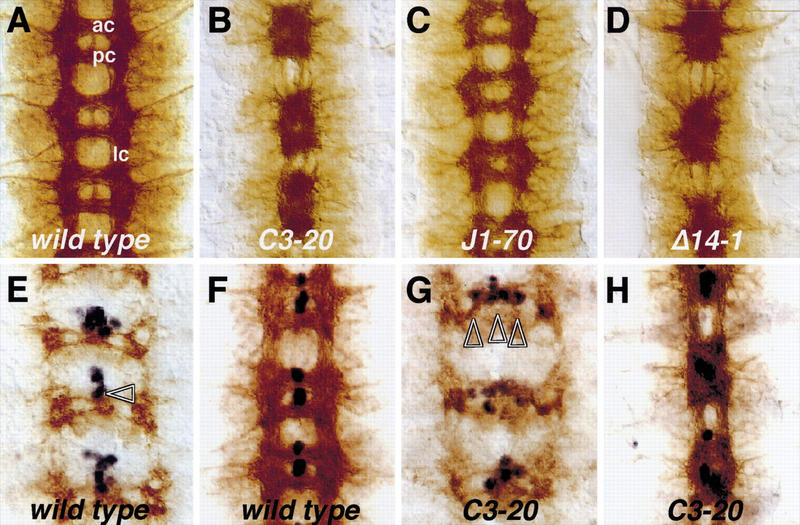
kette affects the migration of the midline glial cells. Frontal views of dissected embryonic nerve cords stained for the overall axon pattern using the mAb BP102 and subsequent HRP immunohistochemistry. (B–C) On the basis of their mutant phenotypes, the different kette alleles were placed in the following allelic series: ketteC3–020 = ketteJ4–048 > ketteP168 > ketteG1–037 > ketteJ1–070. The genotype is indicated; not all genotypes are shown. The midline glial cells (E–H) are labeled in blue using the enhancer trap insertion AA142. Anterior is up. (A) In a wild-type embryo, most CNS axons are organized in a ladder-like pattern consisting of two longitudinal connectives (lc) and two commissures in each neuromere. Anterior commissure (ac) and posterior commissure (pc) are well separated. (B) Amorphic kette mutants show fused commissures and disrupted longitudinal connectives. (C) The hypomorphic mutation J1–70 results in a subtle commissural and connective phenotype. (D) The excision mutation Δ14–2 shows the amorphic kette phenotype. (E) In stage 12/13 wild-type embryos, the midline glial cells have migrated toward the commissures and start to intercalate between anterior and posterior commissures (arrowhead). (F) In stage 16 wild-type embryos, two midline glial cells have migrated between anterior and posterior commissure. (G) In mutant ketteC3–20 embryos, the midline glial cells migrate in an abnormal path and are found on the entire width of the commissure (arrowheads). (H) In stage 16 mutant kette embryos, the midline glial cells fail to intercalate between the segmental commissure and a fused commissure phenotype develops.
Figure 4.

The kette locus. The extent of the kette gene is shown, transcription is from left to right, the exon-intron structure, the P-element l(3)03335 integration site, and the extend of the deletion Δ14–2 are indicated. The deduced KETTE protein and the location of the kette mutations are shown. Shaded boxes indicate deduced transmembrane domains (Baumgartner et al. 1995). The deduced KETTE protein (1126 aa), the human HEM-2 protein (1134 amino acids, accession no. 3043698), and a deduced C. elegans HEM-2 protein (1141 amino acids, accession no. 3875597) show ∼60% identity throughout the entire ORF. Amino acid changes in different kette alleles are C3-20 (W → stop) change at position 256; G1-37 (H → L) change at position 334; J4-48 (W → stop) change at position 490; J1-70 (V → A) change at position 927. All mutations affect conserved amino acids.
kette disrupts neuron–glial interaction at the CNS midline
Strong kette alleles show a fused commissure phenotype, which is generally due to a defect in the migration of midline glial cells (Klämbt et al. 1991). To analyze the migration of these cells, we used the midline glia marker AA142. In late stage 12 embryos, the midline glial cells are found anterior to where commissural axons cross the midline (Fig. 1E). In wild-type embryos, these cells initiate a posterior-ward migration. In stage 16 embryos, two midline glial cells have migrated between anterior and posterior commissure and separated the two axon bundles (Fig. 1F). In kette mutant embryos, the midline glial cells are specified normally, however, their migration appears abnormal. Contrary to the wild type, the midline glial cells migrate laterally away from the midline along the forming commissural axon bundles (Fig. 1G, arrowheads). This is reminiscent of the phenotype of embryos lacking pointed, which encodes a glial differentiation factor (Klämbt 1993; Klaes et al. 1994). Because kette appears to act in the VUM midline neurons (Hummel et al. 1999b), the cause of this phenotype is different from the one in pointed embryos.
Glial migration defects could be due to alterations in the axonal projection pattern of the VUM neurons. The axonal processes of the VUM neurons are in direct contact with the migrating glial cells and presumably provide the substrate for their migration (Klämbt et al. 1991; Stollewerk and Klämbt 1997). Differentiation of the VUM neurons can be analyzed by use of the mAb 22C10. The VUM neurons reside ventrally in the CNS midline and project through the posterior commissure to the anterior commissure, where they turn and grow toward the periphery (Fig. 2A, arrows). In kette mutant embryos, the projection of the VUM neurons was frequently affected (Fig. 2B). Pathfinding defects were found at positions in which wild-type VUM axons changed their growth directions. Possibly due to maternal kette contribution (see below), this early kette phenotype is variable. In ∼20% of the embryos, the VUM axons project normally to the posterior commissure but do not follow the correct path once they have reached the anterior commissure (Fig. 2B, arrows). In ∼30% of the mutant embryos, severe VUM projection defects are found from early stages onward (Fig. 2B′). In the remaining embryos, the projection defects are such that we cannot discriminate VUM axons from other neuronal processes. However, in all cases, the number of the midline neurons is normal in mutant kette embryos, but their position is slightly altered (Fig. 2C,D).
Figure 2.
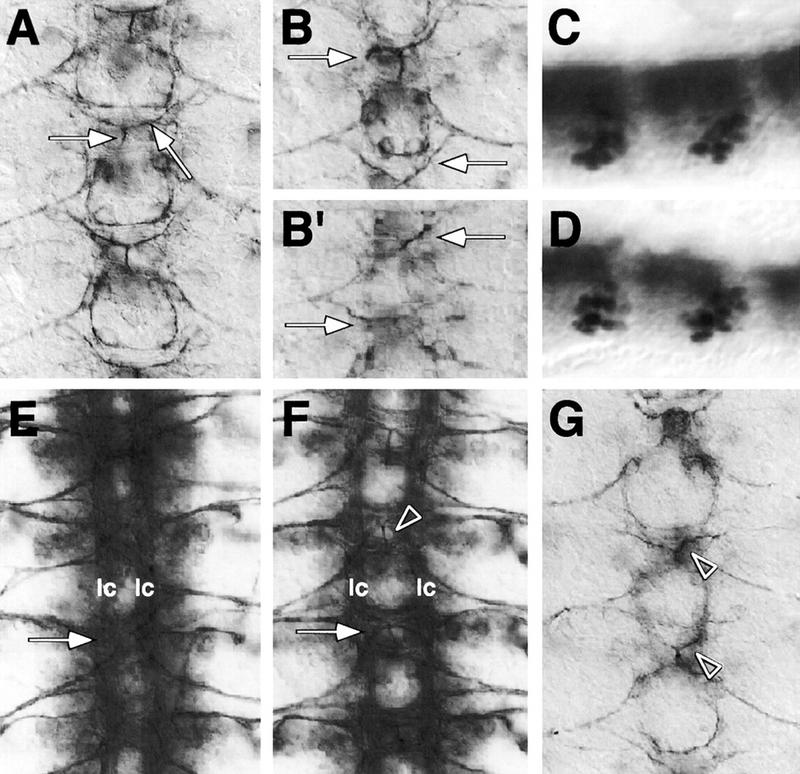
kette functions in midline neurons. Frontal views (A,B,E–G) of dissected embryonic CNS preparations; anterior is up. C and D are whole mount stainings of stage 16 embryos; anterior is to the left. CNS axon tracts are labeled in brown using mAb 22C10. The midline neurons (C,D) are labeled in blue using the enhancer trap insertion X55. (A) In stage 14 wild-type embryos, the VUM axons originate in the midline, project through the posterior commissure to the anterior commissure, where they bifurcate and turn to finally leave the CNS (arrows). (B,B′) In kette mutant embryos, the VUM axons project abnormally. In every neuromere, variable defects in the projection pattern can be observed (arrows, cf. with A). (C) Lateral view of a wild-type stage 16 embryonic CNS. (D) Lateral view of a kette mutant CNS stage 16 embryo. The number and position of the midline neurons has been determined using the marker X55. No changes in number and only slight changes in position were found. (E) In a stage 16 ketteJ4–48 mutant embryo, commissures are fused (arrow) and the two longitudinal connectives (lc) are closer toward the midline. (F) In a stage 16 UAS-kette; simGAL4 mutant ketteJ4–48 embryo, we observe a partial rescue of the kette commissure phenotype (arrow). A space appears between anterior and posterior commissure, and the two longitudinal connectives are located in further distance from the midline. The arrowhead indicates a bifurcating VUM axon. (G) Stage 13 wild-type embryo expressing elevated levels of kette in all midline cells (UAS-kette/simGAL4). Defects in the projection pattern of the VUM neurons can be observed (arrowheads, cf. with A).
kette mutations lead to a variety of defects during embryogenesis
Beside the fusion of commissures, we observed a pronounced reduction of longitudinal connectives in strong kette mutants (Fig. 1). Using an antibody directed against the FASCICLIN II protein, we found that longitudinal connectives do not properly form, and we observed axons occasionally crossing the midline (Fig. 3A,D). Defects can already be detected during early stages of axonal outgrowth (data not shown). No gross alterations in the number as well as fate of cortical neurons were detected using different antibodies (anti-EVE, anti-ELAV, and anti-EAGLE). The longitudinal glial cells that normally are found on the entire longitudinal connective are present in their normal number but are displaced and concentrated at the commissures (Fig. 3B,E).
Figure 3.
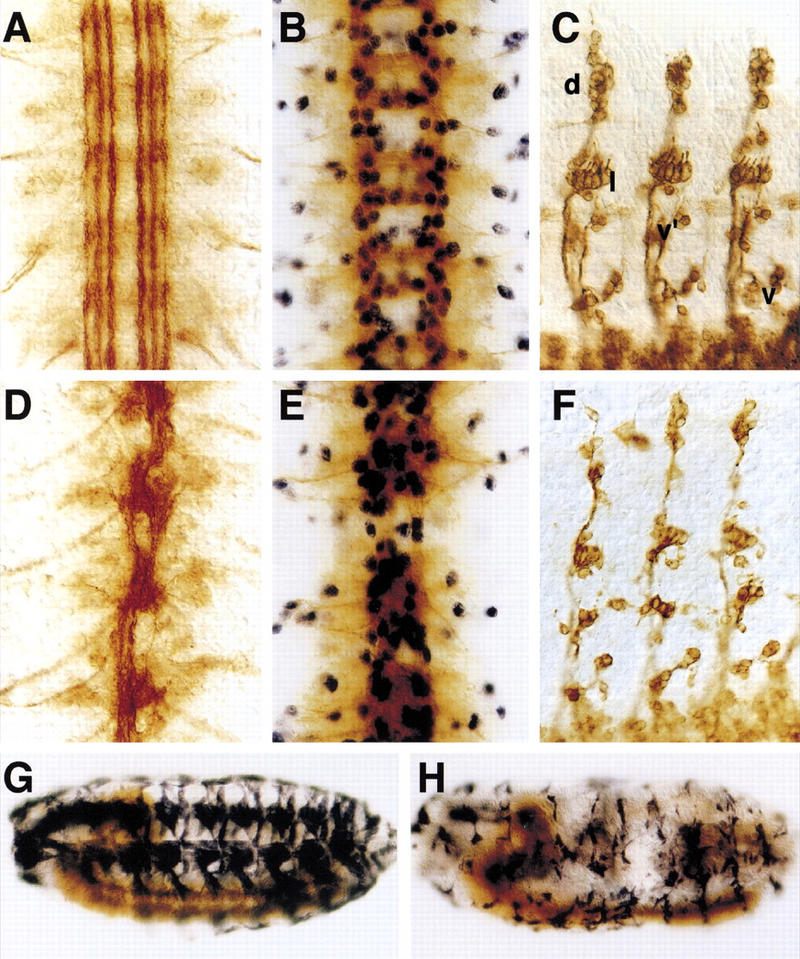
Phenotypic analysis of kette. Flat preparations of stage 16 CNS (A,B,E,D) or PNS (C,F) stained for the presence of different neural markers; (A,D) mAb 1D4, (B,E) mAb BP102 and anti-REPO antibodies, (C,F) mAb 22C10. (G,H) Whole mount embryos carrying a MHC–lacZ reporter, stained for β-Gal expression to label all somatic muscle cells. (A) In wild-type embryos, the FASCICLIN II protein is expressed in discrete fascicles. (D) In kette embryos, individual fascicles cannot be recognized. Often breaks are observed in the connectives. (B) Lateral glial cells are labeled in blue. They are found in a regular pattern in wild-type embryos. (E) In mutant kette embryos, lateral glial cells appear in their normal number. However, they do not migrate along the connectives and are found above the commissures. (C) A wild-type embryo stained for the presence of the 22C10 antigen, which labels the sensory neurons as well as their axonal projections. Sensory neurons are found in four discrete clusters, the dorsal cluster (d), the lateral cluster (l), the ventro-lateral (v′), and the ventral cluster (v). (F) In homozygous kette mutant embryos, reductions in the number of sensory neurons are found in all clusters. Axonal projections into the CNS appear unaffected. (G) In wild-type embryos, a regular pattern of 30 somatic muscle fibers is seen. (H) kette mutant embryos show reduced numbers of muscle fibers. In addition, the remaining muscle fibers appear smaller than in wild-type embryos.
Additional kette phenotypes were observed in the embryonic PNS. We found a slight reduction in the number of all sensory neurons as well as changes in their cellular morphology (Fig. 3C,F). In accordance with the reduction in cell number, the size of the segmental nerves was reduced, the axonal projections toward the ventral nerve cord, however, appeared normal.
A strong disorganization of motoaxons was found (data not shown) that correlates with defects in the somatic musculature. Using a MHC–lacZ reporter construct, a regular pattern of 30 individual polyploid muscles per abdominal hemisegment can be recognized in wild-type embryos (Fig. 3G). In kette mutant embryos, a severe reduction in the number of muscle fibers can be observed. The remaining muscle cells appear much smaller as in wild type (Fig. 3H). In addition, kette also affects midgut formation (data not shown).
Molecular analysis of kette
Genomic kette sequences were cloned by inverse PCR using the P-element insertion l(3)03335. Sequence analysis showed that the transcription unit flanking the P-element insertion site had been described before as the Dhem-2 gene (Baumgartner et al. 1995). The P-element insertion occurred at position 39 within the transcribed region. Dhem2 encodes a putative transmembrane protein homologous to the vertebrate Hem-2/NAP1 protein and a deduced Caenorhabditis elegans hem-2 protein (see Fig. 4 for details) (Baumgartner et al. 1995). The kette excision mutant ketteΔ14–2 removes the Dhem-2 gene. To obtain further proof that kette encodes the DHEM-2 protein, we determined the Dhem-2 DNA sequence of four kette alleles (Fig. 4). The strongest alleles, C3–20 and J4–48, both carry mutations that lead to a truncation of the deduced KETTE protein, which correlates with the amorphic character of these mutations. The hypomorphic kette mutation G1–37 carries a H → L change at a conserved position. The weak J1–70 mutation carries a relatively neutral V → A substitution at the end of the third predicted transmembrane region. These data thus show that kette encodes the DHEM-2 protein.
Expression of kette during embryogenesis
The P-element insertion l(3)03335 into the kette gene shows restricted β-galactosidase (β-Gal) expression in the VUM midline neurons at stage 12 (Fig. 5A). The midline expression persists until stage 14 (Fig. 5B), and thus is present throughout the growth period of the VUM axons, which is in agreement with the predicted function of kette in the midline neurons (Hummel et al. 1999b).
Figure 5.

Expression of kette during embryogenesis. (A,B) The P-element insertion into the kette gene carries a lacZ gene and was used to monitor kette transcription with anti-β-Gal antibodies. First β-Gal expression is detected in the midline neurons of stage 11 embryos and persists until stage 14. (C–F) Show whole mount in situ hybridizations of kette cDNA in wild-type embryos. (C–E) kette mRNA is broadly expressed until stage 14. (D) From stage 13 onward, kette expression can be seen in some CNS midline cells (arrow). (F) Lateral view of a stage 16 embryo. kette expression becomes confined to the CNS.
In contrast to the restricted β-Gal expression pattern of the kette enhancer trap, the endogenous kette gene shows a much broader expression pattern. As determined by use of a digoxygenin-labeled kette cDNA, high levels of kette RNA are provided maternally (data not shown). Ubiquitous kette RNA distribution is found until stage 13, when kette expression becomes restricted to the CNS (Fig. 5C–F). Expression of kette in distinct groups of midline cells, which, on the basis of their position, appear to correspond to the midline neurons, can be detected from stage 13 onward (Fig. 5D). The discrepancy of the RNA distribution and the specific β-Gal expression pattern might indicate a complex regulation of the kette gene.
kette functions in midline neurons
To further analyze the requirement of kette in commissure formation, we used the GAL4 system to express kette in midline cells. The above-described results indicated that kette is required in the midline neurons. In agreement with this notion, we failed to obtain phenotypic rescue of the kette-fused commissure phenotype following expression of kette in the midline glial cells using the slit–GAL4 driver line (Scholz et al. 1997; data not shown). However, when we expressed kette in all midline cells using the single minded–GAL4 driver line (Scholz et al. 1997), we obtained a partial phenotypic rescue of the ketteJ4–048 phenotype (Fig. 2E,F). Following CNS midline expression of kette, commissures were separated and the connectives were found at a further distance from the midline (Fig. 2F, arrow). In particular, the VUM neurons show a normal projection pattern (Fig. 2F, arrowhead). We never obtained a complete rescue of the kette mutant CNS phenotype. The remaining axonal defects could either originate from the function of kette in lateral neurons or could be due to an inappropriate expression of kette in the midline cells. The importance of the exact expression level is also supported by the observation that expression of high levels of kette in all CNS midline cells in a wild-type background resulted in an abnormal axonal projection phenotype of the VUM neurons, similar to the mutant kette phenotype (Fig. 2B,G). In older embryos, the appearance of the overall axon pattern resembles a phenotype of hypomorphic kette or amorphic dock mutations (data not shown).
kette genetically interacts with dock
The vertebrate homolog of KETTE has been shown to interact with the first SH3 domain of the NCK adapter protein (Kitamura et al. 1996). The Drosophila homolog of NCK is encoded by dreadlocks (Garrity et al. 1996). dock was identified in a screen for mutations affecting axonal pathfinding and targeting of the adult photoreceptor neurons (Garrity et al. 1996). In wild-type third instar larvae, the different photoreceptor cells stop their axonal growth in two distinct layers of the optic lobe, the lamina and the medulla (Fig. 6A). In contrast, dock mutant photoreceptor cells fail to establish this specific targeting, leading to a disruption of the lamina neuropile organization (Fig. 6B). In ∼70% of the third instar larvae homozygous for the hypomorphic ketteΔ2–6 allele (n = 25), we found a weak disorganization of the lamina plexus and an abnormal bundling of R-cell axons in the medulla (Fig. 6C). The remaining larvae showed a stronger disorganization of the R-cell axons (20%) or were indistinguishable from wild type (10%). Further reduction of the kette gene function results in an enhancement of this axonal phenotype in 50% of the analyzed transheterozygous mutant kette larva (n = 30; Fig. 6D). If we remove one copy of dock in the background of a hypomorphic kette mutation, we observed a considerable enhancement of the larval projection phenotype in 60% of the individuals (n = 25; Fig. 6E). In addition, we observed a significant enhancement of the homozygous dock phenotype when removing one copy of kette in a dock mutant background (n = 18; Fig. 6F).
Figure 6.
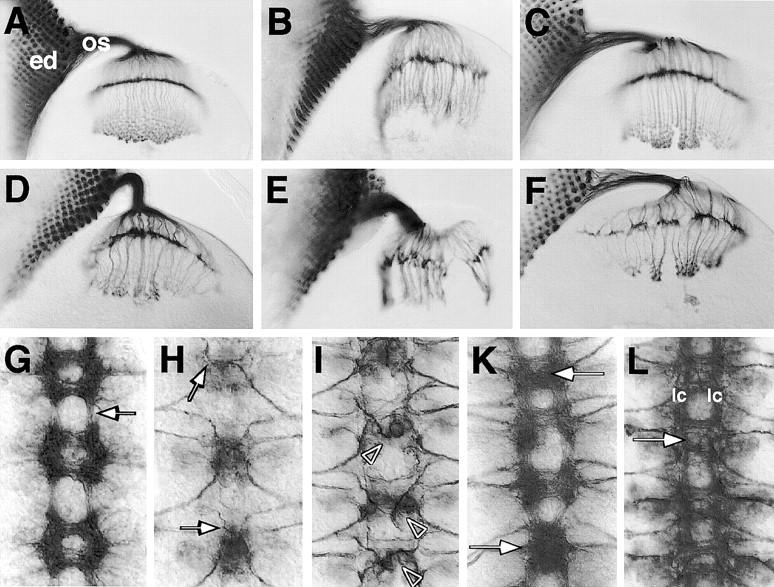
kette genetically interacts with dock and Drac1. (A) In wild-type larvae, the 22C10 antigen labels the photoreceptor axons as they grow from the eye disc (ed) through the optic stalk (os) into the brain. Here they terminate in the lamina or the medulla. (B) In homozygous mutant dockP2 larvae and (C) in homozygous ketteΔ2–6 larvae, the growth of retinula axons into the brain is disrupted. (D) Following further reduction of kette function in ketteΔ2–6/ketteC3–20 larvae, this axonal phenotype becomes more pronounced. Following removal of one copy of dock in a homozygous ketteΔ2–6 larvae (E) or one copy of kette in homozygous mutant dockP2 larvae (F), we observed a much more severe axonal projection phenotype. (G–L) Frontal views of dissected embryonic CNS preparations. Anterior is up. (G) We used mAb BP102 to visualize the overall CNS axon pattern in mutant dock embryos. The zygotic dock phenotype is characterized by a reduction in the longitudinal connectives (arrow) and a slight fusion of the segmental commissures. (H) To determine the morphology of the VUM neurons, we used the mAb 22C10.As in mutant kette embryos, variable projection defects of the VUM axons were found in mutant dock embryos (arrows). (I) Stage 14 embryo of the genotype UAS–Drac1V12; simGAL4 expressing activated DRAC1 in all midline cells. Frequent defects in the projection pattern of the VUM neurons are observed (arrowheads). (K) In stage 16 embryos, a fused commissure phenotype can be seen (arrows). (L) Expression of Drac1 in all CNS midline cells of mutant kette embryos partially rescued the mutant CNS phenotype. The longitudinal connectives (lc) appeared further apart and the commissures appear separated. In addition, we often observed VUM axons projecting normally at the midline (arrow).
Similar to the findings of Desai et al. (1999), we observed a reduction in the size of the longitudinal connectives in the embryonic CNS (Fig. 6G). This phenotype resembles a hypomorphic kette connective phenotype. In mutant dockP2 embryos, commissure separation is also affected, comparable with the hypomorphic phenotype seen in ketteJ1–70 embryos (Fig. 1). In correlation with the commissural phenotype, the VUM axons do not project properly in mutant dock embryos (Fig. 6H, arrows).
In summary, these data show that both kette and dock mutants genetically interact and share a number of phenotypic traits. This suggests that these genes might be acting in the same genetic pathway during axonal pathfinding.
The role of small GTPases in the CNS midline
The RHO family of small GTPases has been described previously to constitute important regulatory factors also interacting with NCK (Hall 1998). To analyze the functional interaction of KETTE with members of the RHO family, we expressed activated as well as dominant-negative versions of DCDC42 and DRAC1 (kindly provided by L. Luo, Stanford University, CA) in the midline cells of wild-type and kette mutant embryos. Expression of both of these mutant proteins in all midline cells using the simGAL4 driver line results in similar axonal defects (Fig. 6I). The projection of the VUM neurons resembled the phenotype observed in kette mutant embryos. In addition, the cell bodies of the VUM neurons appeared sometimes displaced (not shown). In stage 16 embryos, the segmental commissures appear fused, which again indicates the importance of the midline neurons for the migration of the midline glia (Fig. 6K). We observed only a weak commissural disorganization when we expressed the different DCDC42 or DRAC1 proteins in the midline glial cells only (data not shown). In all experiments, the expression of DRAC1 appears to have more pronounced effects on the axonal morphology.
To further test the interaction of kette and RAC1, we expressed activated DRAC1 in all midline cells of mutant ketteJ4–48 embryos. The commissures appeared separated, indicating that the midline glial cells were able to migrate between anterior and posterior commissures. Concomitantly, the connectives are further distant from the midline (Fig. 6L, arrow), indicating that expression of activated Drac1 can partially rescue the kette phenotype.
kette affects the organization of the cytoskeleton
Among others, the NCK adapter protein transduces signals via CDC42 and RAC1 to the ACTIN cytoskeleton. To analyze the ACTIN cytoskeleton of mutant kette embryos, we used a GFP–moesin transgene (Edwards et al. 1997). This protein binds to the F-actin fibers and thus allows us to determine their subcellular distribution using confocal microscopy. In wild-type embryos, F actin is found in axonal processes that are arranged in the typical ladder-like pattern (Fig. 7A). Prominent expression is also detected in the epidermis and the somatic musculature (Fig. 7A, arrow). In similar focal planes, kette embryos appear very different. Within the CNS, the typical fused commissure phenotype of mutant kette embryos is evident (Fig. 7B). We observed frequent intense granular staining in the CNS and in the lateral body wall (Fig. 7B, arrowheads). Furthermore, the regular appearance of the cytoskeleton is disrupted in both mesoderm and ectoderm (Fig. 7B, arrow). In a tangential section of the dorsal epidermis, individual cells can be seen in wild-type embryos. Some cells form hairs, characterized by thin F-actin bundles (Fig. 7C). In mutant ketteC3–20 embryos, pronounced F-actin bundles were found, which often have a wavy appearance (Fig. 7D, arrow). In addition, the cortical actin cytoskeleton appears to stain weaker compared with wild-type embryos.
Figure 7.

kette affects the cytoskeleton. GFP–MOESIN was used to monitor the organization of the F-actin bundles. (A–D) Confocal images obtained from whole mount stage 16 embryos, anterior is to the left. (A) Distinct F-actin expression can be detected in the CNS axons, the ectoderm and the somatic musculature (arrow). (B) In mutant kette embryos, the CNS axon tracts are disorganized as seen in Fig. 1. The organization of F-actin fibers in the lateral body wall is abnormal. No clear concentration of staining can be found at the apical surface of the embryo. In addition, a granular appearance of F-actin staining can be observed in the nervous system as well as in the lateral body wall (arrowheads). (C) Tangential view of the dorsal epidermis. F-actin bundles line up the cell boundaries. Some cells project fine hairs (arrowhead). (D) Similar view of a kette mutant embryo. The cell borders are not staining as clear as in wild type. In addition, long hair-like structures are visible that are not found in the wild type (arrow).
Discussion
Here we presented a detailed analysis of the kette gene. Mutations in kette affect the organization of the cytoskeleton. kette is expressed in neurons and is needed for correct axonal pathfinding. The KETTE protein seems to interact with the SH2–SH3 adapter DOCK and at least part of the kette function might be mediated via small GTPases such as DRAC1.
kette affects the actin cytoskeleton
First evidence that kette causes axonal defects by affecting the organization of the cytoskeleton stem from detailed phenotypic analyzes. Beside its function in axonal pathfinding, we observed defects in the morphology of trichomes and bristles in flies homozygous for the weak hypomorphic ketteΔ2–6 allele. Around 10% of the bristles appear wavy or do bend sharply and wing trichomes are enlarged and sometimes split (data not shown). These phenotypes resemble those observed for mutations affecting the organization of the F-actin bundles or following expression of mutated DCDC42 or DRAC1 (Verheyen and Cooley 1994; Eaton et al. 1995, 1996). Similarly, elevated levels of GTPase function in the developing eye cause late developmental defects as observed in hypomorphic kette mutations (Hariharan et al. 1995; Sawamoto et al. 1999; data not shown). cdc42 mutations have been isolated (Fehon et al. 1997), but, presumably due to maternal contribution, loss of cdc42 function does not lead to an embryonic CNS phenotype (data not shown). Both CDC42 and RAC1 are important regulators of the actin cytoskeleton (Hall 1998). The transduction of extracellular signaling to small GTPases is believed to involve NCK-type adapter proteins. Here we showed that several phenotypic traits of kette are shared by mutations in the Drosophila gene dock, which encodes a NCK homolog. Furthermore, dock and kette genetically interact. The genetic data in combination with the kette loss-of-function and kette overexpression phenotypes lead us to propose a model on the Dhem2/NAP1 function in cytoskeleton organization as discussed below.
Does KETTE act as a membrane bound receptor?
The DHEM-2 protein is predicted to be an integral transmembrane protein with six transmembrane domains (Baumgartner et al. 1995). The amorphic mutations ketteJ4–48 and ketteC3–20 delete the carboxy-terminal half of the protein. Thus, an important function must reside in this part. In the CNS, the membrane protein KETTE could be participating directly in the neuron-glial interaction at the midline, where it could act as a signal to direct glial migration. Alternatively, KETTE could serve as a receptor of possibly glial-derived signals during VUM growth cone guidance. The experimental data suggest that KETTE transduces information to the neuronal cytoskeleton, which is in agreement with a receptor function.
Binding on a hypothesized ligand would lead to a concentration of KETTE at the interaction zone, where it could initiate further signaling to induce cytoskeletal reorganization. Interestingly, the loss-of-function phenotype of kette resembles the gain-of-function phenotype, which could indicate that the correct amount of KETTE is important.
KETTE could be a more general factor that helps to recruit NCK-like adapter proteins to the membrane, where it interacts with different receptor tyrosine kinases via its SH2 domain (McCarty 1998). In this scenario, the specific interaction between the midline glia and the midline neurons would be detected by a still-unknown receptor also interacting with the NCK adapter homolog. The FGF receptor is capable to signal via NCK (Ryan and Gillespie 1994; Rockow et al. 1996; Gupta and Mayer 1998). In Drosophila, the FGF receptor homolog BREATHLESS is expressed in neuronal midline cells and mutations in this gene result in a mild migration defect of the midline glial cells (Klämbt et al. 1992). Whether this phenotype—which is reminiscent of hypomorphic kette or amorphic dock mutations—results from an interference with NCK/DOCK signaling remains to be shown.
Does KETTE signal via a NCK adapter protein?
The vertebrate KETTE homolog is HEM-2/NAP1 with 86% amino acid identity over the entire ORF, indicating that presumed protein–protein interactions are also conserved. To date, no hem-2 mutation has been described in vertebrates. The first SH3 domain of NCK was used to isolate NCK-associated proteins (NAP) and led to the identification of HEM-2/NAP1. Binding of HEM-2/NAP1 to NCK appears to be mediated by a 140-kD protein (Kitamura et al. 1996). Interestingly, in a screen for proteins interacting with activated RAC1, a complex consisting of HEM-2/NAP1 and a 140-kD protein was isolated (Kitamura et al. 1997). Thus, the 140-kD protein might be a novel adapter linking HEM-2/NAP1 signaling along two routes to the small GTPases. It will be of interest to identify Drosophila genes interacting with kette. One such candidate was found in a recent screen for modifiers of the phenotype of hypomorphic kette mutations (T. Hummel, unpubl.).
The Drosophila NCK homolog is encoded by dock (Garrity et al. 1996). dock function appears highly specialized for growth-cone guidance as no mutant phenotypes have been reported in the mesoderm or the ectoderm (Garrity et al. 1996; Desai et al. 1999). Because kette shows more pleiotropic defects, other adapter proteins may interact with the KETTE protein.
Neurite extension and remodeling of the cytoskeleton
During axonal pathfinding, coordinated cytoskeletal remodeling occurs at the tip of the extending neurites, the growth cone (Gordon-Weeks 1993; Bentley and O'Connor 1994). The Rho family of GTPases mediates the regulation of the reorganization of the actin cytoskeleton induced by extracellular signals: CDC42, RAC1, and RHOA (Nobes and Hall 1995; Hall 1998). In fibroblast cells, the different GTPases induce different cellular responses (Ridley and Hall 1992; Ridley et al. 1992; Nobes and Hall 1995). Similarly, different functions appear to be associated with the different Drosophila GTPases. RHO as well as CDC42 function is needed for cell shape changes during gastrulation, dorsal closure, bristle, and hair formation (Eaton et al. 1995, 1996; Hariharan et al. 1995; Murphy and Montell 1996; Barrett et al. 1997; Strutt et al. 1997). Bristle and hair formation are similarly affected by kette.
Expression of activated DRAC1 in the CNS midline cells can rescue the kette mutant phenotype. Perturbations of DRAC1 activity blocks axonal growth (Luo et al. 1994; Kaufmann et al. 1998) and cell migrations during fly development (Harden et al. 1995; Murphy and Montell 1996). Following expression of mutant DRAC1 proteins, abnormal actin condensation occurs (Luo et al. 1994). Similarly, actin organization appears affected in mutant kette embryos. We are currently analyzing whether kette and Drac1 are acting in parallel or in a common pathway.
Conclusions
The data presented in this work suggest that KETTE provides a novel mechanism linking extracellular signals to the neuronal cytoskeleton. Central relay proteins are SH2–SH3 adapter proteins, which control the organization of the actin cytoskeleton via a number of downstream proteins. Biochemical data suggest that additional proteins (p140 kD) may bypass the function of SH2–SH3 adapter proteins (Kitamura et al. 1996, 1997), but a detailed analysis awaits its isolation. The KETTE protein might interact with extracellular signals, which, in the CNS, might possibly be presented by glial cells. To gain further insights in the neuron-glial interaction at the midline, future work will be directed toward the identification of these components.
Materials and methods
Genetics
In a large-scale EMS mutagenesis, we identified the following kette alleles C3–20; G1–37; J4–48; P168; J1–70 (Hummel et al. 1999a). All mutations were induced on a st, e background. To monitor the midline glial cells, we used the P[white, lacZ] enhancer trap insertion AA142 (Klämbt et al. 1991) in the C3–20 and P168 mutant background. Transposase-induced excision mutagenesis of kette03335 was performed as described (Klämbt et al. 1992). Flies homozygous for the excision allele ketteΔ2–6 eclosed at reduced rates, whereas ketteC3–20/ketteΔ2–6 animals almost never eclosed. This indicates that the allele ketteΔ2–6 reduces kette function only moderately. Double mutant analyses and rescue experiments were performed using blue balancers (Hummel et al. 1999a).
DNA work
DNA sequences flanking the P-element insertion were cloned by inverse PCR. Details are available on request. Sequencing was performed using a ABI310 sequencer according to the manufacturer's instructions. Genomic as well as cDNA clones were kindly provided by S. Baumgartner. During the sequencing, we noted several differences to the published sequences (see accession no. X80028). An intron reported by Baumgartner et al. (1995) was not detected.
Misexpression studies
To determine the gain-of-function phenotype of kette, we used the GAL4 system (Brand and Perrimon 1993). A kette full-length cDNA clone was inserted into the pUAST vector and used for germ-line transformation. Eighteen independent transgenes with different expressivity of the mini white gene were established.
Histology
Immunohistochemistry and whole mount in situ hybridizations were done as described (Tautz and Pfeifle 1989; Hummel et al. 1997). Confocal microscopy was performed according to Edwards et al. (1997).
Acknowledgments
We thank D. Kiehart, L. Luo, T. Raabe, Y. Rao, D. Schmucker, and L. Zipursky for fly stocks. S. Baumgartner for providing us with cDNA and genomic clones. C.S. Goodman and A. Travers for antibodies. L. Meadows and N. Deshpande for the anti-EAGLE staining. K. Schimmelpfeng for many discussions, I. Bunse for help with sequencing. G. Edenfeld, S. Granderath, E. Knust, U. Lammel, and J. Pielage for critically reading of the manuscript, and unknown reviewers for helpful comments. This work was supported through grants of the DFG and the HFSPO to C.K.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL klaembt@uni-muenster.de; FAX 49 251 832 4686.
References
- Barrett K, Leptin M, Settleman J. The Rho GTPase and a putative RhoGEF mediate a signaling pathway for the cell shape changes in Drosophila gastrulation. Cell. 1997;91:905–915. doi: 10.1016/s0092-8674(00)80482-1. [DOI] [PubMed] [Google Scholar]
- Baumgartner S, Martin D, Chiquet-Ehrismann R, Sutton J, Desai A, Huang I, Kato K, Hromas R. The HEM proteins: A novel family of tissue specific transmembrane proteins expressed from invertebrates through mammals with an essential function in oogenesis. J Mol Biol. 1995;251:41–49. doi: 10.1006/jmbi.1995.0414. [DOI] [PubMed] [Google Scholar]
- Bentley D, O'Connor PT. Cytoskeletal events in growth cone steering. Curr Opin Neurobiol. 1994;4:43–48. doi: 10.1016/0959-4388(94)90030-2. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Desai CJ, Garrity PA, Keshishian H, Zipursky SL, Zinn K. The Drosophila SH2 SH3 adapter protein Dock is expressed in embryonic axons and facilitates synapse formation by the RP3 motoneuron. Development. 1999;126:1527–1535. doi: 10.1242/dev.126.7.1527. [DOI] [PubMed] [Google Scholar]
- Eaton S, Auvinen P, Luo L, Jan YN, Simons K. CDC42 and Rac1 control different actin dependent processes in the Drosophila wing disc epithelium. J Cell Biol. 1995;131:151–164. doi: 10.1083/jcb.131.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton S, Wepf R, Simons K. Roles for Rac1 and Cdc42 in planar polarization and hair outgrowth in the wing of Drosophila. J Cell Biol. 1996;135:1277–1289. doi: 10.1083/jcb.135.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards KA, Demsky M, Montague RA, Weymouth N, Kiehart DP. GFP moesin illuminates actin cytoskeleton dynamics in living tissue and demonstrates cell shape changes during morphogenesis in Drosophila. Dev Biol. 1997;191:103–117. doi: 10.1006/dbio.1997.8707. [DOI] [PubMed] [Google Scholar]
- Fehon RG, Oren T, LaJeunesse DR, Melby TE, McCartney BM. Isolation of mutations in the Drosophila homologues of the human Neurofibromatosis 2 and yeast CDC42 genes using a simple and efficient reverse genetic method. Genetics. 1997;146:245–252. doi: 10.1093/genetics/146.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrity PA, Rao Y, Salecker I, McGlade J, Pawson T, Zipursky SL. Drosophila photoreceptor axon guidance and targeting requires the dreadlocks SH2/SH3 adapter protein. Cell. 1996;85:639–650. doi: 10.1016/s0092-8674(00)81231-3. [DOI] [PubMed] [Google Scholar]
- Goodman CS, Doe CQ. Embryonic development of the Drosophila central nervous system. In: Bate CM, Martinez-Arias A, editors. The development of Drosophila melanogaster. Vol. 1. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1993. pp. 1131–1206. [Google Scholar]
- Gordon-Weeks PR. Organization of microtubules in axonal growth cones: A role for microtubule associated protein MAP 1B. J Neurocytol. 1993;22:717–725. doi: 10.1007/BF01181317. [DOI] [PubMed] [Google Scholar]
- Gupta RW, Mayer BJ. Dominant negative mutants of the SH2/SH3 adapters Nck and Grb2 inhibit MAP kinase activation and mesoderm specific gene induction by eFGF in Xenopus. Oncogene. 1998;17:2155–2165. doi: 10.1038/sj.onc.1202158. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Harden N, Loh HY, Chia W, Lim L. A dominant inhibitory version of the small GTP binding protein Rac disrupts cytoskeletal structures and inhibits developmental cell shape changes in Drosophila. Development. 1995;121:903–914. doi: 10.1242/dev.121.3.903. [DOI] [PubMed] [Google Scholar]
- Hariharan IK, Hu KQ, Asha H, Quintanilla A, Ezzell RM, Settleman J. Characterization of rho GTPase family homologues in Drosophila melanogaster: Overexpressing Rho1 in retinal cells causes a late developmental defect. EMBO J. 1995;14:292–302. doi: 10.1002/j.1460-2075.1995.tb07003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris R, Sabatelli LM, Seeger MA. Guidance cues at the Drosophila midline: Identification and characterization of two Drosophila netrin/unc6 homologs. Neuron. 1996;17:217–228. doi: 10.1016/s0896-6273(00)80154-3. [DOI] [PubMed] [Google Scholar]
- Hing H, Xiao J, Harden N, Lim L, Zipursky SL. Pak functions downstream of Dock to regulate photoreceptor axon guidance in Drosophila. Cell. 1999;97:853–863. doi: 10.1016/s0092-8674(00)80798-9. [DOI] [PubMed] [Google Scholar]
- Hu S, Reichardt LF. From membrane to cytoskeleton: Enabling a connection. Neuron. 1999;22:419–422. doi: 10.1016/s0896-6273(00)80696-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Milfay D, Williams LT. Binding of NCK to SOS and activation of ras dependent gene expression. Mol Cell Biol. 1995;15:1169–1174. doi: 10.1128/mcb.15.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel T, Schimmelpfeng K, Klämbt C. Commissure formation in the embryonic CNS of Drosophila: I Identification of the required gene functions. Dev Biol. 1999a;208:381–398. doi: 10.1006/dbio.1999.9235. [DOI] [PubMed] [Google Scholar]
- ————— Commissure formation in the embryonic CNS of Drosophila: II Function of the different midline cells. Development. 1999b;126:771–779. doi: 10.1242/dev.126.4.771. [DOI] [PubMed] [Google Scholar]
- Hummel T, Schimmelpfeng K, Klämbt C. Fast and efficient egg collection and antibody staining from large numbers of Drosophila strains. Dev Genes & Evol. 1997;207:131–135. doi: 10.1007/s004270050100. [DOI] [PubMed] [Google Scholar]
- Karpen G, Spradling AC. Analysis of subtelomeric heterochromatin in the Drosophila minichromosome Dp1187 by single P element insertional analysis. Genetics. 1992;132:737–753. doi: 10.1093/genetics/132.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann N, Wills ZP, Van Vactor D. Drosophila Rac1 controls motor axon guidance. Development. 1998;125:453–461. doi: 10.1242/dev.125.3.453. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Kitamura Y, Yonezawa K, Totty NF, Gout I, Hara K, Waterfield MD, Sakaue M, Ogawa W, et al. Molecular cloning of p125Nap1, a protein that associates with an SH3 domain of Nck. Biochem Biophys Res Commun. 1996;219:509–514. doi: 10.1006/bbrc.1996.0264. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Kitamura T, Sakaue H, Maeda T, Ueno H, Nishio S, Ohno S, Osada S, Sakaue M, Ogawa W, Kasuga M. Interaction of Nck associated protein 1 with activated GTP binding protein Rac. Biochem J. 1997;322:873–878. doi: 10.1042/bj3220873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaes A, Menne T, Stollewerk A, Scholz H, Klämbt C. The Ets transcription factors encoded by the Drosophila gene pointed direct glial cell differentiation in the embryonic CNS. Cell. 1994;78:149–160. doi: 10.1016/0092-8674(94)90581-9. [DOI] [PubMed] [Google Scholar]
- Klämbt C. The Drosophila gene pointed encodes two ETS like proteins which are involved in the development of the midline glial cells. Development. 1993;117:163–176. doi: 10.1242/dev.117.1.163. [DOI] [PubMed] [Google Scholar]
- Klämbt C, Jacobs JR, Goodman CS. The midline of the Drosophila central nervous system: A model for the genetic analysis of cell fate, cell migration, and growth cone guidance. Cell. 1991;64:801–815. doi: 10.1016/0092-8674(91)90509-w. [DOI] [PubMed] [Google Scholar]
- Klämbt C, Glazer L, Shilo BZ. breathless, a Drosophila FGF receptor homolog, is essential for migration of tracheal and specific midline glial cells. Genes & Dev. 1992;6:1668–1678. doi: 10.1101/gad.6.9.1668. [DOI] [PubMed] [Google Scholar]
- Kolodziej PA, Jan LY, Jan YN. Mutations that affect the length, fasciculation, or ventral orientation of specific sensory axons in the Drosophila embryo. Neuron. 1995;15:273–286. doi: 10.1016/0896-6273(95)90033-0. [DOI] [PubMed] [Google Scholar]
- Li W, Hu P, Skolnik EY, Ullrich A, Schlessinger J. The SH2 and SH3 domain containing Nck protein is oncogenic and a common target for phosphorylation by different surface receptors. Mol Cell Biol. 1992;12:5824–5833. doi: 10.1128/mcb.12.12.5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Liao YJ, Jan LY, Jan YN. Distinct morphogenetic functions of similar small GTPases: Drosophila Drac1 is involved in axonal outgrowth and myoblast fusion. Genes & Dev. 1994;8:1787–1802. doi: 10.1101/gad.8.15.1787. [DOI] [PubMed] [Google Scholar]
- Luo L, Hensch TK, Ackerman L, Barbel S, Jan LY, Jan YN. Differential effects of the Rac GTPase on Purkinje cell axons and dendritic trunks and spines. Nature. 1996;379:837–840. doi: 10.1038/379837a0. [DOI] [PubMed] [Google Scholar]
- McCarty JH. The Nck SH2/SH3 adaptor protein: A regulator of multiple intracellular signal transduction events. BioEssays. 1998;20:913–921. doi: 10.1002/(SICI)1521-1878(199811)20:11<913::AID-BIES6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Mitchell KJ, Doyle JL, Serafini T, Kennedy TE, Tessier-Lavigne M, Goodman CS, Dickson BJ. Genetic analysis of Netrin genes in Drosophila: Netrins guide CNS commissural axons and peripheral motor axons. Neuron. 1996;17:203–215. doi: 10.1016/s0896-6273(00)80153-1. [DOI] [PubMed] [Google Scholar]
- Murphy AM, Montell DJ. Cell type specific roles for Cdc42, Rac, and RhoL in Drosophila oogenesis. J Cell Biol. 1996;133:617–630. doi: 10.1083/jcb.133.3.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Quilliam LA, Lambert QT, Mickelson-Young LA, Westwick JK, Sparks AB, Kay BK, Jenkins NA, Gilbert DJ, Copeland NG, Der CJ. Isolation of a NCK associated kinase, PRK2, an SH3 binding protein and potential effector of Rho protein signaling. J Biol Chem. 1996;271:28772–28776. doi: 10.1074/jbc.271.46.28772. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Hall A. The small GTP binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP binding protein rac regulates growth factor induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Rivero-Lezcano OM, Marcilla A, Sameshima JH, Robbins KC. Wiskott Aldrich syndrome protein physically associates with Nck through Src homology 3 domains. Mol Cell Biol. 1995;15:5725–5731. doi: 10.1128/mcb.15.10.5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockow S, Tang J, Xiong W, Li W. Nck inhibits NGF and basic FGF induced PC12 cell differentiation via mitogen activated protein kinase independent pathway. Oncogene. 1996;12:2351–2359. [PubMed] [Google Scholar]
- Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, Kirschner MW. The interaction between N WASP and the Arp2/3 complex links Cdc42 dependent signals to actin assembly. Cell. 1999;97:221–231. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- Ryan PJ, Gillespie LL. Phosphorylation of phospholipase C gamma 1 and its association with the FGF receptor is developmentally regulated and occurs during mesoderm induction in Xenopus laevis. Dev Biol. 1994;166:101–111. doi: 10.1006/dbio.1994.1299. [DOI] [PubMed] [Google Scholar]
- Sawamoto K, Yamada C, Kishida S, Hirota Y, Taguchi A, Kikuchi A, Okano H. Ectopic expression of constitutively activated Ral GTPase inhibits cell shape changes during Drosophila eye development. Oncogene. 1999;18:1967–1974. doi: 10.1038/sj.onc.1202522. [DOI] [PubMed] [Google Scholar]
- Scholz H, Sadlowski E, Klaes A, Klämbt C. Control of midline glia development in the embryonic Drosophila CNS. Mech Dev. 1997;64:137–151. doi: 10.1016/s0925-4773(97)00078-6. [DOI] [PubMed] [Google Scholar]
- Seeger M, Tear G, Ferres-Marco D, Goodman CS. Mutations affecting growth cone guidance in Drosophila: Genes necessary for guidance toward or away from the midline. Neuron. 1993;10:409–426. doi: 10.1016/0896-6273(93)90330-t. [DOI] [PubMed] [Google Scholar]
- Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J. Human p21 activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol. 1997;7:202–210. doi: 10.1016/s0960-9822(97)70091-5. [DOI] [PubMed] [Google Scholar]
- Stein E, Huynh-Do U, Lane AA, Cerretti DP, Daniel TO. Nck recruitment to Eph receptor, EphB1/ELK, couples ligand activation to c Jun kinase. J Biol Chem. 1998;273:1303–1308. doi: 10.1074/jbc.273.3.1303. [DOI] [PubMed] [Google Scholar]
- Stollewerk A, Klämbt C. The midline glial cells are required for compartmentalization of axon commissures in the embryonic CNS of Drosophila. Dev Genes & Evol. 1997;207:401–409. doi: 10.1007/s004270050129. [DOI] [PubMed] [Google Scholar]
- Strutt DI, Weber U, Mlodzik M. The role of RhoA in tissue polarity and Frizzled signalling. Nature. 1997;387:292–295. doi: 10.1038/387292a0. [DOI] [PubMed] [Google Scholar]
- Symons M, Derry JM, Karlak B, Jiang S, Lemahieu V, McCormick F, Francke U, Abo A. Wiskott Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell. 1996;84:723–734. doi: 10.1016/s0092-8674(00)81050-8. [DOI] [PubMed] [Google Scholar]
- Tanaka E, Sabry J. Making the connection: Cytoskeletal rearrangements during growth cone guidance. Cell. 1995;83:171–176. doi: 10.1016/0092-8674(95)90158-2. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Lu W, Gupta R, Mayer BJ. Expression of mutated Nck SH2/SH3 adaptor respecifies mesodermal cell fate in Xenopus laevis development. Proc Natl Acad Sci. 1997;94:4493–4498. doi: 10.1073/pnas.94.9.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Feng GS, Li W. Induced direct binding of the adapter protein Nck to the GTPase activating protein associated protein p62 by epidermal growth factor. Oncogene. 1997;15:1823–1832. doi: 10.1038/sj.onc.1201351. [DOI] [PubMed] [Google Scholar]
- Tapon N, Hall A. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr Opin Cell Biol. 1997;9:86–92. doi: 10.1016/s0955-0674(97)80156-1. [DOI] [PubMed] [Google Scholar]
- Tautz D, Pfeifle C. A non radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma. 1989;98:81–85. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- Verheyen EM, Cooley L. Profilin mutations disrupt multiple actin dependent processes during Drosophila development. Development. 1994;120:717–728. doi: 10.1242/dev.120.4.717. [DOI] [PubMed] [Google Scholar]