

Abstract
A conditional allele of type 1 protein phosphatase (glc7-129) in Saccharomyces cerevisiae causes first cycle arrest in G2/M, characterized by cells with a short spindle and high H1 kinase activity. Point-of-execution experiments indicate Glc7p function is required in G2/M just before anaphase for the completion of mitosis. Loss of the spindle/kinetochore checkpoint in glc7-129 cells abolishes the G2/M cell cycle arrest with a concomitant increase in chromosome loss and reduced viability. These results support a role for Glc7p in regulating kinetochore attachment to the spindle, an event monitored by the spindle/kinetochore checkpoint.
Keywords: Protein phosphatase I, yeast, mitotic spindle, kinetochore, checkpoint
A critical event in the mitotic cell cycle is the attachment of every chromosome to the mitotic spindle before the onset of anaphase. Cells have evolved a surveillance mechanism, termed the spindle/kinetochore checkpoint, which monitors the attachment of chromosomes to the spindle in mitosis (Rudner and Murray 1996; Hardwick 1998). This regulatory process was first defined at the molecular level by identification of mutations in six genes, bub1, bub2, bub3 and mad1, mad2, mad3, which resulted in the failure of cells to arrest in mitosis in the presence of the microtubule depolymerizing drug benomyl (Hoyt et al. 1991; Li and Murray 1991). Four of the MAD/BUB genes have orthologs in mammals, of which two have been shown to be required for the spindle/kinetochore checkpoint and whose gene products associate with the kinetochores of pre-anaphase chromosomes (Chen et al. 1996; Li and Benezra 1996; Taylor and McKeon 1997). Results from yeast and metazoans indicate that the kinetochore is not only the site at which the spindle/kinetochore checkpoint originates, but also the sensor for proper chromosomal attachment to the spindle (for review, see Straight 1997). Activation of this checkpoint results in inactivation of the anaphase-promoting complex (APC), a ubiquitin E3-like complex required for the initiation of anaphase (Zachariae et al. 1996; Fang et al. 1998).
Protein phosphatase type 1 (PP1) has an essential function in mitotic progression in organisms ranging from yeast to mammals. Loss-of-function mutations in Schizosaccharomyces pombe (Ishii et al. 1996), Saccharomyces cerevisiae (Hisamoto et al. 1994; MacKelvie et al. 1995; Baker et al. 1997), Aspergillus nidulans (Doonan and Morris 1989), and Drosophila (Axton et al. 1990) cause mitotic arrest. In mammalian cells, microinjection of anti-PP1 antibodies into late G2 cells also induces metaphase arrest (Fernandez et al. 1992). What is the nature of the cell cycle defect that results in mitotic arrest? PP1 could be required directly for progression through mitosis, for example, by inactivating an anaphase inhibitor or by activating the APC. Alternatively, PP1 has an activity that when absent or defective activates a checkpoint that induces G2/M arrest. In this study we show that a conditional allele, glc7-129, of the gene encoding PP1 in S. cerevisiae causes arrest by the latter mechanism.
Results
The Glc7p point of execution is in G2/M
Yeast cells containing the glc7-129 allele arrest at the nonpermissive temperature (12°C) as large-budded cells with 2N DNA content (Baker et al. 1997). However, this terminal phenotype may not reflect the cell cycle stage at which Glc7p activity is required for progression through the cell cycle. To identify the execution point of Glc7p, cells containing a single chromosomal copy of glc7-129 (YAB128) were grown at the permissive temperature (30°C) to log phase and then placed on an agarose pad containing synthetic complete (SC) medium for microscopic observation. After images were collected, the microscope slide was incubated for 20 hr at the nonpermissive temperature (12°C) before images of the same cells were collected again. Representative images of cells before and after the shift are displayed in Figure 1. Cells were classified morphologically as unbudded, small-budded, medium-budded, large-budded with a short spindle, and large-budded with an anaphase spindle (for definition of each class, see Materials and Methods). An integrated NUF2–GFP fusion (Kahana et al. 1995) in the strain allowed us to locate the spindle pole bodies (SPBs). As shown in Figure 1, all cells that were in a cell cycle stage prior to anaphase at the time of the shift arrested as large-budded cells with a short spindle. However, many cells in anaphase at the time of the shift proceeded into the next cell cycle at the nonpermissive temperature. These results indicate that Glc7p function is required at or before anaphase to promote cell cycle progression.
Figure 1.

glc7-129 point of execution is in G2/M. Images of glc7-129 NUF2-GFP (YAB128) cells logarithmically growing at 30°C were captured using DIC (left) and fluorescence (right) microscopy. The fluorescence images represent localization of the spindle pole body(s) (as detected by a Nuf2p–GFP fusion). After shift to 12°C for ∼20 hr, images were captured of the same cells. The numbers below each panel indicate the number of cells that arrest in the first cell cycle as large-budded cells with a short spindle.
The results of reciprocal shifts confirmed the point of execution of Glc7p. glc7-129 cells (YAB128) were arrested in S phase with hydroxyurea (HU) or in G2/M with nocodazole at the permissive temperature (30°C). HU and nocodazole were then washed out, and individual cells were observed microscopically to confirm cell cycle arrest before shifting to the nonpermissive temperature. After ∼20 hr at the nonpermissive temperature (12°C), the terminal arrest phenotype of individual cells was quantified. All HU-treated cells (100%) arrested in the first cell cycle as large-budded cells with a short spindle (n = 19). Of nocodazole-treated cells, 42% of the cells completed mitosis, divided, and arrested in the next cell cycle (n = 24). The remaining 58% arrested as large-budded cells with a short spindle. In contrast, wild-type cells released from HU or nocodazole arrest were found to undergo at least one cell division after 20 hr at 12°C (data not shown). Two conclusions can be drawn from these results: (1) glc7-129 cells arrest in the first cell cycle after shift to the nonpermissive temperature; and (2) the execution point of glc7- 129 lies between S phase and anaphase, most likely very near anaphase.
glc7-129 cells delay in G2/M with a short spindle
To characterize the cell cycle defect(s) of the glc7-129 mutant in more detail, time-lapse microscopy was performed at the semipermissive temperature (22°C) on glc7-129 cells expressing Nuf2p–GFP. Cells were monitored by differential interference contrast (DIC) and fluorescence microscopy at 2-min intervals to monitor cell morphology and spindle dynamics, respectively. A representative montage of images from a wild-type cell and several glc7-129 cells is presented in Figure 2A. From our analysis of cell morphology and spindle length, as judged by the distance between the two Nuf2p–GFP spots, we formed three conclusions about the cell cycle delay in glc7-129 cells. First, glc7-129 cells retain short spindles for very long periods of time. The glc7-129 cell marked with the small black arrow in Figure 2A contained a clearly defined short spindle at the 16-min time point but did not proceed through anaphase until the 160-min time point. Other glc7-129 cells in the montage maintained the short spindle for the duration of the experiment. In contrast, a wild-type cell, marked with a large arrow in Figure 2A, proceeded from a single, unseparated SPB into anaphase B within the 16-min interval of the montage. The distance between SPBs is presented graphically in Figure 2B for three wild-type and three glc7-129 cells. The GLC7 cells retained a short spindle (defined as 2 discernible SPBs no more than 2.5 μm apart) for 7 ± 2 min (n = 5) before proceeding into anaphase, whereas the glc7-129 cells retained a short spindle for 86 ± 34 min (n = 5). The average time glc7-129 cells retained short spindles is likely an underestimate because many cells had a short spindle for the entire duration of the experiments. We note that the average time that cells of our wild-type maintained short spindles is shorter than that reported previously (Yeh et al. 1995; Straight et al. 1997). Because Nuf2p–GFP accumulates on the nuclear side of the spindle (Osborne et al. 1994), our estimate of spindle length may be an underestimate of the true length. Our analysis of two-dimensional instead of three-dimensional projections will also cause underestimates of spindle length. We also must propose that the short spindle in the glc7-129 cells is longer than wild type because we can observe a short spindle in these cells. Further analysis using a probe such as GFP–tubulin will resolve differences in short spindle length. These factors make it difficult to accurately determine the length of time wild-type cells have a short spindle, but the conclusion remains the same, that glc7-129 cells retain a short spindle for excessively long times.
Figure 2.
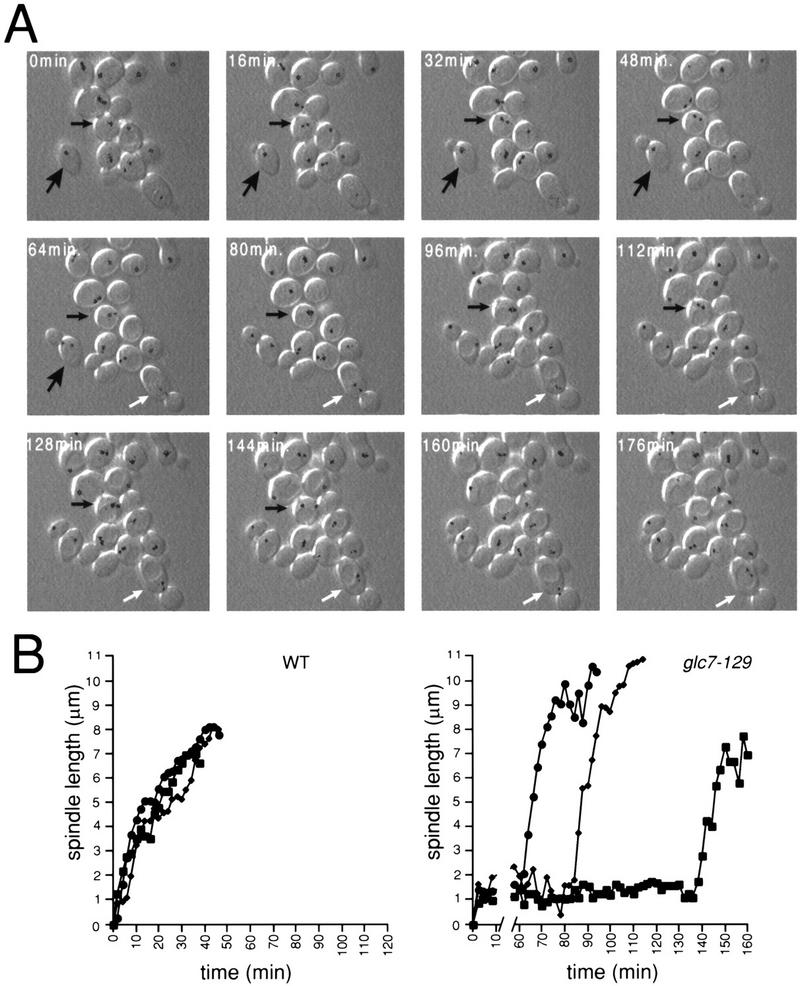
glc7-129 cells delay with a short spindle. (A) A montage of GLC7 NUF2–GFP (YAB 131) (large arrow) and glc7-129 NUF2–GFP (YAB 128) cells growing logarithmically at 30°C, using combined fluorescence and DIC time-lapse microscopy. The Nuf2p–GFP fluorescence at each spindle pole body was pseudocolored black. (Small black arrow) A glc7-129 cell that goes through anaphase; (white arrow) a glc7-129 cell arrested with a short spindle oscillating between the mother and bud. (B) The distance (μm) between spindle pole bodies was calculated for three wild-type cells (left) and three glc7-129 cells (right) at 2-min time points.
The second conclusion we can make from our analysis is that the mitotic delay is not the result of some general defect in microtubule dynamics. As in wild-type cells, the short spindle in glc7-129 cells oscillates dynamically in both orientation and position between the 2-min time points of the time-lapse experiment. For example, the spindle in the glc7-129 cell marked with the white arrow (Fig. 2A) moved from the mother cell into the bud and back into the mother cell during the course of this experiment. Once anaphase begins, the rate of spindle elongation (from ∼2 to ∼6 μm) in glc7-129 cells is more rapid (0.61 ± 0.12 μm/min, n = 5) than in wild-type cells (0.18 ± 0.04 μm/min, n = 5).
The third conclusion from our time-lapse experiments is that cytokinesis is delayed in glc7-129 cells. The time between mid-anaphase (spindle length ∼6 μm) and cytokinesis was found to be 49 ± 12 min in wild-type cells (n = 5), similar to results previously published (Yeh et al. 1995). In contrast, glc7-129 cells took much longer to complete cytokinesis. One glc7-129 cell underwent cytokinesis 70 min after mid-anaphase. Four other cells had not undergone cytokinesis at the completion of the time-lapse data acquisition (>54, 92, 116, and 124 min after mid-anaphase). Interestingly, a defect or delay in cytokinesis is caused by another allele of GLC7 (Stark 1996).
The glc7-129-mediated G2/M arrest requires the spindle/kinetochore checkpoint
To test the possibility that glc7-129-mediated G2/M arrest is due to activation of a cell cycle checkpoint, the glc7-129 strain was crossed to strains containing null alleles of mad1 and bub3, and rad9 (required for the DNA-damage checkpoint) to recover the double mutant strains mad1 glc7-129 (YAB123), bub3 glc7-129 (YAB92), and rad9 glc7-129 (YAB82). Analysis of tetrads from these crosses revealed that spore clones containing glc7-129 and either mad1 or bub3 grew more slowly than spore clones containing glc7-129 alone. In contrast, rad9 glc7-129 double mutants showed no synthetic growth defect (data not shown). The mad1 glc7-129 or bub3 glc7-129 strains exhibit DNA content (Fig. 3A) and morphology (Fig. 3B), typical of cycling cells. At the nonpermissive temperature, the glc7-129 strain had extremely elevated levels of H1 kinase activity (Fig. 3C), indicative of cells arrested in mitosis (Surana et al. 1993). The mean kinase activity in the glc7-129 strain was ∼13× higher than wild-type kinase activity (Fig. 3D). In contrast, the mad1 glc7-129 strain had wild-type levels of kinase activity, consistent with the flow cytometry data. Cells were treated with nocodazole, a microtubule-depolymerizing drug known to activate the spindle/kinetochore checkpoint. The elevated levels of H1 kinase activity observed in nocodazole-treated cells was reproducibly less than that observed for glc7-129 cells (about four times higher than wild type).
Figure 3.

DNA content, cell morphology, and H1 kinase activity indicate that the glc7-129 induced G2/M arrest requires a functional spindle checkpoint. (A) Flow cytometry for DNA content of wild-type (KT1112), mad1 (YAB127), glc7-129 (YAB27), mad1 glc7-129 (YAB123), bub3 glc7-129 (YAB92), and rad9 glc7-129 (YAB82) strains at 30°C and 12°C for 12 hr. (B) The morphological types of cells used in A were quantified microscopically, n > 200. (C) Kinase activity towards histone H1 was assayed for cells incubated at the nonpermissive temperature (12°C) for 12 hr. Labeled histone H1 was monitored using SDS-PAGE and autoradiography. (D) Labeled histone H1 was quantified using a PhosphorImager in three independent kinase assays and the average activity and standard error for each of the strains are represented.
Time-lapse microscopy was performed on mad1 glc7-129 containing NUF2–GFP (YAB152). As shown in Figure 4A, the mad1 mutation completely eliminates the anaphase delay of glc7-129 at 22°C. mad1 glc7-129 maintains a short spindle for 6 ± 1 min similar to wild-type cells (n = 5). The rate of spindle elongation in the mad1 glc7-129 strain, as in the glc7-129 strain, is actually faster than that of wild-type cells (0.33 ± 0.20 μm/min, n = 5). However, the time from mid-anaphase to cytokinesis appears longer in the double mutant compared to wild type. Two cells underwent cytokinesis 48 and 54 min after mid-anaphase. Three other cells had not undergone cytokinesis at the completion of the time-lapse data acquisition (>106, 130, and 166 min after mid-anaphase).
Figure 4.
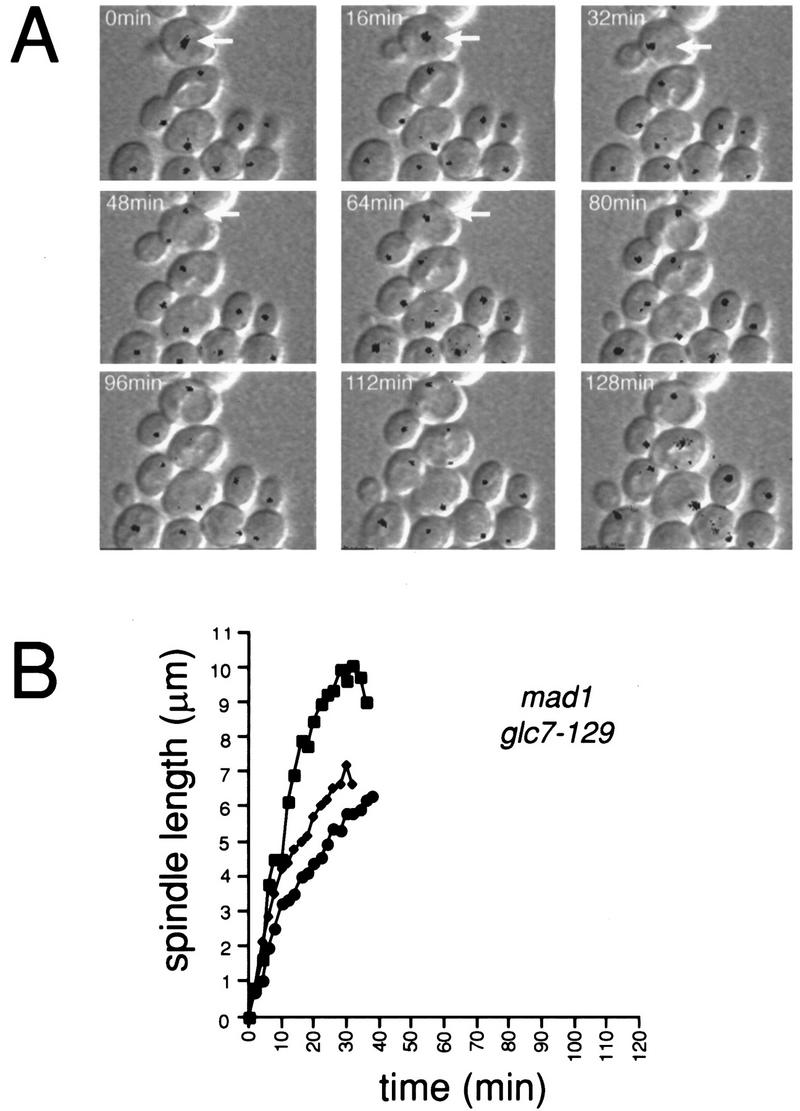
MAD1 is required for the G2/M delay of cells containing glc7-129. (A) mad1 glc7-129 NUF2–GFP (YAB152) cells were treated and analyzed by DIC and fluorescence time-lapse microscopy as in Fig. 2A. In the first panel, an arrow marks a mad1 glc7-129 cell in approximately the same stage of the cell cycle (G1/S) as the wild-type (Fig. 2A; large black arrow) and glc7-129 (Fig. 2A; small black arrow) cells. Note that by 64 min, the double mutant has proceeded into anaphase. (B) The distance (μm) between spindle pole bodies in three cells was plotted vs. time.
glc7-129 cells require a functional spindle/kinetochore checkpoint for accurate chromosome segregation and viability
To test the possibility that the slower growth rate of mad1 glc7-129 strains is due to increased rates of chromosome loss and reduced viability, we assayed the percentages of viable cells in glc7-129 and mad1 glc7-129 strains at the permissive temperature and after 12 and 24 hr at the nonpermissive temperature. As shown in Table 1, only 79% of the mad1 glc7-129 and 68% of the bub3 glc7-129 cells were viable at the permissive temperature. In contrast, the glc7-129 strain maintained high viability even after 24 hr at the nonpermissive temperature (85%). After 24 hr at the nonpermissive temperature, the viability of the mad1 glc7-129 and bub3 glc7-129 strains was greatly reduced (17% and 22%, respectively). These strains also exhibited a rate of chromosome loss 2–3 orders of magnitude higher than in wild-type cells (Table 1). In contrast, glc7-129 and mad1 strains displayed wild-type rates of chromosome loss at 30°C and rates approximately 10-fold above wild-type at 22°C. Together, these data indicate that glc7-129 cells that lack the spindle/kinetochore checkpoint exhibit a high rate of cell death due to chromosome instability.
Table 1.
Percentage viability and chromosome loss
| Straina
|
Relevant genotype
|
Percent viability
|
||||
|---|---|---|---|---|---|---|
| 30°C
|
12°C
|
Chromosome loss
|
||||
| (12 hr)
|
(24 hr)
|
30°C
|
22°C
|
|||
| KT1112/1385 | wild type | 98 | 98 | 98 | 1.8 × 10−5 | 5.5 × 10−5 |
| YAB127/144 | mad1 | 97 | 94 | 96 | 4.4 × 10−5 | 8.3 × 10−4 |
| YAB27/86 | glc7–129 | 93 | 92 | 85 | 2.0 × 10−5 | 1.7 × 10−4 |
| YAB123/148 | mad1 glc7–129 | 79 | 37 | 17 | 5.0 × 10−3 | 5.0 × 10−2 |
| YAB92 | bub3 glc7–129 | 68 | 38 | 22 | N.D. | N.D. |
| YAB82 | rad9 glc7–129 | 87 | 80 | 66 | N.D. | N.D. |
Percent viability (n = 300) and chromosome loss were determined as described in Materials and Methods (N.D.) Not determined.
Strain numbers listed first (e.g., KT1112) were haploids used for determining percent viability; second strain numbers (e.g., 1385) were homozygous diploids used for determining chromosome loss.
Discussion
We show here that cells containing the recessive cold-sensitive allele glc7-129 delay in mitosis because of activation of the spindle/kinetochore checkpoint. Sassoon et al. (1999) have likewise found that the temperature-sensitive allele glc7-10 activates the checkpoint. The fact that loss of PP1 function in other organisms also causes mitotic arrest suggests that the role of PP1 and, thus, the consequences of its deficiency, might be highly conserved. Precedence for the conservation of PP1 function is found in glycogen metabolism where PP1 holoenzymes, composed of the PP1 catalytic subunit and a glycogen-targeting subunit, is thought to dephosphorylate glycogen synthase in both yeast (Stark 1996) and mammals (Hubbard and Cohen 1993).
The specificity of PP1 is thought to be regulated by subunits that target PP1 to its substrate and/or regulate its activity (Hubbard and Cohen 1993). Studies in yeast indicate that the physiological defects of a number of glc7 mutants are caused by the failure of the variant Glc7 proteins to associate with a specific targeting subunit rather than to defects in catalytic activity (Stuart et al. 1994; Tu and Carlson 1995). The allele described in this work, glc7-129, contains two point mutations altering residues that lie on the outer surface of the protein (D137A, E138A) (Baker et al. 1997). glc7-129 mutants have defects in glycogen biosynthesis, sporulation, as well as in mitosis but have a normal glucose response (Baker et al. 1997). Consistent with its glycogen biosynthesis defect, Glc7-129p does fail to bind Gac1p (D.L. Frederick and K. Tatchell, in prep.). It is, therefore, possible that the defect responsible for the mitotic arrest of glc7-129 is caused by a specific defect in its interaction with a mitosis-specific regulatory subunit rather than a general defect in catalytic activity. However, the nature of the Glc7p holoenzyme required for the mitotic role is unknown to us as yet.
Assuming that the primary event monitored by the spindle/kinetochore checkpoint is the attachment of kinetochores to the spindle, we envision two potential roles for PP1 in this process. First, PP1 could dephosphorylate components of the mitotic spindle and the failure to do so could cause changes in spindle dynamics that activate the checkpoint. Supporting this idea is the observation that introduction of PP1-specific phosphatase inhibitors into mammalian cells causes defects in spindle dynamics (Tournebize et al. 1997). However, our results do not support a change in spindle dynamics as a cause of the mitotic defect in glc7-129 cells. We detected no obvious defects in microtubule morphology in glc7-129 cells. Also, unlike mutants known to affect microtubule dynamics, we have observed that glc7-129 cells are not hypersensitive to the microtubule-depolymerizing drug benomyl. Finally, the dynamically oscillating short spindle and apparently normal anaphase in glc7-129 cells argue against a general role in spindle dynamics.
The second possibility is that Glc7p must dephosphorylate kinetochore proteins to allow spindle attachment. The phosphorylation state of kinetochore proteins may affect their binding to CEN DNA and/or their competence to bind microtubules. Two essential components of the S. cerevisiae kinetochore, Cbf2p/Ndc10p and Ctf13p, have been shown to be phosphorylated in vitro or as recombinant proteins in insect cells, respectively (Jiang et al. 1995; Kaplan et al. 1997). Also, treatment of yeast kinetochore proteins with phosphatase in vitro prevents binding to centromere sequences (Lechner and Carbon 1991). Therefore, the phosphorylation state of kinetochore complexes may affect CEN DNA binding. Using the transcription readthrough assay (Perier and Carbon 1992), an in vivo assay that presumably measures kinetochore protein complex formation and/or structure on CEN DNA, we did not observe significant differences in kinetochore/centromere complex formation and/or structure in wild-type and glc7-129 strains (data not shown). This corroborates the results of Sassoon et al. (1999), who found that centromere binding activity is normal in extracts prepared from glc7-10 strains. Thus, defects in Glc7p do not appear to alter kinetochore complex formation and structure.
If glc7 mutants that activate the spindle/kinetochore checkpoint have normal kinetochore/CEN DNA complex formation and structure but lose chromosomes at a high rate in the absence of the checkpoint, then the most likely defect is the binding of kinetochores to microtubules. This scenario is consistent with the point-of-execution experiments placing Glc7p function at G2/M just prior to anaphase and is directly supported by the results of Sassoon et al. (1999) that extracts prepared from glc7-10 cells are deficient in microtubule binding and that addition of recombinant Cbf2p/Ndc10p can restore this activity. It remains to be seen if Cbf2p/Ndc10p is a substrate of Glc7p in vivo.
Materials and methods
Yeast strains, media, and general methods
The yeast strains, with the exception of KT81 (MATa hom3) and KT82 (matα hom3 ilv1), are congenic to strain KT1112 (MATα ura-52 leu2 his3) (Stuart et al. 1994). To integrate the glc7-129 allele into the yeast genome at the GLC7 locus, a BglII–KpnI fragment from pNC160–glc7-129 (Baker et al. 1997) was inserted into pRS306 (Sikorski and Hieter 1989) at the BamHI and KpnI sites, which results in an amino-terminal truncation of Glc7-129p. The resulting plasmid was linearized within glc7-129 with SalI and transformed into diploid strain KT1112/1113. Ura+ transformants were sporulated, and MATa and MATα Ura+ haploids were recovered. The Ura+ haploids arrested in mitosis at 12°C and failed to accumulate glycogen, two traits observed previously from the plasmid-borne glc7-129 mutant. The URA3 locus in these strains showed tight genetic linkage to the GLC7 locus. The cold-sensitive Ura+ haploids were then grown on 5-fluoro-orotic acid (5-FOA) media to select for cells that had lost the URA3 gene through recombination. A resulting Ura−, cold-sensitive 5-FOA revertant was designated strain YAB27. The NUF2–GFP gene fusion on plasmid pJK67 (Kahana et al. 1995) was linearized with BstE1 and integrated into the chromosomal NUF2 locus of strains KT1112, YAB27, and YAB123. Loss of viability was determined as described (Wang and Burke 1995). Inviability was scored as the inability of cells to undergo more than three divisions (i.e., ≤eight cells) within 24 hr after plating cells at the permissive temperature. At least 300 cells were scored for each strain.
Point-of-execution experiments
Yeast strain YAB128 was grown to log phase at 30°C, and cells were placed on a slide with a thin 2% agarose pad in SC medium. DIC and fluorescent images were captured from several fields of cells. Individual cells were classified as unbudded, small bud (bud size <0.3× mother cell diameter), medium bud (bud size 0.3×–0.6× mother cell diameter), large bud (bud size >0.6× mother cell diameter) with short spindle (<2 μm), and large bud (bud size >0.6× mother cell diameter) with anaphase spindle (>2 μm). The slide was then incubated at 12°C for ∼20 hr and images of the same fields were captured again.
Logarithmically growing YAB128 cells at 30°C were arrested in S phase or G2/M by incubating for 3 hr in YPD containing 0.1 m HU or 15 μg/ml nocodazole, respectively, HU or nocodazole was then washed out, images of cells were captured immediately, and cells were then shifted to 12°C for ∼20 hr. Cells were scored for their terminal phenotype as above. As a control in the preceding experiments, wild-type GLC7 cells, upon release from arrest, were found to undergo at least one cell division after 20 hr at 12°C.
Analysis of chromosome III mis-segregation
MATa/α diploids can become mating competent through either the loss of one copy of chromosome III, resulting in hemizygosity, or mitotic recombination, resulting in homozygosity for the MAT locus. To quantitate the mating efficiency of MATa/α diploids, single colonies from each strain were inoculated into 5 ml of liquid YPD and grown to early log phase (∼1 × 107 cells/ml) at 30°C or 22°C. Cells (5 × 106) from each strain were then mixed with 2 × 107 logarithmically growing haploid cells KT81 (MATa) and KT82 (MATα), filtered onto a 25-mm × 0.45 μm filter, and incubated on a YPD plate at 30°C for 3.5 hr. The cells were then washed off the filters and plated onto minimal media to select for triploids. After 2 days, colonies were counted and used to calculate the rate of chromosome III mis-segregation according to the following equation: No. of colonies from MATa mating + no. of colonies from MATα mating/2 divided by 5 × 106.
Microscopic analysis
Images were collected through an Olympus UPlanFl 100×/1.3 NA objective with a Photometrics CH250/A cooled CCD camera (KAF1400 Kodak chip) using Scanalytics IPLab Spectrum (Fairfax, VA) software. To collect time-lapse images of cells containing NUF2–GFP, cells were grown to mid-log phase in YPD, harvested by centrifugation, and placed on microscope slides on a pad of 2% agarose in SC medium as described (Waddle et al. 1996). Images of living cells were collected using a DAGE VE1000 SIT camera and DSP2000 signal processor. At each time interval, a two-dimensional projection of eight fluorescent images in different Z-axis planes was acquired using a Chroma (Brattleboro, VT) 41001 filter set. A separate DIC image was captured from a central Z-axis plane. A custom script written for IPLab Spectrum software was used to acquire both fluorescence and DIC images. For time-lapse microscopy of wild-type and glc7-129 strains shown in Figure 2, the wild-type cells were distinguished from the glc7-129 cells by briefly staining the wild-type cells with the chitin-specific stain Calcofluor at 1 μg/ml and then imaging the field of cells with UV light using a DAPI filter cube to distinguish wild type from glc7-129. Neither the Calcofluor staining or the brief UV light treatment altered growth rates.
Protein kinase assay
Whole-cell extracts were made as described by Stuart et al. (1994) and protein concentrations determined by the Bradford assay (Bradford 1976) using BSA as a standard. The protein kinase assays were performed essentially as described by Hisamoto et al. (1994) except that 100 μg of total protein from each of the strains was incubated with 5 μg of suc1–Sepharose (Upstate New York Biotechnology) that was diluted to a volume of 100 μl in lysis buffer (50 mm Tris-HCl at pH 7.5, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1 mm sodium PPi) containing protease inhibitors (0.5 mm PMSF and 1 μg each of chymostatin, leupeptin, antipain, and pepstatin A/1 ml).
Flow cytometry
Cells were processed for flow cytometry as described (Baker et al. 1997). Quantitation of morphology of the cells used for flow cytometry was performed using DIC microscopy. The position of the nucleus was inferred from the location of DNA stained with propidium iodide. For each strain, n > 300.
Acknowledgments
We thank Jim Waddle for help with our microscopic analysis; Jason Kahana and Pam Silver for the gracious gift of the NUF2–GFP fusion; Dan Burke for yeast strains and helpful discussion; Lucy Robinson, Eric Aamodt, and Eric First for critically reading our manuscript; Francois Perier and John Carbon for plasmids; and Ingrid Sassoon and Tony Hyman for sharing their data. This work was supported by National Institutes of Health grant GM-477899.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL ktatch@mail.sh.lsumc.edu; FAX (318) 675-5180.
References
- Axton JM, Dombradi V, Cohen PTW, Glover DM. One of the protein phosphatase 1 isoenzymes in Drosophila is essential for mitosis. Cell. 1990;63:33–46. doi: 10.1016/0092-8674(90)90286-n. [DOI] [PubMed] [Google Scholar]
- Baker SH, Frederick DL, Bloecher A, Tatchell K. Alanine scanning mutagenesis of protein phosphatase type 1 in the yeast Saccharomyces cerevisiae. Genetics. 1997;145:615–626. doi: 10.1093/genetics/145.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Chen RH, Waters JC, Salmon ED, Murray AW. Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science. 1996;274:242–246. doi: 10.1126/science.274.5285.242. [DOI] [PubMed] [Google Scholar]
- Doonan JH, Morris NR. The bimG gene of Aspergillus nidulans, required for completion of anaphase, encodes a homolog of mammalian phosphoprotein phosphatase 1. Cell. 1989;57:987–996. doi: 10.1016/0092-8674(89)90337-1. [DOI] [PubMed] [Google Scholar]
- Fang G, Yu H, Kirschner MW. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes & Dev. 1998;12:1871–1883. doi: 10.1101/gad.12.12.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez A, Brautigan DL, Lamb NJC. Protein phosphatase type 1 in mammalian cell mitosis: Chromosome localization and involvement in mitotic exit. J Cell Biol. 1992;116:1421–1430. doi: 10.1083/jcb.116.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbsky GJ, Ricketts WA. Differential expression of a phosphoepitope at the kinetochores of moving chromosomes. J Cell Biol. 1993;122:1311–1321. doi: 10.1083/jcb.122.6.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick KG. The spindle checkpoint. Trends Genet. 1998;14:1–4. doi: 10.1016/S0168-9525(97)01340-1. [DOI] [PubMed] [Google Scholar]
- Hisamoto N, Sugimoto K, Matsumoto K. The Glc7 type 1 protein phosphatase of Saccharomyces cerevisiae is required for cell cycle progression in G2/M. Mol Cell Biol. 1994;14:3158–3165. doi: 10.1128/mcb.14.5.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt MA, Totis L, Roberts BT. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 1991;66:507–517. doi: 10.1016/0092-8674(81)90014-3. [DOI] [PubMed] [Google Scholar]
- Hubbard MJ, Cohen P. On target with a new mechanism for the regulation of protein phosphorylation. Trends Biochem Sci. 1993;18:172–177. doi: 10.1016/0968-0004(93)90109-z. [DOI] [PubMed] [Google Scholar]
- Ishii K, Kumada K, Toda T, Yanagida M. Requirement for PP1 phosphatase and 20S cyclosome/APC for the onset of anaphase is lessened by the dosage increase of a novel gene sds23+ EMBO J. 1996;15:6629–6640. [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Lim MY, Yoon HJ, Thorner J, Martin GS, Carbon J. Overexpression of the yeast MCK1 protein kinase suppresses conditional mutations in centromere-binding protein genes CBF2 and CBF5. Mol Gen Genet. 1995;246:360–366. doi: 10.1007/BF00288609. [DOI] [PubMed] [Google Scholar]
- Kahana JA, Schnapp BJ, Silver PA. Kinetics of spindle pole body separation in budding yeast. Proc Natl Acad Sci. 1995;92:9707–9711. doi: 10.1073/pnas.92.21.9707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan KB, Hyman AA, Sorger PK. Regulating the yeast kinetochore by ubiquitin-dependent degradation and Skp1p-mediated phosphorylation. Cell. 1997;91:491–500. doi: 10.1016/s0092-8674(00)80435-3. [DOI] [PubMed] [Google Scholar]
- Lechner J, Carbon J. A 240 kd multisubunit protein complex, CBF3, is a major component of the budding yeast centromere. Cell. 1991;64:717–725. doi: 10.1016/0092-8674(91)90501-o. [DOI] [PubMed] [Google Scholar]
- Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell. 1991;66:519–531. doi: 10.1016/0092-8674(81)90015-5. [DOI] [PubMed] [Google Scholar]
- Li Y, Benezra R. Identification of a human mitotic checkpoint gene: hsMAD2. Science. 1996;274:246–248. doi: 10.1126/science.274.5285.246. [DOI] [PubMed] [Google Scholar]
- MacKelvie SH, Andrews PD, Stark MJR. The Saccharomyces cerevisiae gene SDS22 encodes a potential regulator of the mitotic function of yeast type 1 protein phosphatase. Mol Cell Biol. 1995;15:3777–3785. doi: 10.1128/mcb.15.7.3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne MA, Schlenstedt G, Jinks T, Silver PA. Nuf2, a spindle pole body-associated protein required for nuclear division in yeast. J Cell Biol. 1994;125:853–866. doi: 10.1083/jcb.125.4.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangilinan F, Spencer F. Abnormal kinetochore structure activates the spindle assembly checkpoint in budding yeast. Mol Biol Cell. 1996;7:1195–1208. doi: 10.1091/mbc.7.8.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perier F, Carbon J. A colony color assay for Saccharomyces cerevisiae mutants defective in kinetochore structure and function. Genetics. 1992;132:39–51. doi: 10.1093/genetics/132.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudner AD, Murray AW. The spindle assembly checkpoint. Curr Opin Cell Biol. 1996;8:773–780. doi: 10.1016/s0955-0674(96)80077-9. [DOI] [PubMed] [Google Scholar]
- Sassoon, I., F.F. Severin, P.A. Andrews, M. Taba, K.B. Kaplan, A.J. Ashford, M.J.R. Stark, P.K. Sorger, and A.A. Hyman. 1999. Regulation of yeast Saccharomyces cerevisiae kinetochores by the type 1 phosphatase Glc7p. Genes & Dev. (this issue). [DOI] [PMC free article] [PubMed]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark MJR. Yeast protein serine/threonine phsophatases: Multiple roles and diverse regulation. Yeast. 1996;12:1647–1675. doi: 10.1002/(SICI)1097-0061(199612)12:16%3C1647::AID-YEA71%3E3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Straight AF. Cell cycle: Checkpoint proteins and kinetochores. Curr Biol. 1997;7:613–616. doi: 10.1016/s0960-9822(06)00315-0. [DOI] [PubMed] [Google Scholar]
- Straight AF, Marshall WF, Sedat JW, Murray AW. Mitosis in living budding yeast: Anaphase A but no metaphase plate. Science. 1997;277:574–578. doi: 10.1126/science.277.5325.574. [DOI] [PubMed] [Google Scholar]
- Stuart JS, Frederick DL, Varner CM, Tatchell K. The mutant type 1 protein phosphatase encoded by glc7-1 from Saccharomyces cerevisiae fails to interact productively with the GAC1-encoded regulatory subunit. Mol Cell Biol. 1994;14:896–905. doi: 10.1128/mcb.14.2.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surana U, Amon A, Dowzer C, McGrew J, Byers B, Nasmyth K. Destruction of the CDC28/CLB mitotic kinase is not required for the metaphase to anaphase transition in budding yeast. EMBO J. 1993;12:1969–1978. doi: 10.1002/j.1460-2075.1993.tb05846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, McKeon F. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 1997;89:727–735. doi: 10.1016/s0092-8674(00)80255-x. [DOI] [PubMed] [Google Scholar]
- Tournebize R, Andersen SS, Verde F, Doree M, Karsenti E, Hyman AA. Distinct roles of PP1 and PP2A-like phosphatases in control of microtubule dynamics during mitosis. EMBO J. 1997;16:5537–5549. doi: 10.1093/emboj/16.18.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu J, Carlson M. REG1 binds to protein phosphatase type 1 and regulates glucose repression in Saccharomyces cerevisiae. EMBO J. 1995;14:5939–5946. doi: 10.1002/j.1460-2075.1995.tb00282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddle JA, Karpova TS, Waterston RH, Cooper JA. Movement of cortical actin patches in yeast. J Cell Biol. 1996;132:861–870. doi: 10.1083/jcb.132.5.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Burke DJ. Checkpoint genes required to delay cell division in response to nocodazole respond to impaired kinetochore function in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:6838–6844. doi: 10.1128/mcb.15.12.6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E, Skibbens RV, Cheng JW, Salmon ED, Bloom K. Spindle dynamics and cell cycle regulation of dynein in the budding yeast, Saccharomyces cerevisae. J Cell Biol. 1995;130:687–700. doi: 10.1083/jcb.130.3.687. [DOI] [PMC free article] [PubMed] [Google Scholar]