

Abstract
Under the conditions of ruthenium catalyzed transfer hydrogenation, 2-butyne couples to benzylic and aliphatic alcohols 1a–1i to furnish allylic alcohols 2a–2i, constituting a direct C-H vinylation of alcohols employing alkynes as vinyl donors. Under related transfer hydrogenation conditions employing formic acid as terminal reductant, 2-butyne couples to aldehydes 4a, 4b, and 4e to furnish identical products of carbonyl vinylation 2a, 2b, and 2e. Thus, carbonyl vinylation is achieved from the alcohol or the aldehyde oxidation level in the absence of any stoichiometric metallic reagents. Nonsymmetric alkynes 6a–6c couple efficiently to aldehyde 4b to provide allylic alcohols 2m–2o as single regioisomers. Acetylenic aldehyde 7a engages in efficient intramolecular coupling to deliver cyclic allylic alcohol 8a.
Carbonyl vinylation is a convergent protocol for the preparation of allylic alcohols. Following the seminal work of Oguni (1984) and Noyori (1986),1 enantioselective catalytic addition of vinylzinc reagents to aldehydes were reported by Oppolzer (1992) and Wipf (1994).2,3,4 Although such transformations exhibit high stereoselectivity, vinylzinc generation relies upon stoichiometric alkyne hydrometallation (R2BH or Cp2ZrHCl) with subsequent transmetallation to zinc using ZnMe2. Thus, alkyne activation requires successive use of four stoichiometric organometallic reagents (Scheme 1).
Scheme 1.
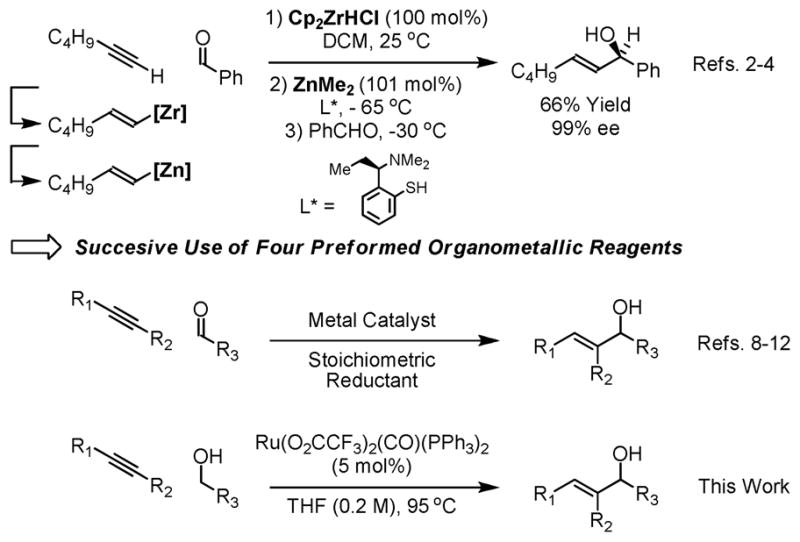
Selected milestones in carbonyl vinylation.
Direct metal catalyzed alkyne-carbonyl reductive coupling bypasses the use of multiple stoichiometric organometallic reagents. This reactivity pattern was first observed in cyclizations of acetylenic aldehydes catalyzed by rhodium, titanium and nickel, as reported by Ojima (1994),5 Crowe (1995)6 and Montgomery (1997),7 respectively. Intermolecular variants of the nickel catalyzed reactions soon followed.8,9 However, while reductive couplings of this type signal a departure from stoichiometric organometallics, they employ reductants that generate molar equivalents of metallic byproducts.
Completely atom economical alkyne-carbonyl and imine-carbonyl reductive couplings are achieved under the conditions rhodium and iridium catalyzed hydrogenation.10,11,12 This concept was extended to C-C bond forming transfer hydrogenation, wherein hydrogen embedded within an alcoholic reactant, typically isopropanol, serves as terminal reductant.13,14 Most significantly, an alcohol may serve dually as hydrogen donor and precursor to the carbonyl electrophile, enabling byproduct-free carbonyl addition from the alcohol oxidation level.10d,13,14a,c,d,15
Under the conditions of ruthenium catalyzed transfer hydrogenation employing RuHCl(CO)(PPh3)3 as catalyst, carbonyl allylation and propargylation are achieved from the alcohol or aldehyde oxidation level using conjugated dienes and enynes as surrogates to preformed allyl and allenyl metal reagents, respectively.14a,c Here, we report the first direct C-H vinylation of alcohols, which is achieved by way of alkyne-alcohol C-C bond-forming transfer hydrogenation employing Ru(O2CCF3)2(CO)(PPh3)2 as catalyst.
Recently, we disclosed a method for carbonyl propargylation from the alcohol or aldehyde oxidation level via enyne-carbonyl transfer hydrogenative coupling employing RuHCl(CO)(PPh3)3 as catalyst (eqn. 1).14c In subsequent studies, it was found that the regiochemistry of C-C coupling is altered the upon the use of Ru(O2CCF3)2(CO)(PPh3)2 as catalyst in the absence of added ligand (eqn. 2). Interestingly, both regioselectivities differ from those observed under the conditions of rhodium12 or nickel catalysis,16 wherein coupling at the acetylenic terminus of the enyne is observed.
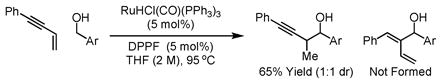 |
(eq. 1) |
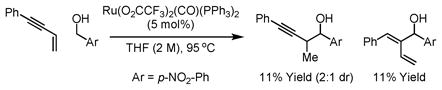 |
(eq. 2) |
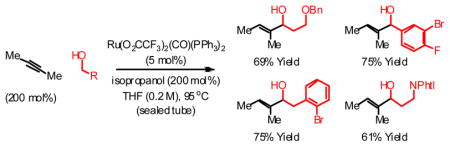
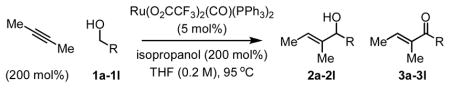
These results suggested the feasibility of using non-conjugated alkynes in transfer hydrogenative C-C coupling, which would constitute a direct C-H vinylation of alcohols employing alkynes as vinyl donors. After extensive optimization, it was found that 2-butyne (200 mol%) and p-nitrobenzyl alcohol 1b (100 mol%) combine to form the desired product of C-H vinylation, allylic alcohol 2b, in 78% isolated yield simply upon heating in THF solvent at 95 °C (sealed tube) in the presence of Ru(O2CCF3)2(CO)(PPh3)2 (5 mol%) and isopropanol (200 mol%). Enone 3b also forms in 12% isolated yield. Under these conditions, diverse benzylic and aliphatic alcohols 1a–1l are converted to the corresponding allylic alcohols 2a–2l, accompanied by variable quantities of the corresponding enones 3a–3l (Table 1). Added isopropanol (200 mol%) was found to minimize formation of enones 3a–3l.
Table 1.
Allylic alcohols 2a–2l via ruthenium catalyzed transfer hydrogenative coupling of butyne to alcohols 1a–1l.a
 | |||||
|---|---|---|---|---|---|
| Entry | Alcohol | Product | R | Time (h) | Yield 2 (3) |
| 1 | 1a | 2a (3a) | Ph | 9 | 72% (4%)b |
| 2 | 1b | 2b (3b) | p-NO2-Ph | 13 | 78% (12%) |
| 3 | 1c | 2c (3c) | p-Br-Ph | 13 | 81% (7%) |
| 4 | 1d | 2d (3d) | p-CO2Me-Ph | 13 | 81% (10%) |
| 5 | 1e | 2e (3e) | m-MeO-Ph | 13 | 78% (6%) |
| 6 | 1f | 2f (3f) | m-F-Ph | 13 | 79% (11%) |
| 7 | 1g | 2g (3g) | 3,5-Cl2-Ph | 13 | 76% (14%) |
| 8 | 1h | 2h (3h) | 3-Br, 4-F-Ph | 9 | 75% (< 1%) |
| 9 | 1i | 2i (3i) | (CH2)2OBn | 13 | 69% (< 1%) |
| 10 | 1j | 2j (3j) | (CH2)3OBn | 18 | 65% (< 1%) |
| 11 | 1k | 2k (3k) | (CH2)2NPhtl | 18 | 61% (< 1%) |
| 12 | 1l | 2l (3l) | CH2(o-Br-Ph) | 13 | 75% (< 1%)b |
Cited yields are of material isolated by silica gel chromatography and refer to pure 2a–2l free of any enone byproduct.
The reaction was conducted at 0.6 M concentration.
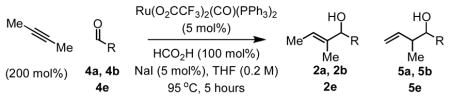
Carbonyl vinylation from the aldehyde oxidation level also was explored. Using isopropanol as terminal reductant, low conversion was observed. However, in reactions mediated by formic acid (100 mol%), aldehydes 4a, 4b and 4e were converted to allylic alcohols 2a, 2b and 2e in good yield, accompanied by products of olefin isomerization 5a, 5b and 5e. Here, sodium iodide (5 mol%) was found to suppress over-oxidation leading to enone side-products (Table 2).
Table 2.
Ruthenium catalyzed transfer hydrogenative coupling of butyne to aldehydes 4a, 4b and 4e.a
 | ||||
|---|---|---|---|---|
| Entry | Aldehyde | Product | R | Yield (2:5) |
| 1 | 4a | 2a (5a) | Ph | 88% (5:1) |
| 2 | 4b | 2b (5b) | p-NO2-Ph | 78% (10:1) |
| 3 | 4e | 2e (5e) | m-MeO-Ph | 91% (7:1) |
See supporting information for detailed procedures.
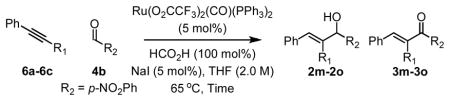
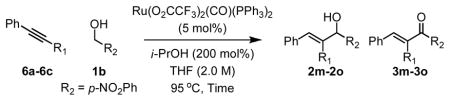
The coupling of nonsymmetric alkynes 6a–6c also was explored from the aldehyde oxidation level employing aldehyde 4b. Using formic acid as reductant, efficient vinylation occurs to provide allylic alcohols 2m–2o as single regioisomers. Over-oxidation of 2m–2o to form enones 3m–3o was not observed. Under the standard conditions cited in Table 1, the coupling of nonsymmetric alkynes 6a–6c to p-nitrobenzyl alcohol 1b to form allylic alcohols 2m–2o was less efficient (Table 2). Finally, whereas cyclization of acetylenic alcohols failed, the reductive cyclization of acetylenic aldehyde 7a proceeds efficiently to deliver 8a in 84% isolated yield (eqn. 1).
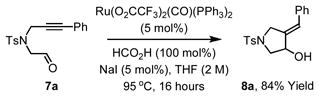 |
(eqn. 1) |
In summary, through C-C bond forming transfer hydrogenation, direct vinylation of alcohols or aldehydes is achieved using alkynes as vinyl donors in the absence of any stoichiometric metallic reagents. Future studies will focus on the development of improved second generation catalysts for the transformations reported herein and related alcohol-unsaturate C-C couplings.
Supplementary Material
Table 3.
Ruthenium catalyzed transfer hydrogenative coupling of alkynes 6a–6c to aldehyde 4b (top) and alcohol 1b (bottom).a
 | ||||
|---|---|---|---|---|
| Entry | Alkyne (200 mol%) | Product | Time (hr) | Yield (2:3) |
| 1 | 6a, R1 = Ph | 2m (3m) | 24 hr | 91% (>20:1) |
| 2 | 6b, R1 = (CH2)2OBn | 2n (3n) | 16 hr | 84% (>20:1) |
| 3 | 6c, R1 = CH2NHBoc | 2o (3o) | 13 hr | 75% (>20:1) |
 | ||||
|---|---|---|---|---|
| Entry | Alkyne (200 mol%) | Product | Time (hr) | Yield 2 (3) |
| 1 | 6a, R1 = Ph | 2m (3m) | 37 hr | 62% (12%) |
| 2 | 6b, R1 = (CH2)2OBn | 2n (3n) | 13 hr | 58% (>1%) |
| 3 | 6c, R1 = CH2NHBoc | 2o (3o) | 13 hr | 15% (>1%) |
See supporting information for detailed procedures. Isolated yields refer to pure 2m–2o free of any enone byproduct.
Acknowledgments
Acknowledgment is made to the NIH (RO1-GM069445) and the ACS-GCI Pharmaceutical Roundtable.
Footnotes
Supporting information available: Experimental procedures and spectral data for new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.(a) Oguni N, Omi T. Tetrahedron Lett. 1984;25:2823. [Google Scholar]; b) Kitamura M, Suga S, Kawai K, Noyori R. J Am Chem Soc. 1986;108:6071. doi: 10.1021/ja00279a083. [DOI] [PubMed] [Google Scholar]
- 2.For enantioselective catalytic addition of vinylzinc reagents to aldehydes, see: Oppolzer W, Radinov RN. Helv Chim Acta. 1992;75:170.Oppolzer W, Radinov RN. J Am Chem Soc. 1993;115:1593.Soai K, Takahashi J. Chem Soc, Perkin Trans 1. 1994:1257.Wipf P, Xu W. Tetrahedron Lett. 1994;35:5197.Oppolzer W, Radinov RN, De Brabander J. Tetrahedron Lett. 1995;36:2607.Wipf P, Ribe S. J Org Chem. 1998;63:6454.Oppolzer W, Radinov RN, El-Sayed E. J Org Chem. 2001;66:4766. doi: 10.1021/jo000463n.Dahmen S, Bräse S. Org Lett. 2001;3:4119. doi: 10.1021/ol016954r.Chen YK, Lurain AE, Walsh PJ. J Am Chem Soc. 2002;124:12225. doi: 10.1021/ja027271p.Ji JX, Qiu LQ, Yip CW, Chan ASC. J Org Chem. 2003;68:1589. doi: 10.1021/jo026551k.Lurain AE, Walsh PJ. J Am Chem Soc. 2003;125:10677. doi: 10.1021/ja035213d.Jeon SJ, Chen YK, Walsh PJ. Org Lett. 2005;7:1729. doi: 10.1021/ol050255n.Lauterwasser F, Gall J, Hoefener S, Bräse S. Adv Synth Catal. 2006;348:2068.Jeon SJ, Fisher EL, Carroll PJ, Walsh PJ. J Am Chem Soc. 2006;128:9618. doi: 10.1021/ja061973n.Salvi L, Jeon SJ, Fisher EL, Carroll PJ, Walsh PJ. J Am Chem Soc. 2007;129:16119. doi: 10.1021/ja0762285.
- 3.For reviews on catalytic enantioselective aldehyde vinylation using organozinc reagents, see: Wipf P, Kendall C. Chem Eur J. 2002;8:1778. doi: 10.1002/1521-3765(20020415)8:8<1778::aid-chem1778>3.0.co;2-h.Wipf P, Nunes RL. Tetrahedron. 2004;60:1269.
- 4.For catalytic enantioselective ketone vinylation using organozinc reagents, see: Li H, Walsh PJ. J Am Chem Soc. 2004;126:6538. doi: 10.1021/ja049206g.Li H, Walsh PJ. J Am Chem Soc. 2005;127:8355. doi: 10.1021/ja0425740.Jeon SJ, Li H, Garcia C, La Rochelle LK, Walsh PJ. J Org Chem. 2005;70:448. doi: 10.1021/jo048683e.
- 5.Ojima I, Tzamarioudaki M, Tsai CY. J Am Chem Soc. 1994;116:3643. [Google Scholar]
- 6.Crowe WE, Rachita MJ. J Am Chem Soc. 1995;117:6787.For an aligned study, see: Kablaoui NM, Buchwald SL. J Am Chem Soc. 1995;117:6785.
- 7.For intramolecular nickel catalyzed alkyne-carbonyl reductive coupling, see: Oblinger E, Montgomery J. J Am Chem Soc. 1997;119:9065.Tang XQ, Montgomery J. J Am Chem Soc. 1999;121:6098.Tang XQ, Montgomery J. J Am Chem Soc. 2000;122:6950.Knapp-Reed B, Mahandru GM, Montgomery J. J Am Chem Soc. 2005;127:13156. doi: 10.1021/ja054590i.
- 8.For intermolecular Ni-catalyzed alkyne-carbonyl reductive coupling, see: Huang WS, Chan J, Jamison TF. Org Lett. 2000;2:4221. doi: 10.1021/ol006781q.Miller KM, Huang WS, Jamison TF. J Am Chem Soc. 2003;125:3442. doi: 10.1021/ja034366y.Takai K, Sakamoto S, Isshiki T. Org Lett. 2003;5:653. doi: 10.1021/ol0272996.Mahandru GM, Liu G, Montgomery J. J Am Chem Soc. 2004;126:3698. doi: 10.1021/ja049644n.
- 9.A review of Ni-catalyzed alkyne-carbonyl reductive coupling: Montgomery J, Sormunen GJ. Top Curr Chem. 2007;279:1.
- 10.For reviews of hydrogen-mediated C-C coupling, see: Ngai MY, Kong JR, Krische MJ. J Org Chem. 2007;72:1063. doi: 10.1021/jo061895m.Skucas E, Ngai MY, Komanduri V, Krische MJ. Acc Chem Res. 2007;40:1394. doi: 10.1021/ar7001123.Shibahara F, Krische MJ. Chem Lett. 2008;37:1102. doi: 10.1246/cl.2008.1102.
- 11.For hydrogenative coupling of non-conjugated alkynes to carbonyl compounds and imines, see: Rhee JU, Krische MJ. J Am Chem Soc. 2006;128:10674. doi: 10.1021/ja0637954.Skucas E, Kong JR, Krische MJ. J Am Chem Soc. 2007;129:7242. doi: 10.1021/ja0715896.Barchuk A, Ngai MY, Krische MJ. J Am Chem Soc. 2007;129:8432. doi: 10.1021/ja073018j.Ngai MY, Barchuk A, Krische MJ. J Am Chem Soc. 2007;129:12644. doi: 10.1021/ja075438e.Han SB, Kong JR, Krische MJ. Org Lett. 2008;10:4133. doi: 10.1021/ol8018874.
- 12.For hydrogenative coupling of 1,3-enynes to carbonyl compounds and imines, see: Jang HY, Huddleston RR, Krische MJ. J Am Chem Soc. 2004;126:4664. doi: 10.1021/ja0316566.Kong JR, Cho CW, Krische MJ. J Am Chem Soc. 2005;127:11269. doi: 10.1021/ja051104i.Kong JR, Ngai MY, Krische MJ. J Am Chem Soc. 2006;128:718. doi: 10.1021/ja056474l.Komanduri V, Krische MJ. J Am Chem Soc. 2006;128:16448. doi: 10.1021/ja0673027.Hong YT, Cho CW, Skucas E, Krische MJ. Org Lett. 2007;9:3745. doi: 10.1021/ol7015548.
- 13.For Ir-catalyzed transfer hydrogenative C-C coupling, see: Bower JF, Skucas E, Patman RL, Krische MJ. J Am Chem Soc. 2007;129:15134. doi: 10.1021/ja077389b.Bower JF, Patman RL, Krische MJ. Org Lett. 2008;10:1033. doi: 10.1021/ol800159w.
- 14.For Ru-catalyzed transfer hydrogenative C-C coupling, see: Shibahara F, Bower JF, Krische MJ. J Am Chem Soc. 2008;130:6338. doi: 10.1021/ja801213x.Ngai MY, Skucas E, Krische MJ. Org Lett. 2008;10:2705. doi: 10.1021/ol800836v.Patman RL, Williams VM, Bower JF, Krische MJ. Angew Chem Int Ed. 2008;47:5220. doi: 10.1002/anie.200801359.Shibahara F, Bower JF, Krische MJ. J Am Chem Soc. 2008;130:14120. doi: 10.1021/ja805356j.
- 15.Rh-catalyzed alcohol-vinylarene C-C coupling has been described. The requirement of BF3 and trends in substrate scope suggest these processes involve alcohol dehydrogenation-reductive Prins addition: Shi L, Tu YQ, Wang M, Zhang FM, Fan CA, Zhao YM, Xia WJ. J Am Chem Soc. 2005;127:10836. doi: 10.1021/ja0528331.
- 16.Ni-catalyzed reductive coupling of 1,3-enynes to carbonyl compounds: Miller KM, Luanphaisarnnont T, Molinaro C, Jamison TF. J Am Chem Soc. 2004;126:4130. doi: 10.1021/ja0491735.Miller KM, Jamison TF. Org Lett. 2005;7:3077. doi: 10.1021/ol051075g.Miller KM, Colby EA, Woodin KS, Jamison TF. Adv Synth Catal. 2005;347:1533.Also see reference 8d.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.