

Abstract
Members of the mammalian mismatch repair protein family of MutS and MutL homologs have been implicated in postreplicative mismatch correction and chromosome interactions during meiotic recombination. Here we demonstrate that mice carrying a disruption in MutS homolog Msh5 show a meiotic defect, leading to male and female sterility. Histological and cytological examination of prophase I stages in both sexes revealed an extended zygotene stage, characterized by impaired and aberrant chromosome synapsis, that was followed by apoptotic cell death. Thus, murine Msh5 promotes synapsis of homologous chromosomes in meiotic prophase I.
Keywords: Meiosis, recombination, mismatch repair, synapsis, mouse
Only recently, it was appreciated that in yeast and mammalian cells, DNA mismatch repair proteins are crucial to the fidelity of both DNA replication and chromosome interactions during the first meiotic cell division. These proteins are classified as MutS or MutL homologs, referring to the paradigmatic Escherichia coli mutS,L postreplicative mismatch repair (MMR) system (Kolodner 1996). Bacterial MutS protein recognizes mis- or unpaired bases (mismatches) that arise by erroneous DNA replication, whereas MutL is believed to act as a bridging factor that binds to the DNA–MutS complex and is required for triggering excision and resynthesis of the error-containing DNA strand (Modrich 1991). In eukaryotes, six MutS homologs, designated MSH1–6, and at least three MutL homologs, MLH1, PMS1, and PMS2 have been identified (Kolodner 1996). MutS function is represented by a dimer composed of MSH2 and MSH6 and a dimer of MSH2 and MSH3, which have specific and redundant mismatch recognition capacities (Drummond et al. 1995; Palombo et al. 1995; Johnson et al. 1996; Marsischky et al. 1996; Genschel et al. 1998; Umar et al. 1998). The mammalian equivalent of bacterial MutL is a heterodimer of MutL homologs MLH1 and PMS2 (Li and Modrich 1995).
Germ-line defects in human mismatch repair genes were found to underly hereditary nonpolyposis colorectal cancer, a familial cancer predisposition syndrome characterized by an early onset of tumors of the gastrointestinal and genitourinary tracts (Lynch et al. 1995). Several mouse strains were generated that carry disruptions in MMR genes. Mice homozygous for loss-of-function alleles of Msh2, Msh6, Mlh1, or Pms2 were healthy at birth but appeared to be highly predisposed to tumorigenesis (De Wind et al. 1995, 1998; Reitmair et al. 1996; Edelmann et al. 1997; Prolla et al. 1998). These observations clearly corroborate the pivotal role of MMR in mutation avoidance.
Disruptions of the murine mutL homologs caused an additional phenotype: Pms2-deficient males and both male and female Mlh1-deficient mice were infertile (Baker et al. 1995, 1996; Edelmann et al. 1996). Whereas female Pms2−/− mice were fertile, male Pms2-deficient mice produced a strongly reduced number of spermatozoa that were grossly abnormal. About 80% of Pms2-deficient spermatocytes showed abnormalities in meiotic prophase I, characterized by extensive single axial element formation with either very little normal synaptonemal complex formation, incomplete synapsis, or aberrant synapsis between nonhomologous chromosomes. Pms2-deficient oocytes have not been studied in this respect (Baker et al. 1995). Mlh1-deficient male mice did not produce spermatozoa at all, and in females, ovulations were rare. In Mlh1-deficient spermatocytes, chromosome pairing and synapsis appeared normal, however, desynapsis gave rise to primarily univalent homologs that were not associated via chiasmata as in normal diplotene (Baker et al. 1996). Clearly, the two murine MutL-like proteins are implicated in proper meiosis, although they act at different stages. Pms2 appears to be involved in synapsis of homologous chromosomes early in prophase I; Mlh1 acts late in prophase I and seems to be required for the formation of crossing-over between homologous chromosomes. A similar function was recently ascribed to Saccharomyces cerevisiae MLH1 (Hunter and Borts 1997).
Loss-of-function mutations in the murine mutS homologs Msh2 and Msh6 did not cause fertility problems (De Wind et al. 1995; Edelmann et al. 1997). This suggested that another MutS homolog may serve as a partner of Pms2 and/or Mlh1 in homology search and/or crossing-over during meiotic recombination. Likely candidates for this function are the MSH4 and MSH5 proteins that have been identified in S. cerevisiae (Ross-Macdonald and Roeder 1994; Hollingsworth et al. 1995). Although both exhibit considerable sequential and structural similarity to the family of MutS-like proteins and probably act as a heterodimer (Pochart et al. 1997), MSH4 and MSH5 are not involved in DNA mismatch repair. Instead, these proteins are specific for meiosis and, similar to yeast and murine MLH1/Mlh1, appear to promote crossing-over during meiotic recombination.
To study the role of the MutS protein family in mammalian meiotic recombination, we introduced an inactivating mutation in the murine Msh5 gene and found that this caused both male and female sterility. Our data reveal a function of Msh5 early in meiosis I.
Results
Generation of Msh5-deficient mice
A cDNA fragment was obtained encoding the carboxy-terminal half of mouse Msh5 (see Materials and Methods). Comparison of the predicted amino acid sequence with MutS homologs in S. cerevisiae, identified MSH5 as its closest relative (41% identity and 63% similarity, Hollingsworth et al. 1995). Moreover, the mouse amino acid sequence showed 93% identity and 96% similarity to human MSH5 (GenBank accession no. AF034759).
Figure 1A shows Msh5 expression to be high in the mouse testis, but virtually absent in the other tissues examined. Low expression was observed in embryonic stem (ES) cells (Fig. 1D). This may indicate that mouse Msh5 has a meiosis-specific function similar to that described previously for MSH5 in S. cerevisiae (Hollingsworth et al. 1995). To investigate this possibility, mice were generated carrying a disruption in the Msh5 gene.
Figure 1.




Identification and disruption of murine Msh5. (A) Msh5 expression in mouse tissues. In each lane 2 μg of mRNA was loaded. A 560-bp Msh5 cDNA fragment was used as a probe. (B) Schematic representation of the mouse Msh5 locus and targeting construct used to replace a 2.3-kb NcoI fragment for the hygromycin-resistance marker. The positions of the external probes used for detection of homologous recombination events are indicated (1 and 2). (C) Southern blot showing the diagnostic ScaI fragment indicative of Msh5 gene disruption in Msh5+/+, Msh5+/−, Msh5−/− ES cells using probe 2. Arrows indicate the positions of the wild-type (14.7 kb) and mutant (5.5 kb) alleles. (D) Northern blot showing Msh5 RNA expression in Msh5+/+, Msh5+/−, Msh5−/− ES cells using a 560-bp Msh5 cDNA fragment as a probe, of which 211 bp correspond to sequences upstream of the NcoI deletion. In each lane 1.5 μg of mRNA was loaded. (E) RT–PCR of poly(A) RNA from Msh5+/+, Msh5+/−, Msh5−/− ES cells using primers upstream (a), within (b), and downstream (c) of the NcoI deletion. Arrows indicate amplified Msh5 sequences (Msh5, 237, 269, and 281 bp, respectively) and Hprt sequences (Hprt, 185 bp) that were coamplified as an internal control.
The mouse Msh5 cDNA sequence was used to isolate genomic DNA fragments carrying Msh5-coding sequences. These were used to generate a targeting vector, in which the putative ATPase domain of the gene (Hollingsworth et al. 1995) was replaced by a hygromycine-resistance marker (Fig. 1B). 129/OLA-derived ES cells were electroporated with the linearized targeting vector. Gancyclovir- and hygromycin-resistant ES cell clones were isolated and screened for homologous recombination events by Southern blot analysis. Thirty percent of the resistant clones were found to be heterozygous for Msh5 (Fig. 1C). A homozygous Msh5 mutant ES cell line was obtained by selecting a heterozygous line at high hygromycin concentration for duplication of the disrupted allele and loss of the wild-type allele. Northern blot analysis and RT–PCR, using primer sequences upstream, within, and downstream of the deleted Msh5 sequence, revealed that a normal RNA transcript, encoding the putative ATPase domain was absent in the homozygous Msh5 mutant line. Only residual transcripts of upstream and downstream sequences were present (Fig. 1D,E). The mutated Msh5 allele will therefore be indicated as Msh5−.
Two independent targeted ES cell clones (Msh5+/−) were used to generate chimeric mice, which transmitted the disrupted allele through the germ line. Intercrossing of Msh5 heterozygotes produced progeny in the expected Mendelian distribution, indicating that Msh5 deficiency is not associated with embryonic or neonatal lethality.
Msh5 deficiency causes infertility
Msh5-deficient mice were healthy at birth and appeared to develop normally into adulthood. However, despite normal mating behavior of Msh5−/− males and females towards wild-type animals, they failed to give rise to pregnancies, indicating male and female sterility. To determine the cause of infertility, the reproductive organs of homozygous mutant males and females, sacrificed at 2–3 months of age, were examined. The testis weight of Msh5−/− males was only 30% of normal and no sperm were present in the caput epididymis. In Msh5−/− females, ovaries appeared rudimentary and were devoid completely of follicles and oocytes.
Histological analysis of mutant testis
In testis sections of Msh5-deficient mice, the only germ cells present were spermatogonia and spermatocytes (Fig. 2A,B). Spermatogonial multiplication proceeded apparently as normal in these mice, with many spermatocytes being formed. However, tubular cross-sections with and without spermatocytes were observed. Also, tubular cross-sections were present in which the spermatocytes massively entered into apoptosis. Apoptotic cell death was confirmed by TUNEL (data not shown). In tubular cross-sections showing apoptotic spermatocytes, the spermatogonia present on the basal membrane were recognized as Intermediate (In) spermatogonia just before or carrying out their division into B spermatogonia (Fig. 2C–E). In the highly organized and rigidly timed spermatogenic process, the division of In spermatogonia into B spermatogonia takes place in epithelial stage IV (Russell et al. 1990). Hence, in the Msh5-deficient mouse, the spermatogonial compartment was apparently normal and spermatocytes were present in seemingly normal numbers up to epithelial stage IV, when they all disappeared through apoptosis. In epithelial stage IV, spermatocytes normally are in pachytene stage of the meiotic prophase. Comparing the spermatocytes in Msh5-deficient mice just before apoptosis with spermatocytes in stage IV of wild-type mice, the chromosome threads in the spermatocytes were thinner than those in wild-type mice (Fig. 2F,G). At the light microscopic level, the stage IV spermatocytes in Msh5-deficient mice had a more zygotene-like appearance rather than pachytene. In stage V, when B spermatogonia are present, all spermatocytes had disappeared (Fig. 2E). In wild-type and heterozygous mice no apoptotic spermatocytes were observed in stage IV.
Figure 2.
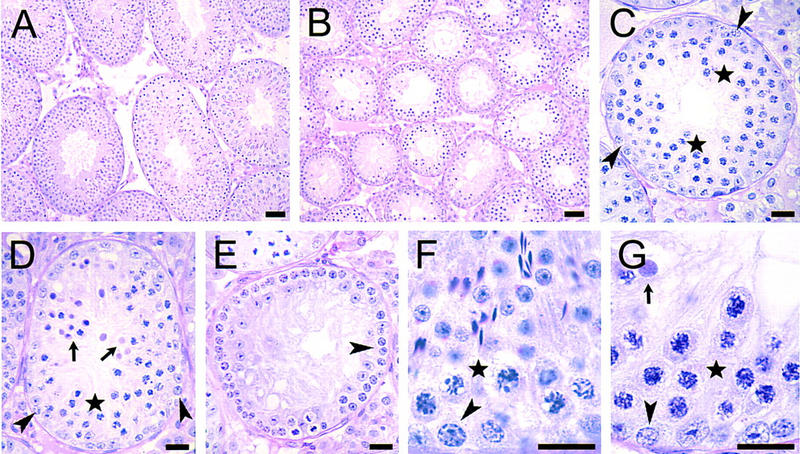
Testis histology of Msh5-deficient mice. (A) Testis of a wild-type mouse, showing normal spermatogenesis. (Magnification, 190×.) (B) Testis of an Msh5-deficient mouse. The seminiferous tubules have a much smaller diameter as spermatids, and in many tubules, spermatocytes are missing. (Magnification, 190×.) (C) Seminiferous tubular cross-section in an Msh5-deficient mouse in which spermatocytes (cells surrounding asterisks) are abundantly present. At the basal membrane A spermatogonia (arrowheads) are present. (Magnification, 230×.) (D) Similar, but a little more advanced stage in the epithelial cycle, i.e., in stage IV as the In spermatogonia (arrowheads) are just about to enter mitosis. The spermatocytes are just before (cells surrounding asterisk) or in apoptosis (arrows). (Magnification, 230×.) (E) Similar, but still more advanced, i.e. in stages V–VI as B spermatogonia (arrowhead) are present (Russell et al. 1990). Spermatocytes are completely absent now. (Magnification, 230×.) (F) Higher power photograph of early stage IV of the cycle of the seminiferous epithelium in a wild-type mouse. Here round and elongated spermatids are present, pachytene spermatocytes (e.g., cells down left and right of asterisk) and In spermatogonia (arrowhead) about to divide into B spermatogonia. (Magnification, 570×.) (G) Area in the same stage of the epithelial cycle as evidenced by the presence of In spermatogonia about to divide (arrowhead) in an Msh5-deficient mouse. Spermatids are completely missing. The spermatocytes (cells surrounding asterisk) are just about to enter apoptosis and one of them is already apoptotic (arrow). In the spermatocytes the condensed chromosomes are less thick, and have a finer appearance than in the pachytene spermatocytes in stage IV in wild type mice. (Magnification, 570×.) Bar, 20 μm.
Aberrant synaptonemal complex formation in Msh5-deficient spermatocytes
To more accurately define the meiotic defect in Msh5-deficient mice, meiocytes were analyzed for the formation of synaptonemal complexes (SCs). Preparation for SC formation starts in leptotene, when each chromosome forms an axial element that originates at the anchorage sites of the telomeric regions on the nuclear membrane and ultimately extends over its entire length (Dietrich and De Boer 1983; Goetz et al. 1984; Sherthan et al. 1996). Before completion of this process, axial elements of homologous chromosomes begin to synapse (and are now called lateral elements) and a central element is formed in between (Offenberg et al. 1991; Heyting 1996; Sherthan et al. 1996) confirming the formation of the tripartite SC. When central elements start to arise, zygotene is underway. After complete SC formation, meiotic prophase enters the pachytene stage. At subsequent desynapsis, the central element is lost and the meiocyte enters the diplotene stage. Sites of crossing-over are indicated by axial elements that cannot be pulled apart easily. At diakinesis, bivalent chromatin spiralizes and chiasmata, the sites in which crossing-over occurred, become unambiguously visible. After metaphase I when the bivalents are aligned in the meiotic spindle, actual segregation, i.e., the reduction in chromosome number follows in anaphase I. Figure 3A and B illustrate chromosome behavior during zygotene and mid-pachytene in wild-type spermatocytes as visualized with a polyclonal antibody that recognizes predominantly the Scp3 protein component of axial/lateral elements. In Figure 3A SC formation starts when the axial elements have not yet fully formed. Consistent with the histological data, the seminiferous tubules in Msh5-deficient mice completely lacked pachytene and diplotene-stage spermatocytes. Instead, two deviant types of zygotene were distinguished, zygotene I and zygotene II. In type I cells (Figs. 3C, 4C), axial element formation was often discontinuous and synapsis was largely incomplete. By subjective assessment chromosome synapsis could vary from close to zero (V-shaped contacts between axial elements) to brightly fluorescent bars indicating SC formation over up to 25% of the genome. Figure 3C shows a zygotene I in which some intensely fluorescent SC segments are clearly visible. However, a single zygotene I nucleus can contain complete bivalents together with entirely univalent chromosomes. Attachment plaques at the telomeric regions, normally only seen during late pachytene (in males) and diplotene, can start to become pronounced. Zygotene type II (Figs. 3D, 4E) is largely similar, but axial elements were always continuous and synapsis had progressed to ∼50% of the genome. Occasionally, nonhomologous synapsis (partner exchange) was observed (Fig. 3C,D).
Figure 3.
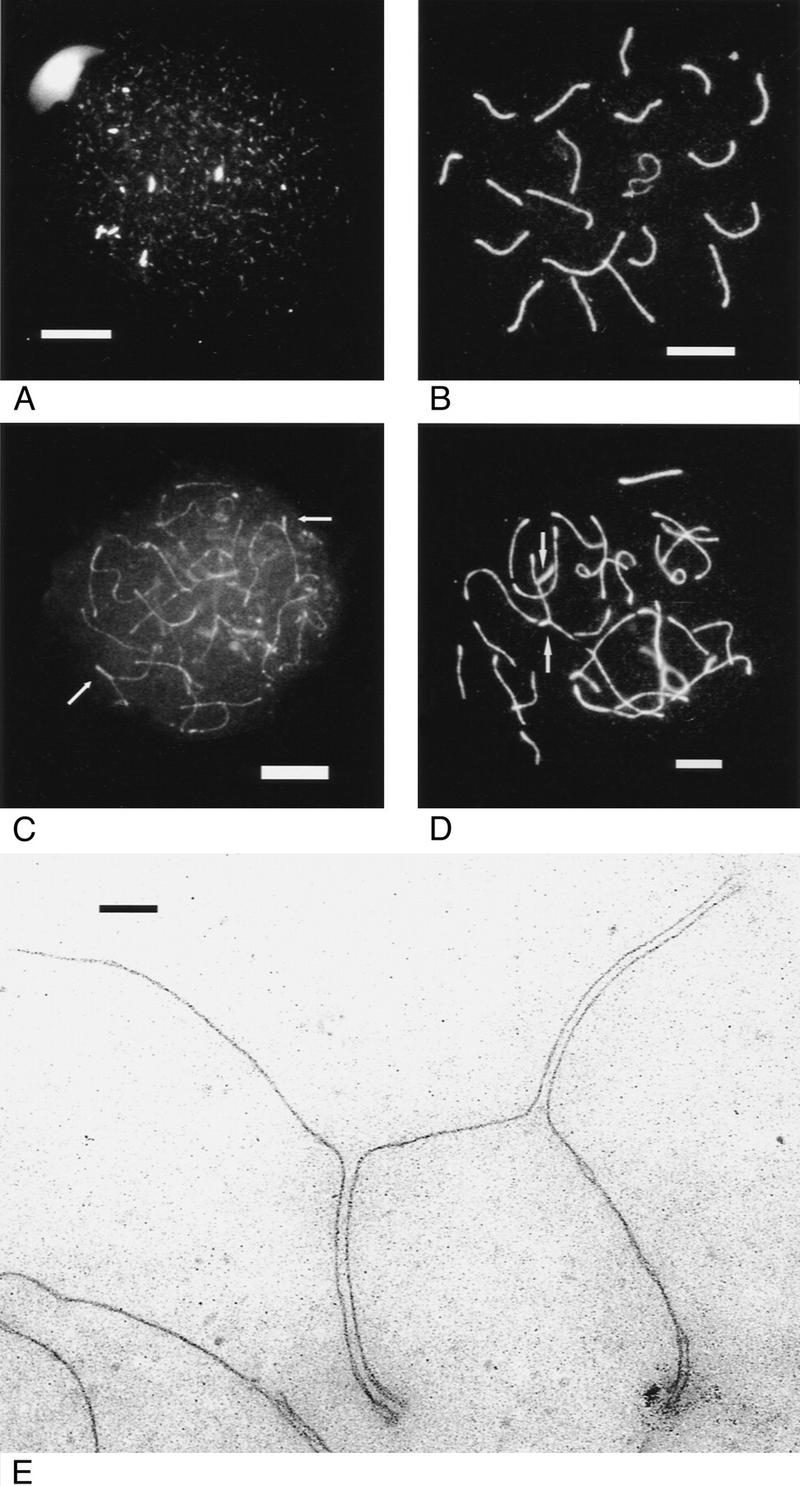
Immunofluorescence of axial/lateral SC elements of spread primary spermatocytes from wild-type and Msh5−/− males. (A) Wild-type early zygotene, bright spots indicate SC formation. (B) Wild-type mid pachytene. (C) Msh5−/− zygotene I. Arrows indicate SC formation. (D) Msh5−/− zygotene II. Arrows indicate partner exchange. Bar, 8 μm. (E) Msh5−/− zygotene I visualized by electron microscopy, showing partner exchange. Bar, 200 nm.
Figure 4.
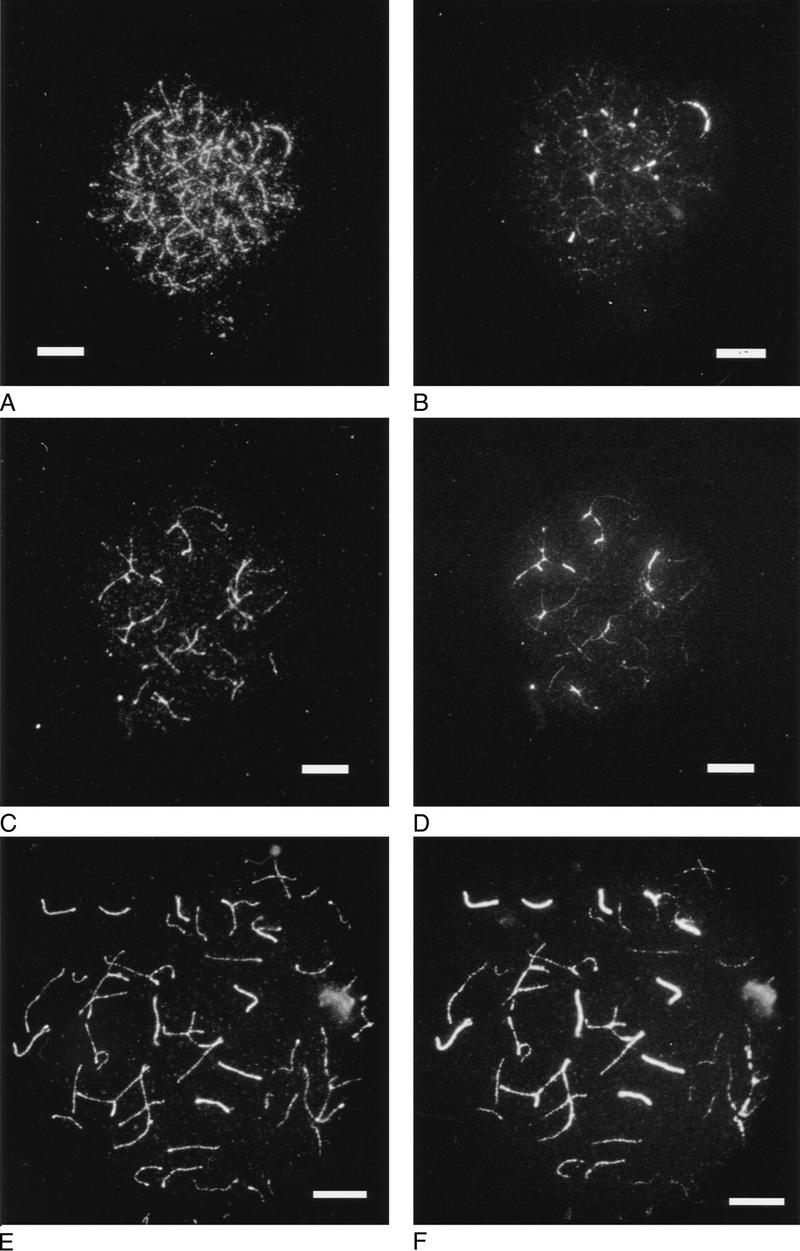
Axial/lateral element (A,C,E) and central element immunofluorescence (B,D,F) of spermatocytes from wild-type and Msh5−/− males. (A,B) Wild-type early zygotene; (C,D) Msh5−/− zygotene I; (E,F) Msh5−/− zygotene II. Bar, 8 μm.
To further characterize synapsis in type I and type II zygotene, Msh5-deficient spermatocytes were stained doubly for axial/lateral elements and the central element protein Scp1, indicative of SC formation (Fig. 4). Figure 4A and B show a wild-type early zygotene nucleus in which axial element formation is extensive and central element formation has just started. Axial/lateral and central element formation in mutant zygotene is shown in Figure 4C and D (zygotene I) and E and F (zygotene II). In the latter, complete SC formation can be seen for six bivalents. Although synapsis was never complete over the genome, the presence of anti-Scp1 activity suggests the formation of genuine SC.
To establish the width of SCs in zygotene I and II, spermatocytes from a homozygous mutant male were used for electron microscopy. SCs were largely of normal structure and could extend over the length of an entire bivalent. Regularly, the bivalents had SC formation over part of their length and demonstrated attempts to synapse. In the 50 nuclei investigated, four contained only univalents; in all other cells, at least one subchromosomal length of SC was present, next to several univalents. Univalents and unsynapsed axial elements had a thickened telomeric region, likely at the centromeric end. Most strikingly, half of the cells exhibited partner exchange, involving three to eight chromosomes (Fig. 3E).
The frequency distribution in spreaded spermatocytes of the various prophase I stages during male meiosis in wild-type and Msh5-deficient males is given in Figure 5. Data from both spreaded and fibrin-clotted spermatocyte preparations have been collected. In general, the two techniques agreed well in the classification of the first meiotic prophase stages (data not shown). Zygotene I was the dominant cell type in the mutant mice.
Figure 5.

Histogram of male first meiotic prophase stages in Msh5+/+ (█, N = 121) and Msh5−/− (□, N = 127) spreaded primary spermatocyte preparations after immunofluorescence staining of axial/lateral elements. The data of two males have been pooled per genotype.
In summary, Msh5 deficiency causes arrest of male meiosis in a zygotene stage that is characterized by limited normal SC formation and frequent synapsis between nonhomologous chromosomes.
Meiotic arrest in Msh5-deficient oocytes
Axial/lateral immunofluorescence was used to identify meiotic prophase I stages in wild-type and Msh5-deficient fetuses at days 15, 16, and 18 of gestation. Table 1 shows that in wild-type fetuses, oocytes moved gradually from zygotene/pachytene at embryonic day 15 (E15) to pachytene at E16 and diplotene at E18. In Msh5-deficient oocytes, a clear block was discernible at zygotene (Table 1). Figure 6, A and B, give examples of mutant nuclei present at E15 and E16. Note that normal female leptotene/zygotene differs from male leptotene/zygotene in that axial core formation is more extensive before SC formation takes place. In Figure 6A, complete axial element formation is shown, and possibly some SC formation but development of this nucleus was delayed as judged by the increased intensity of axial fluorescense. This pattern represents the vast majority of nuclei present at E15 and E16 (Table 1). Figure 6B shows a nucleus at E16 that had many characteristics of pachytene and extensive but still incomplete SC formation. This pattern, which was found in 10% of the nuclei at E16 and appeared somewhat more advanced than zygotene II, was designated incomplete pachytene.
Table 1.
Frequency distribution of first meiotic prophase stages in oocytes
|
Msh5+/+ (age postcoitum)
|
Msh5−/− (age postcoitum)
|
|||||
|---|---|---|---|---|---|---|
| Meiotic stage
|
15
|
16a
|
18
|
15
|
16a
|
18
|
| Leptotene | 5.6 | 0 | 2.6 | 14.5 | 4.6 | 0 |
| Zygotene I | 20.8 | 25 | 5.1 | 61.3 | 47.7 | 0 |
| Zygotene II | 32 | 11 | 0 | 24.2 | 35.4 | 0 |
| <Pachyteneb | 0 | 0 | 0 | 0 | 9.2 | 0 |
| Pachytene | 41.6 | 48.3 | 25.7 | 0 | 0 | 0 |
| Diplotene | 0 | 9.4 | 61.5 | 0 | 0 | 0 |
| N.C.c | 0 | 6.3 | 5.1 | 0 | 3.1 | 100 |
| Number of cells | 72 | 64 | 39 | 62 | 65 | 8 |
Dominant stages are shaded.
Meiocytes were spread at ages 15 and 18 postcoitum and fibrin clotted at age 16.
Incomplete pachytene.
Not categorized.
Figure 6.

Axial/lateral element immunofluorescence in female Msh5−/− nuclei at first meiotic prophase. (A) Zygotene I at E15 with complete formation of axial elements; (B) incomplete pachytene at E16 in a fibrin clot; (C) oocyte at E18 with only univalents (arrows indicate points of attraction between homologous axial elements); (D) oocyte at E18 with asynaptic/desynaptic morphology. Bar, 8 μm.
The few meiotic cells that were recovered from a homozygous mutant fetus at E18, were of an asynaptic/desynaptic nature and were labeled not categorized (Table 1; Fig. 6C,D). Occasionally, foci of attraction between axial elements were visible, suggesting recombinational activity between homologous chomosomes (Fig. 6C).
These results demonstrate that also in female meiosis, Msh5 deficiency caused an arrest at the stage of SC formation. Some cells survived to a (by real time) postpachytene stage, carrying univalent chromosomes without chiasmata.
Discussion
Mouse models carrying loss-of-function alleles of members of the eukaryotic mismatch repair gene family of MutS and MutL homologs have established a dual function of MutL homologs Mlh1 and Pms2: Both act in postreplicative mismatch repair and meiotic recombination. In contrast, no meiotic requirement was observed for Msh2 and Msh6, suggesting the involvement of another MutS homolog in meiotic recombination. A possible candidate is S. cerevisiae MSH5, which stimulated crossing-over during meiotic recombination but had no apparent function in DNA mismatch repair. To test this possibility, we disrupted the murine Msh5 gene and found severe meiotic defects that parallelled those in Pms2 and possibly Mlh1-deficient mice.
Meiotic defects in Msh5-deficient mice
S. cerevisiae msh4 and msh5 mutant strains show a meiotic defect characterized by reduced spore viability, increased levels of metaphase I nondisjunction, and decreased levels of reciprocal exchange (Ross-Macdonald and Roeder 1994; Hollingsworth et al. 1995). A similar phenotype has been described in yeast mlh1 (Hunter and Borts 1997). MSH4 and MSH5, probably functioning as a heterodimeric protein complex (Pochart et al. 1997), and MLH1 were therefore implicated in resolution of meiotic recombination intermediates, favoring crossing-over formation between homologous chromosomes in meiosis I. Whereas the meiotic mlh1 phenotype appeared to be reproducible in Mlh1-deficient mice (Baker et al. 1996), we demonstrate here that Msh5 deficiency in mice interferes with an earlier stage of meiotic prophase I, i.e., with chromosome pairing and synapsis. Although in both male and female Msh5-deficient meiocytes, large variations in the extent of chromosome synapsis were observed, SC formation was never complete, hence cytologically normal pachytene stages were never observed.
Histological analysis of the Msh5 knockout phenotype in males indicated a short death phase at the seminiferous tubule epithelial stage that is defined by division of In to B spermatogonia (stage IV, De Rooij 1998). At this stage, spermatocytes are normally in the transition from mid to late pachytene, which at the SC level correlates with the growth of attachment plaques, expansion of the sex vesicle, shortening of the XY synaptonemal complex region, and appearance of the curl in the axial element of the X chromosome. None of these features were part of the abnormal zygotene I and II morphology. Thus, whereas zygotene in normal mouse spermatogenesis only extends over 1.5–2 days (stages IX–XI), (abnormal) zygotene in Msh5-deficient mice was elongated by 4–5 days into stage IV, without any sign of progression into pachytene. We speculate that at this stage of the seminiferous epithelium a pseudo G2/M checkpoint mechanism is operational for clearing the seminiferous tubule from incompetent meiotic cells. The same checkpoint mechanism may account for spermatocyte arrest in Is(7;1)40H heterozygotes (Searle et al. 1983) and in Atm−/−, Atm−/−;p53−/−, Atm−/−;p21−/−, and Dmc1−/− mice, which all correspond histologically with the pattern seen in Msh5-deficient mice, that is, death at stage IV (Xu et al. 1996; T. Ashley, pers. comm.; and deduced from Yoshida et al. 1998). In Msh5-deficient females, meiocytes disappeared between embryonic day 16 and 18, suggesting a similar checkpoint to operate in female meiotic prophase I. The alleged pachytene checkpoint identified here appears to act earlier than the synapsis checkpoint that was postulated on the basis of, for example, chromosome X monosomy or heterozygosity for the X autosome translocation T(X;16)16H and which results in the elimination of metaphase I or late pachytene spermatocytes, respectively (Odorisio et al. 1998). The designation synapsis checkpoint used by these authors now seems somewhat unfortunate as elimination of cells occurred around the prophase I to metaphase I transition for monosomy X males and shows variegation for T16H.
The radical elimination of Msh5-deficient meiocytes with defects in SC formation, precluded to study the effect of Msh5 deficiency at later stages. However, at embryonic day 18, some homozygote mutant oocytes were recovered in which most chromosomes appeared as univalents. These patterns may either represent leptotene/zygotene stage nuclei, or may have resulted from abnormal postpachynemal desynapsis leading to univalents. Occasional foci of attraction between homologs is suggestive of the latter interpretation. The absence of chiasmata in these nuclei could have been the result of the earlier abnormalities in chromosome pairing and synapsis. This scenario, however, is unlikely to explain the phenotype in yeast msh4/5, in which apparently normal DNA-exchange interactions occurred as deduced from the normal levels of gene conversion. Also, yeast MSH4 was localized to pachytene chromosomes indicating a function that succeeded the initiation of recombination and synapsis (Ross-Macdonald and Roeder 1994; Hollingsworth et al. 1995). Furthermore, the phenotype of Mlh1-deficient mice (Baker et al. 1996; see below), also suggested cross-over formation to be a step that is distinct temporarily and mechanistically from initiation of homologous recombination. The chromosome pattern in Msh5 mutant oocytes at E18 may therefore indicate that mouse Msh5 has an additional function later in meiotic prophase I, i.e., in formation of chiasmata and cross-overs.
MutS/MutL function in mouse meiosis
The early aberrations in meiotic chromosome behavior identified in Msh5 mutant mice correspond closely to the defects observed in Pms2-deficient mice. Although Pms2 deficiency apparently had a leaky phenotype in the sense that it did not affect numbers of ovulated oocytes at the beginning of fecundity and allowed some production of (abnormal) spermatozoa, the majority of spermatocytes showed abnormalities in zygotene that were largely reminiscent of those in Msh5-deficient meiocytes. These included incomplete SC formation (in Pms2−/− somewhat less severe than in Msh5−/−) and extensive partner exchange. Although degeneration of nuclei characterized by fragmentation of axes was observed, it should be noted that a histologically defined stage of apoptotic cell death in Pms2-deficient mice was not identified (Baker et al. 1995). The abnormal chromosome behavior in Mlh1-deficient mice, that is, XY univalency in pachytene and the absence of chiasmata in diplotene was interpreted to indicate a function of Mlh1 in formation of crossing-overs. The chromosome pattern in Msh5-deficient oocytes recovered at E18 may indicate a similar function of Msh5. It is tempting to speculate that Msh5 exerts its early and (possibly) late meiotic functions as a complex with Pms2 and Mlh1, respectively, conforming to MutS/MutL interactions that are involved in DNA mismatch repair.
The phenotype of yeast msh5 does not seem to include a defect in chromosome synapsis as observed in Msh5-deficient mice. This may indicate functional divergence of yeast MSH5 and mouse Msh5 or reflect differences in yeast and mouse meiosis. For example, the enzymatic requirements for homologous chromosome synapsis in yeast may be less stringent than in mice. This is also suggested by comparison of the meiotic defects in Dmc1- or Pms2-deficient mice and dmc1 or pms1 mutant yeast strains (murine Pms2 is the homolog of yeast PMS1). In mice, Dmc1 (Pittman et al. 1998; Yoshida et al. 1998), Pms2 (Baker et al. 1995), and Msh5 deficiency cause defects in synapsis of homologous chromosomes. In yeast dmc1, chromosome synapsis, although strongly delayed, ultimately reaches wild-type levels (Rockmill et al. 1995); msh5 appears to have no effect on synapsis (although this has not been tested directly; Hollingsworth et al. 1995); and pms1 does not interfere with meiotic chromosome segregation at all (Hunter and Borts 1997). The absolute requirement for the enzymatic machinery that promotes proper homolog recognition and synapsis in mouse meiosis, may be relaxed in yeast owing to its much smaller genome size.
In summary, our results demonstrate that Msh5 function in mouse meiosis coincides with that of Pms2, that is, promotion of chromosome synapsis with possibly concomitant formation of recombination intermediates. Mlh1 is involved in resolution of these intermediates leading to crossing-over. Msh5 may also be involved in this process, but further experiments are required to ascertain this point.
Materials and methods
Identification of murine Msh5
A mouse cDNA sequence was obtained by screening a teratocarcinoma cDNA library (λZAP, Stratagene) with the human EST clone 115902. Two clones were obtained that contained an open reading frame of 570 bp. By using a 3′ RACE protocol (Frohman 1990) this sequence could be extended to 1230 bp encoding a 365-amino-acid carboxy-terminal protein sequence that showed extensive similarity to yeast and human MSH5 (GenBank accession nos. L42517 and F034759, respectively).
Generation of ES cell lines with a disrupted Msh5 allele
Two Msh5 genomic clones were isolated by screening a mouse 129/Sv λgt12 genomic library with murine Msh5 cDNA used as a probe. Restriction and hybridization mapping and subsequent sequencing revealed three exons that comprise the putative ATPase domain present in the carboxy-terminal part of MutS homologs.
A targeting vector was constructed by subcloning a 13-kb ScaI–NotI genomic fragment, and replacing a 2.3-kb NcoI fragment with the hygromycine-resistance cassette (Fig. 1B). The targeting fragment was flanked by a thymidine kinase gene cassette. ES cells (2 × 107) derived from 129/OLA mice were electroporated in the presence of 100 μg of targeting vector linearized with SfiI, as described previously (te Riele et al. 1992). The cells were selected with hygromycin B (150 μg/ml) for 4 days and hygromycin B plus Gancyclovir (5 μm) for an additional 6 days. Genomic DNA from individual doubly-resistant colonies was digested with ScaI and analyzed by Southern blotting using both 5′ and 3′ intronic sequences flanking the targeting construct as a probe (Fig. 1B). To obtain an ES-cell line carrying the disruption in both copies of Msh5, heterozygous mutant cells were cultured in the presence of high concentrations of hygromycin B (0.9 and 1.0 mg/ml) as described before (De Wind et al. 1995).
Expression analysis
A mouse multiple tissue Northern blot (MTN, Clontech) was probed with Msh5 cDNA to determine Msh5 expression.
Total RNA was isolated from Msh5+/+, Msh5+/−, and Msh5−/− ES cells (grown on BRL-conditioned medium) according to the RNAzol B protocol (Campro). Subsequently, poly(A) RNA was isolated (PolyATtract, Promega), and 1 μg was used for dT-primed cDNA synthesis (SuperScript II, GIBCO-BRL). cDNA was PCR amplified using primers located upstream, within, and downstream of the deleted Msh5 NcoI sequence. Coamplification of an Hprt sequence served as an internal control.
Generation of Msh5 mutant mice
Two independently targeted clones were checked for diploidy by karyotype analysis, and subsequently microinjected into C57BL/6 blastocysts. Resulting male chimeric mice were mated with FVB females, and both gave germ-line transmission of the disrupted Msh5 allele. Msh5−/− mice were obtained by intercrossing heterozygous F1 mice. Genotyping of the mice was performed by PCR analysis on tail DNA.
Histology
Testes of three wild-type, two heterozygous, and three homozygous Msh5 mutant mice (2–3 months of age) were fixed in Bouin’s fluid and embedded in glycol methacrylate (Technovit, Kulzer & Co., Wehrheim, Germany). Sections (5 μm) were stained by the periodic acid Schiff reaction (PAS) and counterstained with Gill’s hematoxylin (Polysciences, Warrington, PA). The stages of the cycle of the seminiferous epithelium and the various cell types were distinghuished according to Russell et al. (1990). Apoptotic nuclei were detected with TdT-mediated dUTP nick-end labeling (In Situ Cell Death Detection Kit, Boehringer Mannheim).
Cytological analysis of male and female meiosis
For cytological analysis, five males 2–3 months of age (two wild type and three Msh5−/−) and twenty-one 15–18 days old fetuses obtained from Msh5+/− intercrosses were used (the morning of finding the vaginal plug was designated day zero). Before processing, testes were weighed and sperm from the caput epididymis was counted according to Searle and Beechey (1974). Sedimention spreading of primary spermatocyte nuclei to study meiotic prophase followed the protocol of Peters et al. (1997) [paraformaldehyde (pfa) fixation, with some minor modifications]. Part of the cell suspension was pfa fixed in a fibrin clot, using the method of Hunt et al. (1995). Fibrin-clotted and spread meiotic prophase stages were immunostained with the rabbit polyclonal serum 175. Serum 175, elicited against whole SCs as described by Offenberg et al. (1991) predominantly recognizes the Mr 30,000–33,000 Scp3 axial and lateral SC element components and was diluted 1:1000 to 1:1500. FITC-labeled swine anti-rabbit (Monosan/Sanbio PS 117F, 1:100) was used as the second antibody. Alternatively, a mouse ascites fluid monoclonal against the lateral/axial elements (anti-Cor1 recognizes the 30,000–33,000 SC component, Dobson et al. 1995), detected by Alexa 594-labeled goat anti-mouse (Molecular probes, 1:100) was used in conjuncture with a rabbit polyclonal against Scp1, a dominant central element SC protein (Meuwissen et al. 1992; dilution 1:100, detected by FITC-labeled swine anti-rabbit as used before).
From the third Msh5−/− male, surface-spread spermatocytes were processed for electron microscopy using silver staining (Peters et al. 1997), and inspected with a Jeol 1200 EKII electron microscope.
Acknowledgments
We thank Niels de Wind for identifying and characterizing EST clone 115902, Karin van Veen and René Bobeldijk for help in generating chimeric mice, members of the departments of animal and histology of the Netherlands Cancer Institute (NKI) for animal care and examination, Martin van de Valk for help with histology, Karin van’t Wout for help with apotosis assays, Frits van der Hoeven for performing chiasma and chromosome counts, R. Scriwanek for preparing the histology photographs, Christa Heyting for the anti-SC and anti-Scp1 polyclonal antibodies, and Peter Moens for the anti-Cor 1 monoclonal antibody. We appreciate comments on the manuscript from René Bernards and Nanna Claij. This work was supported financially by the Dutch Cancer Foundation (NKI 95-958 to H.t.R.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL hriele@nki.nl; FAX 31-20 512 1954.
References
- Baker SM, Bronner CE, Zhang L, Plug AW, Robatzek M, Warren G, Elliott EA, Yu J, Ashley T, Arnheim N, Flavell RA, Liskay RM. Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell. 1995;82:309–319. doi: 10.1016/0092-8674(95)90318-6. [DOI] [PubMed] [Google Scholar]
- Baker SM, Plug AW, Prolla TA, Bronner CE, Harris AC, Yao X, Christie D-M, Monell C, Arnheim N, Bradley A, Ashley T, Liskay RM. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat Genet. 1996;13:336–342. doi: 10.1038/ng0796-336. [DOI] [PubMed] [Google Scholar]
- De Rooij DG. Stem cells in the testis. Int J Exp Pathol. 1998;79:67–80. doi: 10.1046/j.1365-2613.1998.t01-1-00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wind N, Dekker M, Berns A, Radman M, te Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82:321–330. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- De Wind N, Dekker M, van Rossum A, van der Valk M, te Riele H. Mouse models for hereditary nonpolyposis colorectal cancer. Cancer Res. 1998;58:248–255. [PubMed] [Google Scholar]
- Dietrich AJJ, de Boer P. A sequential analysis of the development of the synaptonemal complex in spermatocytes of the mouse by electron microscopy using hydroxyurea and agar filtration. Genetica. 1983;61:119–129. [Google Scholar]
- Dobson MJ, Pearlman RE, Karaiskakis A, Spyropoulos B, Moens PB. Synaptonemal complex proteins: Occurrence, epitope mapping and chromosome disjunction. J Cell Sci. 1995;107:2749–2760. doi: 10.1242/jcs.107.10.2749. [DOI] [PubMed] [Google Scholar]
- Drummond JT, Li G-M, Longley MJ, Modrich P. Isolation of an hMSH2–p160 heterodimer that restores DNA mismatch repair to tumor cells. Science. 1995;268:1909–1912. doi: 10.1126/science.7604264. [DOI] [PubMed] [Google Scholar]
- Edelmann W, Cohen PE, Kane MF, Lau K, Morrow B, Bennett S, Umar A, Yang K, Kunkel T, Cattoretti G, Chaganti R, Pollard JW, Kolodner R, Kucherlapati R. Meiotic pachytene arrest in Mlh1-deficient mice. Cell. 1996;85:1125–1134. doi: 10.1016/s0092-8674(00)81312-4. [DOI] [PubMed] [Google Scholar]
- Edelmann W, Yang K, Umar A, Heyer J, Lau K, Fan K, Liedtke W, Cohen PE, Kane MF, Lipford JR, Yu N, Crouse GF, Pollard JW, Kunkel T, Lipkin M, Kolodner R, Kucherlapati R. Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell. 1997;91:467–477. doi: 10.1016/s0092-8674(00)80433-x. [DOI] [PubMed] [Google Scholar]
- Frohman MA. RACE: Rapid amplification of cDNAs ends. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. PCR protocols: A guide to methods and applications. San Diego, CA: Academic Press; 1990. pp. 28–38. [Google Scholar]
- Genschel J, Littman SJ, Drummond JT, Modrich P. Isolation of MutSβ from human cells and comparison of the mismatch repair specificities of MutSβ and MutSα. J Biol Chem. 1998;273:19895–19901. doi: 10.1074/jbc.273.31.19895. [DOI] [PubMed] [Google Scholar]
- Goetz P, Chandley AC, Speed RM. Morphological and temporal sequence of meiotic prophase development at puberty in the male mouse. J Cell Sci. 1984;65:249–263. doi: 10.1242/jcs.65.1.249. [DOI] [PubMed] [Google Scholar]
- Heyting C. Synaptonemal complexes: Structure and function. Curr Opin Cell Biol. 1996;29:389–396. doi: 10.1016/s0955-0674(96)80015-9. [DOI] [PubMed] [Google Scholar]
- Hollingsworth NM, Ponte L, Halsey C. MSH5, a novel MutS homolog, facilitates meiotic recombination between homologs in Saccharomyces cerevisiae but not mismatch repair. Genes & Dev. 1995;9:1728–1739. doi: 10.1101/gad.9.14.1728. [DOI] [PubMed] [Google Scholar]
- Hunt PA, LeMaire R, Embury P, Mroz K, Sheean L. Analysis of chromosome behavior in intact mammalian oocytes: Monitoring the segregation of a univalent chromosome during mammalian female meiosis. Hum Mol Genet. 1995;4:2007–2012. doi: 10.1093/hmg/4.11.2007. [DOI] [PubMed] [Google Scholar]
- Hunter N, Borts RH. Mlh1 is unique among mismatch repair proteins in its ability to promote crossing-over during meiosis. Genes & Dev. 1997;11:1573–1582. doi: 10.1101/gad.11.12.1573. [DOI] [PubMed] [Google Scholar]
- Johnson RE, Kovvali GK, Prakash L, Prakash S. Requirement of the yeast MSH3 and MSH6 genes for MSH2-dependent genomic stability. J Biol Chem. 1996;271:7285–7288. doi: 10.1074/jbc.271.13.7285. [DOI] [PubMed] [Google Scholar]
- Kolodner R. Biochemistry and genetics of eukaryotic mismatch repair. Genes & Dev. 1996;10:1433–1442. doi: 10.1101/gad.10.12.1433. [DOI] [PubMed] [Google Scholar]
- Li GM, Modrich P. Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proc Natl Acad Sci. 1995;92:1950–1954. doi: 10.1073/pnas.92.6.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch HT, Smyrk T, Lynch J, Fitzgibbons R, Jr, Lanspa S, McGinn T. Update on the differential diagnosis, surveillance and management of hereditary nonpolyposis colorectal cancer. Eur J Cancer. 1995;31A:1039–1046. doi: 10.1016/0959-8049(95)00126-4. [DOI] [PubMed] [Google Scholar]
- Marsischky GT, Filosi N, Kane MF, Kolodner R. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes & Dev. 1996;10:407–420. doi: 10.1101/gad.10.4.407. [DOI] [PubMed] [Google Scholar]
- Meuwissen RLJ, Offenberg HH, Dietrich AJJ, van Iersel M, Heyting C. A coiled-coil related protein specific for synapsed regions of meiotic prophase chromosomes. EMBO J. 1992;11:5091–5100. doi: 10.1002/j.1460-2075.1992.tb05616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrich P. Mechanisms and biological effects of mismatch repair. Annu Rev Genet. 1991;25:229–253. doi: 10.1146/annurev.ge.25.120191.001305. [DOI] [PubMed] [Google Scholar]
- Odorisio T, Rodriguez TA, Evans EP, Clarke AR, Burgoyne PS. The meiotic checkpoint monitoring synapsis eliminates spermatocytes via p53-independent apoptosis. Nat Genet. 1998;18:257–261. doi: 10.1038/ng0398-257. [DOI] [PubMed] [Google Scholar]
- Offenberg HH, Dietrich AJJ, Heyting C. Tissue distribution of two major components of synaptonemal complexes of the rat. Chromosoma. 1991;101:83–91. doi: 10.1007/BF00357057. [DOI] [PubMed] [Google Scholar]
- Palombo F, Gallinary P, Iaccarino I, Lettieri T, Hughes M, D’Arrigo A, Truong O, Hsuan JJ, Jiricny J. GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science. 1995;268:1912–1914. doi: 10.1126/science.7604265. [DOI] [PubMed] [Google Scholar]
- Peters AHFM, Plug AW, van Vugt M, de Boer P. A drying down technique for spreading of mammalian meiocytes from the male and female germline. Chrom Res. 1997;5:66–68. doi: 10.1023/a:1018445520117. [DOI] [PubMed] [Google Scholar]
- Pittman DL, Cobb J, Shimenti KJ, Wilson LA, Cooper DM, Brignull E, Handel MA, Schimenti JC. Meiotic prophase arrest with failure of chromosome synapsis in mice deficient for Dmc1, a germline-specific RecA homolog. Mol Cell. 1998;1:697–705. doi: 10.1016/s1097-2765(00)80069-6. [DOI] [PubMed] [Google Scholar]
- Pochart P, Woltering D, Hollingsworth NM. Conserved properties between functionally distinct MutS homologs in yeast. J Biol Chem. 1997;272:30345–30349. doi: 10.1074/jbc.272.48.30345. [DOI] [PubMed] [Google Scholar]
- Prolla TA, Baker SM, Harris AC, Tsao J-L, Yao X, Bronner CR, Zheng B, Gordon M, Reneker J, Arnheim N, Shibata D, Bradley A, Liskay RM. Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat Genet. 1998;18:276–279. doi: 10.1038/ng0398-276. [DOI] [PubMed] [Google Scholar]
- Reitmair AH, Redston M, Chun Cai J, Chuang TCY, Bjerknes M, Cheng H, Hay K, Gallinger S, Bapat B, Mak T. Spontaneous intestinal carcinomas and skin neoplasms in Msh2-deficient mice. Cancer Res. 1996;56:3842–3849. [PubMed] [Google Scholar]
- Russell LD, Ettlin RA, Hikim APS, Clegg ED. Histological and histopathological evaluation of the testis. Clearwater, FL: Cache River Press; 1990. [Google Scholar]
- Rockmill B, Sym M, Scherthan H, Roeder GS. Roles for two RecA homologs in promoting meiotic chromosome synapsis. Genes & Dev. 1995;9:2684–2695. doi: 10.1101/gad.9.21.2684. [DOI] [PubMed] [Google Scholar]
- Ross-Macdonald P, Roeder GS. Mutation of a meiosis-specific MutS homolog decreases crossing over but not mismatch correction. Cell. 1994;79:1069–1080. doi: 10.1016/0092-8674(94)90037-x. [DOI] [PubMed] [Google Scholar]
- Scherthan H, Weich S, Schwegler H, Heyting C, Harle M, Cremer T. Centromere and telomere movements during early meiotic prophase of mouse and man are associated with the onset of chromosome pairing. J Cell Biol. 1996;134:1109–1125. doi: 10.1083/jcb.134.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle AG, Beechey CV. Sperm-count, egg-fertilization and dominant lethality after X-irradiation of mice. Mutat Res. 1974;1:63–72. doi: 10.1016/0027-5107(74)90009-8. [DOI] [PubMed] [Google Scholar]
- Searle AG, Beechey CV, de Boer P, de Rooij DG, Evans EP, Kirk M. A male-sterile insertion in the mouse. Cytogenet Cell Genet. 1983;36:617–626. doi: 10.1159/000131985. [DOI] [PubMed] [Google Scholar]
- Te Riele H, Robanus Maandag E, Berns A. Highly efficient gene targeting in embryonic stem cells through homologous recombination with isogenic DNA constructs. Proc Natl Acad Sci. 1992;89:5128–5132. doi: 10.1073/pnas.89.11.5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umar A, Risinger JI, Glaab WE, Tindall KR, Barrett JC, Kunkel TA. Functional overlap in mismatch repair by human MSH3 and MSH6. Genetics. 1998;148:1637–1646. doi: 10.1093/genetics/148.4.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes & Dev. 1996;10:2411–2422. doi: 10.1101/gad.10.19.2411. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Kondoh G, Matsuda Y, Habu T, Nishimune Y, Morita T. The mouse RecA-like gene Dmc1 is required for homologous chromosome synapsis during meiosis. Mol Cell. 1998;1:707–718. doi: 10.1016/s1097-2765(00)80070-2. [DOI] [PubMed] [Google Scholar]