

Abstract
Epilepsy associated with preterm birth is often refractory to anticonvulsants. Children who are born preterm are also prone to cognitive delay and behavioral problems. Brains from these children often show diffuse abnormalities in cerebral circuitry that is likely caused by disrupted development during critical stages of cortical formation. To test the hypothesis that prenatal injury impairs the developmental switch of γ-amino butyric acid (GABA)ergic synapses from excitatory to inhibitory, thereby disrupting cortical circuit formation and predisposing to epilepsy, we used immunohistochemistry to compare the expression of cation-chloride transporters that developmentally regulate postsynaptic GABAergic discharges in postmortem cerebral samples from infants born preterm with known white matter injury (n = 11) with that of controls with minimal white matter gliosis (n = 7). Controls showed the expected developmental expression of cation-chloride transporters NKCC1 and KCC2 and of calretinin, a marker of a GABAergic neuronal subpopulation. Samples from infants with white matter damage showed a significant loss of expression of both NKCC1 and KCC2 in subplate and white matter. By contrast, there were no significant differences in total cell number or glutamate transporter VGLUT1 expression. Together, these novel findings suggest a molecular mechanism involved in the disruption of a critical stage of cerebral circuit development after brain injury from preterm birth that may predispose to epilepsy.
Keywords: Brain development, Epilepsy, KCC2, NKCC1, Perinatal brain injury, Preterm, Subplate
INTRODUCTION
During the past 15 years, the incidence of preterm births has increased by 20% in the United States (1). Perinatal brain damage from preterm birth causes chronic neurological deficits including cerebral palsy, epilepsy, cognitive delay, and behavioral abnormalities; those born earlier in gestation (<32 weeks) are at a particularly high risk (2). The cumulative impact of very preterm birth on these children and their families, as well as society, is only beginning to be appreciated as more survivors require medical care, special education, and assistance into adulthood (3). The affected children are also at significant risk for cognitive delay and behavioral problems such as autism and attention-deficit disorder (4-6). Among adults who were born at less than 32 weeks’ gestation, executive function was impaired even when scores were normalized for differences in intelligent quotient scores (7). The severity of deficits tends to inversely vary with gestational age at birth. Up to 50% of extremely low–birth weight infants with white matter lesions develop epilepsy as children (8). Epilepsy in children born preterm is frequently caused by multiple independent seizure foci and is poorly controlled. The formation of excess local circuits at the expense of typical regional circuits (9) and loss of subplate neurons (10) are among the theories that have been proposed to explain this predisposition to epilepsy. Understanding the pathogenesis of epilepsy in this population may lead not only to more effective therapies but may also provide insights into the mechanisms involved in the associated cognitive and behavioral deficits.
In addition to white matter injury, events associated with preterm birth frequently induce pathological damage to the overlying cortex, termed encephalopathy of prematurity (11). This involves damage to the subplate, a transient structure critical to the development of cerebral cortical circuits (10, 12, 13); subplate neurons guide the development of cortical inhibitory circuits during late gestation (14). A specific progression from purely tonic γ-amino butyric acid (GABA)ergic extrasynaptic signaling to activity-dependent phasic GABAergic synapses followed by glutamatergic synapses occurs during central nervous system (CNS) development (15, 16). Alterations in the spatiotemporal progression of this developmental sequence could disrupt circuit formation. For example, when thalamocortical axons arrive in cortical layer IV, depolarizing responses occur at both the projection neuron and the fast-spiking parvalbumin-positive interneuron, establishing the basic circuit pattern (17). With upregulation of the neuron-specific potassium-chloride cotransporter KCC2, the interneuron switches to hyperpolarizing, thereby effectively forming a precisely timed disynaptic feed-forward inhibitory circuit and narrowing the window to integrate excitatory inputs (18). A narrow window reinforces specific circuits and permits synaptic remodeling while avoiding excitotoxicity (19). If the initial excitatory synapses form incorrectly or if the addition for the feed-forward inhibition is deficient, abnormally diffuse poorly integrated circuits could lower the seizure threshold and impair cognition and behavior. This pattern of synaptic switching by interneurons from excitation to inhibition occurs during CNS development and is recapitulated during neurogenesis in the mature CNS (20) and after injury in the mature brain (21, 22).
Synaptogenesis and circuit formation are spatially and temporally regulated in the cerebral cortex by the sequential expression and activation of the cation-chloride ion transporters sodium-potassium-chloride cotransporter 1 (NKCC1) and KCC2 (16). Ion transporters, along with ligand-receptor channels, regulate membrane potential and, thus, the flow of electrical information in the CNS (23). The NKCC1 increases the intracellular chloride concentration and is essential for the formation of initial activity-dependent excitatory circuits (24, 25). Depolarization potentiated by NKCC1 activates calcium channels essential for neuronal maturation (23). Either excess or loss of NKCC1 expression can lower the seizure threshold (26-29). In contrast, KCC2 extrudes chloride ions, lowers the equilibrium potential for GABAA and glycine receptors, and thus increases the likelihood of hyperpolarization with ligand binding (27). The KCC2 is essential for dendritic spine formation (30), cortical interneuron migration termination (31), and synaptogenesis (32). Both NKCC1 and KCC2 are expressed on neurons and oligodendroglial lineage cells (33), although their function in oligodendrocytes remains obscure. During cerebral development, NKCC1 expression decreases whereas that of KCC2 expression increases (27, 28). This pattern of opposing expression is not as prominent in other regions of the CNS such as the brainstem (34) and spinal cord (35). Alterations of the expression pattern of these transporters are observed in human epilepsy resection samples (36, 37) and after injury in the mature CNS (38-40).
In human infants born preterm, injury often starts in utero (41, 42) and involves the developing white matter, subplate, and overlying cortex (11). Compared with human infants with minimal brain injury, infants with white matter injury after preterm birth lose expression of GABAergic markers including parvalbumin, found in fast-spiking interneurons, and GABAA receptors (12, 43). The subplate is essential for development of KCC2 expression and GABAergic circuit formation in the cortex (14); it is damaged in infants born preterm (12, 44). We propose that prenatal injury and subsequent injury to the subplate alter NKCC1 and KCC2 expression, and that this subsequently disrupts formation of crucial cerebral circuits. Therefore, we investigated cotransporter expression in samples from human infants born preterm with and without severe white matter injury. Our findings may lead to novel therapeutic targets for this population of children and adults who have refractory epilepsy and cognitive deficits associated with preterm birth.
MATERIALS AND METHODS
Human Cerebral Tissue Samples
This study was performed in compliance with the regulations of the Institutional Review Board of University Hospitals Case Medical Center. Samples from the brains of postmortem infants who were born preterm were retrospectively identified from the Rainbow Babies & Children’s Hospital Pathology Registry. Paraffin sections were obtained from the previously embedded specimens. Every effort was made to anatomically match comparative samples, but this was limited in some cases by tissue availability. Two pediatric neuropathologists (Irina Mikolaenko and Mark Cohen) performed routine neuropathologic analyses, and brain damage was graded as minimal (mild white matter gliosis) or significant (perinatal telencephalic leukoencephalopathy [PTL], periventricular leukoencephalopathy [PVL], or cystic PVL). Perinatal telencephalic leukoencephalopathy was characterized by diffuse astrocytic hypercellularity, whereas PVL showed marked astrocytic hypercellularity with focal necrosis. There were no differences in postmortem interval between the minimal and significant injury groups.
Immunohistochemistry
Eight-micrometer-thick paraffin sections were depar-affinized, rehydrated, and treated for antigen retrieval. Serial incubations with H2O2 block, primary antibodies, appropriate biotin-conjugated secondary antibodies, and Vectastain (Vector Laboratories, Burlingame, CA) were performed, and the antibodies were visualized with 3-3′diaminobenzidine (Sigma, St Louis, MO). Primary antibodies included NKCC1 (1:50; Chemicon, Temecula, CA), KCC2 (1:300; Upstate Biochemicals, Lake Placid, NY), calretinin (1:500; Sigma), and VGLUT1 (1:200; Chemicon). Every effort was made to label all samples with all 4 antibodies, but this was not possible in all cases. Adjacent sections were stained with hematoxylin or cresyl violet (Fisher, Pittsburgh, PA). Sections were dehydrated and coverslipped. For a few samples, adjacent sections were immunolabeled without and with primary antibodies to clarify the specificity of the immunostaining (data not shown). Anatomically matched sections were photographed at the same magnification, and clearly immunolabeled cells with nuclei were counted in at least 3 fields from the white matter and subplate for each infant by an investigator blinded to the clinical factors and pathological diagnosis.
Quantification and Statistics
All observations and counts were performed with the observer blinded to the pathological diagnosis and clinical factors. Results are expressed as the mean ± SEM. Differences between the means for minimal versus significant white matter damage were compared using the Student 2-tailed t-test, with p < 0.05 considered significant.
RESULTS
Cation-Chloride Cotransporter Expression Is Developmentally Regulated in the Human Cerebrum
The developmental progression of NKCC1 and KCC2 expression was assessed in relation to more established neuro-anatomical markers at 3 time points in infants with minimal evidence of brain damage (Fig. 1). Cresyl violet staining demonstrated gradual expansion of the cortex with an increase in neuropil. Immunolabeling of calretinin, a calcium-binding protein that is expressed in a subpopulation of GABAergic neurons relatively early in gestation, and NKCC1 and KCC2 were compared in the same region in the upper part of Cortical Layer II. Calretinin was expressed in a minority of cells, and its expression gradually increased throughout development. The NKCC1 and KCC2 were expressed in most of the cortical cells. The expression of NKCC1 was present at 20 weeks, peaked at approximately 35 weeks, and gradually decreased in the postnatal period. There was a gradual increase in KCC2 expression beginning at 20 weeks’ gestation, and continuing through the neonatal period, a pattern similar to that of calretinin (Fig. 1). Although their expression patterns differed, both NKCC1 and KCC2 were expressed after 20 weeks’ gestation to term equivalent, the time when perinatal brain injury from preterm birth occurs.
FIGURE 1.
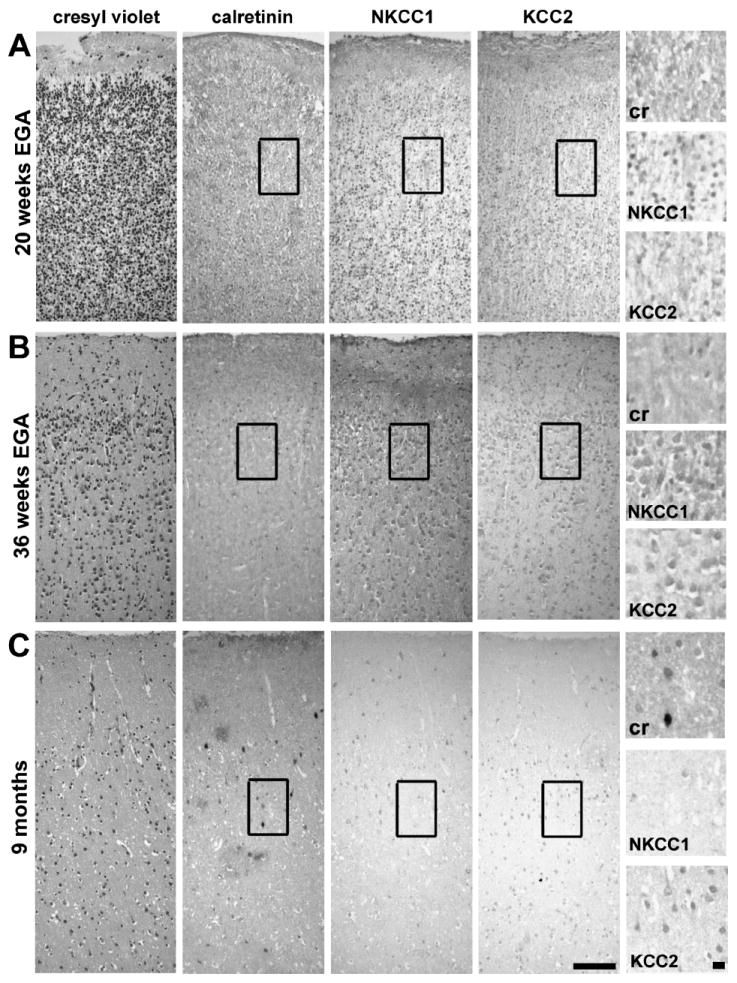
Samples from infants born preterm with relatively spared white matter demonstrate the developmental expression of calretinin and the cation-chloride cotransporters sodium-potassium-chloride cotransporter 1 (NKCC1) and potassium-chloride cotransporter 2 (KCC2) in the cerebral cortex. Infant A was born at 20 weeks′ gestation and died immediately. Infant B was born at 30 weeks′ gestation and died after 6 weeks. Infant C was born at 23 weeks′ gestation and died at 9 months from non-CNS complications secondary to preterm birth. Cresyl violet–stained sections show the expansion of the superficial layers of the cortex with maturation. Immunostaining for calretinin is minimal at 20 weeks and gradually increases through development. The expression of NKCC1 is present at 20 weeks′ gestation, peaks during late gestation (36 weeks), and diminishes during the postnatal period. The expression of KCC2 is present but limited in the cerebral cortex at 20 weeks′ gestation. The expression of KCC2 continues to increase through the third trimester and postnatal period. Scale bars = 100 μm (low magnification) and 10 μm (high magnification). EGA, estimated gestational age.
Loss of Expression of Cation-Chloride Cotransporters in Infants With White Matter Injury
In contrast to the labeling pattern in samples from infants with minimal signs of white matter damage (controls), loss of NKCC1 and KCC2 expression was found in the white matter, subplate, and cortex of infants with damage to the developing white matter (PTL, PVL, cystic PVL; n = 11). The amount of immunolabeling per cell was less for NKCC1 in infants with white matter injury (Fig. 2). In addition, significantly fewer cells were NKCC1 positive in the subplate and white matter of age-matched samples from infants with white matter damage (24–38 weeks, n = 10) compared with those with minimal damage (controls, n = 4; p < 0.03 and p = 0.047, respectively) (Figs. 4, 5). A similar pattern of loss of NKCC1 expression was observed in the cortex, but this difference was not reliably quantifiable because of changes in cell density during rapid cortical growth. Loss of NKCC1 expression in the subplate seemed to correlate with white matter damage severity. In 2 of 5 samples with PTL, there was a marked decrease in the number of NKCC1-positive subplate cells; in 4 of 5 with more severe PVL, there was a marked loss of NKCC1-positive subplate cells. In contrast to the subplate, NKCC1 loss in white matter did not correlate with the severity of white matter injury. In the white matter, 2 of 3 samples with PTL and 2 of 5 with PVL showed marked loss of NKCC1 expression. These results suggest that CNS injury manifesting as white matter damage in preterm infants is associated with loss of NKCC1 expression during the third trimester, a critical period for early circuit development.
FIGURE 2.
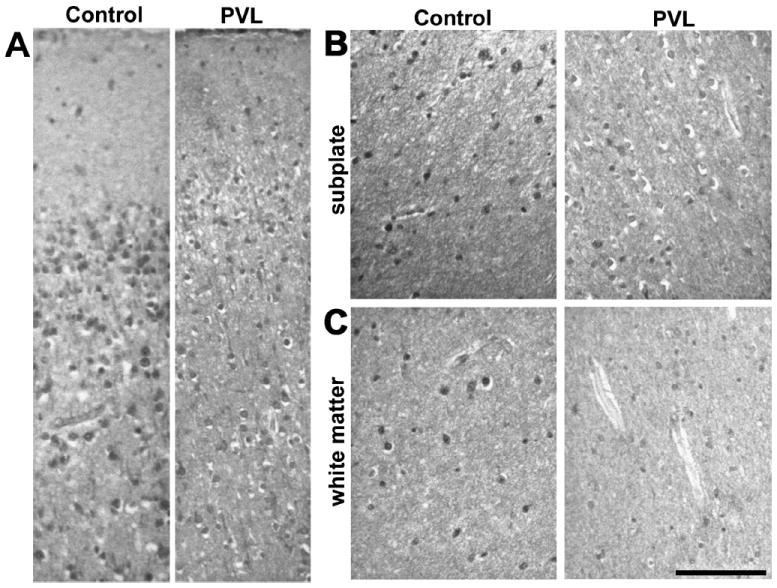
Sodium-potassium-chloride cotransporter 1 (NKCC1) immunolabeling shows less staining for NKCC1 in each cell and overall density of cells labeled in samples of the superficial cortex (A), subplate (B), and deep white matter (C) in an infant with white matter injury (periventricular leukoencephalopathy [PVL]) compared with an age-matched infant with minimal white matter gliosis (control). Scale bars = 100 μm.
FIGURE 4.
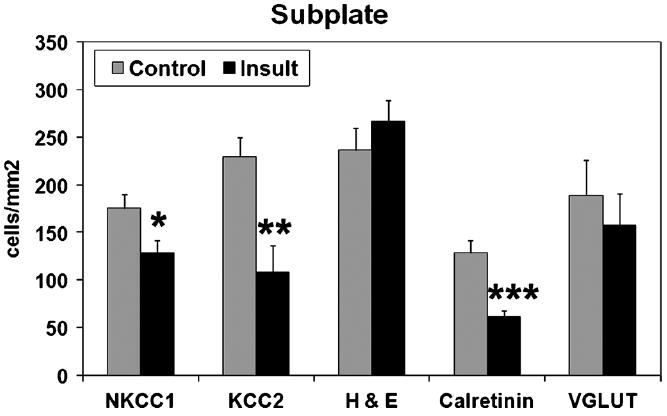
Cell densities in the subplate. There were fewer cells immunolabeled for the cotransporters sodium-potassium-chloride cotransporter 1 (NKCC1) and potassium-chloride cotransporter 2 (KCC2) in samples from infants with white matter damage (insult, n = 10, estimated gestational age, 24–38 weeks) compared with infants in an age-matched cohort without white matter damage (control, n = 4). Differences for both NKCC1-positive cells and KCC2-positive cells were significant (2-tailed t-test, *p < 0.03 and **p = 0.005). There was no difference in total cell density, as shown by hematoxylin and eosin (H&E). There were significantly fewer calretinin-labeled cells in infants with white matter injury (n = 6, ***p = 0.003) compared with “control” infants (n = 4); there was no difference in glutamate transporter (VGLUT) immunostaining.
FIGURE 5.
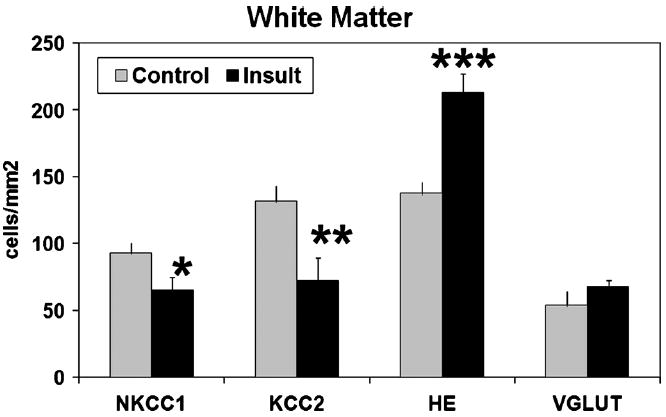
Cell densities in white matter. There were fewer cells immunolabeled for the cotransporters sodium-potassium-chloride cotransporter 1 (NKCC1) and potassium-chloride cotransporter 2 (KCC2) in white matter of infants with evidence of injury (insult, estimated gestational age, 24–38 weeks; n = 8) compared with those in specimens from age-matched infants with minimal white matter injury (n = 4; *p = 0.044 and **p = 0.019, respectively). More cells were present on hematoxylin and eosin (H&E) staining in infants with white matter damage (***p = 0.001), consistent with the criterion of astrocytic hypercellularity that defines white matter damage. There was no difference in glutamate transporter (VGLUT)–labeled cells.
There was also marked loss of KCC2-positive cells in the cortex, subplate (p = 0.005), and white matter (p = 0.019) of infants who have white matter injury compared with the controls (Figs. 3–5). Diffuse loss of KCC2-labeled cells occurred in both the subplate and white matter of almost all infants with PTL and PVL, although there was no apparent correlation with the degree of white matter damage. Infants tended to show diffuse loss of KCC2 immunoreactivity in both the subplate and white matter rather than focal areas of decreased expression. Because samples from relatively older gestation infants with white matter damage did not show the expected developmental increase in KCC2 expression, loss of KCC2 expression seemed to be prolonged and persistent rather than a transient delay.
FIGURE 3.
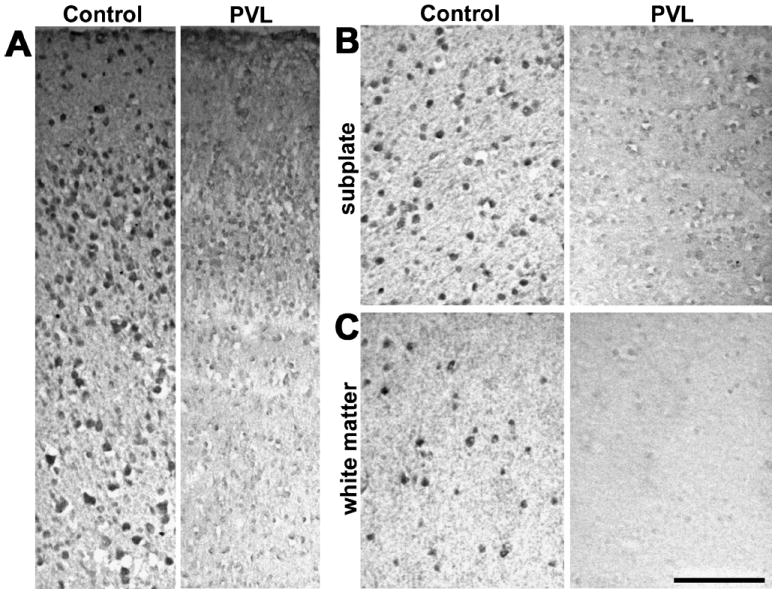
Potassium-chloride cotransporter 2 (KCC2) immunolabeling is markedly diminished in the superficial cortex (A), subplate (B), and deep white matter (C) from an infant with white matter injury (periventricular leukoencephalopathy [PVL]) compared with a sample from an age-matched infant with minimal white matter gliosis (control). Scale bars = 100 μm.
To determine whether the loss of chloride cotransporter expression reflected a general or specific loss of immunolabeled cells, cells in the subplate and white matter of cresyl violet–stained sections were counted. No significant difference in subplate total cell density was found between samples with minimal or significant white matter damage (Fig. 4). A marked increase in the number of cells was present in the white matter of infants with PTL or PVL compared with controls with minimal white matter gliosis (p = 0.001), which is expected because hypercellularity is a criterion for white matter injury (Fig. 5).
To determine whether the loss of NKCC1 and KCC2 expression was specific to infants with white matter injury, we also examined the expression of 2 other neuronal markers. Calretinin is present during the third trimester in a subset of GABAergic interneurons, cells that are vulnerable to injury from preterm birth (12). Loss of calretinin-positive cells was found in the subplate of infants with white matter injury (p = 0.003; Fig. 4), similar to the loss found in a prior study (12). By contrast, there was no difference in the density of cells immunolabeled for glutamate transporter VGLUT1, a marker of glutamate signaling (45, 46), in the white matter between samples with white matter damage and controls (Fig. 5). These results suggest that the loss of NKCC1 and KCC2 immunolabeling in infants with white matter injury is attributable to decreased expression of these cotransporters and not simply a reflection of a generalized cell loss.
DISCUSSION
Children born before 32 weeks’ gestation are at a high risk for epilepsy, cognitive delay, and behavioral problems (2). Currently, the only treatments are symptomatic and of limited efficacy. Loss of subplate neurons and GABAergic cortical neurons has been described in brains from infants born preterm (12) and in animal models (13, 47). Recent advances in neurodevelopment suggest that cation-chloride cotransporters NKCC1 and KCC2 may be at risk in the developing brains of these infants.
Before initial synapse formation, a tonic level of GABA is the first patterned type of network activity; extrasynaptic GABA facilitates neuronal development (15). During cerebral circuit formation, the developing brain begins to respond to stimuli. The NKCC1 is expressed during gestation and the early neonatal period and raises the intracellular chloride concentration above equilibrium, thereby increasing the likelihood of depolarization with ligand binding by GABA and glycine. The NKCC1 is essential for dendritic growth and synapse formation (20, 48, 49), and excess or limited NKCC1 expression in the developing brain facilitates epilepsy (26, 29). In contrast to NKCC1, KCC2 extrudes chloride, and its expression increases postnatally in the cerebrum. The KCC2 is initially localized to the somata of immature neurons and subsequently moves into dendrites of mature neurons (50). When KCC2 expression is deficient, rodents demonstrate abnormalities consistent with defective fast-spiking inhibition, including increased anxiety, a lower seizure threshold, and impaired learning (51). Levels of KCC2 activation are influenced by growth factors (52, 53), oligomer formation (54), localization in lipid rafts (55, 56), and cell surface expression time (22). The cation-chloride cotransporter–interacting protein inactivates NKCC1 and activates KCC2, likely through heterodimer formation (57). Regulation of the NKCC1 and KCC2 expression in the injured developing CNS has not yet been studied in animal models.
In human temporal lobe epilepsy samples, a subset of pyramidal neurons showed depolarization in response to GABA stimulation, suggesting alterations in chloride equilibrium (58, 59). Analyses of temporal lobe epilepsy tissue have shown downregulation of both NKCC1 and KCC2 in the subiculum (37, 60, 61). It has also been proposed that a reversion to the immature depolarized state occurs in focal cortical dysplasia (62, 63); samples from human focal cortical dysplasia resections showed elevated NKCC1 and diminished KCC2 levels in specific regions (37, 64). The expression of KCC2 was also shifted from dendritic to somata prominence in focal cortical dysplasia samples (36), similar to the pattern observed in immature neurons. Changes in NKCC1 and KCC2 are also observed in rodent models of epilepsy (65). Levels of KCC2 remain depressed for an extended period after CNS injury, perhaps explaining the predilection to develop epilepsy after insults (40, 66). Taken together, these data suggest that loss of chloride cotransporter expression may contribute to epilepsy and other cortical circuit abnormalities in children born preterm.
The present study has several limitations. The samples studied were necessarily limited to those from infants who died as neonates. Thus, the postmortem samples may not accurately reflect changes in the brains of infants with perinatal brain injury who survive. The varied ages and clinical courses of the infants also confound the analysis. Despite these limitations, the novel alterations in cation-chloride expression levels after brain injury from preterm birth identified may eventually lead to therapeutic interventions and improved outcomes for these children. For example, bu-metanide, an inhibitor of NKCC1, enhances the efficacy of phenobarbital in the treatment of neonatal seizures (67). Neuroprotective agents administered during the neonatal period to infants at high risk for deficits may minimize loss of NKCC1 and KCC2, optimize synaptogenesis and circuit formation, and thus decrease chronic neurological impairments in these unfortunate children.
Acknowledgments
This project was supported by a stipend from the Research Training in Heart, Lung, Blood and Sleep Diseases Training Grant T35 HL082544 to Ian Thompson, and a Rainbow Babies & Children’s Hospital Board of Trustees award to Shenandoah Robinson.
Footnotes
Online-only figures are available at http://www.jneuropath.com.
References
- 1.Martin J, Hamilton B, Sutton P, et al. Births: Final data for 2005. National Vital Statistics Reports. 2007;56:1–3. 21, 65, 79–80. [PubMed] [Google Scholar]
- 2.Wilson-Costello D, Friedman H, Minich N, et al. Improved survival rates with increased neurodevelopmental disability for extremely low birth weight infants in the 1990s. Pediatrics. 2005;115:997–1003. doi: 10.1542/peds.2004-0221. [DOI] [PubMed] [Google Scholar]
- 3.Petrou S. The economic consequences of preterm birth during the first 10 years of life. BJOG. 2005;112:10–15. doi: 10.1111/j.1471-0528.2005.00577.x. [DOI] [PubMed] [Google Scholar]
- 4.Allin M, Rooney M, Cuddy M, et al. Personality in young adults who are born preterm. Pediatrics. 2006;117:309–16. doi: 10.1542/peds.2005-0539. [DOI] [PubMed] [Google Scholar]
- 5.Hack M, Youngstrom E, Cartar L, et al. Behavioral outcomes and evidence of psychopathology among very low birth weight infants at age 20 years. Pediatrics. 2004;114:932–40. doi: 10.1542/peds.2003-1017-L. [DOI] [PubMed] [Google Scholar]
- 6.Saigal S, Doyle L. An overview of mortality and sequelae of preterm birth from infancy to adulthood. Lancet. 2008;371:261–69. doi: 10.1016/S0140-6736(08)60136-1. [DOI] [PubMed] [Google Scholar]
- 7.Nosarti C, Giouroukou E, Micali N, et al. Impaired executive functioning in young adults born very preterm. J Int Neuropsychol Soc. 2007;13:571–81. doi: 10.1017/S1355617707070725. [DOI] [PubMed] [Google Scholar]
- 8.Gurses C, Gross D, Andermann F, et al. Periventricular leukomalacia and epilepsy: Incidence and seizure pattern. Neurology. 1999;52:341–45. doi: 10.1212/wnl.52.2.341. [DOI] [PubMed] [Google Scholar]
- 9.Marin-Padilla M. Perinatal brain damage, cortical reorganization (acquired cortical dysplasias), and epilepsy. Adv Neurol. 2000;84:153–72. [PubMed] [Google Scholar]
- 10.Volpe J. Subplate neurons—missing link in brain injury of the premature infant. Pediatrics. 1996;97:112–13. [PubMed] [Google Scholar]
- 11.Volpe J. Brain injury in premature infants: A complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009;8:110–24. doi: 10.1016/S1474-4422(08)70294-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robinson S, Li Q, DeChant A, et al. Neonatal loss of gamma amino butyric acid pathway expression after human perinatal brain injury. J Neurosurg. 2006;104:396–408. doi: 10.3171/ped.2006.104.6.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McQuillen P, Sheldon R, Shatz C, et al. Selective vulnerability of sub-plate neurons after early neonatal hypoxia-ischemia. J Neurosci. 2003;23:3308–15. doi: 10.1523/JNEUROSCI.23-08-03308.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanold P, Shatz C. Subplate neurons regulate maturation of cortical inhibition and outcome of ocular dominance plasticity. Neuron. 2006;51:627–38. doi: 10.1016/j.neuron.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 15.Ben-Ari Y. Excitatory actions of GABA during development: The nature of the nurture. Nature Neurosci Rev. 2002;3:728–39. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- 16.Farrant M, Kaila K. The cellular, molecular and ionic basis of GABAA receptor signalling. Progress Brain Res. 2007;160:59–87. doi: 10.1016/S0079-6123(06)60005-8. [DOI] [PubMed] [Google Scholar]
- 17.Daw M, Ashby M, Issac J. Coordinated developmental recruitment of latent fast spiking interneurons in layer IV barrel cortex. Nat Neurosci. 2007;10:453–61. doi: 10.1038/nn1866. [DOI] [PubMed] [Google Scholar]
- 18.Hull C, Scanziani M. It’s about time for thalamocortical circuits. Nat Neurosci. 2007;10:400–2. doi: 10.1038/nn0407-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akerman C, Cline H. Refining the roles of GABAergic signaling during neural circuit formation. Trends Neurosci. 2007;30:382–89. doi: 10.1016/j.tins.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 20.Ge S, Goh E, Sailor K, et al. GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature. 2006;439:589–93. doi: 10.1038/nature04404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rivera C, Voipio V, Kaila K. Two developmental switches in GABAergic signalling: The K+-Cl− cotransporter KCC2 and carbonic anhydrase CAVII. J Physiol. 2005;562:27–36. doi: 10.1113/jphysiol.2004.077495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wake H, Watanabe M, Moorhouse A, et al. Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J Neurosci. 2007;27:1642–50. doi: 10.1523/JNEUROSCI.3104-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blaesse P, Airaksinen M, Rivera C, et al. Cation-chloride transporters and neuronal function. Neuron. 2009;61:820–38. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 24.Nakanishi K, Yamada J, Takayama C, et al. NKCC1 activity modulates formation of functional inhibitory synapses in cultured neocortical neurons. Synapse. 2007;61:138–49. doi: 10.1002/syn.20352. [DOI] [PubMed] [Google Scholar]
- 25.Sipila S, Huttu K, Soltesz I, et al. Depolarizing GABA acts on intrinsically bursting pyramidal neurons to drive giant depolarizing potentials in the immature hippocampus. J Neurosci. 2005;25:5280–89. doi: 10.1523/JNEUROSCI.0378-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dzhala V, Talos D, Sdrulla D, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–13. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- 27.Rivera C, Voipio J, Payne J, et al. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–55. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 28.Yamada J, Okabe A, Toyoda H, et al. Cl− uptake promoting depolarizing GABA actions in immature rat neocortical neurons is mediated by NKCC1. J Physiol. 2004;557:829–41. doi: 10.1113/jphysiol.2004.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sipila S, Huttu K, Yamada J, et al. Compensatory enhancement of intrinsic spiking upon NKCC1 disruption. J Neurosci. 2009;27:6982–88. doi: 10.1523/JNEUROSCI.0443-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Khirug S, Cai C, et al. KCC2 interacts with the dendritic cyto-skeleton to promote spine development. Neuron. 2007;56:1019–33. doi: 10.1016/j.neuron.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 31.Bortone D, Polleux F. KCC2 expression promotes the termination of cortical interneuron migration in a voltage-sensitive calcium-dependent manner. Neuron. 2009;62:53–71. doi: 10.1016/j.neuron.2009.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanis J, Bellemer A, Moresco J, et al. The potassium chloride cotransporter KCC-2 coordinates development of inhibitory neurotransmission and synapse structure in Caenorhabditis elegans. J Neurosci. 2009;29:9943–54. doi: 10.1523/JNEUROSCI.1989-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malek S, Coderre E, Stys P. Aberrant chloride transport contributes to anoxic/ischemic white matter injury. J Neurosci. 2003;23:3826–36. doi: 10.1523/JNEUROSCI.23-09-03826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balakrishnan V, Becker M, Lohrke S, et al. Expression and function of chloride transporters during development of inhibitory neurotransmission in the auditory brainstem. J Neurosci. 2003;23:4134–45. doi: 10.1523/JNEUROSCI.23-10-04134.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delpy A, Allain A-E, Meyrand P, et al. NKCC1 cotransporter inactivation underlies embryonic development of chloride-mediated inhibition in mouse spinal motoneuron. J Physiol. 2008;586:1059–75. doi: 10.1113/jphysiol.2007.146993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munakata M, Watanabe M, Otsuki T, et al. Altered distribution of KCC2 in cortical dysplasia in patients with intractable epilepsy. Epilepsia. 2007;48:837–44. doi: 10.1111/j.1528-1167.2006.00954.x. [DOI] [PubMed] [Google Scholar]
- 37.Sen A, Martinian L, Kikolic M, et al. Increased NKCC1 expression in refractory human epilepsy. Epilepsy Res. 2007;74:220–27. doi: 10.1016/j.eplepsyres.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 38.Bonislawski D, Schwarzbach E, Cohen A. Brain injury impairs dentate gyrus inhibitory efficacy. Neurobiol Dis. 2007;25:163–69. doi: 10.1016/j.nbd.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papp E, Riveria C, Kaila K, et al. Relationship between neuronal vulnerability and potassium-chloride co-transporter 2 immunoreactivity in hippocampus following transient forebrain ischemia. Neuroscience. 2008;154:677–89. doi: 10.1016/j.neuroscience.2008.03.072. [DOI] [PubMed] [Google Scholar]
- 40.Pathak H, Weissinger F, Terunuma M, et al. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. 2007;27:14012–22. doi: 10.1523/JNEUROSCI.4390-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bracci R, Buonocore G. Chorioamnionitis: A risk factor for fetal and neonatal morbidity. Biol Neonate. 2003;83:85–96. doi: 10.1159/000067956. [DOI] [PubMed] [Google Scholar]
- 42.Hansen A, Collins M, Genest D, et al. Very low birth weight infant’s placenta and its relation to pregnancy and fetal characteristics. Pediatr Dev Pathol. 2000;3:419–30. doi: 10.1007/s100240010043. [DOI] [PubMed] [Google Scholar]
- 43.Iai M, Takashima S. Thalamocortical development of parvalbumin neurons in normal and periventricular leukomalacia brains. Neuropediatrics. 1999;30:14–18. doi: 10.1055/s-2007-973450. [DOI] [PubMed] [Google Scholar]
- 44.Kostovic I, Judas M. Prolonged coexistence of transient and permanent circuitry elements in the developing cerebral cortex of fetuses and preterm infants. Dev Med Child Neurol. 2006;48:388–93. doi: 10.1017/S0012162206000831. [DOI] [PubMed] [Google Scholar]
- 45.Boulland J-L, Qureshi T, Seal R, et al. Expression of the vesicular glutamate transporters during development indicates the widespread co-release of multiple neurotransmitters. J Comp Neurol. 2004;480:264–80. doi: 10.1002/cne.20354. [DOI] [PubMed] [Google Scholar]
- 46.Eastwood S, Harrison P. Decreased expression of vesicular glutamate transporter 1 and complexin II mRNAs in schizophrenia: Further evidence for a synaptic pathology affecting glutamate neurons. Schizophrenia Res. 2005;73:159–72. doi: 10.1016/j.schres.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 47.McQuillen P, Ferriero D. Perinatal subplate neuron injury: Implications for cortical development and plasticity. Brain Pathol. 2005;15:250–60. doi: 10.1111/j.1750-3639.2005.tb00528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pfeffer CK, Stein V, Keating DJ, et al. NKCC1-dependent GABAergic excitation drives synaptic network maturation during early hippocampal development. J Neurosci. 2009;29:3419–30. doi: 10.1523/JNEUROSCI.1377-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y, Liu L, Xia Z. Brain-derived neurotrophic factor stimulates the transcriptional and neuroprotective activity of myocyte-enhancer factor 2C through an ERK1/2-RSJ2 signaling cascade. J Neurochem. 2007;102:957–66. doi: 10.1111/j.1471-4159.2007.04606.x. [DOI] [PubMed] [Google Scholar]
- 50.Zhang L-L, Fina M, Vardi N. Regulation of KCC2 and NKCC during development: Membrane insertion and differences between cell types. J Comp Neurol. 2006;499:132–43. doi: 10.1002/cne.21100. [DOI] [PubMed] [Google Scholar]
- 51.Tornberg J, Voikar V, Savilahti H, et al. Behavioral phenotypes of hypomorphic KCC2-deficient mice. Eur J Neurosci. 2005;21:1327–37. doi: 10.1111/j.1460-9568.2005.03959.x. [DOI] [PubMed] [Google Scholar]
- 52.Aguado F, Carmona M, Pozas E, et al. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl− co-transporter KCC2. Development. 2003;130:1267–80. doi: 10.1242/dev.00351. [DOI] [PubMed] [Google Scholar]
- 53.Kelsch W, Hormuzdi S, Straube E, et al. Insulin-like growth factor 1 and a cytosolic tyrosine kinase activate chloride outward transport during maturation of hippocampal neurons. J Neurosci. 2001;21:8339–47. doi: 10.1523/JNEUROSCI.21-21-08339.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blaesse P, Guillemin I, Schindler J, et al. Oligomerization of KCC2 correlates with development of inhibitory neurotransmission. J Neurosci. 2006;2641:10407–19. doi: 10.1523/JNEUROSCI.3257-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hartmann A-M, Blaesse P, Krantz T, et al. Opposite effect of membrane raft perturbation on transport activity of KCC2 and NKCC1. J Neurochem. 2009;111:321–31. doi: 10.111/j.1471-4159.2009.06343.x. [DOI] [PubMed] [Google Scholar]
- 56.Watanabe M, Wake H, Moorhouse A, et al. Clustering of neuronal K+-Cl− cotransporters in lipid rafts by tyrosine phosphorylation. J Biol Chem. 2009;284:27980–88. doi: 10.1074/jbc.M109.043620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wenz M, Hartmann A-M, Friauf E, et al. CIP1 is an activator of the K+-Cl− cotransporter KCC2. Biochem Biophys Res Comm. 2009;381:388–92. doi: 10.1016/j.bbrc.2009.02.057. [DOI] [PubMed] [Google Scholar]
- 58.Cohen I, Navarro V, Clemenceau S, et al. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–21. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- 59.Huberfeld G, Wittner L, Clemenceau S, et al. Perturbed chloride ho-meostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–73. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muñoz A, Méndez P, DeFelipe J, et al. Cation-chloride cotransporters and GABA-ergic innervation in the human epileptic hippocampus. Epilepsia. 2007;48:663–73. doi: 10.1111/j.1528-1167.2007.00986.x. [DOI] [PubMed] [Google Scholar]
- 61.Palma E, Amici M, Sobrero F, et al. Anomalous levels of C1− transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc Natl Acad Sci U S A. 2006;103:8465–68. doi: 10.1073/pnas.0602979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cepeda C, Andre V, Levine M, et al. Epileptogenesis in pediatric cortical dysplasia: The dysmature cerebral developmental hypothesis. Epilepsy Behav. 2006;9:219–35. doi: 10.1016/j.yebeh.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 63.Cepeda C, Andre V, Wu N, et al. Immature neurons and GABA networks may contribute to epileptogenesis in pediatric cortical dysplasia. Epilepsia. 2007;48:79–85. doi: 10.1111/j.1528-1167.2007.01293.x. [DOI] [PubMed] [Google Scholar]
- 64.Aronica E, Boer K, Redeker S, et al. Differential expression patterns of chloride transporters, NA+-K+-2Cl−-cotransporter and K+-Cl−-cotransporter, in epilepsy-associated malformations of cortical development. Neurosci. 2007;145:185–96. doi: 10.1016/j.neuroscience.2006.11.041. [DOI] [PubMed] [Google Scholar]
- 65.Shimizu-Okabe C, Okabe A, Kilb W, et al. Changes in the expression of cation-Cl- cotransporters, NKCC1 and KCC2, during cortical malformation induced by neonatal freeze. Neurosci Res. 2007;59:288–95. doi: 10.1016/j.neures.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 66.Jin X, Huguenard J, Prince D. Impaired Cl− extrusion in layer V pyramidal neurons of chronically injured epileptogenic neocortex. J Neurophysiol. 2005;93:2117–26. doi: 10.1152/jn.00728.2004. [DOI] [PubMed] [Google Scholar]
- 67.Dzhala V, Brumback AC, Staley K. Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol. 2008;63:222–35. doi: 10.1002/ana.21229. [DOI] [PubMed] [Google Scholar]