

Summary
Methionine aminopeptidases (MetAPs) remove the N-terminal initiator methionine during protein synthesis, a pre-requisite step for N-terminal myristoylation. N-myristoylation of proto-oncogene c-Src is essential for its membrane association and proper signal transduction. We used bengamides, a family of general MetAP inhibitors, to understand the downstream physiological functions of MetAPs. c-Src from bengamide A-treated cells retained its N-terminal methionine and suffered a decrease in N-terminal myristoylation, which was accompanied by a shift of its subcellular distribution from the plasma membrane to the cytosol. Furthermore, bengamide A decreased the tyrosine kinase activity of c-Src both in vitro and in vivo and eventually delayed cell cycle progression through G2/M. Thus, c-Src is a physiologically relevant substrate for MetAPs whose dysfunction is likely to account for the cell cycle effects of MetAP inhibitors including bengamide A.
Introduction
The proteolytic removal of the N-terminal methionine is catalyzed by a family of enzymes known as methionine aminopeptidases (MetAPs). Prokaryotic organisms typically have only one type of MetAP, whereas eukaryotes have two isoforms, type I and type II. These enzymes have been demonstrated to be essential for host survival in bacteria and yeast [1–3]. However, little is known about their physiological functions, particularly in multicellular organisms. Human MetAP2 was identified as the physiological target for the fumagillin family of natural products (Figure 1), which are potent inhibitors of angiogenesis [4,5]. Further studies have demonstrated that inhibition of MetAP2 by fumagillin/ovalicin leads to the activation of p53 and induction of p21 to arrest cell cycle progression of endothelial cells at late G1 phase [6,7]. Such observations have also been confirmed by a genome-wide functional analysis of human cell cycle regulators using small interfering RNAs (siRNAs) [8]. To date, several inhibitors of MetAP2 have been developed as potential therapeutic agents for cancer [9–12]. Using pyridine-2-carboxylates (Figure 1), which selectively inhibit the enzymatic activities of MetAP1, we have recently found that MetAP1 is involved in the cell cycle progression during G2/M phase, suggesting that MetAP1 may also serve as an anti-cancer drug target [13].
Figure 1.

Structures of the Inhibitors for Methionine Aminopeptidases.
Bengamide A and LAF 389 are non-selective inhibitors for both MetAP1 and MetAP2. Fumagillin, ovalicin and TNP-470 are human MetAP2-specific inhibitors. IV-43 is a human MetAP1-specific inhibitor.
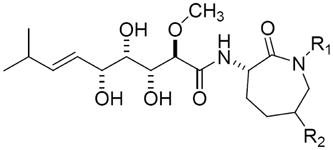
The bengamides are unique, mixed biogenetic sponge-derived natural products that were first reported in the late 1980’s [14]. This compound family (Figure 1) has been a continual focus of research on the isolation of new analogs, chiral total synthesis, and biological evaluation of the structural-activity relationships [14,15]. Extensive studies in vitro have established that few structural changes are tolerated in order to maintain biological activity [16]. Interestingly, the profiles in the NCI 60 cell-line panel for bengamides A, B and P are unique compared to all the standard antitumor compounds in the NCI database [17]. Significant antitumor activity in vivo has been observed for bengamides A and B, which led to the design of a synthetic bengamide analog, LAF389 (Figure 1), for clinical evaluation. Although LAF389 exhibited promising antitumor activity during preclinical studies, its phase I clinical trial has been terminated due in part to unpredictable cardiovascular toxicity [18]. Nonetheless, the mechanism of action for the bengamides remains unclear. Recently, bengamides have been identified as a new class of inhibitors for human MetAPs using proteomic approaches [19]. However, enzymatic studies suggest that the bengamides are not selective for either MetAP1 or MetAP2 (Table 1). The unpredicted clinical toxicity, therefore, is likely a consequence of the global inhibition of the N-terminal methionine processing.
Table 1.
The Inhibitory Profiles for Bengamides on Cell Proliferation and MetAPs Enzyme Activities.
 |
Inhibition of enzyme activity (IC50 in μM) | Inhibition of cell proliferation (IC50 in nM) | |||||
|---|---|---|---|---|---|---|---|
| bengamide | R1 | R2 | MetAP1 | MetAP2 | HeLa | HCT116 | BAEC |
| A | H | -OCO(CH2)12CH3 | 1.9±0.2 | 10.5±3.8 | 0.2±0.04 | 0.4±0.03 | 0.3±0.07 |
| B | CH3 | -OCO(CH2)12CH3 | 29.3±10.4 | 17.9±7.9 | 1.6±0.5 | 5.7±2.5 | 0.5±0.2 |
| G | H | -OCO(CH2)11CH3 | 26.8±18.3 | >50 | 0.4±0.03 | 0.4±0.1 | 1.4±0.5 |
| L | H | -OCO(CH2)11CH(CH3)2 | 37.1±13.4 | >50 | 2.2±0.5 | 0.4±0.1 | 2.4±0.3 |
| M | CH3 | -OCO(CH2)11CH(CH3)2 | 5.4±2.3 | >50 | 17.0±3.0 | 3.0±0.2 | 2.8±0.4 |
| N | H | -OCO(CH2)10CH(CH3)2 | 40.2±14.3 | >50 | 3.4±0.4 | 2.8±0.5 | 11.0±4.0 |
| O | CH3 | -OCO(CH2)10CH(CH3)2 | 2.7±0.4 | >50 | 0.9±0.2 | 0.8±0.2 | 0.9±0.1 |
HeLa, human cervical adenocarcinoma cells; HCT116, human colon adenocarcinoma cells; BAEC, bovine aorta endothelial cells.
Cleavage of the N-terminal initiator methionine is a necessary step for the regulation of protein half life [20], post-translational modifications, such as N-terminal acetylation [21], N-terminal myristoylation [22], and the functions of glutamine-dependent amidotransferases [23]. Using two-dimensional gel electrophoresis and MALDI-TOF techniques, several protein substrates for MetAP enzymes have been identified and confirmed, including but not limited to glyceraldehyde-3-phosphate dehydrogenase (GAPDH), cyclophilin A [24], 14-3-3γ [19], glutathione S-transferase (GST) [25], bovine rhodanese [26], 14kDa migration-inhibitory-factor-related protein (MRP14) [27], and actin [28]. However, the physiological relevance for retention of the N-terminal methionine on these substrate proteins remains unclear.
One of the most studied proto-oncogenes, c-Src, is known to undergo N-terminal myristoylation. This modification is required for the association of c-Src with plasma membrane and for proper signal transduction [29]. Importantly, the Src family kinases have been shown to play pivotal roles in cell cycle progression [30,31], making them potential candidates to mediate the cell cycle effects of MetAP inhibitors. Using bengamide A, along with the MetAP isoform-specific inhibitors (Figure 1), we demonstrate that c-Src is a common substrate for both MetAP1 and MetAP2. Treatment of cells with bengamide A leads to the retention of c-Src N-terminal methionine, and decreases c-Src N-terminal myristoylation. Consequently, c-Src protein relocates from the plasma membrane to cytosol and nucleus. Furthermore, bengamide A decreases the tyrosine kinase activity of c-Src in vitro and in vivo, leading to a slower cell cycle progression through G2/M. The MEF cells deficient in Src/Yes/Fyn are more resistant to bengamide A than wild type MEFs for cell proliferation. Together, these results suggest that c-Src is a physiologically relevant substrate for methionine aminopeptidases. Deregulation of c-Src subcellular localization and its tyrosine kinase activities may, therefore, account for the cell cycle effects by the inhibitors of MetAPs.
Results
Bengamides inhibit both human MetAP1 and MetAP2 in vitro
In a preliminary structure-activity relationship study, we determined the effects of seven bengamide analogs on the enzymatic activity of both recombinant human MetAP1 and MetAP2 in vitro and on the proliferation of the primary bovine aortic endothelial cells (BAEC) and two tumor cell lines. As previously reported [19], most bengamide analogs are non-selective for either of the MetAP enzymes in vitro (Table 1). However, some analogs, such as bengamide M and O, exhibited 10–20-fold selectivity towards MetAP1. Among all analogs tested, bengamide A showed the highest potency for the inhibition of both MetAP enzymes in vitro and cell proliferation. We therefore used bengamide A in all subsequent investigations.
Inhibition of Both MetAP1 and MetAP2 by Bengamide A Causes Retention of the N-terminal Methionine on c-Src
The N-terminal penultimate amino acid of c-Src protein is glycine, which indicates that c-Src is a substrate of MetAPs [32]. We adapted a previously reported assay from Wang et al. [9] to measure the N-terminal initiator methionine level on c-Src. C-terminally myc-tagged c-Src protein was transiently expressed in HEK293 cells for 48 hours with an 18-hour drug-treatment before harvest. Total cellular proteins were labeled metabolically with [35S]-methionine for the last 4 hours. Tagged c-Src was then immunoprecipitated by an anti-myc antibody. The pellets were extensively washed to reduce the non-specific binding of other [35S]-labeled cellular proteins. One aliquot of the immunoprecipitate was used for Western blotting as input control. A mixture of recombinant MetAP1 and MetAP2 (20 μM each) in reaction buffer was added to the other aliquot. This suspension was rotated for 1 hour at room temperature to release the initiator methionines from the immobilized myc-tagged c-Src. As shown in Figure 2B, treatment of immunoprecipitated c-Src with MetAPs released the N-terminal methionine residues from unprocessed protein. Although less c-Src protein was contained in the bengamide-treated sample (Figure 2A), more methionines were released from the immunoprecipitated c-Src N-termini, suggesting that c-Src protein is an in vivo substrate for both methionine aminopeptidases.
Figure 2.

Inhibition of Methionine Aminopeptidases by Bengamide A Changes c-Src N-terminal Myristoylation Status.
Immunoprecipitation of C-terminal myc-tagged c-Src protein from HEK293 cell lysate was aliquoted either for western blotting by anti-Myc antibody (A), or for [35S]-methionine scintillation counting (B) after in vitro processing by both MetAP1 and MetAP2. Bengamide A (10 nM) was added for the last 18 hours. Immunoprecipitation from [3H]-myristic acid-labeled HEK293 cell lysate were aliquoted either for western blot (C, E) or for [3H] scintillation counting as an indication of N-myristoylation (D, F). CHX, cycloheximide (4 μg/mL); Beng A, bengamide A (100 nM) and 5-FU, 5-fluorouracil (100 μM), IV-43 (10 μM), TNP-470 (100 nM) and the combination of IV-43 (10 μM) and TNP-470 (100 nM), were added to cell media for the last 6 hours.
Bengamide A Decreases the N-terminal Myristoylation of c-Src Protein
The requirement of glycine at the +1 position for significant N-terminal myristoylation [22] predicted that retention of the initiator methionine will decrease the N-terminal myristoylation of c-Src. To assess the effect of bengamide A on c-Src N-terminal myristoylation, [3H]-myristic acid was added to cell culture media to label the C-terminal-myc-tagged c-Src proteins in the presence of different inhibitors. Western blot by anti-myc antibody showed no significant difference in c-Src expression level from different samples (Figure 2C). A significant decrease in N-terminal myristoylation of c-Src was observed when cells were treated with bengamide A (Figure 2D). As a positive control, cycloheximidine (CHX), a general protein synthesis inhibitor, was found to reduce the N-myristoylation of c-Src. In contrast, 5-fluorouracil (5-FU), a DNA replication inhibitor, did not have any effect on c-Src myristoylation in this assay (Figure 2D). To assess the contribution to c-Src myristoylation by either MetAP1 or MetAP2, we carried out additional experiments by treating cells with IV-43 (10 μM), TNP-470 (100 nM) or the combination of both drugs. As shown in Figure 2F, inhibition of either MetAP enzyme alone by IV-43 and TNP-470, respectively, had marginal effects the c-Src N-terminal myristoylation. However, concurrent inhibition by the combination of both inhibitors significantly decreased the N-myristoylation level of c-Src, suggesting that MetAP1 and MetAP2 are functionally redundant in processing the initiator methionine of c-Src.
Bengamide A Treatment Changes the Subcellular Distribution of c-Src
Since N-terminal myristoylation is a post-translational modification required for the membrane localization of c-Src, we next examined the effect of bengamide A on the subcellular localization of c-Src protein. Microsomes and cytosolic proteins were fractionated by ultracentrifugation and analyzed by Western blot using E-cadherin and tubulin as the plasma membrane and cytosolic protein markers, respectively (Figure 3A). As expected, c-Src was primarily localized in the membrane fraction from the vehicle-control HeLa cells. This subcellular localization was not affected by the MetAP1-specific inhibitor, IV-43 (10 μM), or the MetAP2-specific inhibitor, TNP-470 (100 nM). However, bengamide A (10 nM) caused a significant amount of c-Src protein to redistribute from the membrane fraction into the cytosolic fraction (Figure 3A). Interestingly, a combination of IV-43 and TNP-470 was also able to increase the cytosolic c-Src protein level, indicating that MetAP1 and MetAP2 are functionally redundant for processing the initiator methionine from c-Src.
Figure 3.

Bengamide A Changes Subcellular Distribution of c-Src Tyrosine Kinase.
(A) HeLa cell lysate was homogenized and separated into cytosol (c) and total membrane (m) fractions, followed by immunoblotting for c-Src, tubulin and E-cadherin. Cells were treated with IV-43 (10 μM), TNP-470 (100 nM), bengamide A (10 nM) and the combination of IV-43 (10 μM) and TNP-470 (100 nM) for 24 hours before harvest. (B) Indirect immunofluorescence microscopy of HeLa cell by c-Src antibody. 10 nM bengamide A was used to treat cells for 24 hours. Images were obtained from a 63× objective (scale bar = 20 μm).
In a complementary approach to determine the subcellular distribution of c-Src, we used indirect immunofluorescence to detect c-Src localization in vivo. As shown in Figure 3B, c-Src protein predominantly localized to the plasma membrane as well as in the perinuclear region, presumably the ER membrane in DMSO-treated cells. Treatment with bengamide A (10 nM) led to the redistribution of c-Src into cytosol. It is noteworthy that a significant amount of c-Src protein was also seen in the nucleus.
Bengamide A Decreases the Tyrosine Kinase Activity of c-Src
Activation of c-Src tyrosine kinase has been demonstrated to be involved in signal transduction pathways that control cell growth and cellular architecture [33]. To assess if bengamide A treatment could affect the tyrosine kinase activities of c-Src, we used acid-denatured rabbit muscle enolase as an exogenous substrate for the in vitro kinase assay. Transiently transfected HEK293 cells were treated with different drugs before C-terminal myc-tagged c-Src protein was immunoprecipitated. The pellets were extensively washed before incubation in a kinase reaction mixture for 30 min at 30°C. The reaction was then stopped by addition of SDS-PAGE sample buffer and boiling in water for 3 min. Phosphorylated enolase was detected by autoradiography on the SDS-PAGE gel. As shown in Figure 4A, immunoprecipitates by anti-myc IgG phosphorylated enolase in vitro, whereas negative control by normal mouse IgG did not. To prove that this phosphorylation is mediated through the tyrosine kinase activity of c-Src, the in vitro kinase assay was carried out in the presence of PP2 (10 nM), an inhibitor for Src family kinases. Disappearance of phosphorylated enolase from PP2-treated sample confirmed that phosphorylation of enolase was catalyzed by the tyrosine kinase activity of c-Src. Using this assay, we observed that the immunoprecipitates from bengamide A-treated cells have much lower kinase activity than those from DMSO-treated control cells. The combination of IV-43 and TNP-470 treatment was also able to decrease c-Src kinase activity, albeit to a lesser extent than bengamide A (Figure 4A). Similar results were obtained when endogeneous c-Src proteins were immunoprecipitated and used in the same in vitro kinase assay (Figure 4B). It is noteworthy that treatment with either IV-43 or TNP-470 alone did not affect c-Src kinase activity, further supporting the functional redundancy of these two MetAP enzymes. Addition of bengamide A to the in vitro kinase assay without any cellular treatment, however, did not change the tyrosine kinase activities of c-Src (Figure 4C).
Figure 4.

Bengamide A Reduces c-Src Tyrosine Kinase Activity.
All cells were treated with DMSO, IV-43 (10 μM), TNP-470 (100 nM), bengamide A (10 nM) and the combination of IV-43 (10 μM) and TNP-470 (100 nM) for 24 hours before harvest. (A). Acid-denatured enolase was used in the in vitro kinase assay for immunoprecipitated extrageneous c-Src autoradiography (upper panel), Commassie blue-stained enolase (middle panel) and anti-myc immunoblot (lower panel) are shown as gel-loading controls. (B) and (C). Immunoprecipitated endogeneous c-Src autoradiography is shown in the upper panel. Commassie blue-stained enolase (middle panel) and anti-Src immunoblot (lower panel) are shown as gel-loading controls. +Bengamide A represents the addition of bengamide A (100 nM) into the immunoprecipitates before the initiation of the kinase assay. (D) HeLa cell lysate was immunoblotted by anti-phospho-Tyr419 and anti-Tyr530. Immunoblots of total c-Src and β-actin are shown as gel loading controls. Na3PO4 (2 mM) was added to the culture medium for 15 min before harvesting the cells. (E) Immunoblotting of HeLa total cell lysate by anti-phosphotyrosine (clone 4G10). Arrows indicated the positions of change with 24-hours bengamide A (100 nM) treatment. Tubulin immunoblot is shown as the gel loading control. (F). Src++ MEF cell lysate is immunoblotted by anti-Tyr530 as an indication of active c-Src in Src++ MEF. Bengamide A (10 nM) was added to the culture medium for 24 hours. Na3PO4 (2 mM) was added to the medium for 15 min before harvesting the cells. Immunoblot of total c-Src is shown as gel loading control.
It has been well documented that dephosphorylation of Tyr530 and auto-phosphorylation of Tyr419 on c-Src protein are the consecutive crucial steps involved in the activation of c-Src tyrosine kinase [34]. We next used phosphorylation-specific antibodies to detect active c-Src levels in vivo. As shown in Figure 4D, the amount of phosphorylated Tyr419 c-Src increased when cells were treated with orthovanadate, a general protein tyrosine phosphatase inhibitor. Treatment with either IV-43 or TNP-470 had minor effects on Tyr419 phosphorylation, whereas bengamide A significantly reduced the phosphorylation of Tyr419. The combination of IV-43 and TNP-470 also decreased Tyr419 phosphorylation. We then used a monoclonal antibody that specifically recognizes dephosphorylated Tyr530 to indirectly measure the levels of active c-Src kinase in vivo. Because Na3VO4 inhibits protein tyrosine phosphatases, it is expected to enhance Tyr530 phosphorylation, as reflected in the reduction of the signal generated from this monoclonal antibody. While no obvious changes in Tyr530 phosphorylation were observed for either IV-43 or TNP-470, bengamide A reduced the level of unphosphorylated Tyr530, indicating an increase in the amount of phosphorylated Tyr530 and hence a less active c-Src kinase. Consistently, we also observed a decline in the presence of both IV-43 and TNP-470. Taken together, these results suggested that inhibition of MetAPs by either bengamide A, or the combination of IV-43 and TNP-470, decrease the c-Src tyrosine kinase activity in vivo.
Bengamide A Does Not Change Global Tyrosine Phosphorylation
To address the possibility that bengamide A function as a non-specific inhibitor for tyrosine kinases, we used a phospho-tyrosine specific antibody (clone 4G10) to detect general protein tyrosine phosphorylation. As shown in Figure 4E, bengamide A did not change protein tyrosine phosphorylation at the global level. However, bengamide A reduced tyrosine phosphorylation on a few proteins, with estimated molecular weights of 37 kDa, 30 kDa and 28 kDa, respectively (Figure 4E, arrows). These results suggested that bengamide A is not a general tyrosine kinase inhibitor.
Src Family Kinases Are Required for the Anti-proliferative Effects of Bengamide A
Since bengamides potently inhibit tumor cell proliferation, it remains possible that the observed inhibitory effects on c-Src are secondary to the anti-proliferative properties of bengamides. To address this possibility, we tested bengamide A on mouse embryonic fibroblasts (MEFs) derived from Src/Yes/Fyn (SYF)-deficient mice. As shown in Table 2, the IC50 for bengamide A on the proliferation of wild type MEFs was about 1.6 nM, while the IC50 for SYF cells was over 38-fold higher (61 nM). These results suggested that Src family kinases play an important role in mediating the anti-proliferative activity of bengamide A. This conclusion was further strengthened when bengamide A was found to inhibit Src++ MEFs, which express endogenous wild type c-Src protein but lack the expression of c-Yes and c-Fyn proteins, with an IC50 of 8.1 nM (Table 2). Western blot analysis with anti-Tyr530 antibody demonstrated that bengamide A inhibited the c-Src tyrosine kinase activity in Src++ MEFs (Figure 4F). As a control, all three cell lines are almost equally sensitive to Pateamine A, a potent inhibitor of eukaryotic translation initiation and cell proliferation [35,36]. The resistance observed in SYF MEFs suggested that inhibition of cell proliferation by bengamide A was not due simply to the inactivation of c-Src but was likely caused by the mislocalization of an inactive form of c-Src that \ had a dominant negative effect.
Table 2.
Src family kinases activities are required for cell proliferation inhibition by bengamide A.
| IC50s for MEF cell proliferation (nM) | |||
|---|---|---|---|
| Wild-Type | SYF | Src++ | |
| Bengamide A | 1.6 ± 0.2 | 61.1 ± 3.1 | 8.1 ± 1.0 |
| Pateamine A | 0.5 ± 0.08 | 0.2 ± 0.03 | 0.6 ± 0.1 |
SYF, mouse embryonic fibroblast (MEF) cells from Src/Yes/Fyn triple knock out mice. Src++, MEF cells from Yes/Fyn double knock out mice with the endogenous wildtype c-Src expression.
Bengamides Cause a Delay in Cell Division Cycle
Because Src family kinases are not only transiently activated during signal transduction [30], but also required for cell cycle progression from G2 phase to mitosis [31], we determined the effects of the bengamides on cell cycle by Fluorescent Activated Cell Sorting (FACS). HeLa cells were synchronized at G1/S transition by a double-thymidine block before fresh medium was added to resume the normal division cycle. As shown in Figure 5, there was no difference between vehicle- and bengamide A (10 nM) -treated cells at the 9-hour time point. However, a significant number of control cells progressed through mitosis after 12 hours, whereas most bengamide A-treated cells remained in G2/M phase. Similar effects had been observed for bengamide B and N (data not shown). Eventually, bengamide A-treated cells were able to complete mitosis 24-hours post release (data not shown). These results clearly indicated that the bengamides could delay the cell cycle progression at the G2/M phase.
Figure 5.

Bengamide A Delays Cell Cycle Progression During G2/M Phase.
G1/S-synchronized HeLa cells were released for 9 hours and 12 hours in the presence of bengamide A (100 nM) before fixed and stained by Propidium Iodide for FACS analysis.
Discussion
The reverse chemical genetic approach to identify molecular targets of bioactive small molecules has proven fruitful in advancing our understanding of a wide variety of fundamental cellular processes from signal transduction to cell cycle regulation [36,37]. Originally identified as a common target for the fumagillin/ovalicin family of natural products, human MetAP2 has been demonstrated to play an important role in endothelial cell proliferation [6,7]. Our recent chemical and genetic approaches have revealed that human MetAP1 is also essential in cell growth, in particular at the G2/M transition during cell division cycle [13]. However, the functional redundancy and overlapping substrates for the MetAPs have hindered the efforts to delineate the relevant downstream substrates that mediate the cellular effects of MetAP inhibitors. In this study, we have used several MetAP inhibitors, particularly the non-specific inhibitor bengamide A and the combination of MetAP1-specific and MetAP2-specific inhibitors (Table 1), to identify the physiologically relevant MetAP substrates and demonstrated that c-Src tyrosine kinase is one of the substrates involved in cell proliferation. We have shown that inhibition of both MetAP1 and MetAP2 by bengamide A led to the retention of the initiator methionine on the N-terminus of c-Src. This results in a decrease in c-Src N-terminal myristoylation, which changes the c-Src subcellular localization from the membrane to cytosol and nucleus. These changes are accompanied by a significant decrease in the tyrosine kinase activity of c-Src both in vitro and in vivo. The impaired N-terminal methionine removal for c-Src as a consequence of MetAP inhibition is likely to underlie the delay of cell cycle at G2/M phase caused by bengamide A.
The phosphorylation state of Tyr530 dictates c-Src activity and is dependent on a balance between protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs) [29]. Although PTKs, such as Csk and Chk, are predominantly localized in the cytosol of cells, the countering PTPs, such as PTPα, PTPλ and CD45, are membrane-bound. Therefore, c-Src tyrosine kinase is dynamically regulated both spatially and temporally. When nascent c-Src is synthesized, sequential processing by MetAPs and NMTs (N-myristoyl transferases) ensures its proper membrane localization. Meanwhile, cytosolic Csk phosphorylates Tyr530, keeping c-Src in a low-activity conformation. Our observation of basal c-Src tyrosine kinase activity in DMSO-treated cells is consistent with recent findings using a chemical rescue approach [38]. Once localized on the cellular membrane, c-Src can be activated by PTPs upon different receptor-activated signaling events. Inhibition of MetAPs will, however, alter the subcellular localization of c-Src and change the balance of its accessibility to PTKs and PTPs, thereby reducing c-Src activation and eventually arrest cell cycle prior to the entry into mitosis.
An estimated one hundred proteins are N-terminally myristoylated [22]. These proteins function in oncogenesis, signal transduction, and viral infection. Since N-terminal myristoylation is essentially specific to the N-terminal glycine in proteins, inhibition of methionine aminopeptidases will likely prevent the N-myristoylation of almost all these proteins. TNP-470 has been shown to block the N-myristoylation of endothelial nitric oxide synthase (eNOS) in endothelial cells [10,24], decreasing their production of nitric oxide [39,40]. Because bengamide A inhibits both isoforms of MetAP, it is possible that it also decreases N-myristoylation of other proteins, including the other Src family kinases members, c-Yes and c-Fyn. The disruption of N-myristoylation of other cellular proteins may explain the inhibition, albeit with lower potency, of SYF MEF cell proliferation by bengamide A.
Bengamide A and its analogs exhibit different inhibitory potencies for MetAP enzymes and cellular proliferation. This discrepancy is due in part to the relatively high concentrations of MetAP enzymes required for the in vitro enzymatic assay. Another contributing factor is that MetAP enzymes may not be the only targets for bengamides. Nonetheless, inhibition of MetAP enzymes does occur at the applied concentrations of bengamide A, as judged by the processing of endogenous MetAP substrates [19] and c-Src itself (Figure 1). We note that there is a difference in potency between the inhibition of Tyr419 autophosphorylation (Figure 4D) and the inhibition of c-Src tyrosine kinase activity by using enolase as a substrate (Figure 4A and 4B) for the combination of IV-43 and TNP-470. This difference is likely to arise from the conditions in the in vitro tyrosine kinase assay where saturating concentrations of both protein substrate and ATP were used. Results from such an in vitro assay may not quantitatively correlate with the Tyr419 phosphorylation status of c-Src in vivo.
The bengamides and MetAP1-specific inhibitors, especially pyridine-2-carboxylates (e.g., IV-43) [13,41], both lead to a delay in the progression through the G2/M phase. However, there are at least two significant differences between bengamide A and IV-43 in the underlying mechanisms. First, IV-43 alone is not sufficient to change the subcellular distribution of c-Src protein (Figure 3A), nor is it capable of reducing the activation of c-Src tyrosine kinase (Figure 4). Therefore, the cell cycle effect caused by IV-43 is not likely to be mediated through inhibition of c-Src. Second, bengamide A causes a much longer delay than IV-43. At the 12-hour time point, ~75% of IV-43-treated cells have already re-entered G0/G1 phase [13], whereas only ~25% of bengamide A-treated cells have passed through mitosis (Figure 5). It takes 24 hours for all bengamide A-treated cells to complete mitosis upon release from the double thymidine block, while only 16 hours are required for IV-43-treated cells to finish the cell cycle. It is likely that another MetAP1-specific substrate(s) mediates the G2/M phase delay observed for IV-43 and other MetAP1-specific inhibitors.
Previously, inhibition of MetAP2 by TNP-470 has been shown to activate p53 for cell cycle arrest [6,7]. In fact, the primary MEFs were demonstrated to be sensitive to TNP-470 and other MetAP2-specific inhibitors in a p53-dependent fashion [6, 7]. In contrast, the majority of transformed tumor cells are largely resistant to TNP-470, likely due to the pre-existing p53 mutations in them. Given that MetAP1 and MetaP2 are functionally redundant for processing N-terminal methionine of c-Src and other family members, inhibition of c-Src myristoylation is not likely to be involved in the inhibition of primary endothelial cells and MEFs by TNP-470. A recent study showed that such inhibition also blocks the non-canonical Wnt signaling pathway [42]. It has been shown that the Src family kinases, Fyn and Yes, and non-canonical Wnt signaling converge on RhoA in vertebrate gastrulation cell movements [43], suggesting a potential connection between the Src family kinases and non-canonical Wnt signaling. It is possible that bengamide A can modify both signaling pathways. The lack of effects on c-Src post-translational modification by either MetAP1- or MetAP2-specific inhibitors suggests that isoform-specific inhibitors may be capable of overcoming the toxicity observed for bengamide analogs as anticancer agents [18].
Significance
Methionine aminopeptidases are a family of essential enzymes in both prokaryotes and eukaryotes and represent attractive pharmacological targets. They have been shown to play important roles in a myriad of physiological processes. Few physiologically relevant substrates, however, have been identified to date. In this study, we identify and validate the proto-oncogene, c-Src, as a substrate for both MetAP1 and MetAP2 in vitro and in vivo. We show that inhibition of MetAPs by the non-selective inhibitor bengamide A alters the subcellular distribution of c-Src, significantly decreases its tyrosine kinase activity, and causes a remarkable delay in cell cycle progression through the G2/M transition. Together, these results establish a link between c-Src and MetAPs and suggest that inhibition of MetAPs could indirectly impair the functions of c-Src and likely other oncogenes that are essential for tumor growth.
Experimental Procedures
Cellular Treatment with Human Methionine Aminopeptidases Inhibitors
Cells in all experiments were treated for 24 hours unless indicated otherwise. DMSO was used as vehicle control. Human MetAP1-specific inhibitor IV-43 was added to a final concentration of 10 μM, which is the maximal soluble concentration in DMEM culture medium. Human MetAP2-specific inhibitor TNP-470 was added to a final concentration of 100 nM, the maximal concentration with minimum non-specific cellular toxicity. The MetAP non-selective inhibitor bengamide A was added to a final concentration of 10 nM. A combination of IV-43 and TNP-470 at 10 μM and 100 nM final concentrations, respectively, was used herein. The following concentrations were used in this study: 4 μg/mL for cycloheximide (Sigma, MO), 100 μM for 5-fluorouracil (Sigma, MO), 10 nM for PP2 (Calbiochem, CA) and 2 mM for Na3VO4 (Sigma, MO).
Measurement of N-terminal Methionine
This experiment is adapted from previously developed assay by Wang et al [9]. Briefly, wildtype c-Src cDNA (generous gift from Dr. Akhilesh Pandey, JHMI) was subcloned into pcDNA3.1 Myc/His A vector (Invitrogen, CA) by PCR for C-terminal tagging. The construct was transfected into HEK293 cells (ATCC) for 48 hours with drug treatment for the last 18 hours before harvest. Metabolic labeling of cells was carried out using L[35S]-methionine [>800 Ci/mmol] (PerkinElmer), which was added at 100 μCi/mL to cell media to label cellular proteins for the last 4 hours. Myc-tagged c-Src was immunoprecipitated by anti-myc antibody (9E10, Santa Cruz Biotech, Santa Cruz CA). Pelleted beads were washed with lysis buffer (modified RIPA, Upstate) extensively until the [35S] scintillation counting from each wash was reduced to background. Equal amount (20 μM) of recombinant human MetAP1 and MetAP2, purified as previously described [24,44], in 500 μL enzymatic reaction buffer (50 mM HEPES, pH 7.5, 150 mM NaCl and 100 μM CoCl2), was added to the pelleted beads for 1 hour and incubated at room temperature. The supernatant was scintillation-counted for [35S]-methionine (Microbeta, PerkinElmer, MA). Numeric reading is plotted by GraphPad Prism 4.0 software (San Diego, CA)
Measurement of N-terminal Myristoylation
To measure the N-terminal methionine level, C-terminally tagged c-Src was transfected into HEK293 cells for 48 hours before lysis by modified RIPA buffer. Cycloheximide (4 μg/mL), bengamide A (100 nM) and 5-Fluorouracil (100 μM) were added to cell media for the last 6 hours. Metabolic [3H]-myristic acid [54 Ci/mmol, 10 mCi/mL in EtOH] (PerkinElmer) was added to final concentration of 100 μCi/mL to label cellular proteins for the last 4 hours. Myc-tagged c-Src was immunoprecipitated and washed extensively with lysis buffer until [3H] counting from each wash was reduced to background. The pelleted beads were then incubated with 1 M hydroxylamine (pH 7.0) for 24 hours to remove any non-specific ester bonding with [3H]-myristic acid. The beads were then washed to reduce background reading, before [3H] scintillation counting was finally performed (Microbeta, PerkinElmer, MA). Numeric reading was plotted by GraphPad Prism 4.0 software.
Subcellular Fractionation
Experimental procedures were adapted from Gaschet and Hsu [45]. Cells (1 × 107) were collected from culture flasks by scraping and washed once with PBS. Cells were then suspended in hypotonic buffer consisting of 20 mM Tris·HCl pH 7.5, 2 mM EDTA, 5 mM EGTA and 10 mM β-mercaptoethanol, for 15 minutes before being sheared by Dounce homogenizer (30 strokes) on ice. The resulting homogenate was centrifuged at 1000 × g for 10 min at 4 °C to obtain a post-nuclear supernatant. This supernatant was further centrifuged at 200,000 × g for 30 min (TL-100 ultracentrifuge, Beckman) to obtain the cytosol (supernatant) and membrane (pellet) fractions. The pellet was washed with hypotonic buffer and the 200,000 × g centrifugation was repeated for 30 min. The membrane pellet was then dissolved in hypotonic buffer supplemented with 1% NP-40. Equal fractions of both were analyzed by SDS-PAGE followed by immunoblotting using appropriate antibodies.
Cell Culture and Immunofluorescent Staining
HeLa cell line was obtained from ATCC and cultured according to vendors instructions. Procedures for indirect immunofluorescent staining were adapted from Dang et al [46]. Briefly, cells were plated on cover slips and allowed to recover for 16–24 hours before treated with bengamide A (10 nM) for 24 hours. Cells were then fixed with 4% para-formaldehyde for 15 min, washed in PBS, permeabilized by 0.5% Triton X-100 and blocked with 10% donkey serum in PBS prior to 1 hour incubation with primary anti-Src antibody (sc-5266), purchased from Santa Cruz Biotech. (Santa Cruz, CA). Cells were subsequently incubated in three changes of PBS for 5 min each before incubation with FITC-conjugated secondary antibody for 1 hour, washed in PBS 3 times for 5 min each and finally mounted. Vectashield mounting medium (Vector Laboratories) was used and images were captured using Zeiss LSM510 confocal microscope with C-Apochromat 63× objective. Images were processed by LSM5 Image Examiner and/or Adobe Photoshop CS2. Data obtained from the green/FITC channel are shown in green.
In vitro Tyrosine Kinase Assay
The in vitro tyrosine kinase assay is adapted from Current Protocols in Protein Science (1997) 13.7.1–13.7.22, using acid-denatured rabbit muscle enolase (SigmaAldrich, MO) as a substrate. Briefly, C-terminally tagged c-Src was transfected into HEK293 for 48 hours. Cells were treated with different drugs for the last 24 hours before lysis in modified RIPA buffer for immunoprecipitation by either anti-Myc (9E10) or anti-c-Src (GD11, Upstate) monoclonal antibodies. The immunoprecipitate was washed 3 times with lysis buffer and once with assay buffer (20 mM HEPES, 1 mM MnCl2, 1 mM DTT). The pelleted beads were then suspended in assay buffer and divided into 2 aliquots. One aliquot was used to determine the c-Src abundance by immunoblotting, the other aliquot was used for the in vitro kinase assay. For the kinase assay, the pellet was suspended in 20 μL assay buffer plus 2.5 μg acid-denatured enolase, 5 μCi [32P]-γ-ATP and 5 μM ATP, incubated at 30 °C for 30 min. PP2 (10 nM) or +bengamide A (100 nM) was added before the 30 °C incubation. Reaction was stopped by the addition of 20 μL 2× SDS loading buffer and boiling for 5 min. The mixture was then briefly centrifuged and the supernatant was resolved on 10% SDS-PAGE gel. Phosphorylated enolase was detected by autoradiography.
In vivo c-Src Phosphorylation and Global Tyrosine Phosphorylation
Src [pY419] polyclonal antibody (#44–660G) and active-Src [Y530] monoclonal antibody (#AHO0051) were purchased from BioSource (Invitrogen, CA). Global tyrosine phosphorylation status was determined by anti-phosphotyrosine monoclonal antibody (4G10) (a generous gift from Dr. Akhilesh Pandey, JHMI). Na3VO4 (2 mM) was added 15 min before HeLa cells were scraped and washed with PBS before lysis by modified RIPA buffer (including proteases inhibitors). Total cell lysates were centrifuged at 1000 × g for 10 min to obtain a post-nuclear supernatant. Protein concentrations for the supernatants were measured and normalized. 4× SDS loading buffer was added to the sample and boiled for 5 min. The samples were resolved by a 10% SDS-PAGE gel before transferred to nitrocellulose membrane for Western Blot.
[3H]-Thymidine Uptake Assay, Cell Cycle Synchronization and Analysis
Assay for [3H]-thymidine uptake was carried out as previously described [12]. [3H]-Thymidine was purchased from PerkinElmer (Wellesley, MA). HCT116, BAEC and Src++ cells were purchased from ATCC (Manassas, VA). SYF MEF cells were generously provided by the Phil A. Cole lab. HeLa cell synchronization by double–thymidine block and subsequent FACS analysis for cell cycle progression were conducted as previously described [13].
Acknowledgments
We would like to thank Dr. Akhilesh Pandey (JHMI) and Jun Zhong for the cDNA of wild type c-Src and 4G10 antibody. We are grateful for access of the microplate reader from Dr. Theresa A. Sharpiro’s lab (JHMI). We also thank Drs. Yingfeng Qiao and Ron Bose from the Cole lab for their help. We appreciate many helpful discussions with Dr. Woon-Kai Low and Jing Xu, and the critical reading of the manuscript by members of the Liu lab. X.H. was a Predoctoral Fellow from Department of Defense Breast Cancer Program. This work was supported by NIH (CA078743) to J.O.L.; NIH (CA074305) to P.A.C.; NIH (CA047135 and CA052955) to P.C.; and by Keck Center to J.O.L. and P.A.C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Lowther WT, Matthews BW. Structure and function of the methionine aminopeptidases. Biochim Biophys Acta. 2000;1477:157–167. doi: 10.1016/s0167-4838(99)00271-x. [DOI] [PubMed] [Google Scholar]
- 2.Chang SY, McGary EC, Chang S. Methionine aminopeptidase gene of Escherichia coli is essential for cell growth. J Bacteriol. 1989;171:4071–4072. doi: 10.1128/jb.171.7.4071-4072.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X, Chang YH. Amino-terminal protein processing in Saccharomyces cerevisiae is an essential function that requires two distinct methionine aminopeptidases. Proc Natl Acad Sci U S A. 1995;92:12357–12361. doi: 10.1073/pnas.92.26.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffith EC, Su Z, Turk BE, Chen S, Chang YH, Wu Z, Biemann K, Liu JO. Methionine aminopeptidase (type 2) is the common target for angiogenesis inhibitors AGM-1470 and ovalicin. Chem Biol. 1997;4:461–471. doi: 10.1016/s1074-5521(97)90198-8. [DOI] [PubMed] [Google Scholar]
- 5.Sin N, Meng L, Wang MQ, Wen JJ, Bornmann WG, Crews CM. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proc Natl Acad Sci U S A. 1997;94:6099–6103. doi: 10.1073/pnas.94.12.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y, Griffith EC, Sage J, Jacks T, Liu JO. Cell cycle inhibition by the anti-angiogenic agent TNP-470 is mediated by p53 and p21WAF1/CIP1. Proc Natl Acad Sci U S A. 2000;97:6427–6432. doi: 10.1073/pnas.97.12.6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeh JR, Mohan R, Crews CM. The antiangiogenic agent TNP-470 requires p53 and p21CIP/WAF for endothelial cell growth arrest. Proc Natl Acad Sci U S A. 2000;97:12782–12787. doi: 10.1073/pnas.97.23.12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mukherji M, et al. Genome-wide functional analysis of human cell-cycle regulators. Proc Natl Acad Sci U S A. 2006;103:14819–14824. doi: 10.1073/pnas.0604320103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J, et al. Tumor suppression by a rationally designed reversible inhibitor of methionine aminopeptidase-2. Cancer Res. 2003;63:7861–7869. [PubMed] [Google Scholar]
- 10.Satchi-Fainaro R, et al. Inhibition of vessel permeability by TNP-470 and its polymer conjugate, caplostatin. Cancer Cell. 2005;7:251–261. doi: 10.1016/j.ccr.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 11.Lu J, Chong CR, Hu X, Liu JO. Fumarranol, a rearranged fumagillin analogue that inhibits angiogenesis in vivo. J Med Chem. 2006;49:5645–5648. doi: 10.1021/jm060559v. [DOI] [PubMed] [Google Scholar]
- 12.Hu X, Addlagatta A, Matthews BW, Liu JO. Identification of Pyridinylpyrimidines as Inhibitors of Human Methionine Aminopeptidases. Angew Chem Int Ed Engl. 2006;45:3772–3775. doi: 10.1002/anie.200600757. [DOI] [PubMed] [Google Scholar]
- 13.Hu X, Addlagatta A, Lu J, Matthews BW, Liu JO. Elucidation of the function of type 1 human methionine aminopeptidase during cell cycle progression. Proc Natl Acad Sci U S A. 2006;103:18148–18153. doi: 10.1073/pnas.0608389103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quinoa E, Adamczeski M, Crews P, Bakus GJ. Bengamides, heterocyclic anthelmintics from a Jaspidae marine sponge. J Org Chem. 1986;51:4494–4497. [Google Scholar]
- 15.Adamczeski M, Quinoa E, Crews P. Novel sponge-derived amino acids. 11 The entire absolute stereochemistry of the bengamides. J Org Chem. 1990;55:240–242. [Google Scholar]
- 16.Kinder FR, Jr, et al. Synthesis and antitumor activity of ester-modified analogues of bengamide B. J Med Chem. 2001;44:3692–3699. doi: 10.1021/jm010188c. [DOI] [PubMed] [Google Scholar]
- 17.Thale Z, et al. Bengamides revisited: new structures and antitumor studies. J Org Chem. 2001;66:1733–1741. doi: 10.1021/jo001380+. [DOI] [PubMed] [Google Scholar]
- 18.Dumez H, Gall H, Capdeville R, Dutreix C, van Oosterom AT, Giaccone G. A phase I and pharmacokinetic study of LAF389 administered to patients with advanced cancer. Anticancer Drugs. 2007;18:219–225. doi: 10.1097/CAD.0b013e328010ef5b. [DOI] [PubMed] [Google Scholar]
- 19.Towbin H, et al. Proteomics-based target identification: bengamides as a new class of methionine aminopeptidase inhibitors. J Biol Chem. 2003;278:52964–52971. doi: 10.1074/jbc.M309039200. [DOI] [PubMed] [Google Scholar]
- 20.Varshavsky A. The N-end rule: functions, mysteries, uses. Proc Natl Acad Sci U S A. 1996;93:12142–12149. doi: 10.1073/pnas.93.22.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perrier J, Durand A, Giardina T, Puigserver A. Catabolism of intracellular N-terminal acetylated proteins: involvement of acylpeptide hydrolase and acylase. Biochimie. 2005;87:673–685. doi: 10.1016/j.biochi.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 22.Boutin JA. Myristoylation. Cell Signal. 1997;9:15–35. doi: 10.1016/s0898-6568(96)00100-3. [DOI] [PubMed] [Google Scholar]
- 23.Massiere F, Badet-Denisot MA. The mechanism of glutamine-dependent amidotransferases. Cell Mol Life Sci. 1998;54:205–222. doi: 10.1007/s000180050145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turk BE, Griffith EC, Wolf S, Biemann K, Chang YH, Liu JO. Selective inhibition of amino-terminal methionine processing by TNP-470 and ovalicin in endothelial cells. Chem Biol. 1999;6:823–833. doi: 10.1016/s1074-5521(99)80129-x. [DOI] [PubMed] [Google Scholar]
- 25.Chen S, Vetro JA, Chang YH. The specificity in vivo of two distinct methionine aminopeptidases in Saccharomyces cerevisiae. Arch Biochem Biophys. 2002;398:87–93. doi: 10.1006/abbi.2001.2675. [DOI] [PubMed] [Google Scholar]
- 26.Miller DM, Kurzban GP, Mendoza JA, Chirgwin JM, Hardies SC, Horowitz PM. Recombinant bovine rhodanese: purification and comparison with bovine liver rhodanese. Biochim Biophys Acta. 1992;1121:286–292. doi: 10.1016/0167-4838(92)90158-a. [DOI] [PubMed] [Google Scholar]
- 27.Raftery MJ, Harrison CA, Alewood P, Jones A, Geczy CL. Isolation of the murine S100 protein MRP14 (14 kDa migration-inhibitory-factor-related protein) from activated spleen cells: characterization of post-translational modifications and zinc binding. Biochem J. 1996;316(Pt 1):285–293. doi: 10.1042/bj3160285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmitz S, Clayton J, Nongthomba U, Prinz H, Veigel C, Geeves M, Sparrow J. Drosophila ACT88F indirect flight muscle-specific actin is not N-terminally acetylated: a mutation in N-terminal processing affects actin function. J Mol Biol. 2000;295:1201–1210. doi: 10.1006/jmbi.1999.3407. [DOI] [PubMed] [Google Scholar]
- 29.Bjorge JD, Jakymiw A, Fujita DJ. Selected glimpses into the activation and function of Src kinase. Oncogene. 2000;19:5620–5635. doi: 10.1038/sj.onc.1203923. [DOI] [PubMed] [Google Scholar]
- 30.Chackalaparampil I, Shalloway D. Altered phosphorylation and activation of pp60c-src during fibroblast mitosis. Cell. 1988;52:801–810. doi: 10.1016/0092-8674(88)90422-9. [DOI] [PubMed] [Google Scholar]
- 31.Roche S, Fumagalli S, Courtneidge SA. Requirement for Src family protein tyrosine kinases in G2 for fibroblast cell division. Science. 1995;269:1567–1569. doi: 10.1126/science.7545311. [DOI] [PubMed] [Google Scholar]
- 32.Vaughan MD, Sampson PB, Honek JF. Methionine in and out of proteins: targets for drug design. Curr Med Chem. 2002;9:385–409. doi: 10.2174/0929867023371102. [DOI] [PubMed] [Google Scholar]
- 33.Erpel T, Courtneidge SA. Src family protein tyrosine kinases and cellular signal transduction pathways. Curr Opin Cell Biol. 1995;7:176–182. doi: 10.1016/0955-0674(95)80025-5. [DOI] [PubMed] [Google Scholar]
- 34.Cole PA, Shen K, Qiao Y, Wang D. Protein tyrosine kinases Src and Csk: a tail’s tale. Curr Opin Chem Biol. 2003;7:580–585. doi: 10.1016/j.cbpa.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 35.Northcote PT, John W, Murray HG. Pateamine: a potent cytotoxin from the New Zealand Marine sponge, mycale sp. Tetrahedron Letters. 1991;32:6411–6414. [Google Scholar]
- 36.Low WK, Dang Y, Schneider-Poetsch T, Shi Z, Choi NS, Merrick WC, Romo D, Liu JO. Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol Cell. 2005;20:709–722. doi: 10.1016/j.molcel.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 37.Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 38.Qiao Y, Molina H, Pandey A, Zhang J, Cole PA. Chemical rescue of a mutant enzyme in living cells. Science. 2006;311:1293–1297. doi: 10.1126/science.1122224. [DOI] [PubMed] [Google Scholar]
- 39.Yoshida T, Kaneko Y, Tsukamoto A, Han K, Ichinose M, Kimura S. Suppression of hepatoma growth and angiogenesis by a fumagillin derivative TNP470: possible involvement of nitric oxide synthase. Cancer Res. 1998;58:3751–3756. [PubMed] [Google Scholar]
- 40.Mauriz JL, Linares P, Macias RI, Jorquera F, Honrado E, Olcoz JL, Gonzalez P, Gonzalez-Gallego J. TNP-470 inhibits oxidative stress, nitric oxide production and nuclear factor kappa B activation in a rat model of hepatocellular carcinoma. Free Radic Res. 2003;37:841–848. doi: 10.1080/1071576031000136577. [DOI] [PubMed] [Google Scholar]
- 41.Luo QL, et al. Discovery and structural modification of inhibitors of methionine aminopeptidases from Escherichia coli and Saccharomyces cerevisiae. J Med Chem. 2003;46:2631–2640. doi: 10.1021/jm0300532. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, et al. A chemical and genetic approach to the mode of action of fumagillin. Chem Biol. 2006;13:1001–1009. doi: 10.1016/j.chembiol.2006.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jopling C, den Hertog J. Fyn/Yes and non-canonical Wnt signalling converge on RhoA in vertebrate gastrulation cell movements. EMBO Rep. 2005;6:426–431. doi: 10.1038/sj.embor.7400386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Addlagatta A, Hu X, Liu JO, Matthews BW. Structural basis for the functional differences between type I and type II human methionine aminopeptidases. Biochemistry. 2005;44:14741–14749. doi: 10.1021/bi051691k. [DOI] [PubMed] [Google Scholar]
- 45.Gaschet J, Hsu VW. Distribution of ARF6 between membrane and cytosol is regulated by its GTPase cycle. J Biol Chem. 1999;274:20040–20045. doi: 10.1074/jbc.274.28.20040. [DOI] [PubMed] [Google Scholar]
- 46.Dang Y, Kedersha N, Low WK, Romo D, Gorospe M, Kaufman R, Anderson P, Liu JO. Eukaryotic initiation factor 2alpha-independent pathway of stress granule induction by the natural product pateamine A. J Biol Chem. 2006;281:32870–32878. doi: 10.1074/jbc.M606149200. [DOI] [PubMed] [Google Scholar]