

Abstract
Hippuristanol is a natural product that has recently been shown to inhibit eukaryotic translation initiation and tumor cell proliferation. To investigate the structure-and-activity relationship of hippuristanol, we synthesized a series of analogs by expanding the size of its F ring and determined their effects on the proliferation of cancer cell lines. All changes to the F ring of hippuristanol resulted in 3-fold to >100-fold decrease in activity.
Hippuristanol, a polyoxygenated steroid with potent cytotoxicities against tumor cell lines, was discovered from the gorgonian Isis hippuris.1 Recently, it was reported that this natural product appeared to work through eukaryotic initiation factor (eIF)4A I and II, an ATP-dependent RNA helicase, blocking its binding to mRNA hence inhibiting translational initiation, making it the second small molecule, after the marine natural product pateamine A, that modulate the function of eIF4A protein family.2 Given the critical roles of translation in cell proliferation, hippuristanol is considered a promising lead for the development of anti-tumor agents. Moreover, translation is also necessary for viral infection, suggesting that hippuristanol has potential as an antiviral agent.
Recently, we disclosed the first synthesis of hippuristanol using the commercially available hydrocortisone as a starting material.3 A key step in the synthesis entailed the addition of 2,3-dihydrofuran to a β-hydroxymethylketone precursor to construct the spiroketal functionality (Fig. 1). This chemistry rendered it possible to attain several analogs with structural alteration at the F-ring of hippuristanol. The preliminary structure/activity relationship data suggested that besides the three hydroxyls at C3, C11 and C20, the “R” configuration at C22 and the three methyl groups on the F ring were critical for activity. Herein, we report the synthesis of a new series of analogs of hippuristanol with altered structures and stereochemistry of the E and F rings and the determination of their antiproliferative activity.
Figure 1.

Hippuristanol and its synthetic strategy.
In our previous preliminary SAR study, we adopted a structural “contraction” strategy by making analogs with the removal of a subset or all of the methyl groups on the F-ring of hippuristanol in addition to altering the stereochemistry at C22, C20 and C24.3 In the present work, we decided to proceed, for most analogs, with structural “expansion”, i.e., increasing the ring size from five to six, adding an extra methyl group or enlarging the methyl groups to ethyl groups.
For the F-ring building blocks, we began with two commercially available α, α-dimethyl-γ-butyrolactone 2 and γ-butyrolactone 3 (Scheme 1). Nucleophilic attack of the γ-butyrolactone derivatives with the Grignard reagents (prepared from magnesium with iodomethane or bromoethane) in Et2O at 30 °C led to the corresponding diols, which were then oxidized to 3,3,4,4-tetramethylbutyrolactone 4 or 4,4-diethylbutyrolactone 5 with the required methyl or ethyl groups by PCC, in 83% and 86% yield, respectively.3,4 Treatment of the resulting lactones with DIBAL-H at −68 °C in CH2Cl2 offered pairs of racemic acetals that were stable below 20 °C. Finally, the acetals were dehydrated at high temperature (250 °C) without any solvent to provide the desired 2,2,3,3-tetramethyl-2,3-dihydrofuran 6 and 2,2-diethyl-2,3-dihydrofuran 7 in 62% and 65% yield, respectively.3,5
Scheme 1.

Reagents and conditions: (a) MeMgI or EtMgBr, Et2O, 30 °C; (b) PCC, CH2Cl2, rt, 83% (for 4, two steps) and 86% (for 5, two steps); (c) DIBAL-H, CH2Cl2, −68 °C; (d) Al2O3, 250 °C, 62% (for 6, two steps) and 65% (for 7, two steps).
To prepare an F-ring analog lacking one of the methyl groups, the commercial (−)-D-pantolactone 8 was chosen as the starting precursor (Scheme 2). Activation of the α-hydroxyl group at pantolactone 8 with an Ms group in CH2Cl2, followed by reduction in the presence of Zn and NaI, provided 3,3-dimethylbutyrolactone 9 in 86% yield.6 Subsequently, reduction and dehydration of 9 gave rise to the corresponding 3,3-dimethyl-2,3-dihydrofuran 10 in 60% yield.
Scheme 2.

Reagents and conditions: (a) MsCl, Et3N, DMAP, CH2Cl2, 0 °C; (b) Zn, NaI, DME, 86% (for two steps); (c) DIBAL-H, CH2Cl2, −68 °C; (d) Al2O3, 200 °C, 60% (for two steps).
With the F-ring precursors in hand, we synthesized their corresponding hippuristanol analogs using similar chemistry as that for the synthesis of hippuristanol (Scheme 3).3 Treatment of the three 2,3-dihydrofuran derivatives prepared above with t-BuLi in THF resulted in the corresponding 5-lithio-2,3-dihydrofuran, which were then added to ketone 1 at 0 °C to provide mainly the required 20β-OH adducts. Otherwise, low temperature (−68 °C) led mainly to the adducts with 20α-OH configuration, which we previously found dramatically decreased the antiproliferative activity. The crude products with 20β-OH were acidified directly with the addition of 5% HCl to open the 2,3-dihydrofuran ring to introduce the side chain with a 22-carbonyl and a 25-hydroxyl groups.7 Cyclization of the resulting intermediate containing 16-hydroxyl, 22-carbonyl and 25-hydroxyl substituents was then achieved to provide the desired 22-spiro-ketal functionalities. Subsequent deprotection of the remaining Bz groups with LiAlH4 furnished the desired analogs 11a/11b, 12a/12b, 13a/13b in satisfactory yields.3,8 Analog 14 with 20α-OH were also found in a 17% yield. We note that the epimerization of 22-spiro-ketal could still be observed on acid conditions, suggesting that substitutions in the F ring did not influence the epimerization significantly.
Scheme 3.

Reagents and conditions: (a) t-BuLi, THF, 0 °C; (b) 0.1 M HCl, THF, rt; (c) LiAlH4, THF, 40 °C.
Generally, ketals with six-membered rings are more difficult to epimerize, compared to five-membered rings. Thus, the same procedure used for the preparation of analogs with five-membered F rings was employed on the commercial 3,4-dihydropyran 15 with 1 to synthesize analogs 19a/19b with six-membered F rings in good yields (Scheme 4).3,7 To obtain ring-expansion analogs with methyl substitutions, the commercially available ketone 16 was converted to lactone 17 using Baeyer-Villiger rearrangement in 67% yield.9 Subsequent reduction and dehydration of lactone 17 provided 2,2-dimethyl-3,4-dihydropyran precursor 18. However, the same procedure employed on 2,2-dimethy-3,4-dihydropyran with ketone 1 offered the desired 20a/20b in only 3% and 7% yield, respectively, for the 6-lithio-2,2-dimethyl-3,4-dihydropyran (prepared from 2,2-dimethyl-3,4-dihydropyran with t-BuLi in THF) mainly caused the elimination of 16β-OH to result in a α,β-unsaturated ketone, instead of the expected 20β-OH adduct. Despite the low yields, sufficient amount of each analog was obtained upon scaling up the reaction to enable their evaluation in cell proliferation assays. However, the final congeners with the six-membered F rings (19 and 20) could still epimerize at 22-spiro-ketal upon acidic treatment.
Scheme 4.

Reagents and conditions: (a) t-BuLi, THF, 0 °C; (b) 0.1 M HCl, THF, rt; (c) LiAlH4, THF, 40 °C; (d) 30% H2O2, (CF3CO)2O, rt, 67%; (e) DIBAL-H, CH2Cl2, −68 °C; (f) Al2O3, 250 °C, 37% (two steps).
We determined the activity of the new analogs in HeLa cell proliferation assay using incorporation of [3H]thymidine as a readout (Table 1).3 In the previous study, it was found that R stereochemistry at C22 is essential for activity. The same stereochemical requirement at C22 is seen in the new series of analogs among several pairs of diastereomers that differ only in the stereochemistry at C22. Thus, analogs 11a and 12a are both significantly more active than 11b and 12b. The same stereochemical requirement also applies to the analogs with six-membered F rings with analogs 19a and 20a being much more active than 19b and 20b. For the substituents on the F ring, replacement of the gem-dimethyl3 with gem-diethyl substituents led to a 10-fold decrease in activity (12a). Together with the previous observation that an analog that lacks the two methyl group is inactive,3 these results suggest that the gem-dimethyl substition is optimal for activity. It is interesting to note that the analogs with expanded six-membered F rings retained antiproliferative activity. In particular, 19a whose counterpart with five-membered F ring is inactive,3 is moderately active. This may be due to the potential compensation of the missing gem-dimethyl groups by the methylene carbon inserted into the F ring, partially compensating for the lost hydrophobic interaction with the binding pocket in eIF4A. Restoration of the gem-dimethyl group in 19a led to further enhancement of the activity (20a). Together, these results helped to further define the boundary for future design and synthesis of analogs of hippuristanol with improved stability and high biological activity as leads for developing anticancer and antiviral agents.
Table 1.
Antiproliferative Activity of Hippuristanol Analogs.10
| Cpd
|
Structures
|
IC50 (μM)
|
|---|---|---|
| Hip |

|
0.07 |
| 11a |
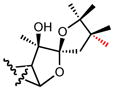
|
0.2 |
| 11b |
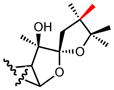
|
2.2 |
| 12a |

|
3.6 |
| 12b |
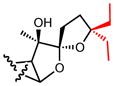
|
>10 |
| 13a |
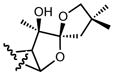
|
>10 |
| 13b |
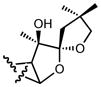
|
>10 |
| 14 |
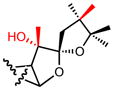
|
>10 |
| 19a |
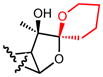
|
1.6 |
| 19b |
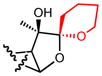
|
>10 |
| 20a |
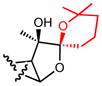
|
0.6 |
| 20b |
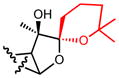
|
4.9 |
Acknowledgments
We thank Prof. Carlos Jiménez of the Universidade de A Coruña (Spain) for providing NMR spectra of the authentic hippuristanol and 22-epi-hippuristanol. Financial support from the National Natural Science Foundation of China (20728202 and 20621062) and the Chinese Academy of Sciences (KJCX2-YW-H08) are gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Jun O. Liu, Email: joliu@jhu.edu.
Biao Yu, Email: byu@mail.sioc.ac.cn.
Reference and notes
- 1.(a) Higa T, Tanaka J-i, Tsukitani Y, Kikuchi H. Chem Lett. 1981:1647. [Google Scholar]; (b) Rao CB, Ramana KV, Rao DV, Fahy E, Faulkner DJ. J Nat Prod. 1988;51:954. [Google Scholar]; (c) Gonzalez N, Barral MA, Rodriguez J, Jimenez C. Tetrahedron. 2001;57:3487. [Google Scholar]
- 2.(a) Bordeleau M-E, Mori A, Oberer M, Lindqvist L, Chard LS, Higa T, Wagner GJ, Belsham G, Tanaka J, Pelletier J. Nat Chem Biol. 2006;2:213. doi: 10.1038/nchembio776. [DOI] [PubMed] [Google Scholar]; (b) Lindqvist L, Oberer M, Reibarkh M, Cencic R, Bordeleau M-E, Voqt E, Marintchev A, Tanaka J, Fagotto F, Altmann M, Wagner G, Pelletier J. PLoS One. 2008;3:e1583. doi: 10.1371/journal.pone.0001583. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Low WK, Dang Y, Schneider-Poetsch T, Shi Z, Choi NS, Merrick WC, Romo D, Liu JO. Mol Cell. 2005;20:709. doi: 10.1016/j.molcel.2005.10.008. [DOI] [PubMed] [Google Scholar]; (d) Bordeleau M-E, Matthews J, Wojnar JM, Lindqvist L, Novac O, Jankowsky E, Sonenberg N, Northcote P, Teesdale-Spittle P, Pelletier J. Proc Natl Acad Sci U S A. 2005;102:10460. doi: 10.1073/pnas.0504249102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Clardy J. ACS Chem Biol. 2006;1:17. doi: 10.1021/cb0600029. [DOI] [PubMed] [Google Scholar]; (f) Pestova TV, Hellen CU. Nat Chem Biol. 2006;2:176. doi: 10.1038/nchembio0406-176. [DOI] [PubMed] [Google Scholar]
- 3.Li W, Dang Y, Liu JO, Yu B. Chem--Eur J. 2009;15:10356. doi: 10.1002/chem.200901732. [DOI] [PubMed] [Google Scholar]
- 4.(a) Barua NC, Schmidt RR. Synthesis. 1986:891. [Google Scholar]; (b) Martin D, Marcos D, Basabe ISP, Romero RE, Moro RF, Lumeras W, Rodríguez L, Urones JG. Synthesis. 2001:1013. [Google Scholar]; (c) Baklan VF, Antipenkova LS, Zakharchenko LI, Fesenko TE, Kukhar VP. Russ J Org Chem. 1984;20:1212. [Google Scholar]; (d) Fukuzawa S–i, Sumimoto N, Fujinami T, Sakai S. J Org Chem. 1990;55:1628. [Google Scholar]
- 5.(a) Botteghi C, Consiglio G, Ceccarelli G, Stefani A. J Org Chem. 1972;37:1835. [Google Scholar]; (b) Paquette LA, Lanter JC, Johnston JN. J Org Chem. 1997;62:1702. [Google Scholar]; (c) Hennessy AJ, Connolly DJ, Malone YM, Guiry PJ. Tetrahedron Lett. 2000;41:7757. [Google Scholar]
- 6.(a) Akbutina FA, Sadretdinov IF, Vasil'eva EV, Miftakhov MS. Russ J Org Chem. 2001;37:695. [Google Scholar]; (b) Ueki T, Kinoshita T. Org Biomol Chem. 2004;2:2777. doi: 10.1039/B407820A. [DOI] [PubMed] [Google Scholar]; (c) Doyle MP, Dyatkin AB. J Org Chem. 1995;60:3035. [Google Scholar]
- 7.(a) Szendi Z, Sweet F. Steroids. 1991;56:458. doi: 10.1016/0039-128x(91)90001-c. [DOI] [PubMed] [Google Scholar]; (b) Hedtmann U, Klintz R, Hobert K, Frelek J, Vlahov I, Welzel P. Tetrahedron. 1991;47:3753. [Google Scholar]; (c) Hobert U, Hedtmann K, Klintz R, Welzel P, Frelek J, Strangmann-Diekmann M, Klöne A, Pongs O. Angew Chem, Int Ed. 1989;28:1515. [Google Scholar]
- 8.Procedure for the synthesis of analogs 12a and 12b: To a solution of 2,2-diethyl-2,3-dihydrofuran 7 (200 μL, 1.46 mmol) in anhydrous THF (3 mL) was added t-BuLi (1.5 M in pentane, 1.3 mL, 1.95 mmol) at −68 °C. The reaction was allowed to warm to 0 °C and the stirring continued for another 15 min at rt. The mixture was cooled to 0 °C again and ketone 1 (91 mg, 0.16 mmol) in anhydrous THF (1 mL) was added. After stirring for 1 h, the reaction was quenched with H2O. The resulting mixture was diluted with EtOAc (30 mL), and washed with brine. The organic layer was dried, filtered, and concentrated. The residue was dissolve in THF (4 mL) and 0.1 M HCl (1 mL). After stirring for 2 h, the mixture was diluted with EtOAc (30 mL), washed with brine, dried, and concentrated. The crude product was dissolved in anhydrous THF (2 mL), and LiAlH4 (33 mg, 0.87 mmol) was added. After stirring for 20 min at 40 °C, the reaction was quenched with H2O slowly. To the resulting mixture was added EtOAc (10 mL), NaHCO3 (1 g) and Na2SO4 (1 g), and the stirring continued for another 3 h. The mixture was then filtered and concentrated to give a residue, which was purified by silica gel column chromatography (petroleum ether-EtOAc, 2:1) to afford analogs 12a (34 mg, 44%) and 12b (10 mg, 13%).
- 9.(a) Lane KJ, Pinder AR. J Org Chem. 1982;47:3171. [Google Scholar]; (b) Kaneda K, Ueno S, Imanaka T. Chem Commun (Cambridge, U K) 1994:797. [Google Scholar]
- 10.Analytical data for the analogs 11a/11b, 12a/12b, 13a/13b, 14, 19a/19b, 20a/20b. Compound 11a: [α]D28 39.9 (c 0.87, CHCl3); 1H NMR (400 MHz, CDCl3): δ 4.32–4.27 (m, 2 H), 4.05 (br s, 1 H), 3.10 (s, 1 H), 2.21–2.17 (m, 1 H), 2.16–2.12 (m, 1 H), 2.06–2.00 (m, 1 H), 1.39 (s, 3 H), 1.29 (s, 3 H), 1.21 (s, 3 H), 1.12 (s, 3 H), 1.08 (s, 3 H), 1.03(s, 3 H), 0.94 (s, 3 H), 0.80–0.76 (m, 1 H); 13C NMR (100 MHz, CDCl3): δ 113.7, 86.2, 79.8, 78.8, 67.7, 66.0, 65.4, 57.8, 56.7, 49.3, 47.4, 41.7, 41.6, 39.5, 35.9, 34.9, 33.8, 32.1, 31.2, 29.8, 28.4, 28.2, 27.5, 25.2, 24.1, 23.9, 23.8, 18.2, 13.6; HR-ESIMS calcd for C29H48O5Na [M + Na]+ 499.33940, found 499.33924. Compound 11b: [α 27 −16.1 (c 0.31, 1H NMR (400 ] DCHCl3); MHz, CDCl3): δ 4.44–4.39 (m, 1 H), 4.30 (br s, 1 H), 4.05 (br s, 1 H), 2.44 (d, J = 14.4 Hz, 1 H), 2.16–2.12 (m, 1 H), 2.07–2.01 (m, 1 H), 1.37 (s, 3 H), 1.31 (s, 3 H), 1.20 (s, 3 H), 1.08 (s, 3 H), 1.04 (s, 3 H), 1.03 (s, 3 H), 0.93 (s, 3 H), 0.81–0.77 (m, 1 H); 13C NMR (100 MHz, CDCl3): δ 117.4, 85.5, 82.7, 78.7, 67.7, 66.2, 66.0, 57.77, 57.75, 48.5, 46.9, 41.8, 41.5, 39.5, 35.8, 34.9, 31.9, 31.8, 31.3, 29.9, 28.2, 27.4, 27.2, 24.6, 24.3, 23.8, 23.7, 18.5, 13.7; HR-ESIMS calcd for C29H48O5Na [M + Na]+ 499.33940, found 499.33975. Compound 12a: [α]D28 +38.0 (c 0.52, CHCl3); 1H NMR (400 MHz, CDCl3): δ 4.36–4.30 (m, 2 H), 4.05 (br s, 1 H), 3.08 (s, 1 H), 2.23–2.19 (m, 1 H), 2.08–2.01 (m, 1 H), 1.42 (s, 3 H), 1.31 (s, 3 H), 1.04 (s, 3 H), 0.89 (t, J = 7.6 Hz, 3 H), 0.84 (t, J = 7.6 Hz, 3 H); 13C NMR (100 MHz, CDCl3): δ 116.1, 88.5, 80.6, 78.9, 68.3, 66.6, 66.4, 58.4, 57.4, 49.9, 42.3, 40.1, 36.5, 35.5, 34.3, 33.5, 33.3, 32.7, 32.1, 31.8, 30.4, 29.8, 28.8, 28.6, 28.1, 18.5, 14.2, 8.9, 8.8; HR-ESIMS calcd for C29H48O5Na [M + Na]+ 499.33940, found 499.34072. Compound 12b: [α]D28 −17.7 (c 0.29, CHCl3); 1H NMR (400 MHz, CDCl3): δ 4.42 (dd, J = 12.8, 7.2 Hz, 1 H), 4.31 (br s, 1 H), 4.05 (br s, 1 H), 2.18–2.14 (m, 1 H), 1.35 (s, 3 H), 1.32 (s, 3 H), 1.03 (s, 3 H), 0.84 (t, J = 7.6 Hz, 6 H); 13C NMR (100 MHz, CDCl3): δ 120.6, 87.9, 82.7, 78.9, 68.1, 66.4, 66.2, 58.4, 58.1, 48.9, 42.1, 39.9, 36.3, 35.3, 32.7, 32.4, 31.9, 31.8, 31.4, 30.3, 30.2, 28.7, 27.9, 26.8, 19.7, 14.2, 8.7, 8.4; HR-ESIMS calcd for C29H48O5Na [M + Na]+ 499.33940, found 499.34087. Compound 13a: [α]D27 +42.8 (c 0.44, CHCl3); 1H NMR (400 MHz, CDCl3): δ 4.38–4.30 (m, 2H), 4.05 (br s, 1 H), 3.78 (d, J = 8.0 Hz, 1 H), 3.53 (d, J = 8.0 Hz, 1 H), 3.10 (s, 1 H), 2.22–2.18 (m, 1 H), 2.07–2.01 (m, 1 H), 1.38 (s, 3 H), 1.30 (s, 3 H), 1.21 (s, 3 H), 1.07 (s, 3 H), 1.03 (s, 3 H), 0.80–0.76 (m, 1 H); 13C NMR (100 MHz, CDCl3): δ 117.3, 80.7, 80.5, 79.8, 68.1, 66.4, 65.6, 58.3, 57.1, 49.8, 47.5, 42.1, 39.9, 37.9, 36.3, 35.2, 34.1, 32.5, 31.6, 30.1, 29.1, 28.6, 27.9, 27.1, 26.3, 18.4, 14.1; HR-ESIMS calcd for C27H44O5Na [M + Na]+ 471.30810, found 471.30893. Compound 13b: [α]D27 −42.0 (c 0.83, CHCl3); 1H NMR (400 MHz, CDCl3): δ 4.43–4.38 (m, 1 H), 4.31 (br s, 1 H), 4.05 (br s, 1 H), 3.64 (d, J = 7.2 Hz, 1 H), 3.51 (d, J = 8.0 Hz, 1 H), 2.20–2.14 (m, 2 H), 2.09–2.02 (m, 1 H), 1.98–1.88 (m, 1 H), 1.37 (s, 3 H), 1.32 (s, 3 H), 1.14 (s, 3 H), 1.05 (s, 3 H), 1.03 (s, 3 H), 0.81–0.78 (m, 1 H); 13C NMR (100 MHz, CDCl3): δ 121.1, 82.7, 79.8, 79.4, 68.0, 66.6, 66.3, 58.3, 58.1, 48.7, 46.4, 42.0, 39.8, 37.9, 36.2, 35.2, 32.3, 31.76, 31.75, 30.3, 28.6, 27.8, 27.7, 26.9, 26.0, 19.0, 14.1; HR-ESIMS calcd for C27H44O5Na [M + Na]+ 471.30810, found 471.30901. Compound 14: [α]D27 −36.8 (c 0.32, CHCl3); 1H NMR (400 MHz, CDCl3): δ 4.60–4.55 (m, 1 H), 4.42 (s, 1 H), 4.31 (br s, 1 H), 4.04 (br s, 1 H), 2.22–2.18 (m, 1 H), 2.12–2.08 (m, 1 H), 1.43 (s, 3 H), 1.24 (s, 3 H), 1.12 (s, 3 H), 1.11 (s, 3 H), 1.07 (s, 3 H), 1.03 (s, 3 H), 0.95 (s, 3 H), 0.82–0.78 (m, 1 H); 13C NMR (100 MHz, CDCl3): δ 114.9, 85.6, 80.1, 79.7, 69.7, 67.7, 65.9, 58.2, 57.6, 47.8, 47.6, 42.8, 39.9, 39.4, 35.8, 34.8, 31.7, 31.4, 30.2, 28.2, 27.4, 24.3, 24.0, 23.8, 23.7, 23.0, 17.8, 13.9; HR-ESIMS calcd for C29H48O5Na [M + Na]+ 499.33940, found 499.33963. Compound 19a: [α]D27 +11.4 (c 0.70, CHCl3); 1H NMR (400 MHz, CDCl3): δ 4.43–4.37 (m, 1 H), 4.30 (br s, 1 H), 4.05 (br s, 1 H), 3.98–3.92 (m, 1 H), 3.76–3.72 (m, 1 H), 3.29 (s, 1 H), 2.22–2.18 (m, 1 H), 2.09–2.03 (m, 1 H), 1.32 (s, 3 H), 1.25 (s, 3 H), 1.03 (s, 3 H), 0.80–0.77 (m, 1 H); 13C NMR (100 MHz, CDCl3): δ 105.0, 81.4, 81.0, 68.1, 66.5, 66.4, 61.8, 58.3, 57.1, 50.0, 42.4, 40.0, 36.3, 35.3, 33.8, 32.6, 31.6, 30.21, 30.18, 28.7, 27.9, 27.4, 24.9, 20.1, 18.5, 14.1; HR-MALDI calcd for C26H42O5Na [M + Na]+ 457.2924, found 457.2933. Compound 19b: [α]D27 –34.3 (c 0.94, CHCl3); 1H NMR (400 MHz, CDCl3): δ 4.38–4.33 (m, 1 H), 4.30 (br s, 1 H), 4.05 (br s, 1 H), 3.79–3.72 (m, 1 H), 3.60–3.56 (m, 1 H), 2.18–2.14 (m, 1 H), 2.09–2.03 (m, 1 H), 1.34 (s, 3 H), 1.31 (s, 3 H), 1.03 (s, 3 H), 0.81–0.77 (m, 1 H); 13C NMR (100 MHz, CDCl3): δ 110.2, 83.5, 79.2, 68.0, 66.8, 66.4, 61.1, 58.5, 58.0, 48.7, 42.0, 39.9, 36.3, 35.3, 32.3, 31.8, 31.2, 30.2, 28.6, 28.1, 27.9, 26.5, 25.1, 19.9, 19.7, 14.1; HR-MALDI calcd for C26H42O5Na [M + Na]+ 457.2924, found 457.2928. Compound 20a: 1H NMR (400 MHz, CDCl3): δ 4.35–4.29 (m, 2 H), 4.05 (br s, 1 H), 3.50 (s, 1 H), 3.10 (s, 1 H), 2.23–2.19 (m, 1 H), 1.39 (s, 3 H), 1.38 (s, 3 H), 1.24 (s, 3 H), 1.20 (s, 3 H), 1.04 (s, 3 H), 0.80–0.77 (m, 1 H). Compound 20b: 1H NMR (300 MHz, CDCl3): δ 4.47–4.40 (m, 1 H), 4.30 (br s, 1 H), 4.05 (br s, 1 H), 2.18–2.12 (m, 2 H), 1.32 (s, 3 H), 1.29 (s, 3 H), 1.26 (s, 3 H), 1.11 (s, 3 H), 1.03 (s, 3 H), 0.81–0.77 (m, 1 H).