

Abstract
Intron definition and splice site selection occur at an early stage during assembly of the spliceosome, the complex mediating pre-mRNA splicing. Association of U1 snRNP with the pre-mRNA is required for these early steps. We report here that the yeast U1 snRNP-specific protein Nam8p is a component of the commitment complexes, the first stable complexes assembled on pre-mRNA. In vitro and in vivo, Nam8p becomes indispensable for efficient 5′ splice site recognition when this process is impaired as a result of the presence of noncanonical 5′ splice sites or the absence of a cap structure. Nam8p stabilizes commitment complexes in the latter conditions. Consistent with this, Nam8p interacts with the pre-mRNA downstream of the 5′ splice site, in a region of nonconserved sequence. Substitutions in this region affect splicing efficiency and alternative splice site choice in a Nam8p-dependent manner. Therefore, Nam8p is involved in a novel mechanism by which a snRNP component can affect splice site choice and regulate intron removal through its interaction with a nonconserved sequence. This supports a model where early 5′ splice recognition results from a network of interactions established by the splicing machinery with various regions of the pre-mRNA.
Keywords: Alternative splicing, commitment complexes, intron, RNA processing, Saccharomyces cervisiae, snRNA
Splicing of nuclear introns is an essential step in gene expression. It occurs by a two-step pathway, conserved from yeast to mammals, which takes place in a large complex termed the spliceosome (Moore et al. 1993). In the first step, the phosphodiester bond at the 5′ splice site is attacked by the 2′OH group of an adenosine residue, the branchpoint, located inside the intron. In the second step, the phosphodiester bond located at the 3′ splice site is attacked by the 3′OH group of the last nucleotide of exon 1. This results in exon ligation, generating the spliced mRNA, while the excised intron is released in a lariat configuration.
Accuracy in the selection of the phosphodiester bond at both splice sites is crucial for gene expression. Indeed, most errors would affect the coding capacity of mature RNAs, thereby impairing functional protein production. The precise selection of the reactive residues is based on recognition of conserved sequences surrounding them. In yeast, the 5′ junction is followed by a highly conserved 5′ splice site consensus sequence GUAUGU. A conserved sequence, UACUAAC, surrounds the branch adenosine (underlined). Finally, a terminal YAG, sometimes preceded by a pyrimidine-rich sequence, is conserved at the 3′ junction. Accurate splice site choice results mainly from the numerous interactions established between the conserved pre-mRNA sequences and splicing factors during spliceosome assembly. Accordingly, the spliceosome is thought to serve not only as the catalyst of the splicing reaction steps, but also to translate the information presented by pre-mRNA into splicing specificity and orderliness. Several lines of evidence suggest that nonconserved pre-mRNA sequences can influence the splicing reaction (e.g., Pikielny and Rosbash 1985; Black 1995), especially in the case of regulated and alternative splicing events. Various examples are known in which splicing regulation depends on the presence of sequences located within the intron or in flanking exons (e.g., McKeown 1992; Chabot 1996). In some cases, it has been shown that these sequences are the targets for specific regulatory factors that interact or interfere with the splicing machinery. Alternative splicing regulation can, therefore, be achieved by regulating the production or the activity of these factors. For example, activation of the sex-specific dsx splicing in the sex determination pathway of Drosophila involves the building of a multiprotein complex composed of SR proteins and specific regulators on regulatory exonic enhancer sequences (Lynch and Maniatis 1995). In contrast, negative splicing regulation of the neuron-specific N1 exon of the mouse c-src transcript involves formation of a protein complex on an internal intronic sequence element (Chan and Black 1995).
Spliceosome assembly in vitro is initiated by formation of stable complexes containing U1 snRNP, pre-mRNA, and non-snRNP factors (Rosbash and Séraphin 1991). It is at this stage that a decision to splice an RNA segment is taken and that an intron is defined as such. Therefore, the early spliceosome assembly events can be the target of regulatory factors that alter splice site choice or modulate splicing activity. In yeast, the earliest known splicing complexes are called commitment complexes because their formation targets the pre-mRNA substrate to the splicing pathway (Séraphin and Rosbash 1989a). The mammalian E complex is the likely counterpart of the yeast commitment complexes (Michaud and Reed 1991). Native gel electrophoresis revealed the presence of two subspecies of commitment complexes in yeast, designated CC1 and CC2 (for commitment complex 1 and 2, respectively). Formation of CC1 is only dependent on the presence of a 5′ splice site in the target pre-mRNA, whereas formation of CC2 requires in addition a branchpoint sequence (Séraphin and Rosbash 1991). Both commitment complexes contain the U1 snRNP and the pre-mRNA (Séraphin and Rosbash 1989a), and the small subunit of the cap-binding complex (CBC) Mud13p (Colot et al. 1996). The large subunit of the yeast CBC complex is encoded by the GCR3 gene (Lewis et al. 1996a and references therein). As Mud13p and Gcr3p form a stable complex (Izaurralde et al. 1994), it is likely that Gcr3p is also a component of the commitment complexes. Biochemical experiments have demonstrated that CBC is not essential for commitment complex formation, although it affects the efficiency of this process (Lewis et al. 1996a; Colot et al. 1996). Interestingly, the homologous human CBC contributes at an early step of the splicing process (Lewis et al. 1996b). In addition, two proteins, Mud2p and BBP, were shown to affect specifically the formation of CC2 (Abovich et al. 1994; Abovich and Rosbash 1997). Mud2p was shown to be present in CC2, whereas BBP associates with the U1 snRNA in a pre-mRNA-dependent manner. Mud2p and BBP appear to be the yeast homologs of metazoan U2AF65 and SF1/mBBP, respectively (Abovich et al. 1994; Arning et al. 1996; Abovich and Rosbash 1997). U2AF65 and SF1/mBBP are E complex components (Michaud and Reed 1993; Abovich and Rosbash 1997), suggesting that this stage of the process is evolutionarily conserved. Several interactions among the splicing factors involved in commitment complex formation and the pre-mRNA have been characterized. U1 snRNA base-pairs with the intron and exon sequence flanking the 5′ splice site (Rosbash and Séraphin 1991; Séraphin and Kandels-Lewis 1993), whereas CBC binds to the pre-mRNA cap (Izaurralde et al. 1994). Genetic and biochemical studies indicate that BBP and Mud2p interact with the pre-mRNA branchpoint region (Berglund et al. 1997; Rain and Legrain 1997), consistent with their requirement for CC2 formation.
Some protein subunits of the yeast U1 snRNP have been identified through their sequence homology to mammalian splicing factors (Smith and Barrell 1991; Tang et al. 1997) or in genetic screens (Liao et al. 1993; Lockhart and Rymond 1994; Kao and Siciliano 1996; McLean and Rymond 1998). Recently, we have purified the yeast U1 snRNP and established a comprehensive list of its protein subunits (Neubauer et al. 1997; Gottschalk et al. 1998). Like its metazoan counterparts, the yeast U1 snRNP contains two classes of proteins: the Sm proteins shared with the U2, U4, and U5 snRNPs and the U1 snRNP-specific proteins (Neubauer et al. 1997; Gottschalk et al. 1998). Interestingly, homologs of all three mammalian U1 snRNP-specific proteins (U1-A, U1-C, and U1-70K) can be found in the yeast complex (Mud1p, yU1-C, and Snp1p, respectively). However, the yeast U1 snRNP contains in addition six specific proteins (Snu71p, Snu65p, Snu56p, Prp39p, Prp40p, and Nam8p) (Gottschalk et al. 1998) from which no mammalian homolog has been described so far. The knowledge of the composition of the yeast U1 snRNP now allows to investigate the role of its constituents in formation of early splicing complexes.
We report here the functional characterization of the yeast U1 snRNP-specific protein Nam8p. Nam8p is stably associated with the U1 snRNA, even at high salt concentrations (Gottschalk et al. 1998). Sequence analysis reveals that it contains three putative RNA-binding domains (Gottschalk et al. 1998). The NAM8 gene was originally identified as a multicopy suppressor of mitochondrial splicing deficiencies, and shown to be nonessential for vegetative growth (Ekwall et al. 1992). An independent screen revealed that Nam8p was essential for meiosis and required for efficient splicing of a meiosis-specific pre-mRNA, the MER2 transcript (Ogawa et al. 1995; Nakagawa and Ogawa 1997). Finally, Nam8p was also isolated in a screen for genes synthetic lethal with U1 snRNA (Gottschalk et al. 1998). We demonstrate that, although not essential, Nam8p is required for efficient recognition of weakened 5′ splice sites in vitro and in vivo. We also show that Nam8p interacts with the pre-mRNA downstream of the 5′ splice site, in a region where the sequence is not conserved. Interestingly, substituting the sequence of this region affects splicing efficiency in a Nam8p-dependent manner. Selection of alternative splice sites can be achieved based on this effect. Our data suggest a model of 5′ splice site recognition involving a network of partially redundant interactions between splicing factors and the pre-mRNA. Alternative splice site selection might result from slight changes in this interaction network that integrates information contained both in conserved and nonconserved intronic sequences.
Results
Nam8p is specifically required for efficient 5′ splice site recognition
Two previous studies indicated that Nam8p affects splicing. First, Nam8p was shown to be required for the meiosis-specific splicing of the MER2 transcript (Nakagawa and Ogawa 1997). The MER2 transcript contains an intron carrying a nonconsensus 5′ splice site that is required for meiosis-specific splicing activation (Engebrecht et al. 1991). On the other hand, Nam8p had been found to be required for efficient splicing of a reporter with a point mutation in the 5′ splice site (Gottschalk et al. 1998). These studies suggested an effect of Nam8p on 5′ splice site recognition. However, it was not clear whether this effect was 5′ splice site specific, or like other splicing mutants (Séraphin and Rosbash 1989b), Nam8p was required for efficient splicing of any weak pre-mRNA. To address this question, we introduced a set of splicing reporters harboring the RP51A intron fused to the β-galactosidase gene (Teem and Rosbash 1983) in a wild-type strain and in a strain lacking the NAM8 gene (Δnam8) (Gottschalk et al. 1998). These various constructs were selected to contain mutations in the 5′ splice site, branchpoint, or 3′ splice site, and to cover a wide variety of splicing efficiencies. Splicing of these reporters was assayed by quantitating the resulting β-galactosidase activity (Fig. 1) and by primer extension (data not shown). Splicing of the wild-type reporter was identical in both wild-type and Δnam8 strains (Fig. 1, bars 1,2). However, a 3- to 10-fold reduction in the β-galactosidase activity was observed for 5′ splice site reporters in the Δnam8 strain compared to the wild-type strain (Fig. 1, bars 3–10). Similar results were obtained with other 5′ splice site mutants (data not shown). In contrast, no significant difference was observed for the 3′ splice site and branchpoint mutants between the wild-type and Δnam8 strains (Fig. 1, bars 11–18). Primer extension analysis supported these findings (data not shown). These results indicate that Nam8p was required specifically for the efficient splicing of pre-mRNA carrying mutated 5′ splice sites, in contrast to what happens with other splicing mutants (Séraphin and Rosbash 1989b). Mutations in the 5′ splice site sequence are known to weaken the base-pairing interaction between the 5′ splice site and the 5′ end of the U1 snRNA (Rosbash and Séraphin 1991). Therefore, these results suggest that Nam8p could be implicated specifically in the stabilization of the U1 snRNP–pre-mRNA interaction.
Figure 1.

Nam8p affects specifically 5′ splice site recognition. Splicing of reporters carrying a wild-type or mutant RP51A intron interrupting the β-galactosidase-coding sequence was analyzed in wild-type (solid bars) and Δnam8 (open bars) strains. Mutations are underlined.
Nam8p is essential for efficient formation of stable commitment complexes on an uncapped pre-mRNA
The presence of Nam8p in U1 snRNP (Gottschalk et al. 1998) suggested that it could influence 5′ splice site selection during commitment complex formation. Therefore, we first tested whether Nam8p was a commitment complex component. We used a strain expressing a tagged version of Nam8p, Nam8p–protA, in which two IgG-binding domains of the Staphylococcus aureus protein A protein were fused to Nam8p (Gottschalk et al. 1998; Puig et al. 1998). Nam8p–protA binds specifically to rabbit IgGs through its protein A tag. Extracts were prepared from the strain expressing Nam8p–protA and used in commitment complex assembly reactions. Addition of rabbit IgGs to commitment complexes assembled in the Nam8p–protA extract retarded the migration of the CC1 and CC2 complexes (data not shown). However, it did not affect the mobility of the commitment complexes assembled in extracts from either a wild-type strain or a strain containing an unrelated tagged protein (data not shown). These results demonstrated that Nam8p is a component of both commitment complexes.
We then investigated the role of Nam8p in commitment complex formation and stability. Commitment complexes were assembled on labeled GpppG-capped pre-mRNA substrates in extracts derived from either the wild-type or Δnam8 strains. We used labeled uncapped pre-mRNA in parallel reactions, because under these conditions commitment complex assembly is less efficient, making the assay more sensitive (Colot et al. 1996). Commitment complexes were assembled and their stability was assayed by following the level of labeled complex remaining at several time points after the addition of a 50-fold excess cold pre-mRNA (Fig. 2; data not shown). Consistent with previous results (Gottschalk et al. 1998), CC2 is formed efficiently and is stable using the capped substrate in both Δnam8 and wild-type strains (Fig. 2, lanes 1–4). Stable CC2 also assembled on the uncapped pre-mRNA in the wild-type extract (Fig. 2, lanes 5,6). In contrast, only traces of CC2 was detected after incubation of the uncapped pre-mRNA in the Δnam8 extract (Fig. 2, lane 7, note that for technical reasons time 0 truly corresponds to a few seconds after the addition of an excess of unlabeled pre-mRNA) and this complex was then very unstable (Fig. 2, lane 8). Similar results were obtained by assaying the formation and stability of CC1 (data not shown). These results indicate that Nam8p is not necessary for commitment complex formation or stability consistent with the dispensability of the NAM8 gene in vivo. However, Nam8p becomes indispensable for this process when another interaction involved in commitment complex formation, namely that between CBC and pre-mRNA cap, is weakened or absent.
Figure 2.

Nam8p stabilizes the commitment complexes assembled on uncapped pre-mRNA. Commitment complexes were assembled on labeled GpppG-capped (lanes 1–4) or uncapped (lanes 5–8) substrates. An excess of cold pre-mRNA was added and the incubation was resumed for 40 min. (Lanes 1,2 and 5,6) Wild-type extract; (lanes 3,4 and 7,8) Δnam8 extract.
Nam8p is required for efficient splicing of uncapped RNA precursor in vivo
The results presented above suggest that splicing of uncapped pre-mRNA should be less efficient in the absence of Nam8p in vivo. To test this possibility, we used a splicing reporter consisting of the SUP6 pre-tRNATyr interrupted by the 52-nucleotide second intron of the MATa1 gene (construct SUP6-1 pre-tRNATyr). This construct was previously built and characterized by Köhrer et al. (1990) and is transcribed by RNA polymerase III. However, this transcript still splices through the normal splicing pathway generating a functional tRNA. Because RNA polymerase III transcripts have been reported to lack a cap structure, this reporter allowed us to test splicing of an uncapped transcript in both wild-type and Δnam8 strains. Plasmid SUP6-1 pre-tRNATyr was transformed in wild-type and Δnam8 strains. Mature tRNAs resulting from the endogenous pre-tRNATyr and the SUP6-1 pre-tRNATyr were distinguished by performing a primer extension reaction on total RNA in the presence of dATP, dGTP, dTTP, and ddCTP. This assay discriminates between the endogenous and reporter tRNAs because of the presence of the suppressor mutation in the latter one. The mature SUP6-1 tRNATyr was detected in both the wild-type and Δnam8 cells (data not shown). However, the corresponding band was not detected in nontransformed cells or in cells expressing the nonspliceable SUP6-2 pre-tRNATyr (Köhrer et al. 1990; data not shown), confirming that it represented the spliced product of the plasmid encoded tRNA. Interestingly, the mature SUP6-1 tRNATyr signal was reproducibly decreased fourfold in the Δnam8 compared to the wild-type strain, whereas at the same time the mature tRNATyr signal, used as internal loading control, was unaffected (data not shown). The absence of Nam8p did not affect splicing of RNA polymerase II transcripts (Fig. 1, bars 1,2; data not shown). This indicates that Nam8p is required for efficient splicing of uncapped RNA precursor in vivo.
Nam8p interacts with the pre-mRNA
Nam8p is essential for stability of commitment complexes assembled on uncapped pre-mRNA in vitro and for efficient splicing of the uncapped SUP6-1 tRNATyr transcript in vivo. In addition it is specifically required for efficient splicing of pre-mRNAs carrying a mutated 5′ splice site in vivo. Taken together these results suggest that Nam8p is involved in the stabilization of the 5′ splice site-U1 snRNP interaction in commitment complexes. The presence of three RRM motifs in the Nam8p sequence suggested that this could be mediated by a direct Nam8p–pre-mRNA contact. To test this possibility, we investigated whether Nam8p could be cross-linked to labeled pre-mRNA. Commitment complexes assembled on a labeled wild-type RP51A-derived pre-mRNA (WT-B) were irradiated with UV light and treated with RNase T1. Proteins attached to labelled RNA fragments were then fractionated by gel electrophoresis and detected by autoradiography. Numerous radioactive bands were observed when analyzing complete reactions (Fig. 3, lanes 1–3). However, a band of the expected size for the Nam8p–protA fusion was specifically detected in the corresponding extract (Fig. 3, lane 1) and was absent from the wild-type extract (Fig. 3, lane 2), suggesting that Nam8p cross-linked to the pre-mRNA. This band was absent from a similar reaction using a mutated substrate unable to assemble commitment complexes (data not shown, see also below) indicating that it was related to splicing. (Note that the untagged Nam8p cannot be detected in Fig. 3, lane 2, because it comigrates with the main radiolabeled cross-linked products as a result of its smaller size; see also below.) Immunoprecipitation of the cross-linked material before electrophoresis confirmed the identity of Nam8p as the cross-linked protein: (1) The size of the cross-linked product differed between an isogenic wild-type strain and the strain carrying a protein A-tagged version of Nam8p (Fig. 3, lanes 10–13). The apparent molecular weight of these two cross-linked species is, furthermore, consistent with the calculated molecular weight of the corresponding polypeptides. (2) The cross-linked Nam8p–protA was specifically precipitated with rabbit IgGs (Fig. 3, cf. lane 4 with lane 7). (3) Both the cross-linked wild-type and Nam8p-tagged proteins were immunoprecipitated by specific anti-Nam8p antibodies (Gottschalk et al. 1998) (Fig. 3, lanes 10–13). Note that in addition to Nam8p–protA, an unidentified species of lower molecular weight is precipitated by rabbit IgG-coupled agarose beads from both tagged and wild-type extracts (Fig. 3, lanes 4–9). Cross-linking was specific for splicing-competent RNA as only background signal could be detected with a substrate carrying a 5′ splice site mutation and a branchpoint deletion that is unable to form commitment complexes (5′ SS mut + ΔBP, Fig. 3, lanes 6,11,13). In contrast, cross-linked products could still be detected with a pre-mRNA mutated only at the branchpoint sequence (ΔBP, Fig. 3, lane 5). This demonstrates that cross-linking occurs in CC1 as this is the only complex assembling on that RNA (Séraphin and Rosbash 1991). However, the level of cross-linking was consistently lower with the substrate lacking the branchpoint compared to a wild-type substrate (Fig. 3, lanes 4,5) suggesting that, in the latter case, cross-linking took place in addition in CC2. Nam8p–protA also cross-linked to an actin pre-mRNA, indicating that the interaction was not intron specific (data not shown).
Figure 3.

Nam8p specifically cross-links to pre-mRNA. Nam8p–protA-containing extract (lanes 1, 4–6, 10,11) and wild-type extract (lanes 2, 7–9, 12–13) were used to assemble commitment complexes on pre-mRNA, UV cross-linked, treated with RNase T1 and immunoprecipitated. Pellets were analyzed by SDS-PAGE. (Lanes 1–3) No immunoprecipitation; (lanes 4–9) immunoprecipitation with IgG-coupled beads; (lanes 10–13) immunoprecipitation with rabbit anti-Nam8p antibodies and Sepharose beads coupled to Staphyloccocus aureus protein A protein. The pre-mRNAs used as substrates were WT-B, ΔBP; and 5′ SSmut + ΔBP (see text). The band visible below Nam8p–protA in lanes 4–9 is nonspecifically precipitated, corresponds to the unidentified major cross-linked product seen in lanes 1,2, and comigrates in this gel conditions with the untagged version of Nam8p. In lane 3, extract buffer rather than extract was used to demonstrate that the products are not generated by intramolecular RNA cross-linking.
Nam8p contacts the pre-mRNA downstream of the 5′ splice site
To identify the region of the pre-mRNA contacting Nam8p, we first generated 3′ truncated pre-mRNAs by linearizing the template DNA with various restriction enzymes before transcription. Nam8p–protA cross-linked with the same efficiency to a control full-length pre-mRNA lacking the branchpoint sequence (ΔBP) and a substrate generated from a cleaved template ending upstream of the branchpoint (data not shown). This result suggested that cross-linking occurred close to the 5′ splice site. To map more precisely the location of the Nam8p–pre-mRNA interaction, we generated site specifically labeled WT-B pre-mRNA substrates (Moore and Sharp 1992). 32P labels were introduced upstream of the guanosine residues at positions −1, +5, and +13 (+1 being the first nucleotide of the intron and −1 the last exon 1 nucleotide, Fig. 4A). These three substrates formed commitment complexes efficiently (data not shown) and therefore, were used in cross-linking reactions (Fig. 4A). After RNase T1 treatment and immunoprecipitation, the cross-linked Nam8p–protA was detected using the substrate labeled at position +13 but not using substrates labeled at position +5 or −1 (Fig 4A, lanes 2–4). These results indicated that Nam8p interacts with nonconserved intronic sequences that are present downstream of residue +5. To confirm this finding, two similarly sized oligonucleotides overlapping the 5′ splice site were synthesized: oligonucleotide A extended into exon 1 upstream of the 5′ splice site, whereas oligonucleotide B covered the 5′ splice site and downstream intronic residues (Fig. 4B). An oligonucleotide covering the branchpoint was used as a specificity control. These three oligonucleotides were 5′ end-labeled and used for cross-linking experiments, omitting RNase T1 digestion. Nam8p–protA cross-linked efficiently to oligonucleotide B (Fig. 4B, lane 2), whereas background signal could be detected using oligonucleotide A or the control oligonucleotide spanning the branchpoint (Fig. 4B, lanes 1,3, respectively). To demonstrate that the cross-linked products detected with these oligonucleotides were related to splicing, we compared the cross-linked products formed using the wild-type oligonucleotide B and a mutant version in which the G located at the position equivalent to intron position 5 was substituted by A. This mutation will weaken U1 snRNP, and consequently Nam8p, binding to the 5′ splice site (Séraphin and Rosbash 1991). Although the wild-type oligonucleotide B was cross-linked efficiently to Nam8p–protA, cross-linking was strongly reduced with the mutant oligonucleotide (Fig 4B, lanes 4,5). The absence of cross-linking did not result from specific degradation of a particular oligonucleotide, as control experiments demonstrated that all four labeled oligonucleotides had similar stability in yeast extracts (data not shown). These results demonstrate that Nam8p contacts the pre-mRNA in a region located between intron position +6 and +15 and covering essentially nonconserved sequences. Given that RRM motifs interact with short stretches of RNA (Nagai et al. 1990; Oubridge et al. 1994; Price et al. 1998) detailed structural studies will be required to understand the precise mode of interaction of Nam8p with this sequence. Our results also demonstrate that the Nam8p–pre-mRNA interaction occurs in CC1, probably also in CC2, and possibly in higher splicing complexes.
Figure 4.
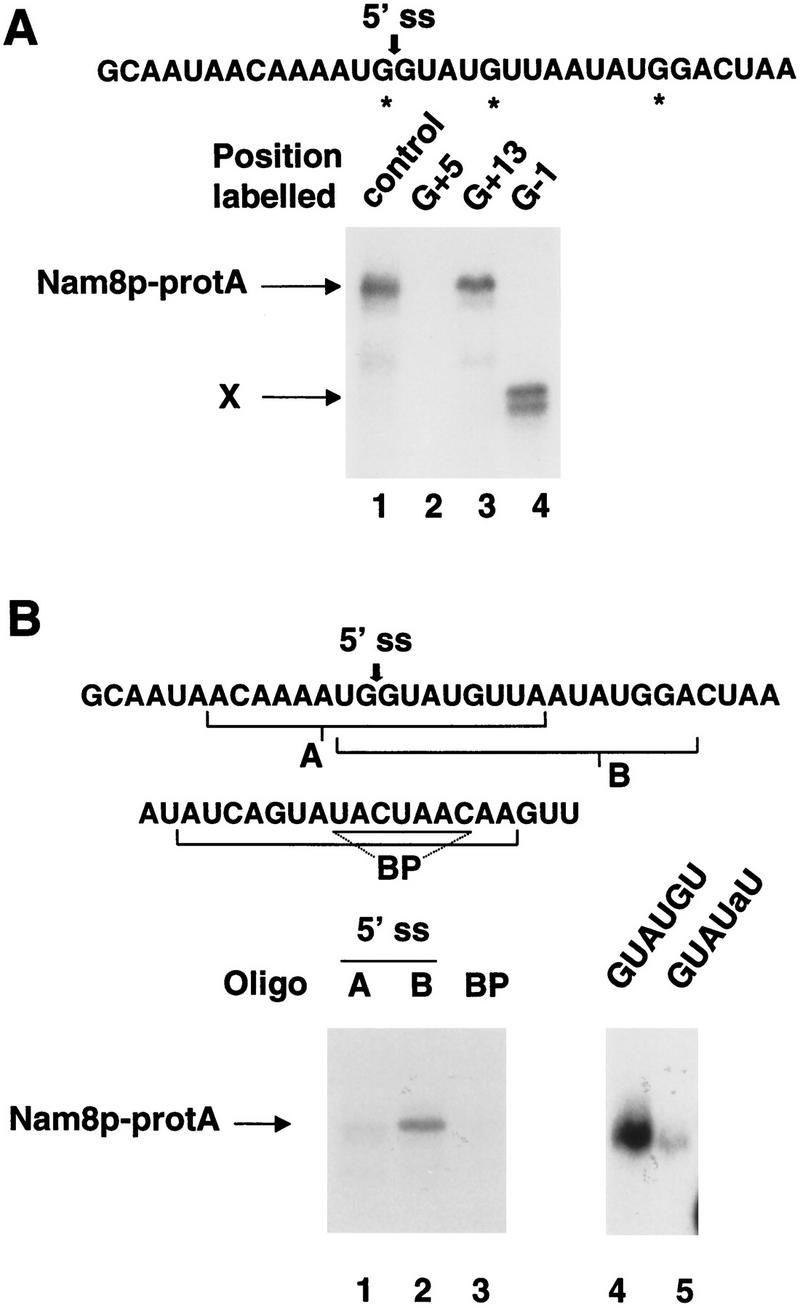
(A) Cross-linking of the Nam8 protein to site-specific-labeled pre-mRNAs. WT-B pre-mRNA was labeled in position −1, +5, and +13, respectively, and used in cross-linking reactions with Nam8p–protA. (Lane 1) Internally labeled pre-mRNA used as a positive control; (lanes 2–4) pre-mRNAs labeled in positions +5, +13, and −1, respectively. X is an unidentified cross-linked product. (B) Nam8p cross-links to an oligonucleotide spanning the 5′ splice site and following residues. RNA oligonucleotides protected at their 5′ end by two deoxynucleotides were used to cross-link Nam8p–protA. (Lane 1) oligonucleotide A overlapping the 5′ splice site and upstream exon sequence; (lane 2) oligonucleotide B spanning the 5′ splice site and following residues; (lane 3) oligonucleotide spanning the branchpoint region; (lane 4) oligonucleotide B; (lane 5) mutant oligonucleotide B carrying a substitution in the 5′ splice site sequence (GUAUaU).
Nonconserved sequences downstream of the 5′ splice site affect splicing in a Nam8p-dependent manner
The data reported above indicate that Nam8p may affect 5′ splice recognition by interacting with the region following the 5′ splice site. A previous study had suggested an effect of the sequence following the 5′ splice site on the efficiency of pre-mRNA splicing (Pikielny and Rosbash 1985). However, no systematic study of this region of the pre-mRNA has been reported and factors implicated in these events have not been described so far. Therefore, we decided to investigate whether the nonconserved sequence located downstream of the 5′ splice site affected pre-mRNA splicing and whether Nam8p was involved in this process. For this purpose, we used a reporter identical to the one described above (see Fig. 1), but carrying a synthetic intron harboring restriction sites around the 5′ splice site, thereby allowing the easy substitution of the corresponding sequence (see Materials and Methods; Fig. 5A). This pre-mRNA is spliced efficiently in a wild-type strain (Fig. 5B, bar 1). As the optimal binding sequence of Nam8p is unknown, we constructed reporter genes carrying various sequences in the Nam8p-binding region, that is, downstream of the 5′ splice site (positions +7 to +26 relative to the cleavage site, Fig. 5A). G-rich or U-rich sequences, or a sequence of average composition that could potentially form a hairpin structure were substituted in our synthetic reporter (Fig. 5A; see Materials and Methods). The four different sequences are named as follows: G (G-rich sequence), U (U-rich sequence), H (potential hairpin), and A (the AU-rich sequence present in the original synthetic intron). These constructs were introduced into isogenic wild-type and Δnam8 yeast strains. The effect of cis-acting sequences and trans-acting factors on 5′ splice site choice was then examined by direct RNA analysis or by following β-galactosidase activity. The quantification of the primer extension reaction is shown in Figure 5B. In the situation where the A or G sequences are located downstream of the 5′ splice site, splicing of this reporter is at most mildly affected by the NAM8 genotype (Fig. 5B, bars 1–4). Comparison of the A and U constructs, which differ only by the sequence of the 20 nonconserved nucleotides after the 5′ splice site, reveals that the U-rich sequence enhanced splicing of the corresponding reporter (threefold increase) in a wild-type yeast strain (Fig. 5B, bars 1,5). Interestingly, the enhancer effect of the U-rich sequence was lost in the Δnam8 strain (Fig. 5B, bars 2,6). The H sequence is better spliced (twofold) than the A sequence in the wild-type strain (Fig. 5B, bars 1,7), but nearly a threefold reduction in the splicing efficiency can be observed in the Δnam8 strain (Fig. 5B, bars 2,8). Our results confirm that the sequence located downstream of the 5′ splice site affects splicing efficiency (see also Pikielny and Rosbash 1985). Our results indicate further that Nam8p, at least in part, mediates this effect and demonstrate that a U-rich sequence located downstream of the 5′ splice site enhances splicing in a Nam8p-dependent manner.
Figure 5.

Nam8p interaction with the sequence located downstream of the 5′ splice site affects splicing and splice site choice. (A) Representation of the reporters used in the experiments shown in B and C. An arrowhead indicates the 5′ splice site. (B) Plasmids carrying A, G, U, and H sequences were transformed in the wild-type (black bars) and Δnam8 (white bars) strains. Splicing efficiency, measured as mRNA versus pre-mRNA ratio (M/P; Pikielny and Rosbash 1985), was determined by primer extension and quantification with a PhosphorImager. All the measurements were normalized using the endogenous RP51A signal. (C) Primer extension analysis of the RNA produced by the different constructs in wild-type (WT) and nam8-disrupted (Δ) strains. Owing to multiple transcription initiation sites of the inducible GAL-CYC1 promoter, pre-mRNA and mRNAs appear as multiple bands. Numbers below each lane represent the splicing efficiency of the upstream site versus the downstream site quantified with a PhosphorImager. Samples 1–4 and 5–10 were run in different gels. (D) Nam8p–pre-mRNA interaction is modulated by the sequence following the 5′ splice site. (Top) Oligonucleotides BG and BU used to compete with oligonucleotide B for the cross-linking with Nam8p–protA in yeast extracts. (Bottom) Labeled oligonucleotide B was premixed with oligonucleotides BU (lanes 1–3) or BG (lanes 4–6) in molar ratios of 1:1 (lanes 1,4), 1:3 (lanes 2,5), and 1:10 (lanes 3,6). Subsequently, the different oligonucleotide mixtures were used in cross-linking reactions.
Nam8p affects alternative 5′ splice site selection
Previous studies in metazoan have demonstrated an effect of the sequence after the 5′ splice site on alternative splice site choice (e.g., Gallego et al. 1992; Del Gatto and Breathnach 1995). In these two examples, an alternatively spliced exon is included in the final messenger RNA depending on the existence of a pyrimidine-rich sequence located in the downstream intron. Our previous results showed that Nam8p can affect the efficiency of splicing depending on the sequence located downstream of the 5′ splice site. Therefore, we wanted to know whether an alternative selection of 5′ splice sites could be achieved in yeast based on their downstream sequence, and whether this effect was dependent on Nam8p. For this purpose we turned to a sensitive assay based on the alternative choice between two 5′ splice sites competing for a single 3′ splice site (Fig. 5A) (Goguel et al. 1991; Séraphin and Kandels-Lewis 1993). The reporters used in this experiment had two duplicated 5′ splice sites and downstream of each of them, the sequences A, U, G, or H used in the previous experiment were introduced independently (Fig. 5A; see also Materials and Methods). The resulting reporters were named using a two-letter code, where the first letter represents the sequence following the upstream 5′ splice site, and the second letter represents the region following the downstream 5′ splice site (Fig. 5A). According to this nomenclature, therefore, the construct named UA has a U-rich sequence after the upstream 5′ splice site and an AU-rich sequence after the downstream one (Fig. 5A). These constructs were introduced into isogenic wild-type and Δnam8 strains. Splicing efficiency was then examined by direct RNA analysis or by analyzing β-galactosidase activity that reports usage of the upstream 5′ splice site (Goguel et al. 1991; Séraphin and Kandels-Lewis 1993). In the AA construct, the downstream site is used with slightly better efficiency independently of the NAM8 genotype (Fig. 5C, lanes 1,2). Comparison of the AA and UA constructs, which differ only by the sequence following the upstream site, revealed that the U-rich sequence strongly enhanced usage of this site in a wild-type yeast strain (Fig. 5C, lanes 1,3). Interestingly, the enhancer effect of the U-rich sequence was lost in the Δnam8 strain (Fig. 5C, lane 4). This loss is sequence rather than position specific as the presence or absence of Nam8p had no effect on splicing of the upstream site for the AA reporter (Fig. 5C, lanes 1,2). The Nam8p-dependent activation of the upstream U-rich site was also detected when a G-rich sequence was present after the downstream 5′ splice site (UG construct, Fig. 5C, lanes 9,10). A weaker Nam8p-dependent activation of the upstream site could also be detected in the related HG construct (Fig. 5C, lanes 5,6). Interestingly, however, in that case, removal of Nam8p did not only reduce the activity of the upstream site but also increased the activity of the downstream site (Fig. 5C, lanes 5,6). Substituting the G-rich sequence by a U-rich sequence at the downstream site activated that site in a wild-type strain (Fig. 5C, cf. the HG construct in lane 5 with the HU construct in lane 7). Importantly, U-rich-mediated activation of the downstream site was Nam8p dependent (Fig. 5C, lane 8). Overall, this result indicates that a U-rich sequence located downstream of a 5′ splice site activated that site. This effect is lost in the Δnam8 background indicating that activation was mediated, at least to a significant extent, by Nam8p. These results supported our previous data obtained with the reporter harboring a single 5′ splice site. The sequence potentially forming a hairpin structure (H) also mediated Nam8p-dependent activation of the preceding 5′ splice site (construct HG, Fig. 5C, lanes 5,6). The G-rich sequence was neutral or possibly negative for splicing in presence of Nam8p as a slight activation of the neighboring 5′ splice site was observed in the Δnam8 strain (Fig. 5C, lane 6). Changes in 5′ splice site usage also resulted in parallel variation in β-galactosidase expression (data not shown) confirming that the effects were biologically relevant. It is, however, noteworthy that the various constructs analyzed only represent selected sequences of RNA. It is, therefore, expected that stronger effects would be detected if constructs harboring sites of extremely high and low affinity for Nam8p were tested. In any case, our results demonstrate that the sequence located immediately downstream of the 5′ splice site affects splice site selection. Furthermore, this effect is mediated, to a large extent, by Nam8p.
Nam8p–pre-mRNA interaction is modulated by the sequence after the 5′ splice site
To demonstrate that the effect of the sequence after the 5′ splice site on splicing in vivo relates to interaction of Nam8p with the pre-mRNA, we carried out an additional cross-linking assay. Two unlabeled oligonucleotides were used to compete for cross-linking of Nam8p–protA to labeled oligonucleotide B in yeast extracts. These two RNA competitors contained the 5′ splice site sequence extended at the 3′ end with the G-rich (oligonucleotide BG) or U-rich (oligonucleotide BU) sequences used in the competition assay performed in vivo (Fig. 5D). Both oligonucleotides had similar stability in yeast extracts (data not shown). In all the different molar ratios used oligonucleotide BU competed more efficiently for cross-linking of Nam8p–protA than oligonucleotide BG (Fig. 5D, lanes 1–6). This result indicates that interaction of Nam8p with the nonconserved region downstream of the 5′ splice site is modulated by the sequence in that region.
Discussion
Our results demonstrate that Nam8p is a component of the commitment complexes. Under conditions that compromise U1 snRNP recruitment to the 5′ splice site (e.g., weak 5′ splice site, lack of a pre-mRNA cap), Nam8p is necessary for efficient 5′ splice site recognition in vivo and contributes to the stability of commitment complexes in vitro. However, Nam8p has no effect in branchpoint or 3′ splice site recognition. In commitment complexes Nam8p contacts the pre-mRNA downstream of the 5′ splice site. These data support a model (Fig. 6) where 5′ splice site recognition results from a network of interactions established by the splicing machinery with various regions, conserved and nonconserved, of the pre-mRNA. During the early steps of splicing, the cellular machinery would, therefore, integrate the information contained in different segments of the pre-mRNA before committing, or not, this molecule to the splicing pathway. However, not all of these interactions would be absolutely required at the same time. For a pre-mRNA with canonical 5′ splice site, the presence of Nam8p (or CBC) is not required (see above). In contrast, at least a subset of the U1 snRNA–pre-mRNA base-pairing interactions are essential (Rosbash and Séraphin 1991). Nam8p becomes indispensable for efficient splicing when the U1 snRNA-pre-mRNA pairing is weakened (see Fig. 1). It is likely that this is attributable to lower commitment complex formation or stability. However, this could not be tested directly because commitment complexes assembled on 5′ splice site mutant substrates are not detected after native gel electrophoresis (Séraphin and Rosbash 1991). The involvement of Nam8p in the recognition of nonconsensus 5′ splice sites probably explains why Nam8p is conserved in yeast, although it is dispensable. Indeed, Nam8p allows optimal splicing of cellular transcripts harboring nonconsensus 5′ splice sites like MER2 or RPL32 (Nakagawa and Ogawa 1997; data not shown) and therefore, probably contributes to yeast fitness. Nam8p becomes also indispensable for efficient splicing when the pre-mRNA cap is missing. Biochemically, this could be attributed to reduced commitment complex formation or instability of commitment complexes (Fig. 2). Consistent with this, Nam8p was identified in a screen for mutants lethal in the absence of the cap-binding complex (P. Fortes and I.W. Mattaj, pers. comm.). The existence of a network of partially redundant interactions that are not all essential provides an ideal situation to finely tune pre-mRNA splicing. This could be mediated by specific regulatory molecules interfering with these interactions or through controlled changes in the affinity of constitutive splicing factors for the relevant region of the pre-mRNA.
Figure 6.

Model for the recognition of the 5′ splice site. The 5′ splice recognition results from a network of interactions established by the splicing machinery with various regions of the pre-mRNA namely CBC–cap structure, U1 snRNA–5′ splice site, and Nam8p region of nonconserved sequence in the intron. Therefore, this network of interactions is based on recognition of conserved (5′ splice site and cap) and nonconserved (downstream intronic region bound by Nam8p) elements in the pre-mRNA. Some interactions are essential, such as U1 snRNA–5′ splice site base pairing (Rosbash and Séraphin 1991). Other interactions contribute positively to stability of the complex but are not absolutely needed. For a pre-mRNA with canonical 5′ splice site, the presence of CBC or Nam8p is not required. CBC stabilizes the complex but is not absolutely necessary as the complexes assembled on uncapped RNA, although present in reduced levels, are functional (Colot et al. 1996; Lewis et al. 1996a). Commitment complexes assembled without Nam8p are stable (Fig. 2) and splicing of pre-mRNAs with canonical 5′ splice site occurs with normal efficiency in the Δnam8 strain (Fig. 1). However, Nam8p becomes indispensable for efficient splicing when the U1 snRNA–pre-mRNA pairing is weakened or with commitment complexes assembled on uncapped pre-mRNA.
We have shown that Nam8p contacts the pre-mRNA in a region downstream of the 5′ splice site, in a nonconserved intronic region. Nam8p is a subunit of the yeast U1 snRNP to which it is strongly associated (Gottschalk et al. 1998). Nam8p is, therefore, likely to be juxtaposed with that specific pre-mRNA region as a consequence of the U1 snRNP binding to the 5′ splice site (mediated in part by U1 snRNA–pre-mRNA base pairing) and of the precise three-dimensional structure of the snRNP (Fig. 6). It is likely that Nam8p–pre-mRNA binding stabilizes the U1 snRNP–5′ splice site interaction. This is supported by our results showing that the Nam8p–oligonucleotide B cross-link is competed more effectively by an oligonucleotide bearing the U-rich element than with one carrying a G-rich element, suggesting that the sequence downstream of the 5′ splice site can modulate U1 snRNP–pre-mRNA interaction through its interaction with Nam8p. The biological relevance of this interaction is demonstrated in the in vivo splicing assays. This observation also demonstrates that the sequence located downstream of the 5′ splice site affects the outcome of the splicing reaction. Among the sequences tested, the U-rich sequence was the best stimulator of splice site choice and was essentially Nam8p dependent. Therefore, it plays the role of an intronic splicing enhancer. However, the optimal sequence allowing Nam8p binding and splicing stimulation remains to be determined. Furthermore, Nam8p–pre-mRNA interaction might be modulated by interaction with other proteins (e.g., Mer1p, Engebrecht et al. 1991). Pikielny and Rosbash (1985) already reported a decrease in splicing efficiency after substitutions in the region downstream of the 5′ splice site. They concluded that nonconserved sequences, surrounding the key intron recognition elements (both splice sites and branchpoint) play a significant role in determining the efficiency of splicing. It was, however, unclear whether the effect observed reflected changes in the structure of the pre-mRNA or interaction with trans-acting factors. It is likely that some of the observed effect can be attributed to the binding of Nam8p to the substituted sequence.
Substitutions in the pre-mRNA region bound by Nam8p affect splicing efficiency (Fig. 5B). In addition we have demonstrated that the corresponding sequence affects alternative splice site selection (Fig. 5C). Therefore, this region of the pre-mRNA may have an important role in regulation of gene expression. However, like for yeast, analysis of metazoan introns has not revealed a general role for the sequence after the 5′ splice site in intron removal. Nevertheless, several examples are known in which splicing regulation of alternatively spliced introns implicates participation of sequences located directly downstream of the 5′ splice site. For example, part of the extracellular domain of the human fibroblast growth factor receptor 2 (FGFR-2) is encoded by two alternatively spliced exons K-SAM and BEK, (Del Gatto and Breathnach 1995). Splicing of the intron located downstream of the K-SAM exon is necessary for the inclusion of this exon in the encoded protein, to the detriment of the BEK exon. This event occurs specifically in epithelial cells and is dependent on the presence of a short pyrimidine-rich sequence located immediately downstream of the 5′ splice site of the spliced intron (Del Gatto and Breathnach 1995). When this pyrimidine-rich sequence is substituted, the intron is not removed and the K-SAM exon is skipped. Improving the non-consensus 5′ splice site of this intron enforces inclusion of the K-SAM exon, independently of the presence of the pyrimidine-rich sequence (Del Gatto and Breathnach 1995). A related situation is observed in the chicken β-tropomyosin gene (Gallego et al. 1992). These results indicate that the pyrimidine-rich sequence helps the splicing machinery to recognize a poor 5′ splice site, whereas the pyrimidine-rich sequence is no longer necessary when the 5′ splice site is optimized. Another example where the importance of the region downstream of the 5′ splice site is outlined is the pre-mRNA encoding the human Tau protein. Mutations in the sequence after the 5′ splice site of intron 10 affect alternative usage of that site (Hutton et al. 1998). These mutations have been proposed to alter RNA secondary structure but could as well affect binding of a splicing factor. All these cases provide examples of the importance of nonconserved sequences located near the intron 5′ end for regulated alternative splicing. It was proposed that these sequences might be targets for factors that would interact with the splicing machinery. The parallel with the role of Nam8p in yeast pre-mRNA splicing is striking. A putative mammalian Nam8p homolog could well be responsible for alternative splicing regulation of these introns. These pyrimidine-rich sequences could act by facilitating the binding of this factor to the pre-mRNA, as happens with Nam8p in yeast. Alternatively, factors or RNA structures hindering a Nam8p-like activity could be used to regulate splicing negatively. Further experiments will be required to establish whether Nam8p-like activities exist in other species.
Materials and methods
Plasmid constructs and in vitro transcription
Reporters used to analyze splicing efficiency in Figure 1 have been previously described: wild-type intron (HZ18, Teem and Rosbash 1983); 5′ splice site mutants GUAUaU (HZ12, Jacquier et al. 1985), GUAUcU and GUAUuU (pBS93 and pBS94, Séraphin et al. 1988), GUgUGU (pBS1223, Luukkonen and Séraphin 1998); branchpoint mutants (pBS1084 and pBS909, Pascolo and Séraphin 1997); and 3′ splice site mutants (pBS842 and pBS850, Luukkonen and Séraphin 1997). WT-B is a derivative of the RP51A intron (Séraphin and Rosbash 1991). 5′ SSmut has a substitution in the 5′ splice site (GUAUaU). ΔBP is a mutant substrate that lacks the branchpoint sequence (ΔUACUAAC) (Séraphin and Rosbash 1991). 5′ SSmut + ΔBP combines the latter two mutations. Actin pre-mRNA (Vijayraghavan et al. 1986) and all the RP51A derivatives used in cross-linking reactions were synthesized with a specific activity of 2 × 107 Ci/mole as previously described (Séraphin and Rosbash 1989a). The constructs WT-B, 5′ SSmut, ΔBP, and 5′ SSmut + ΔBP correspond to the plasmids pBS195, pBS196, pBS199, and pBS345, respectively.
Plasmid pBS1599 (O. Puig, unpubl.) is identical to HZ18 (Teem and Rosbash 1983), but contains an artificial intron interrupting the β-galactosidase coding sequence rather than the RP51A intron (Teem and Rosbash 1983). The backbone of this plasmid was used to build the reporters allowing the analysis of the effect of the sequence downstream of the 5′ splice site as convenient restriction sites flank this region. Sequences U, G, and H (Fig. 5A) were generated by PCR and introduced between the AflII and BglII sites of pBS1599. Sequence H can potentially form a hairpin structure with a ΔG of −8.1 Kcal/mole at 25°C. Constructs G, U, and H correspond to the plasmids pBS1600, pBS1601, and pBS1602, respectively. Duplications were obtained by cloning the XhoI–SacI fragments from these various constructs in the SalI–SacI sites of the same plasmids (Goguel et al. 1991; Séraphin and Kandels-Lewis 1993). Constructs AA, UA, HG, HU, and UG correspond to the plasmids pBS1582, pBS1581, pBS1565, pBS1575, and pBS1566, respectively. All the constructs were confirmed by sequencing.
Plasmid SUP6-1 carries the SUP6 tRNATyr gene in which the 14-nucleotide intron had been replaced by the 52-nucleotide second intron of the MATa1 gene (Köhrer et al. 1990). Plasmid SUP6-2 is a mutated version of SUP6-1 with two single base substitutions in the branchpoint sequence (UAgUAuC).
Splicing analysis
RNA analysis and β-galactosidase assays were carried out as described (Séraphin and Kandels-Lewis 1993). β-Galactosidase assays were performed twice independently and each one in duplicate. Variation between duplicate samples was at most 20%. To analyze RP51A derivatives, primer extension was done by using the primer EM38 (Luukkonen and Séraphin 1997). To analyze splicing of the SUP6-1 and SUP6-2 reporters, primer extension was performed with the primer OP92 5′-CGAGTCGAACGCCCGATCTC-3′ under similar conditions except that dCTP was replaced by ddCTP in the nucleotide mixture. Extension products were analyzed in a denaturing 8% polyacrylamide/7 m urea gel for the SUP6 products and in 6% polyacrylamide/7 m urea gels for the RP51A derivatives. Quantification of all the primer extension results was done with a PhosphorImager (Molecular Dinamics). All signals were normalized to avoid loading differences by using the endogenous RP51A extension product.
Extract preparation and stability of commitment complexes
Yeast extracts were prepared as previously described (Séraphin and Rosbash 1989a) using strain MGD353-13D (Séraphin et al. 1988) for wild type extract, and strains BSY642 and BSY593 for Δnam8 and Nam8p–protA extracts, respectively (Gottschalk et al. 1998).
Commitment complexes were assembled as previously described (Séraphin and Rosbash 1991) except that temperature of incubation was 30°C. After this incubation, for the supershift analysis rabbit IgGs were added to final concentrations of 10 or 100 ng/μl and the samples were incubated at room temperature for 10 more min. Reactions were stopped and analyzed in a native gel (Séraphin and Rosbash 1989a). To assay the stability of the commitment complexes, after the 30-min incubation at 30°C, a 50-fold excess of cold pre-mRNA was added and the reactions were incubated for 1, 5, 10, 20, or 40 min more (Fig. 2; data not shown). Reactions were stopped and analyzed in a native gel. In time 0 the reaction was stopped immediately after the addition of the cold competitor.
Cross-linking
Commitment complexes were assembled on labeled pre-mRNA. After stopping the reaction, samples were transferred to a 96-well plate kept on ice, with a maximum volume of 30 μl per well, and were UV-irradiated in a Stratalinker (Stratagene) at 256 nm for 180 μJ at a distance of 5 cm from the bulbs. Samples were incubated for 30 min at 37°C with 10 U/μl of RNase T1. Immunoprecipitations were carried out by incubation at 4°C with IgG-coupled agarose beads in IPP150 buffer [10 mm Tris-Cl (pH 8.0), 150 mm NaCl, 0.1% NP-40] for 2 hr. For immunoprecipitation with the specific anti-Nam8p antibodies (Gottschalk et al. 1998), the serum was preincubated for 1 hr at 4°C with washed protein A–coupled Sepharose beads. After four washes with IPP150 buffer the pellets were analyzed by SDS-PAGE in 10% polyacrylamide gels.
The truncated substrates were generated by cleaving the plasmids pBS195 and pBS196 with EcoRV before transcription. All the cross-linking signals were quantified using a PhosphorImager.
Site-specific labeling and cross-linking with oligonucleotides
Site-specific labeling of pre-mRNAs was performed as described (Moore and Sharp 1992; Teigelkamp et al. 1995), except that ligation was carried out with 200 U/μl of DNA ligase (New England Biolabs) overnight at 25°C. Subsequently the samples were heated at 65°C for 10 min and 200 U/μl of DNA ligase were added. Incubation was performed for 2 additional hr at 37°C. RNA fragments were purified in denaturing polyacrylamide–urea gels. Oligonucleotides used for cross-linking contained two DNA residues at the 5′ end to protect them from degradation. The remaining part of the oligonucleotide was RNA. These oligonucleotides were labeled at the 5′ end and used as substrates in standard commitment complex reactions. Final concentration of each labelled oligonucleotide in the reaction was 1.5 μm. For the competition experiment molar ratios of labeled versus competitor oligonucleotide of 1:1, 1:3, and 1:10 were used. Cross-linking and immunoprecipitation was performed as with the full-length pre-mRNA, except that no treatment with RNase T1 was carried out.
Acknowledgments
We thank P. Askjaer, E. Bragado-Nilsson, F. Caspary, P. Fortes, P. López, M. Luukkonen, R. Lührmann, I. Mattaj, R. Ramírez, G. Rigaut, B. Rutz, K. Sharma, and J. Valcárcel for stimulating discussions and comments on the manuscript. B. Rutz is also acknowledged for valuable help with the supershift assay, and P. Fortes, and I. Mattaj for communicating unpublished data. O. Groudinsky, H. Ogawa, H. Domdey, and S. Braun are acknowledged for sending yeast strains and/or plasmids. European Molecular Biology Laboratory oligonucleotide synthesis and photolab services are especially acknowledged. This work was supported by an European Union Training and Mobility of Researchers fellowship to O. Puig.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL seraphin@embl-heidelberg.de; FAX +49 6221 387 518.
References
- Abovich N, Rosbash M. Cross-intron bridging interactions in the yeast commitment complex are conserved in mammals. Cell. 1997;89:403–412. doi: 10.1016/s0092-8674(00)80221-4. [DOI] [PubMed] [Google Scholar]
- Abovich N, Liao XC, Rosbash M. The yeast MUD2 protein: An interaction with PRP11 defines a bridge between commitment complexes and U2 snRNP addition. Genes Dev. 1994;8:843–854. doi: 10.1101/gad.8.7.843. [DOI] [PubMed] [Google Scholar]
- Arning S, Gruter P, Bilbe G, Krämer A. Mammalian splicing factor SF1 is encoded by variant cDNAs and binds to RNA. RNA. 1996;2:794–810. [PMC free article] [PubMed] [Google Scholar]
- Berglund JA, Chua K, Abovich N, Reed R, Rosbash M. The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell. 1997;89:781–787. doi: 10.1016/s0092-8674(00)80261-5. [DOI] [PubMed] [Google Scholar]
- Black DL. Finding splice sites within a wilderness of RNA. RNA. 1995;1:763–771. [PMC free article] [PubMed] [Google Scholar]
- Chabot B. Directing alternative splicing: Cast and scenarios. Trends Genet. 1996;12:472–478. doi: 10.1016/0168-9525(96)10037-8. [DOI] [PubMed] [Google Scholar]
- Chan RC, Black DL. Conserved intron elements repress splicing of a neuron-specific c-src exon in vitro. Mol Cell Biol. 1995;15:6377–6385. doi: 10.1128/mcb.15.11.6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colot HV, Stutz F, Rosbash M. The yeast splicing factor Mud13p is a commitment complex component and corresponds to CBP20, the small subunit of the nuclear cap-binding complex. Genes & Dev. 1996;10:1699–1708. doi: 10.1101/gad.10.13.1699. [DOI] [PubMed] [Google Scholar]
- Del Gatto F, Breathnach R. Exon and intron sequences, respectively, repress and activate splicing of a fibroblast growth factor receptor 2 alternative exon. Mol Cell Biol. 1995;15:4825–4834. doi: 10.1128/mcb.15.9.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekwall K, Kermorgant M, Dujardin G, Groudinsky O, Slonimski PP. The NAM8 gene in Saccharomyces cerevisiae encodes a protein with putative RNA binding motifs and acts as suppressor of mitochondrial splicing deficiencies when overexpressed. Mol & Gen Genet. 1992;233:136–144. doi: 10.1007/BF00587571. [DOI] [PubMed] [Google Scholar]
- Engebrecht JA, Voelkel-Meiman K, Roeder GS. Meiosis-specific RNA splicing in yeast. Cell. 1991;66:1257–1268. doi: 10.1016/0092-8674(91)90047-3. [DOI] [PubMed] [Google Scholar]
- Gallego ME, Balvay L, Brody E. Cis-acting sequences involved in exon selection in the chicken beta-tropomyosin gene. Mol Cell Biol. 1992;12:5415–5425. doi: 10.1128/mcb.12.12.5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goguel V, Liao XL, Rymond BC, Rosbash M. U1 snRNP can influence 3′-splice site selection as well as 5′-splice site selection. Genes & Dev. 1991;5:1430–1438. doi: 10.1101/gad.5.8.1430. [DOI] [PubMed] [Google Scholar]
- Gottschalk A, Tang J, Puig O, Salgado J, Neubauer G, Colot HV, Mann M, Séraphin B, Roshbash M, Lührmann R, Fabrizio P. Comprehensive biochemical and genetic analysis of the yeast U1 snRNP reveals five novel proteins. RNA. 1998;4:374–393. [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Izaurralde E, Lewis J, McGuigan C, Jankowska M, Darzynkiewicz E, Mattaj IW. A nuclear cap binding protein complex involved in pre-mRNA splicing. Cell. 1994;78:657–668. doi: 10.1016/0092-8674(94)90530-4. [DOI] [PubMed] [Google Scholar]
- Jacquier A, Rodriguez JR, Rosbash M. A quantitative analysis of the effects of 5′ junction and TACTAAC box mutants and mutant combinations on yeast mRNA splicing. Cell. 1985;43:423–430. doi: 10.1016/0092-8674(85)90172-2. [DOI] [PubMed] [Google Scholar]
- Kao HY, Siliciano PG. Identification of Prp40, a novel essential yeast splicing factor associated with the U1 small nuclear ribonucleoprotein particle. Mol Cell Biol. 1996;16:960–967. doi: 10.1128/mcb.16.3.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhrer K, Vogel K, Domdey H. A yeast tRNA precursor containing a pre-mRNA intron is spliced via the pre-mRNA splicing mechanism. EMBO J. 1990;9:705–709. doi: 10.1002/j.1460-2075.1990.tb08163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD, Gorlich D, Mattaj IW. A yeast cap binding protein complex (yCBC) acts at an early step in pre-mRNA splicing. Nucleic Acids Res. 1996a;24:3332–3336. doi: 10.1093/nar/24.17.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD, Izaurralde E, Jarmolowski A, McGuigan C, Mattaj IW. A nuclear cap-binding complex facilitates association of U1 snRNP with the cap-proximal 5′ splice site. Genes & Dev. 1996b;10:1683–1698. doi: 10.1101/gad.10.13.1683. [DOI] [PubMed] [Google Scholar]
- Liao XC, Tang J, Rosbash M. An enhancer screen identifies a gene that encodes the yeast U1 snRNP A protein: Implications for snRNP protein function in pre-mRNA splicing. Genes & Dev. 1993;7:419–428. doi: 10.1101/gad.7.3.419. [DOI] [PubMed] [Google Scholar]
- Lockhart SR, Rymond BC. Commitment of yeast pre-mRNA to the splicing pathway requires a novel U1 small nuclear ribonucleoprotein polypeptide, Prp39p. Mol Cell Biol. 1994;14:3623–3633. doi: 10.1128/mcb.14.6.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luukkonen BG, Séraphin B. The role of branchpoint–3′ splice site spacing and interaction between intron terminal nucleotides in 3′ splice site selection in Saccharomyces cerevisiae. EMBO J. 1997;16:779–792. doi: 10.1093/emboj/16.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Genetic interaction between U6 snRNA and the first intron nucleotide in Saccharomyces cerevisiae. RNA. 1998;4:167–180. [PMC free article] [PubMed] [Google Scholar]
- Lynch KW, Maniatis T. Synergistic interactions between two distinct elements of a regulated splicing enhancer. Genes & Dev. 1995;9:284–293. doi: 10.1101/gad.9.3.284. [DOI] [PubMed] [Google Scholar]
- McKeown M. Alternative mRNA splicing. Annu Rev Cell Biol. 1992;8:133–155. doi: 10.1146/annurev.cb.08.110192.001025. [DOI] [PubMed] [Google Scholar]
- McLean MR, Rymond BC. Yeast pre-mRNA splicing requires a pair of U1 snRNP-associated tetratricopeptide repeat proteins. Mol Cell Biol. 1998;18:353–360. doi: 10.1128/mcb.18.1.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud S, Reed R. An ATP-independent complex commits pre-mRNA to the mammalian spliceosome assembly pathway. Genes & Dev. 1991;5:2534–2546. doi: 10.1101/gad.5.12b.2534. [DOI] [PubMed] [Google Scholar]
- ————— A functional association between the 5′ and 3′ splice site is established in the earliest prespliceosome complex (E) in mammals. Genes & Dev. 1993;7:1008–1020. doi: 10.1101/gad.7.6.1008. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Sharp PA. Site-specific modification of pre-mRNA: The 2′-hydroxyl groups at the splice sites. Science. 1992;256:992–997. doi: 10.1126/science.1589782. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Query CC, Sharp PA. In: The RNA world. Gesteland R, Atkins J, editors. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 303–357. [Google Scholar]
- Nagai K, Oubridge C, Jessen TH, Li J, Evans PR. Crystal structure of the RNA-binding domain of the U1 small nuclear ribonucleoprotein A. Nature. 1990;348:515–520. doi: 10.1038/348515a0. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Ogawa H. Involvement of the MRE2 gene of yeast in formation of meiosis-specific double-strand breaks and crossover recombination through RNA splicing. Genes Cells. 1997;2:65–79. doi: 10.1046/j.1365-2443.1997.d01-283.x. [DOI] [PubMed] [Google Scholar]
- Neubauer G, Gottschalk A, Fabrizio P, Séraphin B, Lührmann R, Mann M. Identification of the proteins of the yeast U1 small nuclear ribonucleoprotein complex by mass spectrometry. Proc Natl Acad Sci. 1997;94:385–390. doi: 10.1073/pnas.94.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa H, Johzuka K, Nakagawa T, Leem SH, Hagihara AH. Functions of the yeast meiotic recombination genes, MRE11 and MRE2. Adv Biophys. 1995;31:67–76. doi: 10.1016/0065-227x(95)99383-z. [DOI] [PubMed] [Google Scholar]
- Oubridge C, Ito N, Evans PR, Teo CH, Nagai K. Crystal structure at 1.92 Å resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature. 1994;372:432–438. doi: 10.1038/372432a0. [DOI] [PubMed] [Google Scholar]
- Pascolo E, Séraphin B. The branchpoint residue is recognised during commitment complex formation before being bulged out of the U2 snRNA-pre-mRNA duplex. Mol Cell Biol. 1997;17:3469–3476. doi: 10.1128/mcb.17.7.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikielny CW, Rosbash M. mRNA splicing efficiency in yeast and the contribution of nonconserved sequences. Cell. 1985;41:119–126. doi: 10.1016/0092-8674(85)90066-2. [DOI] [PubMed] [Google Scholar]
- Price SR, Evans PR, Nagai K. Crystal structure of the spliceosomal U2′′-U2A′ protein complex bound to a fragment of U2 small nuclear RNA. Nature. 1998;394:645–650. doi: 10.1038/29234. [DOI] [PubMed] [Google Scholar]
- Puig O, Rutz B, Luukkonen BGM, Kandels-Lewis S, Bragado-Nilsson E, Séraphin B. New constructs and strategies for efficient PCR-based gene manipulations in yeast. Yeast. 1998;14:1139–1146. doi: 10.1002/(SICI)1097-0061(19980915)14:12<1139::AID-YEA306>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Rain JC, Legrain P. In vivo commitment to splicing in yeast involves the nucleotide upstream from the branch site conserved sequence and the Mud2 protein. EMBO J. 1997;16:1759–1771. doi: 10.1093/emboj/16.7.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosbash M, Séraphin B. Who’s on first? The U1 snRNP–5′ splice site interaction and splicing. Trends Biochem Sci. 1991;16:187–190. doi: 10.1016/0968-0004(91)90073-5. [DOI] [PubMed] [Google Scholar]
- Séraphin B, Rosbash M. Identification of functional U1 snRNA–pre-mRNA complexes committed to spliceosome assembly and splicing. Cell. 1989a;59:349–358. doi: 10.1016/0092-8674(89)90296-1. [DOI] [PubMed] [Google Scholar]
- ————— Mutational analysis of the interactions between U1 small nuclear RNA and pre-mRNA of yeast. Gene. 1989b;82:145–151. doi: 10.1016/0378-1119(89)90039-5. [DOI] [PubMed] [Google Scholar]
- ————— The yeast branchpoint sequence is not required for the formation of a stable U1 snRNA–pre-mRNA complex and is recognised in the absence of U2 snRNA. EMBO J. 1991;10:1209–1216. doi: 10.1002/j.1460-2075.1991.tb08062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Séraphin B, Kandels-Lewis S. 3′ splice site recognition in S. cerevisiae does not require base pairing with U1 snRNA. Cell. 1993;73:803–812. doi: 10.1016/0092-8674(93)90258-r. [DOI] [PubMed] [Google Scholar]
- Séraphin B, Kretzner M, Rosbash M. A U1 snRNA: pre-mRNA base pairing interaction is required early in yeast spliceosome assembly but does not uniquely define the 5′ cleavage site. EMBO J. 1988;7:2533–2538. doi: 10.1002/j.1460-2075.1988.tb03101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith V, Barrell BG. Cloning of a yeast U1 snRNP 70K protein homolog: Functional conservation of an RNA-binding domain between humans and yeast. EMBO J. 1991;10:2627–2634. doi: 10.1002/j.1460-2075.1991.tb07805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Abovich N, Fleming ML, Séraphin B, Rosbash M. Identification and characterization of a yeast homolog of U1 snRNP-specific protein C. EMBO J. 1997;16:4082–4091. doi: 10.1093/emboj/16.13.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teem JL, Rosbash M. Expression of a beta-galactosidase gene containing the ribosomal protein 51 intron is sensitive to the rna2 mutation of yeast. Proc Natl Acad Sci. 1983;80:4403–4407. doi: 10.1073/pnas.80.14.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teigelkamp S, Newman AJ, Beggs J. Extensive interactions of PRP8 protein with the 5′ and 3′ splice sites during splicing suggest a role in stabilization of exon alignment by U5 snRNA. EMBO J. 1995;14:2602–2612. doi: 10.1002/j.1460-2075.1995.tb07258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayraghavan U, Parker R, Tamm J, Iimura Y, Rossi J, Abelson J, Guthrie C. Mutations in conserved intron sequences affect multiple steps in the yeast splicing pathway, particularly assembly of the spliceosome. EMBO J. 1986;5:1683–1695. doi: 10.1002/j.1460-2075.1986.tb04412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]