

Abstract
In the rat β-tropomyosin (β-TM) gene, exons 6 and 7 are spliced alternatively in a mutually exclusive manner. Exon 6 is included in mRNA encoding nonmuscle TM-1, whereas exon 7 is used in mRNA encoding skeletal muscle β-TM. Previously, we demonstrated that a six nucleotide mutation at the 5′ end of exon 7, designated as ex-1, activated exon 7 splicing in nonmuscle cells. In this study, we show that the activating effect of this mutation is not the result of creating an exonic splicing enhancer (ESE) or disrupting a putative secondary structure. The sequence in exon 7 acts as a bona fide exonic splicing silencer (ESS), which is bound specifically by a trans-acting factor. Isolation and peptide sequencing reveal that this factor is hnRNP H, a member of the heterogeneous nuclear ribonucleoprotein (hnRNP) family. Binding of hnRNP H correlates with the ESS activity. Furthermore, addition of antibodies that specifically recognizes hnRNP H to the splicing reactions or partial depletion of hnRNP H from nuclear extract activates exon 7 splicing in vitro and this effect can be reversed by addition of purified recombinant hnRNP H. These results indicate that hnRNP H participates in exclusion of exon 7 in nonmuscle cells. The involvement of hnRNP H in the activity of an ESS may represent a prototype for the regulation of tissue- and developmental-specific alternative splicing.
Keywords: RNA processing, cis-acting element, trans-acting factor, heterogeneous nuclear ribonucleoproteins, RNA–protein interaction
Alternative RNA splicing is a common mechanism for regulating gene expression in eukaryotes (for reviews, see Adams et al. 1996; Wang et al. 1997; König et al. 1998). By this mechanism, a precursor mRNA (pre-mRNA) transcribed from a single gene can be processed to generate multiple mRNAs that are translated into various protein isoforms, often with different functions according to sex, tissue type, developmental stage, or environmental cues. Thus, studies on the regulation of alternative RNA splicing are essential for a complete understanding of gene expression at the molecular level.
One of the best-known examples demonstrating the importance and regulation of alternative RNA splicing is the genes involved in the Drosophila sex determination pathway (for reviews, see Rio 1993; Wang et al. 1997). The Sex lethal (Sxl) protein is produced in female flies, where it binds to the regulatory element in intron 2 of transformer pre-mRNA and leads to the production of functional transformer protein (Tra) (Boggs et al. 1987; Inoue et al. 1990; Valcárcel et al. 1993). Tra, along with the common splicing factor transformer 2 (Tra 2), binds to the cis-acting element in exon 4 of doublesex pre-mRNA (Hedley and Maniatis 1991; Hoshijima et al. 1991; Ryner and Baker 1991; Inoue et al. 1992; Tian and Maniatis 1993). This binding recruits a set of SR proteins, which are general splicing factors, thereby activating the 3′ splice site upstream of exon 4 and giving rise to the production of the female-specific doublesex mRNA (Zahler et al. 1992; Tian and Maniatis 1993; Wu and Maniatis 1993; Lynch and Maniatis 1995).
Although much less is known about the regulation of alternative splicing in vertebrates, cis-acting regulatory elements have been identified in both exon and intron sequences. Regulatory intron sequences that activate or inhibit splicing have been identified in several premRNAs, including β-tropomyosin (β-TM) (Helfman et al. 1990; Libri et al. 1990; Guo et al. 1991; Sirand-Pugnet et al. 1995), c-src (Black 1992), fibroblast growth factor receptor (FGFR) (Gatto and Breathnach 1995), fibronectin (Huh and Hynes 1994), calcitonin/calcitonin gene-related peptide (Lou et al. 1995), and adenoviral pre-mRNA (Kanopka et al. 1996). Exonic splicing enhancers (ESEs) have also been shown to be involved in the regulation of alternative RNA splicing and purine-rich ESEs have been found in pre-mRNAs such as mouse immunoglobulin M exon M2 (Watakabe et al. 1993), chicken cardiac troponin T exon 5 (Xu et al. 1993), human fibronectin EDA exon (Caputi et al. 1994), the last exon of bovine growth hormone (Hampson et al. 1989), and rat β-TM exon 8 (Tsukahara et al. 1994; Selvakumar and Helfman 1998). In contrast, exonic splicing silencers (ESSs) have been identified only in a few pre-mRNAs such as fibronectin EDA exon (Caputi et al. 1994), human immunodeficiency virus (HIV)-tat exon 2 and tat-rev exon 3 (Amendt et al. 1995; Staffa and Cochrane 1995), FGFR-2 K-SAM exon (Gatto and Breathnach 1995), bovine papillomavirus type 1 pre-mRNA (Zheng et al. 1996), and cell surface molecule CD44 exon 5 (König et al. 1998). To date, only a few trans-acting factors have been implicated in the regulation of alternative splicing in vertebrates. SF2/ASF, a member of the SR protein family, stimulates splicing from the proximal 5′ splice sites in some pre-mRNAs (Ge and Manley 1990; Krainer et al. 1990), whereas heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) antagonizes this activity and stimulates splicing from the distal 5′ splice sites (Mayeda and Krainer 1992; Cáceres et al. 1994; Yang et al. 1994). The activation of splicing by SR proteins is presumably by binding to cis-acting elements such as ESEs (Lavigueur et al. 1993; Sun et al. 1993; Ramchatesingh et al. 1995). SR proteins have also been shown to inhibit adenovirus IIIa pre-mRNA splicing by binding to an intronic repressor (Kanopka et al. 1996). Another group of potential splicing regulators is the hnRNP family of proteins. hnRNP I, also known as the polypyrimidine tract binding protein (PTB), has been implicated in the regulation of alternatively spliced pre-mRNAs, such as β-TM (Mulligan et al. 1992), fibronectin (Norton 1994), α-TM (Lin and Patton 1995; Perez et al. 1997; Gooding et al. 1998), r2 subunit of the GABA receptor (Ashiya and Grabowski 1997), and c-src (Chan and Black 1997). Another member of the hnRNP family, hnRNP F, along with a KH-type splicing regulatory protein (KSRP), binds to the downstream control sequence (DCS) of c-src and activates splicing of the N1 exon (Min et al. 1995, 1997).
We have been using rat β-TM pre-mRNA as a model system to study the regulation of alternative RNA splicing (Helfman et al. 1988, 1990; Helfman and Ricci 1989). The rat β-TM gene consists of 11 exons, and 2 exon pairs are alternatively spliced. Exons 6 and 11 are used for generating TM-1 mRNA in nonmuscle cells, which also corresponds to smooth muscle β-TM; exons 7 and 10 are used for forming β-TM mRNA in skeletal muscle and fetal cardiac muscle cells (Fig. 1A). Previous results from our laboratory demonstrated that splicing of the skeletal muscle-specific exon 7 in nonmuscle cells was blocked in its 3′ splice site (Guo and Helfman 1993). Two cis-acting elements that are critical for blocking exon 7 splicing in nonmuscle cells were identified in sequences at the 5′ end of exon 7 and the 3′ end of intron 6 [the intron regulatory element (IRE)] (Helfman et al. 1990; Guo et al. 1991; Guo and Helfman 1993). Mutations or deletions of the IRE led to utilization of exon 7 in nonmuscle cells. Further studies indicated that PTB, as well as other proteins, such as FUSE-binding protein (FBP) and a homolog of human Sam 68 tyrosine phosphoprotein, formed a complex on the IRE that may be involved in blocking the recognition of the 3′ splice site of exon 7 (Mulligan et al. 1992; Grossman et al. 1998). We also showed that a mutation, designated as ex-1, in which six nucleotides UGUGGG at the 5′ end of exon 7 were mutated to GGAUCC (Fig. 1B), activated exon 7 splicing in nonmuscle cells in vivo (Guo et al. 1991). These data demonstrated that the sequence at the 5′ end of exon 7 function as a cis-acting element regulating rat β-TM pre-mRNA alternative splicing. However, the mechanism by which these sequences contribute to the regulation of splice site selection was not known.
Figure 1.

(A) A schematic diagram of rat β-TM gene. Boxes represent exons and lines introns; shaded boxes represent tissue-specific exons as indicated. (B) The wild-type and mutated sequences of exon 7. Lowercase letters represent sequences in adjacent introns, and +1 is the first nucleotide of exon 7. The mutated sequences are indicated below the wild-type sequences. (C) Stem I of the putative secondary structure in the wild-type substrate. (D) The restored stem I of the putative secondary structure when both complementary strands are mutated simultaneously.
In this report, we show that sequences at the 5′ end of exon 7 function as an ESS. The ubiquitously expressed hnRNP H binds to this ESS, and its binding correlates with the silencer activity. We demonstrate further that the addition of antibodies, which recognize hnRNP H to the splicing reactions or depletion of hnRNP H from nuclear extract, activates exon 7 splicing in vitro, and this activation is reversed by recombinant hnRNP H protein. Our data show for the first time that hnRNP H participates in the negative regulation of alternative RNA splicing and that hnRNP H is a trans-acting factor involved in ESS activity in vertebrates.
Results
An in vitro system to study the ex-1 mutation
Our previous data demonstrated that a mutation at the 5′ end of exon 7, designated as ex-1, reversed normal exon selection in nonmuscle cells in vivo, resulting in utilization of the skeletal muscle-specific exon 7 instead of the nonmuscle-specific exon 6 (Guo et al. 1991). This dramatic effect led us to hypothesize that sequences at the 5′ end of exon 7 may be an important cis-acting element in the regulation of rat β-TM pre-mRNA alternative splicing. By studying how this cis-acting element functions, we may be able to reveal some of the general principles of tissue- and developmental-specific regulation of alternative splicing in vertebrates.
To study biochemically how the ex-1 mutation activates exon 7 splicing in nonmuscle cells, we first designed a suitable pre-mRNA. Although we could have used the minigene p2 that contains genomic sequences from exon 5 to exon 9, the complexity of spliced products and the difficulty of detecting the effect of the ex-1 mutation in vitro (data not shown) counteracted the usefulness of this substrate (Helfman et al. 1988; Guo et al. 1991). Because exon 7 is not required for the complex formation in intron 6 (Grossman et al. 1998), we predicted that the cis-acting element in exon 7 uses a different mechanism from the IRE at the 3′ end of intron 6. Thus, it is possible to study the involvement of exon 7 sequences in the regulation of rat β-TM pre-mRNA alternative splicing independent of intron 6 sequences. Consequently, a pair of simple RNA substrates, the wild-type 5(5)7 and the mutant 5(5)7 ex-1, were generated. Substrate 5(5)7 consists of exon 5, intron 5, and exon 7. To simplify the nomenclature in this paper, an exon is indicated by its number and an intron by its number in parenthesis.
To determine the kinetics of splicing, the wild-type and mutant substrates were subjected to in vitro splicing reaction in a time course reaction. When the mutant substrate was tested, the first step of splicing was detected within 30 min of incubation, resulting in cleaved exon 5 and the intermediate lariat (5)7 (Fig. 2, lane 6). As the splicing reaction proceeded, the intensity of the intermediate lariat (5)7 decreased and the final product 5 + 7 increased (Fig. 2, lanes 7–10). The intensity of the final lariat intron (5) decreased because it was degraded. The wild-type substrate was not spliced even after 2.5 hr of incubation (Fig. 2, lanes 1–5). These results suggest that the mechanism involved in the cis-acting element in exon 7 is independent of intron 6. Thus, the mechanism responsible for the activation of the ex-1 mutation can be studied using this pair of simple RNA substrates.
Figure 2.
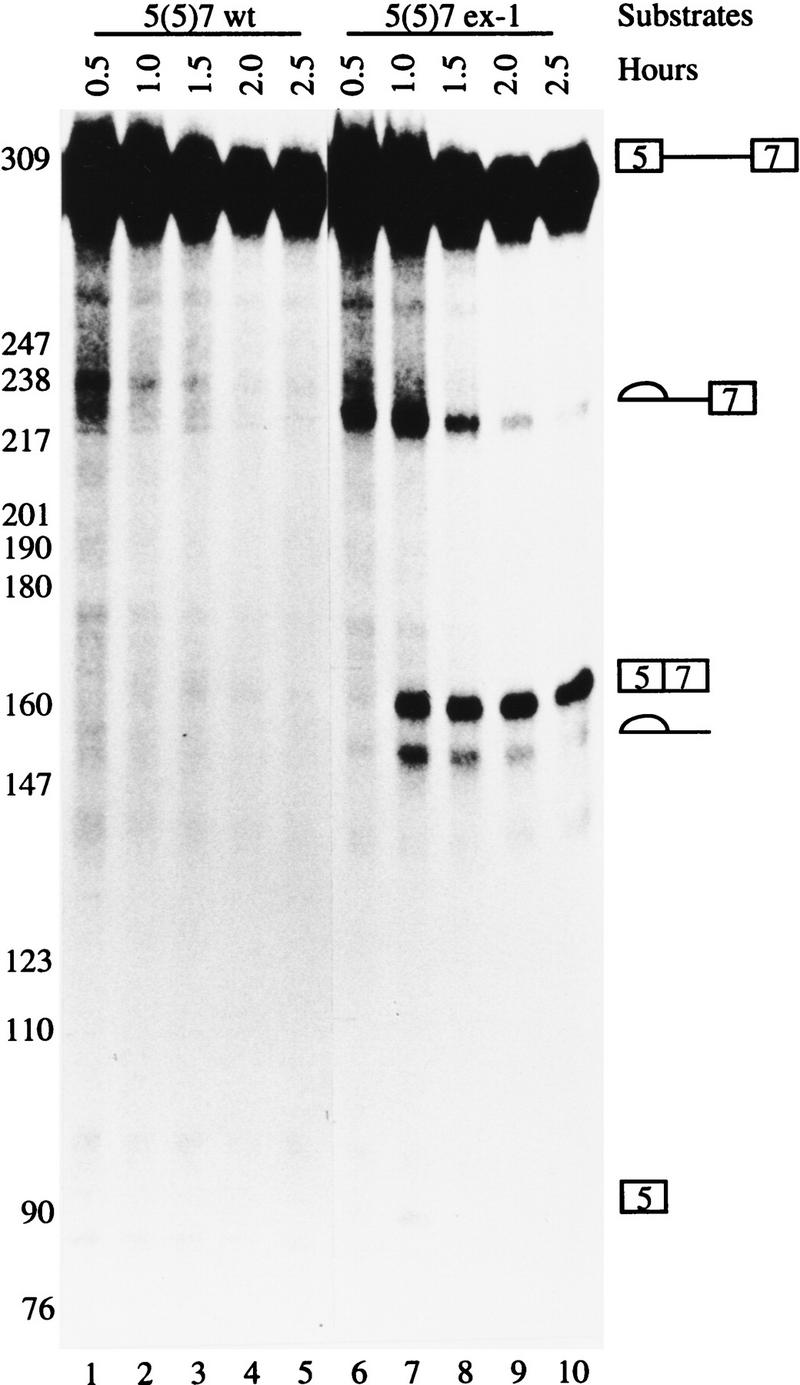
An in vitro system to study the ex-1 mutation. The wild-type and ex-1 mutation 5(5)7 pre-mRNAs containing exon 5, intron 5, and exon 7 were synthesized and subjected to in vitro splicing reactions using the conditions indicated at the top. The RNAs were resolved on 4% polyacrylamide 8 m urea gel and visualized by autoradiography. The precursors, intermediate, and final products are indicated at left.
The sequence at the 5′ end of exon 7 is an exonic splicing silencer
Several possible mechanisms may be responsible for the activation of the ex-1 mutation. One is that the mutation creates an ESE, thereby activating exon 7 splicing in nonmuscle cells. To test this possibility, we made two other random mutations. One mutated UGUGGG to CUACGC (ex-16), and the other mutated AAUGUGGGGA to UGUACGAUCG (ex-110) (see Fig. 1B). We reasoned that if the activation of the ex-1 mutation is a consequence of an ESE, the same effect should not be seen with two other random mutations. However, these two mutated substrates were also spliced (Fig. 3A, lanes 9–16); the ex-110 mutant was spliced more efficiently than the ex-1 mutant (Fig. 3A, cf. lanes 13–16 with lanes 5–8), and the ex-16 mutant was spliced with the highest efficiency (Fig. 3A, lanes 9–12). The band in the wild-type substrate that migrated more slowly than the intermediate lariate (5)7 is a prematurely transcribed RNA (Fig. 3A, lanes 1–4). The possibility remains that all these mutations create ESEs, with the ex-16 mutation being the strongest one. However, combined with the data described later, these data suggest that it is unlikely that the activation of the ex-1 mutation is attributable to creation of an ESE.
Figure 3.



The 5′ end of exon 7 contains an ESS. Transcription and in vitro splicing were carried out as in Fig. 2, and the marker is the same as in Fig. 2. (A) Activation of ex-1 does not result from creation of an ESE. The wild-type and mutant 5(5)7 substrates are indicated at the top. (B) Activation of ex-1 mutation does not result from disruption of the putative secondary structure. The wild-type and mutant 5(5)7 substrates are indicated at the top. (C) Titration experiments. Splicing reactions were carried out in the absence (lanes 1,8,15,19) or presence of 10, 20, or 30 pmoles of the specific competitor ECS, or nonspecific competitor WU as indicated. The substrates are indicated at the top, the precursors and the products on either side. 5(5)6 contains exon 6 instead of exon 7 in 5(5)7; β-globin pre-mRNA consists of exon 1, intron 1, and exon 2 of the human β-globin gene.
The second possible mechanism by which the ex-1 mutation activates exon 7 splicing is by disrupting a secondary structure. A secondary structure was shown in chicken β-TM pre-mRNA (D’Orval et al. 1991; Libri et al. 1991), and stem I (see Fig. 1C) plays a key role in the stabilization of this structure. A similar secondary structure was previously proposed for rat β-TM gene based on computer predictions (Helfman et al. 1990). Because the ex-1 mutation disrupts the formation of stem I, it is possible that this structure also participates in the regulation of rat β-TM pre-mRNA alternative splicing. To test this possibility, we mutated the complementary sequence ACCAACA to GGAAUCC to disrupt the putative secondary structure (ex-m) (see Fig. 1B). If the activation of the ex-1 mutation is attributable to the disruption of the putative secondary structure, the ex-m mutation would also activate exon 7 splicing. However, ex-m did not activate exon 7 splicing (Fig. 3B, lanes 9–12). Furthermore, when the putative secondary structure was reconstituted by simultaneously mutating both complementary strands (ex-1/m) (see Fig. 1B), exon 7 was spliced (Fig. 3B, lanes 13–16). The kinetics and intensities of exon 7 splicing were comparable between the ex-1 and ex-1/m mutants (Fig. 3B, cf. lanes 13–16 with lanes 5–8). These data indicated that the activation of the ex-1 mutation does not result from disruption of the putative secondary structure.
The third possible mechanism responsible for the activation of the ex-1 mutation is disrupting the binding of trans-acting factors. If so, addition of the wild-type sequence to the splicing reaction should titrate away these trans-acting factors and activate exon 7 splicing. When an oligoribonucleotide ECS (ESS-Containing Sequence) that contains a sequence from +2 to +19 of exon 7 was added to the in vitro splicing reaction, the wild-type substrate 5(5)7 was activated, and the activation increased with an increasing amount of ECS (Fig. 3C, lanes 1–4). The activation of exon 7 splicing was specific to ECS because an unrelated oligoribonucleotide WU had no effect on the activation of exon 7 splicing (Fig. 3C, lanes 5–7). To determine whether the effect of ECS was specific to exon 7, we also added ECS to the splicing reactions with a substrate 5(5)6. This substrate has exon 6 as the downstream exon instead of exon 7 as in the substrate 5(5)7. Addition of ECS did not activate exon 6 splicing (Fig. 3C, lanes 15–18) or stimulate the splicing of the human β-globin pre-mRNA (Fig. 3C, lanes 19–22). These results suggest that the binding of transacting factors to sequences within exon 7 may be involved in the regulation of rat β-TM pre-mRNA alternative splicing.
To confirm our conclusion, we tested the splicing of the mutant substrate 5(5)7 ex-1 in titration experiments. As shown in Figure 3A, the ex-1 mutation activates exon 7 splicing, but this activation is not the strongest; both ex-110 and ex-16 activate exon 7 splicing stronger than ex-1. If the inability of exon 7 to be spliced is attributable to the binding of negative trans-acting factors, the failure of the ex-1 mutation to reach maximal activation may be because the ex-1 mutant still weakly associates with the putative trans-acting factors. Therefore, addition of ECS may also enhance splicing of 5(5)7 ex-1, which, indeed, occurred (Fig. 3C, lanes 8–11). This activation was specific for ECS because addition of WU did not activate the splicing of 5(5)7 ex-1 (Fig. 3C, lanes 12–14). We conclude that the sequence UGUGGG at the 5′ end of exon 7 is part of an ESS, and that the binding of trans-acting factors is responsible for the activity of the ESS.
Specific proteins cross-link to the ESS sequences
To identify and characterize the trans-acting factors that bind to the ESS, we performed UV cross-linking experiments with uniformly G- and C-radiolabeled 20-mer RNAs because all the mutations contain an equal amount of G and C. When an oligoribonucleotide containing the wild-type ESS sequence was used in UV cross-linking experiments, a prominent UV cross-linked product was identified (Fig. 4A, lane 1). This cross-linked product was also detected when each of the mutated oligoribonucleotides was used (Fig. 4A, lanes 2–4), but the amount of the product was much less than in the experiment in which the wild-type was used (Fig. 4A, cf. lanes 2–4 with lane 1). The ex-110 mutation probably creates a binding site for an unrelated protein, resulting in the formation of a cross-linked product of <50 kD (Fig. 4A, lane 4, bottom arrow). These results are consistent with the conclusion that the ESS sequence is a binding site for trans-acting factors.
Figure 4.

A protein cross-links to the ESS. (A) 32P-Labeled oligoribonucleotide that contains either the wild-type or mutated ESS as indicated at the top was cross-linked to HeLa cell nuclear extract and resolved on a 10% SDS-polyacrylamide gel. Two arrows indicate two cross-linked products in lane 4; the upper one migrated similarly to the products in other lanes. (B) 32P-Labeled oligoribonucleotide that contains the wild-type ESS was cross-linked to HeLa cell nuclear extract in the absence (lane 1) or presence of 10- , 20- , 40- , 60- , or 80-fold excess of the wild-type competitor or mutant competitor ex-110 as indicated.
To determine whether the cross-linked protein binds specifically to the ESS and whether it binds more strongly to the wild-type ESS than to the mutant sequences, we performed competition assays in the UV cross-linking experiments. The product that cross-linked to the wild-type sequence (Fig. 4B, lane 1) was gradually competed away with an increasing amount of the wild-type competitor (Fig. 4B, lanes 2–6), but not by a comparable amount of either the mutant ex-110, or ex-1, or ex-16 competitor (Fig. 4B, lanes 7–11; data not shown). Taken together, these data suggest that at least one protein specifically binds to the ESS sequence and that its binding may be responsible for the activity of the ESS in exon 7.
hnRNP H binds to the ESS sequence
To identify the binding factor that specifically interacts with the ESS sequence, we fractionated HeLa cell nuclear extracts and assayed each fraction using UV cross-linking to detect the interactions with the radiolabeled oligonucleotide containing the wild-type ESS. The majority of the protein was present in the 20%–50% ammonium sulfate fraction (Fig. 5A, lane 2). The 60%–90% fraction also gave rise to several cross-linked products (Fig. 5A, lane 4), which is not surprising because the natural binding factor was not in this fraction and the binding site of this protein is now available for other proteins.
Figure 5.

Isolation and peptide-sequencing of hnRNP H. (A) HeLa cell nuclear extracts were fractionated by ammonium sulfate precipitation, and each fraction was UV cross-linked to an oligoribonucleotide containing the wild-type ESS. (B) The biotinylated RNA (lane 1) or RNA (lane 2) containing six tandem repeats of the wild-type ESS was incubated with the 20%–50% ammonium sulfate fraction. The RNA–protein complexes were pulled down by streptavidin–agarose. The proteins were eluted with wash buffer containing 0.5 m KCl, resolved on a 10% SDS-polyacrylamide gel, and visualized by silver staining. The arrow indicates the 50-kD protein of interest. (C) Two peptides from the 50-kD protein were obtained by peptide sequencing.
The 20%–50% ammonium sulfate fraction was used to purify the protein by a biotin–streptavidin affinity assay. Accordingly, a biotin-labeled RNA containing six tandem repeats of the ESS sequence was incubated with the 20%–50% ammonium sulfate fraction, and the protein–RNA complexes were recovered by streptavidin–agarose beads. The beads were washed, and the proteins were eluted, resolved on SDS-polyacrylamide gels, and visualized by silver staining. A 50-kD protein was well separated on the gel (Fig. 5B, lane 1). The binding of this protein could be competed away by the wild-type monomer, but not by the ex-110 mutant (data not shown).
The biotin–streptavidin binding assay was scaled up; the recovered proteins were separated by SDS-PAGE and visualized by Coomassie blue G staining. The 50-kD protein band was excised from the gel, and subjected to in gel digestion and microsequencing. One long peptide with 22 amino acid residues (CC50K40) and a short peptide (CC49K20) with 10 amino acid residues were obtained (Fig. 5C). Database search revealed that these two peptides were found in the sequence of hnRNP H (Honoré et al. 1995).
Binding of hnRNP H correlates with the activity of the ESS
To examine whether hnRNP H is involved in the activity of the ESS, we first carried out the biotin–streptavidin affinity assay as described earlier except crude nuclear extract was used in the place of the 20%–50% ammonium sulfate fraction, and followed by Western blot analysis to detect the possible interactions of hnRNP H with the wild-type and different mutant 5(5)7 premRNAs. hnRNP H interacted strongly with the wild-type substrate 5(5)7, as evidenced by recognition of the 50-kD band by a specific antibody against hnRNP H (Fig. 6A, lane 2). Binding of hnRNP H to exon 7 was specific because substituting exon 6 for exon 7 virtually abolished the binding (Fig. 6A, lane 1). hnRNP H also bound to the mutant substrates of 5(5)7, but its apparent affinity with the mutant substrates was weaker than with the wild-type splicing substrate 5(5)7 (Fig. 6A, cf. lane 2 with lanes 3–5).
Figure 6.


hnRNP H specifically binds to the ESS. (A) Biotin–streptavidin binding assay was carried out using the biotinylated wild-type and mutant 5(5)7, and 5(5)6 splicing substrates; the gels were probed with anti-hnRNP H antibody and visualized by ECL. (B) Competitive biotin–streptavidin binding assay. Biotin–streptavidin binding assay was carried out using the biotinylated wild-type 5(5)7 in the presence of 20 or 40 pmoles of oligoribonucleotide containing either the wild-type or mutated ESS, as indicated.
To determine whether the binding of hnRNP H correlates with the activity of the ESS, the wild-type and mutant RNA competitors were used to compete the binding of hnRNP H to the wild-type splicing substrate 5(5)7. Addition of the wild-type RNA reduced the binding of hnRNP H (Fig. 6B, lanes 2,3). The ex-1 competitor also reduced this binding, but its relative competition efficiency was weaker than that of the wild-type competitor (Fig. 6B, lanes 4,5). The competition efficiency of ex-110 was even weaker than that of ex-1 (Fig. 6B, lanes 6,7), and ex-16 was the weakest competitor (Fig. 6B, lanes 8,9). These results indicate that the affinity of hnRNP H with the various substrates (the wild type > ex-1 > ex-110 > ex-16) correlates well with the silencer activities of the different substrates (cf. Fig. 6A, lanes 2–5 with Fig. 3A), and suggest that hnRNP H is involved in the silencer activity.
The hnRNP H antibody activates exon 7 splicing in vitro
To further demonstrate that hnRNP H is involved in the ESS activity, we performed antibody activation experiments. We reasoned that if binding of hnRNP H to the ESS is responsible for its activity, addition of anti-hnRNP H antibody may disrupt binding of hnRNP H to the pre-mRNA or interaction of hnRNP H with other proteins and, consequently, activate exon 7 splicing. As shown in Figure 7, the wild-type substrate 5(5)7 was not spliced under standard splicing conditions (Fig. 7, lane 1). However, addition of anti-hnRNP H antibody activated exon 7 splicing (Fig. 7, lane 2), and the activation was stimulated in a dose dependent manner (Fig. 7, lane 3). This activation was specific to the anti-hnRNP H antibody because addition of the preimmune serum had no effect (Fig. 7, lanes 4,5). Also, the effect of the anti-hnRNP H antibody was specific to exon 7 because substrates 5(5)6 and the human β-globin pre-mRNA were not activated by addition of the antibody (Fig. 7, lanes 11–16).
Figure 7.

Anti-hnRNP H antibody specifically activates exon 7 splicing. In vitro splicing reactions were carried out using the substrates indicated at the top in the absence (lanes 1,5,17) or presence of 5 or 10 μg of anti-hnRNP H (α) or preimmune (p) rabbit sera as indicated below the substrates. The splicing reaction on lane 16 has 20 μg of anti-hnRNP H rabbit serum. Schematic representations of the precursors and products are shown on both sides. For clarity, the precursor and splicing products for the human β-globin pre-mRNA are not indicated (see Fig. 3C for reference).
Because splicing of the ex-1 mutant was not fully activated (see Fig. 3A), we were interested in determining the effect of the anti-hnRNP H antibody on the splicing of the mutant 5(5)7 ex-1. We reasoned that if the dissociation of hnRNP H correlates with the activation of exon 7 splicing, addition of anti-hnRNP H antibody will further stimulate splicing of the ex-1 mutant, which indeed appeared to be the case (Fig. 7A, cf. lanes 7,8 with lane 6). As addition of the preimmune serum had no effect on splicing of the ex-1 mutation (Fig. 7A, lanes 9,10), the stimulation was specific to anti-hnRNP H antibody. These results further support the notion that hnRNP H binding is involved in the silencer activity.
Previously we have demonstrated that utilization of exon 7 in nonmuscle cells is blocked at the upstream 3′ splice site (Guo and Helfman 1993), and that the ex-1 mutation activates exon 7 splicing in nonmuscle cells in vivo (Guo et al. 1991). Because the ex-1 mutation disrupts binding of hnRNP H, we were interested in determining whether hnRNP H is directly involved in the regulation of rat β-TM pre-mRNA alternative splicing. Therefore, we carried out antibody activation experiments using a substrate p2(7/8) that consists of exon 5, intron 5, exon 6, intron 6, and the joined exons 7/8. The splicing of p2(7/8) was inefficient in HeLa cell nuclear extracts because the 3′ splice site upstream of exon 7 is blocked (Fig. 7, lane 17), which is consistent with our previous results (Guo and Helfman 1993). Addition of anti-hnRNP H antibody stimulated exon 7 splicing (Fig. 7, lanes 18,19). This activation probably takes place at the 3′ splice site, as both products 57/8 and 5(5)67/8 were enhanced. Again, addition of the preimmune serum had no effect (Fig. 7, lanes 20,21). From these results, we concluded that hnRNP H is involved in the regulation of rat β-TM pre-mRNA alternative splicing.
Recombinant hnRNP H antagonizes activation of splicing by anti-hnRNP H antibody
To confirm the conclusions derived from the antibody activation experiments that hnRNP H is involved in the ESS activity and regulation of rat β-TM pre-mRNA alternative splicing, we performed rescue experiments. First we tested the splicing of wild-type 5(5)7 substrate, which was activated by the anti-hnRNP H antibody (Fig. 8, lane 2). When recombinant glutathione S-transferase (GST)–hnRNP H fusion protein was added to the splicing reaction, the activation by anti-hnRNP H antibody decreased (Fig. 8, lane 3). Addition of an increasing amount of the recombinant protein resulted in complete repression of exon 7 splicing (Fig. 8, lanes 4,5). This effect was attributable to the hnRNP H protein because addition of bovine serum albumin (BSA) or recombinant GST protein had no effect (Fig. 8, lanes 6–9). Also, recombinant GST–hnRNP H had no effect on splicing of the human β-globin pre-mRNA (Fig. 8, lanes 10–12). We also carried out rescue experiments using p2(7/8) as a splicing substrate. Addition of recombinant GST–hnRNP H protein repressed almost completely the stimulatory effect of anti-hnRNP H antibody on splicing of p2(7/8) (Fig. 8, lanes 14–17). The repression resulted from the hnRNP H protein because addition of BSA or recombinant GST had no effect (Fig. 8, lanes 18–21).
Figure 8.

Recombinant hnRNP H antagonizes the activation of anti-hnRNP H antibody. In vitro splicing reactions were carried out in the absence (lanes 1,13) or presence of 10 μg of anti-hnRNP H rabbit serum, and in the presence of 0.2, 0.4, or 0.6 μg of recombinant GST–hnRNP H (G-H), 0.3 or 0.6 μg of recombinant GST (G), and 0.3 or 0.6 μg of BSA as indicated. The substrates are indicated at the top, and schematic representations of the precursors and splicing products are shown on both sides. For clarity, the precursor and splicing products for the human β-globin pre-mRNA are not indicated (see Fig. 3C for reference).
Depletion of hnRNP H activates splicing of exon 7
The results of antibody activation and recombinant protein add-back experiments strongly suggest that hnRNP H is involved in the suppression of exon 7 usage in nonmuscle cells. However, it is possible that the antibody against hnRNP H titrated away other nuclear components that are critical for the suppression of exon 7 usage, and the addition of recombinant hnRNP H released these components and led to the resuppression of exon 7 splicing in nonmuscle cells.
To establish fully that hnRNP H is involved directly in the suppression of exon 7 usage in nonmuscle cells, we performed a depletion experiment using the polyclonal antibody that was used in the activation experiment, and carried out a splicing experiment using the depleted extract. This antibody is specific to hnRNP H, as it recognized a single band in Western blot when HeLa cell nuclear extract was tested (Fig. 9A, lanes 1–3). About 85% of hnRNP H was depleted from HeLa cell nuclear extract (Fig. 9A, lanes 4,6,8), comparing to the mock depletion (Fig. 9A, lanes 5,7,9). The antibody specifically depleted hnRNP H from HeLa cell nuclear extract, as the concentration of PTB in the depleted extract was not changed (Fig. 9A).
Figure 9.

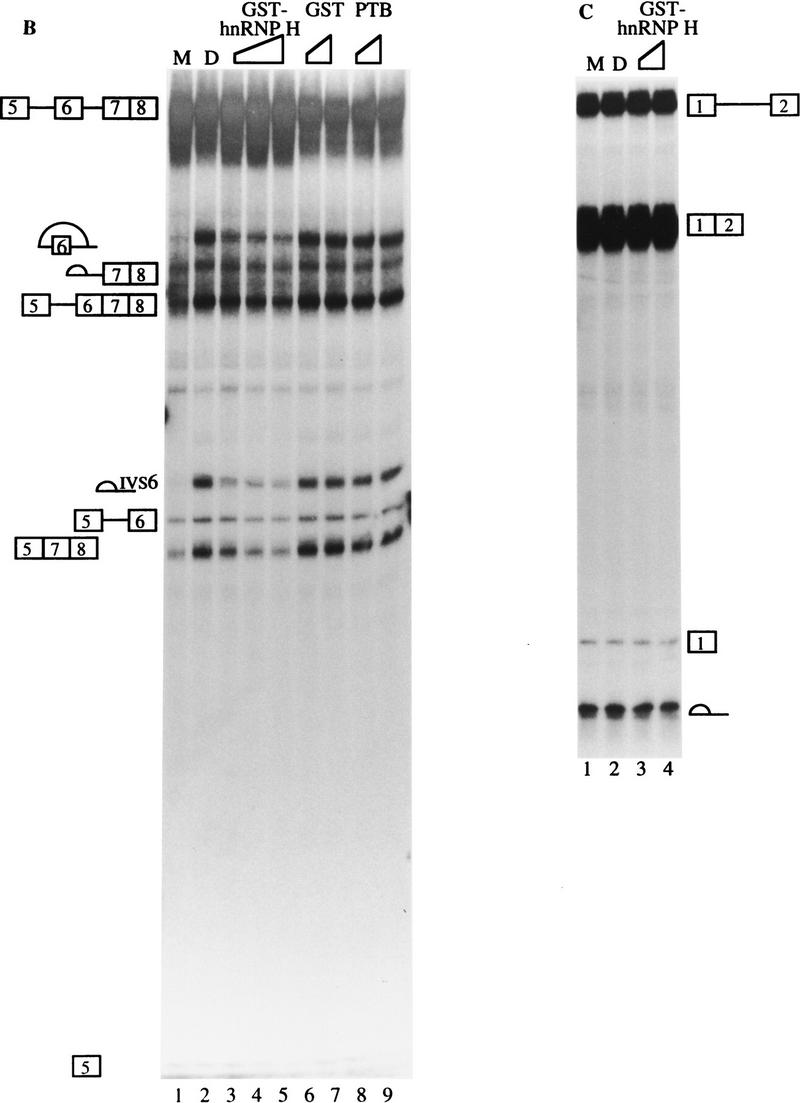
Depletion of hnRNP H activates the splicing of exon 7. (A) Depletion of hnRNP H from HeLa cell nuclear extract. Western blot analysis was carried out with 1, 2, and 4 μg of untreated (lanes 1–3), antibody-depleted (lanes 4,6,8), and mock depleted extract (lanes 5,7,9), probed with anti-hnRNP or anti-PTB antibodies as indicated. (B) Splicing of p2(7/8) in mock-depleted extract (lane 1), or hnRNP H depleted extract without (lane 2) or with increasing amount (0.3, 0.6, and 0.9 μg) of recombinant GST–hnRNP H (lanes 3–5), recombinant GST (0.3 and 0.9 μg, lanes 6,7), or recombinant PTB (0.3 and 0.9 μg, lanes 8,9). (C) Splicing of the human β-globin pre-mRNA in mock-depleted extract (lane 1), hnRNP H-depleted extract without (lane 2), or with 0.3 and 0.9 μg of recombinant GST–hnRNP H (lanes 3,4).
To examine functionally the depleted extracts, we performed an in vitro splicing experiment using p2(7/8) as a substrate. The splicing of p2(7/8) was weak in the mock-depleted extract (Fig. 9B, lane 1). However, the utilization of the 3′ splice site of exon 7 was significantly stimulated in the antibody-depleted extract, as evidenced by the production of an increasing amount of both products 57/8 and 5(5)67/8 (Fig. 9B, lane 2). This stimulation is specific because antibody or mock depletion of hnRNP H had no effect on the splicing of the human β-globin pre-mRNA (Fig. 9C, lanes 1,2). To determine whether the stimulation of the 3′ splice site usage of exon 7 is attributable to the depletion of hnRNP H, we performed a recombinant protein add-back experiment. Addition of 0.3 μg of recombinant GST–hnRNP H to the depleted extract reduced the splicing of p2(7/8) about threefolds (Fig. 9B, lane 3). The resuppression resulted from the addition of hnRNP H because addition of recombinant GST or PTB had no effect (Fig. 9B, lanes 6–9). However, the splicing of exon 7 was not completely resuppressed even when a higher concentration of recombinant hnRNP H was added in splicing reactions (Fig. 9B, lanes 4,5), which suggests that modification of hnRNP H may be required or some other factors may be involved. Addition of GST–hnRNP H to the depleted extract had no effect on the splicing of the human β-globin (Fig. 9C, lanes 3,4). Collectively, these results indicate that hnRNP H is involved directly in the suppression of exon 7 splicing in nonmuscle cells.
Discussion
The 5′ end of the skeletal muscle exon 7 contains an ESS
Previous data from our laboratory showed that a mutation at the 5′ end of exon 7, designated as ex-1, activated exon 7 splicing in nonmuscle cells in vivo (Guo et al. 1991). In this report we show that activation of the ex-1 mutation is neither attributable to creation of an ESE nor to disruption of a putative secondary structure, but rather results from disruption of an ESS that contains the sequence UGUGGG.
Several ESSs have been identified in various mammalian pre-mRNAs (Caputi et al. 1994; Amendt et al. 1995; Gatto and Breathnach 1995; Staffa and Cochrane 1995; Zheng et al. 1996; König et al. 1998). The ESS identified in exon 7 of rat β-TM pre-mRNA probably functions through repression of the 3′ splice site of skeletal muscle-specific exon because that is the major blocking site to repress utilization of exon 7 in nonmuscle cells (Guo and Helfman 1993). Furthermore, addition of anti-hnRNP H antibody to p2(7/8) splicing reactions stimulated the splicing of two spliced products: 57/8 and 5(5)67/8, both of which use the same 3′ splice site upstream of exon 7. The ability of an ESS to exert repression on its upstream 3′ splice site is a common feature for the ESSs identified in mammals, as is the requirement for a weak 3′ splice site. The ESS in exon 7 requires a weak 3′ splice site for its function because only a substrate with intron 5 or intron 6 of rat β-TM pre-mRNA, but not intron 1 of the human β-globin pre-mRNA or the modified intron 2 of α-TM pre-mRNA with an upstream sequence insertion, can mimic the behavior of the ex-1 mutation (Fig. 2; data not shown). Analysis of exon 7 sequence revealed that, adjacent to the ESS, exon 7 contains a purine-rich element, GAGGAGGAG, that is identical to well-characterized ESEs (Watakabe et al. 1993; Xu et al. 1993). This bipartite architecture may represent a general feature for ESSs to participate in the regulation of alternative splicing because most of the ESSs identified to date share this structure (Amendt et al. 1995; Staffa and Cochrane 1995; Zheng et al. 1996; König et al. 1998). In most cases, ESSs are located downstream of ESEs. However, the ESS in exon 7 lies immediately upstream of the putative ESE. This kind of bipartite structure was also recently identified in exon 5 of the cell surface molecule CD44 (König et al. 1998).
hnRNP H is a splicing regulator
By using UV cross-linking method, we identified a trans-acting factor of ∼50 kD that binds to the ESS in exon 7 of rat β-TM pre-mRNA. Isolation and microsequencing showed that this protein is hnRNP H. The correlation between the binding of hnRNP H and the ESS activity suggests that hnRNP H is involved in the regulation of the ESS in exon 7, which was confirmed both by antibody activation/add-back experiments, and by depletion/add-back experiments. The involvement of hnRNP H in the regulation of alternative splicing may not be surprising, as hnRNP F has been shown to participate in the formation of a multiprotein complex in the DCS of c-src and play a critical role in the regulation of c-src alternative splicing (Min et al. 1995), and hnRNP H and F are highly homologous; their protein sequences are 78% identical (Honoré et al. 1995). Other members of the hnRNP family have also been implicated in the regulation of alternative splicing of several mammalian pre-mRNAs. For example, hnRNP A1 was shown to antagonize the effect of SF2/ASF to promote the distal 5′ splice site usage (Mayeda and Krainer 1992; Yang et al. 1994). hnRNP I, also known as PTB, participates in the regulation of pre-mRNA alternative splicing of α-TM, β-TM, and others. In contrast to other members of the hnRNP family that bind to intron sequences, hnRNP H binds to an exon sequence in vertebrates.
hnRNP H was first identified by two-dimensional gel electrophoresis analysis (Dreyfuss et al. 1993), and the corresponding cDNA has been isolated and characterized (Honoré et al. 1995). The hnRNP H cDNA is predicted to encode a protein with three RNA binding domains (RBD). This protein has a high binding affinity for poly(rG) (Matunis et al. 1994; Honoré et al. 1995), which suggests that hnRNP H may be involved in splicing of other transcripts besides rat β-TM pre-mRNA. The ESS in the FGFR-2 K-SAM exon is also a guanine-rich element TAGGGCAGGC (Gatto and Breathnach 1995). An intronic G-rich sequence (A/U)GGG has been implicated in the regulation of chicken β-TM pre-mRNA alternative splicing (Sirand-Pugnet et al. 1995), and an intron splicing enhancer (ISE) containing a G-rich repeat has been shown to facilitate the inclusion of a micro-exon in chicken troponin T pre-mRNA (Carlo et al. 1996). It will be interesting to determine whether hnRNP H also binds to these sequences and is responsible for their activities. Indeed, hnRNP H was recently identified to be involved in the formation of a multiprotein complex that is required for the activation of the N1 exon splicing in c-src (D.L. Black, pers. comm.).
hnRNP H is involved in the regulation of rat β-TM pre-mRNA alternative splicing
The 3′ splice site upstream of exon 7 is the major site to block utilization of exon 7 in nonmuscle cells (Guo and Helfman 1993). Two cis-acting elements are involved in this block: the IRE located at the 3′ end of intron 6 was shown to be critical for blocking the utilization of exon 7 in nonmuscle cells and this blocking may involve binding of a multiprotein complex (Helfman et al. 1990; Guo et al. 1991; Mulligan et al. 1992; Grossman et al. 1998). Another cis-acting element involved is the sequence at the 5′ end of exon 7 because mutation of this element activates exon 7 splicing in nonmuscle cells (Guo et al. 1991). In this report, we demonstrate that the sequence at the 5′ end of exon 7 is a bona fide ESS. Addition of anti-hnRNP H antibody or depletion of hn RNP H activated exon 7 splicing to produce both products 57/8 and 5(5)67/8, and recombinant hnRNP H antagonized this activation. These results make hnRNP H a good candidate for the repressor that recognizes the ESS at the 5′ end of exon 7, and suggest that hnRNP H is involved directly in the regulation of rat β-TM pre-mRNA alternative splicing.
Our previous data demonstrated that the sequences in exon 7 are not required for the formation of the protein complex that appears to be involved in the activity of the IRE (Grossman et al. 1998), and the data in this report indicate that the cis-acting element in exon 7 that blocks utilization of exon 7 is not dependent on the specific sequences of intron 6. These results suggest that two independent mechanisms operate simultaneously to block exon 7 usage in nonmuscle cells; disrupting either of these mechanisms leads to the release of the block and utilization of exon 7. Thus, it is reasonable to speculate that different tissue types or tissues at different developmental stages may use different mechanisms to release the suppression of exon 7. For example, fetal cardiac muscle may disrupt the complex formation in intron 6 and skeletal muscle may reduce binding of hnRNP H to exon 7 sequence, or vice versa. It will be interesting to determine which mechanisms are used in different cell types during embryonic development.
Several possible mechanisms may account for how hnRNP H blocks the utilization of exon 7 in nonmuscle cells. For example, the binding of hnRNP H may displace the association of SR proteins with exon 7 because a putative ESE is located immediately downstream of the ESS. Thus, binding of hnRNP H to the ESS may sterically interfere with binding of SR proteins to the ESE. When the ESS is mutated, binding of hnRNP H is down regulated and SR proteins may now bind to the ESE. This model is consistent with our data that the 60%–90% ammonium sulfate fraction of HeLa cell nuclear extract gave rise to several cross-linked products because SR proteins are present in this fraction (Fig. 5A). It remains to be determined whether these cross-linked products are SR proteins.
How hnRNP H participates in the regulation of tissue- and developmental-specific alternative splicing remains to be answered. Using either nucleic acid or immunological probes, we did not detect any difference in the expression levels of hnRNP H mRNA or protein in various mouse tissues at different developmental stages (data not shown). Thus, tissue-specific changes in the expression levels of hnRNP H do not appear to be involved in the regulation. One possibility is that additional factors are involved; these factors are tissue specific and function together with hnRNP H in regulation of alternative RNA splicing. This kind of mechanism is used in the regulation of doublesex alternative splicing in Drosophila, where both tra, a female-specific factor, and tra 2, which is expressed in both sexes, are required for the female-specific splicing (McKeown 1992). Also, in P-element alternative splicing, both PSI, a somatic cell-specific factor, and hrp 48, an ubiquitously expressed protein, are needed to repress intron 3 splicing (Rio 1993). Furthermore, skeletal muscle cells may possess a different isoform of hnRNP H because several isoforms of this protein has been identified (B. Honoré, pers. comm.). As each of the three RNA-binding domains of hnRNP H has also strong affinity with poly(rG) (Honoré et al. 1995), different isoforms may exhibit different affinities for the ESS in exon 7. Another possibility is tissue-specific phosphorylation, as phosphorylation has been shown to affect binding of hnRNP C to pre-mRNA (Mayrand et al. 1993), and hnRNP H can be phosphorylated (Honoré et al. 1995). Experiments are currently under way to further study how hnRNP H affects the ESS activity and what determines the basis for the tissue-specific regulation of rat β-TM pre-mRNA alternative splicing.
Materials and methods
Oligonucleotides
Oligonucleotides used for PCR to generate the splicing substrates are as follows:
PCR(1), 5′-GATTTAGGTGACACTATAG; PCR(2), 5′-AACTGCAGGCAGGGGGCAGCGGGCAT; PCR(3), 5′-CAGCTGCAGTAAATGTGGGGACC; PCR(4), 5′-CAGCTGC-AGTAAAGGATCCGACCTAGAGGAGGAGCT; PCR(5), 5′-CAGCTGCAGTAAACTACGCGACCTAGAGGAGGAGCT; PCR(6), 5′CAGCTGCAGTATGTACGATCGCCTAGAGGAGGAGCTGAA; PCR(7), 5′-CGGAATTCCTTGTCCGCTTGGGCTTCCAC; PCR(8), 5′-CGGAATTCCTTGTCCGCTTGGGCTTCCAGAGATTTCAAGTGGATTCCAACAATTTTCAGCTCCTCCT.
The oligonucleotides used to generate the oligoribonucleotides for UV cross-linking experiments and competition in biotin–streptavidin binding assays are as follows: UV(1), 5′-AATTTAATACGACTCACTATAG; UV(2), 5′-TCTAGGTCCCCACATTTCCCTATAGTGAGTCGTATTAAATT; UV(3), 5′-TCTAGGTCGGATCCTTTCCCTATAGTGAGTCGTATTAAATT; UV(4), 5′-TCTAGGTCGCGTAGTTTCCCTATAGTGAGTCGTATTAAA-TT; UV(5), 5′-TCTAGGCGATCGTACATCCCTATAGTGAGTCGTATTAAATT.
The oligodeoxynucleotides used to generate the six-tandem repeats of the ESS sequence are as follows: ESS(1), 5′-GATCTGCAGAAATGTGGGGACCTAGAGAAATGTGGGGACCTAGAG; ESS(2), 5′-GATCCTCTAGGTCCCCACATTTCTCTAGGTCCCCACATTTCTGCA.
The oligoribonucleotides used in titration experiments are as follows: ECS, 5′GGAAAUGUGGGGACCUAGA, in which three nucleotides at both ends are 2′-O-methyl-modified; WU, 5′CGUAUACCCUUGAC, in which two nucleotides at both ends are 2′-O-methyl-modified.
Plasmid constructions
The parental plasmid for all of the splicing substrates is pSP64, which contains an SP6 promoter. To make the wild-type and mutant 5(5)7 substrates, we first constructed a plasmid p5(5) that contains exon 5 and intron 5. To maintain a PstI site in the construct, the fragment was PCR amplified using PCR(1) and PCR(2) as primers and p2(7/8) (Helfman and Ricci 1989) as the template, and was then digested with HindIII and PstI. This construct maintains the length of intron 5, but has a mutation from CC to TG in intron 5 four nucleotides from the 5′ end of the downstream exon. The resulting mutation does not affect the recognition of the downstream AG dinucleotide in splicing. For the construction of the plasmids containing the wild-type, ex-1, ex-16, and ex-110 5(5)7s, PCR using p2(7/8) as the template and PCR(7) with either PCR(3), PCR(4), PCR(5), or PCR(6) as primers, respectively, was performed and each PCR product was inserted into the p5(5) plasmid. To construct 5(5)7 ex-m and 5(5)7 ex-1/m, PCR using p2(7/8) as the template and PCR(8) with either PCR(3) or PCR(4) as primers, respectively, was performed, and each PCR product was inserted into the plasmid p5(5). The sequences of all mutations were confirmed by DNA sequence analysis and are shown in Figure 1.
The plasmid for expressing GST–hnRNP H in Escherichia coli was constructed by inserting the full-length hnRNP H cDNA into pRP265, a derivative of pGEX-2T that has a GST tag. The full-length hnRNP H cDNA was obtained from pT7PL-TOT (Honoré et al. 1995) by a partial digestion using BamHI and EcoRI.
Preparation and fractionation of HeLa cell nuclear extracts
HeLa cell nuclear extracts were prepared as described (Dignam et al. 1983; Krainer et al. 1984). For fractionation of HeLa cell nuclear extracts by ammonium sulfate precipitation, 2 ml of extract was first diluted with 8 ml of buffer D without both glycerol and KCl. Ammonium sulfate was gradually added to the nuclear extract to 20% of saturation. After 30 min of stirring at 4°C, the extract was centrifuged in an H-6000A rotor (Sorvall) at 6000g at 4°C for 20 min to obtain 0%–20% ammonium sulfate fraction, and the resulting supernatant was transferred to a fresh beaker. The same procedure was repeated to obtain the subsequent 20%–50%, 50%–60%, and 60%–90% ammonium sulfate fractions. The fractions were resuspended and dialyzed against buffer D. The remaining supernatant was concentrated in a microconcentrator (Amicon) and dialyzed against buffer D. The 2-ml extract yielded 1 ml of 0%–20% fraction, 2 ml of 20%–50% fraction, 1 ml of 50%–60% fraction, 2 ml of 60%–90% fraction, and 200 μl of supernatant.
Purification of recombinant proteins and antibody preparation
A fresh colony of E. coli BL21 transformed with plasmids containing GST or GST–hnRNP H cDNA was grown in 5 ml of Luria–Bertani (LB) medium containing ampicillin (amp) at 37°C for 12 hr. The culture was transferred to 500 ml of LB + amp and grown to OD600 = 0.1 at 37°C. Isopropyl-β-d-thiogalactopyranoside (IPTG) was added to reach concentration of 0.5 mm and the culture was grown for another hour. The bacteria were harvested, resuspended in 10 ml of freshly made 50% urea solution supplemented with 5 mm dithiothreitol (DTT), and incubated at 65°C for 1 hr with vigorous vortexing every 10 min. The bacterial lysate was clarified by centrifugation at 10,000g, and the supernatant was dialyzed in buffer [20 mm Tris (pH 8.0), 0.1 mm EDTA, 1 mm DTT, 5% glycerol, 60 mm KCl]. After clarification, the supernatant was loaded onto a glutathione–Sepharose 4B column and washed with PBS (140 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, and 1.8 mm KH2PO4). The bound protein was eluted with 10 mm glutathione in 10 mm HEPES (pH 8.0), and dialyzed with buffer D.
The purified GST–hnRNP H protein was sent to COVANCE Research Products Inc. for antibody production in rabbits. Sera (491 and 492) from two rabbits were obtained, and each had a protein concentration of ∼60 μg/ml. Both sera recognized recombinant GST–hnRNP H and a major protein corresponding to hnRNP H in HeLa cell nuclear extracts (data not shown). Both sera precipitated a protein corresponding to hnRNP H from HeLa cell nuclear extracts and gave rise to exclusive nuclear signals in immunofluorescence staining (data not shown). Because 492 had a higher titer than 491 in Western blot analysis, it was used for studies in this report.
Splicing substrate preparation, in vitro splicing, and immunodepletion assays
The plasmids used as templates for the splicing substrates were linearized with the appropriate restriction enzymes, and transcribed by SP6 polymerase as described (Helfman et al. 1988). In vitro splicing reactions were carried out at 30°C for 2 hr unless otherwise indicated. Standard splicing conditions used for these studies consist of 15 μl of HeLa cell nuclear extracts in a final reaction volume of 25 μl containing 2 mm MgCl2, 0.5 mm ATP, 20 mm creatine phosphate, 15 ng of pre-mRNA, 12.8 mm HEPES (pH 8), 14% (vol/vol) glycerol, 60 mm KCl, 0.12 mm EDTA, and 0.3 mm DTT. In some reactions, 1 or 3 mm MgCl2 was used instead of 2 mm MgCl2, and some reactions contained 2.5% polyvinyl alcohol (PVA). For titration and antibody activation experiments, the reaction mixtures without pre-mRNA were preincubated with an RNA competitor or an antibody, respectively, at 30°C for 10 min, and then the pre-mRNA was added to proceed the splicing reaction. To antagonize the activation of anti-hnRNP H antibody, the reaction mixtures without pre-mRNA were preincubated with a recombinant protein or BSA and the antibody at 30°C for 10 min, and then the pre-mRNA was added to proceed the splicing reaction.
Immunodepletion and add-back experiments were performed as described (Zuo and Maniatis 1996). Briefly, 0.5 ml of protein A–Sepharose beads (Pharmacia) were incubated with 1 ml of anti-hnRNP H or preimmune serum (mock) for 2 hr at 4°C. The beads were washed twice with 10 ml of buffer D with 0.05% Triton X-100 and without glycerol (wash buffer), packed into a minicolumn, and equilibrated with 10 ml of buffer D. Six hundred microliters of HeLa cell nuclear extract was passed through the column four times at 4°C, and the eluate was passed through a fresh protein A column (0.5 ml in bed volume) twice to remove residual antibody. The antibody- and mock-depleted HeLa cell nuclear extracts were tested for concentration of hnRNP H and PTB by Western blotting, and used directly in standard splicing experiments. For add-back experiments, recombinant proteins were added to the depleted extracts and the splicing reactions were assembled.
Preparation of oligoribonucleotides and UV cross-linking experiments
The wild-type, ex-1, ex-16, and ex-110 oligoribonucleotides were synthesized using oligonucleotides UV(1) with either UV(2), UV(3), UV(4), or UV(5), respectively, as templates; the transcription was conducted as instructed using the MEGAshortscriptTM T7 Kit (Ambion). For UV cross-linking experiments, the RNA probes were synthesized in the presence of [α-32P]CTP and [α-32P]GTP (3000 Ci/mmole). The reactions were assembled in a 96-well plate in a final reaction volume of 12.5 μl containing 20,000 cpm of 32P-labeled probe, 8 μl HeLa cell nuclear extract, either fractionated with ammonium sulfate precipitation or nonfractionated, 2 mm MgCl2, 0.5 mm ATP, 20 mm creatine phosphate, 0.2 mg/ml tRNA, and 0.016 mg/ml BSA. After incubation at 30°C for 10 min, the reaction mixtures were irradiated at 4°C with 254 nm UV light at a distance of 4.5 cm from the source for 10 min. The RNA molecules in the reaction mixtures were digested at 37°C with RNase A (1 mg/ml) and RNase T1 (2.5 U/μl) for 10 min. The reaction mixture was boiled in SDS loading buffer, resolved on a 10% SDS-polyacrylamide gel, and visualized by autoradiography. In competition experiments, the competitor was also included in the reaction mixture.
Preparation of biotinylated RNA and biotin–streptavidin-binding assay
The biotinylated RNAs were synthesized as instructed using the MEGAscript™ SP6 Kit (Ambion). Two micrograms of biotin-labeled RNA was mixed with 10 μl of HeLa cell nuclear extract containing 0.05% Triton X-100 and 1 mg/ml heparin. After incubation on ice for 30 min, 20 μl of solution that contains 10 μl of packed streptavidin–agarose was added. The reaction mixture was shaken at 4°C for another 30 min and washed with buffer D with 0.05% Triton X-100 and without glycerol (wash buffer). The bound proteins were resolved in 10% SDS-polyacrylamide gel, Western blotted using anti-hnRNP H antibody, and visualized by enhanced chemiluminescence (ECL). For competition experiments, the oligoribonucleotide competitor was synthesized as described earlier. The competitor was preincubated with the reaction mixture on ice for 30 min and then the biotin-labeled RNA was added to proceed the reaction.
Isolation and peptide sequencing of hnRNP H
The isolation of hnRNP H followed the protocol of biotin–streptavidin binding assay except that the 20%–50% ammonium sulfate fraction was used instead of HeLa cell nuclear extracts. The bound proteins were eluted with wash buffer containing 0.5 m KCl, concentrated in a microconcentrator (Amicon), resolved on a 10% SDS-polyacrylamide gel, and visualized by either silver or Coomassie Brilliant Blue G staining. The 50-kD protein was carefully excised from the gel and subjected to in-gel digestion using Achromobacter protease I in 50 μg/ml in 50 mm Tris-HCl (pH 9.0) as described previously (Wang et al. 1996). The resulting polypeptides were separated by HPLC using a Vydac C18 column (1.0 × 250 mm, 10 μm, 300 Å) and sequenced by automated protein sequencers (Applied Biosystems 494).
Acknowledgments
We acknowledge Philip Renna and Jim Duffy for their art work. We are grateful to Bent Honoré for providing the hnRNP H cDNA and unpublished data, and Qiang Wu for providing some reagents. We thank Douglas Black for communicating the unpublished paper and useful comments on this manuscript. We also thank Anne Vaahtokari, Adrain Krainer, Paul Bingham, Nouria Hernandez, and Mike Myers for critical reading of this manuscript, as well as Akiya Watakabe, Judy Wang, Mike Murray, and Meena Selvakumar for helpful discussion. D.M.H. was supported by National Institutes of Health (NIH) grant GM43049. R.K. was supported by NIH grant CA95508.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL helfman@cshl.org; FAX (516) 367-8815.
References
- Adams MD, Rudner DZ, Rio DC. Biochemistry and regulation of pre-mRNA splicing. Curr Opin Cell Biol. 1996;8:331–339. doi: 10.1016/s0955-0674(96)80006-8. [DOI] [PubMed] [Google Scholar]
- Amendt BA, Si Z-H, Stoltzfus CM. Presence of exon splicing silencers within human immunodeficiency virus type 1 tat exon 2 and tat-rev exon 3: Evidence for inhibition mediated by cellular factors. Mol Cell Biol. 1995;15:4606–4615. doi: 10.1128/mcb.15.8.4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashiya M, Grabowski PJ. A neuron-specific splicing switch mediated by an array of pre-mRNA repressor sites: Evidence of a regulatory role for the polypyrimidine tract binding protein and a brain-specific PTB counterpart. Neuron. 1997;3:996–1015. [PMC free article] [PubMed] [Google Scholar]
- Black D. Activation of c-src neuron-specific splicing by an unusual RNA element in vitro and in vivo. Cell. 1992;69:795–807. doi: 10.1016/0092-8674(92)90291-j. [DOI] [PubMed] [Google Scholar]
- Boggs RT, Gregor P, Idriss S, Belote JM, McKeown M. Regulation of sexual differentiation in D. melanogaster via alternative splicing of RNA from the transformer gene. Cell. 1987;50:739–747. doi: 10.1016/0092-8674(87)90332-1. [DOI] [PubMed] [Google Scholar]
- Cáceres JF, Stamm S, Helfman DM, Krainer AR. Regulation of alternative splicing in vivo by overexpression of antagonistic splicing factors. Science. 1994;265:1706–1709. doi: 10.1126/science.8085156. [DOI] [PubMed] [Google Scholar]
- Caputi M, Casari G, Guenzi S, Tagliabue R, Sidoli A, Melo CA, Baralle FE. A novel bipartite splicing enhancer modulates the differential processing of the human fibronectin EDA exon. Nucleic Acids Res. 1994;22:1018–1022. doi: 10.1093/nar/22.6.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlo T, Sterner DA, Berget SM. An intron splicing enhancer containing a G-rich repeat facilitates inclusion of a vertebrate micro-exon. RNA. 1996;2:342–353. [PMC free article] [PubMed] [Google Scholar]
- Chan RC, Black DL. The polypyrimidine tract binding protein binds upstream of neural cell-specific c-src exon N1 to repress the splicing of the intron downstream. Mol Cell Biol. 1997;17:4667–4676. doi: 10.1128/mcb.17.8.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Orval BC, Carafa YD, Pugnet PS, Gallego M, Brody E, Marie J. RNA secondary structure repression of a muscle-specific exon in HeLa cell nuclear extracts. Science. 1991;252:1823–1828. doi: 10.1126/science.2063195. [DOI] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss G, Matunis MJ, Pinol-Roma S, Burd CG. hnRNP proteins and the biogenesis of mRNA. Annu Rev Biochem. 1993;62:289–321. doi: 10.1146/annurev.bi.62.070193.001445. [DOI] [PubMed] [Google Scholar]
- Gatto FD, Breathnach R. Exon and intron sequences, respectively, repress and activate splicing of a fibroblast growth factor receptor 2 alternative exon. Mol Cell Biol. 1995;15:4825–4834. doi: 10.1128/mcb.15.9.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge H, Manley JL. A protein factor, ASF, controls cell-specific alternative splicing of SV40 early pre-mRNA in vitro. Cell. 1990;62:25–34. doi: 10.1016/0092-8674(90)90236-8. [DOI] [PubMed] [Google Scholar]
- Gooding C, Roberts GC, Smith CWJ. Role of an inhibitory pyrimidine element and polypyrimidine tract binding protein in repression of a regulated α-tropomyosin exon. RNA. 1998;4:85–100. [PMC free article] [PubMed] [Google Scholar]
- Grossman JS, Meyer MI, Wang Y-C, Mulligan GJ, Kobayashi R, Helfman DM. The use of antibodies to the polypyrimidine tract binding protein (PTB) to analyze the protein components that assemble on alternative spliced pre-mRNAs which use distant branch points. RNA. 1998;4:1–13. doi: 10.1017/s1355838298971448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Helfman DM. Cis-elements involved in alternative splicing in the rat β-tropomyosin gene: The 3′-splice site of the skeletal muscle exon 7 is the major site of blockage in nonmuscle cells. Nucleic Acids Res. 1993;21:4762–4768. doi: 10.1093/nar/21.20.4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Mulligan GJ, Wormsley S, Helfman DM. Alternative splicing of the β-tropomyosin pre-mRNA: cis-Acting elements and cellular factors that block the use of a skeletal muscle exon in non-muscle cells. Genes & Dev. 1991;5:2096–2107. doi: 10.1101/gad.5.11.2096. [DOI] [PubMed] [Google Scholar]
- Hampson RK, Follette LL, Rottman FM. Alternative processing of bovine growth hormone mRNA is influenced by downstream exon sequences. Mol Cell Biol. 1989;9:1604–1610. doi: 10.1128/mcb.9.4.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedley ML, Maniatis T. Sex-specific splicing and polyadenylation of dsx pre-mRNA requires a sequence that binds specifically to tra-2 protein in vitro. Cell. 1991;65:579–586. doi: 10.1016/0092-8674(91)90090-l. [DOI] [PubMed] [Google Scholar]
- Helfman DM, Ricci WM. Branch point selection in alternative splicing of tropomyosin pre-mRNAs. Nucleic Acids Res. 1989;17:5633–5650. doi: 10.1093/nar/17.14.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfman DM, Ricci WM, Finn LA. Alternative splicing of tropomyosin pre-mRNAs in vitro and in vivo. Genes & Dev. 1988;2:1627–1638. doi: 10.1101/gad.2.12a.1627. [DOI] [PubMed] [Google Scholar]
- Helfman DM, Roscigno RF, Mulligan JG, Finn LA, Weber KS. Identification of two distinct intron elements involved in alternative splicing of β-tropomyosin pre-mRNA. Genes & Dev. 1990;4:98–110. doi: 10.1101/gad.4.1.98. [DOI] [PubMed] [Google Scholar]
- Honoré B, Rasmussen HH, Vorum H, Dejgaard K, Liu X, Gromov P, Madsen P, Gesser B, Tommerup M, Celis JE. Heterogeneous nuclear ribonucleoproteins H, H′, and F are members of a ubiquitously expressed subfamily of related but distinct proteins encoded by genes mapping to different chromosomes. J Biol Chem. 1995;270:28780–28789. doi: 10.1074/jbc.270.48.28780. [DOI] [PubMed] [Google Scholar]
- Hoshijima K, Inoue K, Higuchi I, Sakamoto H, Shimura Y. Control of doublesex alternative splicing by transformer and transformer-2 in Drosophila. Science. 1991;252:833–836. doi: 10.1126/science.1902987. [DOI] [PubMed] [Google Scholar]
- Huh GS, Hynes RO. Regulation of alternative pre-mRNA splicing by a novel repeated hexanucleotide element. Genes & Dev. 1994;8:1561–1574. doi: 10.1101/gad.8.13.1561. [DOI] [PubMed] [Google Scholar]
- Inoue K, Hoshijima K, Sakamoto H, Shimura Y. Binding of the Drosophila sex-lethal gene product to the alternative splice site of transformer primary transcript. Nature. 1990;344:461–463. doi: 10.1038/344461a0. [DOI] [PubMed] [Google Scholar]
- Inoue K, Hoshijima K, Higuchi I, Sakamoto H, Shimura Y. Binding of the Drosophila transformer and transformer-2 proteins to the regulatory elements of doublesex primary transcript for sex-specific RNA processing. Proc Natl Acad Sci. 1992;89:8092–8096. doi: 10.1073/pnas.89.17.8092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanopka A, Möhlemann O, Akusjärvi G. Inhibition by SR proteins of splicing of a regulated adenovirus pre-mRNA. Nature. 1996;381:535–538. doi: 10.1038/381535a0. [DOI] [PubMed] [Google Scholar]
- König H, Ponta H, Herrlich P. Coupling of signal transduction to alternative pre-mRNA splicing by a composite splice regulator. EMBO J. 1998;17:2904–2913. doi: 10.1093/emboj/17.10.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krainer AR, Maniatis T, Ruskin B, Green MR. Normal and mutant human β-globin pre-mRNAs are faithfully and efficiently spliced in vitro. Cell. 1984;36:993–1005. doi: 10.1016/0092-8674(84)90049-7. [DOI] [PubMed] [Google Scholar]
- Krainer AR, Conway GC, Kozak D. The essential pre-mRNA splicing factor SF2 influences 5′ splice site selection by activating proximal sites. Cell. 1990;62:135–42. doi: 10.1016/0092-8674(90)90237-9. [DOI] [PubMed] [Google Scholar]
- Lavigueur A, La Branche H, Kornblihtt AR, Chabot B. A splicing enhancer in the human fibronectin alternate ED1 exon interacts with SR proteins and promotes U2 snRNP binding. Genes & Dev. 1993;7:2405–2417. doi: 10.1101/gad.7.12a.2405. [DOI] [PubMed] [Google Scholar]
- Libri D, Goux-Pelletan M, Brody E, Fiszman MY. Exon as well as intron sequences are cis-regulating elements for the mutually exclusive alternative splicing of the β-tropomyosin gene. Mol Cell Biol. 1990;10:5036–5046. doi: 10.1128/mcb.10.10.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libri D, Piseri A, Fiszman MY. Tissue-specific splicing in vivo of the β-tropomyosin gene: Dependence on an RNA secondary structure. Science. 1991;252:1842–1845. doi: 10.1126/science.2063196. [DOI] [PubMed] [Google Scholar]
- Lin CH, Patton JG. Regulation of alternative 3′ splice site selection by constitutive splicing factors. RNA. 1995;1:234–245. [PMC free article] [PubMed] [Google Scholar]
- Lou H, Yang Y, Cote GJ, Berget SM, Gagel RF. An intron enhancer containing a 5′ splice site sequence in the human calcitonin/calcitonin gene-related peptide gene. Mol Cell Biol. 1995;15:7135–7142. doi: 10.1128/mcb.15.12.7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KW, Maniatis T. Synergistic interactions between two distinct elements of a regulated splicing enhancer. Genes & Dev. 1995;9:284–293. doi: 10.1101/gad.9.3.284. [DOI] [PubMed] [Google Scholar]
- Matunis MJ, Xing J, Dreyfuss G. The hnRNP F protein: Unique primary structure, nucleic acid-binding properties, and subcellular localization. Nucleic Acids Res. 1994;22:1059–1067. doi: 10.1093/nar/22.6.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeda A, Krainer AR. Regulation of alternative pre-mRNA splicing by hnRNP A1 and splicing factor SF2. Cell. 1992;68:365–375. doi: 10.1016/0092-8674(92)90477-t. [DOI] [PubMed] [Google Scholar]
- Mayrand SH, Dwen P, Pederson T. Serine/threonine phosphorylation regulates binding of C hnRNP proteins to pre-mRNA. Proc Natl Acad Sci. 1993;90:7764–7768. doi: 10.1073/pnas.90.16.7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown M. Sex differentiation: The role of alternative splicing. Curr Opin Genet Dev. 1992;2:299–303. doi: 10.1016/s0959-437x(05)80288-6. [DOI] [PubMed] [Google Scholar]
- Min H, Chan RC, Black DL. The generally expressed hnRNP F is involved in a neural-specific pre-mRNA splicing event. Genes & Dev. 1995;9:2659–2671. doi: 10.1101/gad.9.21.2659. [DOI] [PubMed] [Google Scholar]
- Min H, Turck CW, Nikolic JM, Black DL. A new regulatory protein, KSRP, mediates exon inclusion through an intronic splicing enhancer. Genes & Dev. 1997;11:1023–1036. doi: 10.1101/gad.11.8.1023. [DOI] [PubMed] [Google Scholar]
- Mulligan GJ, Guo W, Wormsley S, Helfman DM. Polypyrimidine tract binding protein interacts with sequences involved in alternative splicing of β-tropomyosin pre-mRNA. J Biol Chem. 1992;267:25480–25487. [PubMed] [Google Scholar]
- Norton PA. Polypyrimidine tract sequences direct selection of alternative branch sites and influence protein binding. Nucleic Acids Res. 1994;22:3854–3860. doi: 10.1093/nar/22.19.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez I, Lin CH, McAfee JG, Patton JG. Mutation of PTB binding sites causes misregulation of alternative 3′ splice site selection in vivo. RNA. 1997;3:764–778. [PMC free article] [PubMed] [Google Scholar]
- Ramchatesingh J, Zahler AM, Neugebauer KM, Roth MB, Cooper TA. A subset of SR proteins activates splicing of the cardiac troponin T alternative exon by direct interactions with an exonic enhancer. Mol Cell Biol. 1995;15:4898–4907. doi: 10.1128/mcb.15.9.4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio DC. Splicing of pre-mRNA: Mechanism, regulation and role in development. Curr Biol. 1993;3:574–584. doi: 10.1016/0959-437x(93)90093-5. [DOI] [PubMed] [Google Scholar]
- Ryner LC, Baker BS. Regulation of doublesex pre-mRNA processing occurs by 3′-splice site activation. Genes & Dev. 1991;5:2071–2085. doi: 10.1101/gad.5.11.2071. [DOI] [PubMed] [Google Scholar]
- Schmitt P, Gattoni R, Keoavong P, Stevenin J. Alternative splicing of E1A transcripts of adenovirus requires appropriate ion conditions in vitro. Cell. 1987;50:31–39. doi: 10.1016/0092-8674(87)90659-3. [DOI] [PubMed] [Google Scholar]
- Selvakumar, M. and D.M. Helfman. Exonic splicing enhancers contribute to the use of both 3′ and 5′ splice site usage of rat β-tropomyosin pre-mRNA. RNA (in press). [DOI] [PMC free article] [PubMed]
- Sirand-Pugnet P, Durosay P, Brody E, Marie J. An intronic (A/U)GGG repeat enhances the splicing of an alternative intron of the chicken β-tropomyosin pre-mRNA. Nucleic Acids Res. 1995;23:3501–3507. doi: 10.1093/nar/23.17.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staffa A, Cochrane A. Identification of positive and negative splicing regulatory elements within the terminal tat-rev exon of human immunodeficiency virus type 1. Mol Cell Biol. 1995;15:4597–4605. doi: 10.1128/mcb.15.8.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Mayeda A, Hampson RK, Krainer AR, Rottman FM. General splicing factor SF2/ASF promotes alternative splicing by binding to an exonic splicing enhancer. Genes & Dev. 1993;7:3519–3529. doi: 10.1101/gad.7.12b.2598. [DOI] [PubMed] [Google Scholar]
- Tian M, Maniatis T. A splicing enhancer complex controls alternative splicing of doublesex pre-mRNA. Cell. 1993;74:105–114. doi: 10.1016/0092-8674(93)90298-5. [DOI] [PubMed] [Google Scholar]
- Tsukahara T, Casciato C, Helfman DM. Alternative splicing of β-tropomyosin pre-mRNA: Multiple cis-elements can contribute to the use of the 5′- and 3′-splice sites of the non-muscle/smooth muscle exon 6. Nucleic Acids Res. 1994;22:2318–2325. doi: 10.1093/nar/22.12.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcárcel J, Singh R, Zamore PD, Green MR. The protein sex-lethal antagonizes the splicing factor U2AF to regulate the alternative splicing of transformer pre-mRNA. Nature. 1993;362:171–175. doi: 10.1038/362171a0. [DOI] [PubMed] [Google Scholar]
- Wang R, Kobayashi R, Bishop JM. Cellular adherence elicits ligand-independent activation of the Met cell-surface receptor. Proc Natl Acad Sci. 1996;93:8425–8430. doi: 10.1073/pnas.93.16.8425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YC, Selvakumar M, Helfman DM. Alternative pre-mRNA splicing. In: Krainer AR, editor. mRNA processing: Frontiers in molecular biology. Oxford, UK: IRL Press; 1997. pp. 242–279. [Google Scholar]
- Watakabe A, Tanaka K, Shimura Y. The role of exon sequences in splice site selection. Genes & Dev. 1993;7:407–418. doi: 10.1101/gad.7.3.407. [DOI] [PubMed] [Google Scholar]
- Wu JY, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- Xu R, Teng J, Cooper TA. The cardiac troponin T alternative exon contains a novel purine-rich positive splicing element. Mol Cell Biol. 1993;13:3660–3674. doi: 10.1128/mcb.13.6.3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Bani M-R, Lu S-J, Rowan S, Ben-David Y, Chabot B. The A1 and A1b proteins of heterogeneous nuclear ribonucleoparticles modulate 5′ splicing site selection in vivo. Proc Natl Acad Sci. 1994;91:6924–6928. doi: 10.1073/pnas.91.15.6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler AM, Lane WS, Stolk JA, Roth MB. SR proteins: A conserved family of pre-mRNA splicing factors. Genes & Dev. 1992;6:837–847. doi: 10.1101/gad.6.5.837. [DOI] [PubMed] [Google Scholar]
- Zheng Z-M, He P, Baker CC. Selection of the bovine papillomavirus type 1 nucleotide 3225 3′ splice site is regulated through an exonic splicing enhancer and its justaposed exonic splicing suppressor. J Virol. 1996;70:4691–4699. doi: 10.1128/jvi.70.7.4691-4699.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo P, Maniatis T. The splicing factor U2AF mediates critical protein-protein interactions in constitutive and enhancer-dependent splicing. Genes & Dev. 1996;10:1356–68. doi: 10.1101/gad.10.11.1356. [DOI] [PubMed] [Google Scholar]