

Abstract
Mesothelin is a cell-surface tumor-associated antigen expressed in several human cancers. The limited expression of mesothelin on normal tissues and its high expression in many cancers make it an attractive candidate for targeted therapies utilizing monoclonal antibodies, immunoconjugates and immunotoxins. Mesothelin is actively shed from the cell surface and is present in the serum of patients with malignant mesothelioma, which could negatively affect the response to these therapies. We have found that mesothelin sheddase activity is mediated by a tumor necrosis factor-α converting enzyme (TACE), a member of the MMP/ADAM family. We showed that EGF and TIMP-3 act through TACE as endogenous regulators of mesothelin shedding. We also found that reducing shedding significantly improved the in vitro cytotoxicity of immunotoxin SS1P, which targets mesothelin and is currently in clinical trials for the treatment of patients with mesothelioma and lung cancer. Our findings provide a mechanistic understanding of mesothelin shedding and could help improve mesothelin-based targeted therapies.
Keywords: mesothelioma, mesothelin, sheddase, ectodomain shedding, MMP inhibitor, TACE
Introduction
Mesothelin is a differentiation antigen located mainly on the cell surface (1, 2). Its distribution in normal human tissues is essentially limited to the mesothelial cells lining the pleura, peritoneum and pericardium. However, it is highly expressed in many common epithelial cancers including approximately 100% of epithelial malignant mesotheliomas and ductal pancreatic adenocarcinomas, 67–100% of ovarian cancers and 41–53% of lung adenocarcinomas. In addition, mesothelin is expressed to varying degrees by other tumors including cervical, head and neck, gastric, esophageal and bile duct carcinomas (3, 4). The restricted expression of mesothelin on normal tissues and high expression in many cancers makes it a good target for tumor-specific immunotherapy.
Several mesothelin-targeted immunotherapy approaches are under preclinical or clinical development for the treatment of mesothelin-expressing tumors. Among these approaches, immunotoxin SS1P is the most clinically advanced agent. It is composed of the anti-mesothelin variable fragment (Fv), SS1, fused to a truncated Pseudomonas exotoxin A (PE38). It kills cells by binding to mesothelin, entering cells by receptor-mediated endocytosis and inhibiting protein synthesis. Several minor responses were observed in phase I clinical trials of SS1P (5, 6). Based on preclinical studies that show marked synergy between SS1P and chemotherapy (7, 8) a clinical trial of SS1P in combination with pemetrexed and cisplatin is currently ongoing in patients with pleural mesothelioma (5, 6). MORAb-009, a chimeric monoclonal antibody against mesothelin, has completed phase I testing, and about half of patients have stable disease (9). It is now being evaluated in phase II studies for treatment of pancreatic cancer and mesothelioma. Adoptive T-cell strategy that targets mesothelin has also been tested in a mouse model and eradicated large tumor xenografts (10, 11). Additionally, mesothelin is an immunogenic protein, and it represents an attractive target for active immunotherapy using mesothelin-based cancer vaccines. Humoral and cellular immunity against mesothelin-expressing tumors has been observed in preclinical studies (12–14).
Although the normal biological function of mesothelin is not known, its shedding has important clinical significance. Serum mesothelin levels are elevated in mesothelioma and ovarian cancer patients. Mesothelin measurements are useful for the diagnosis of these cancers and to monitor response to treatment (15, 16). However, mesothelin shedding may have a detrimental effect on mesothelin-targeted therapies. The loss of mesothelin sites on tumor cells and increased dissociation of mesothelin-targeted agents could compromise the therapeutic effect. In addition, shed mesothelin accumulates in interstitial fluid of solid tumors, forming a barrier which prevents monoclonal antibodies from reaching the tumor cells and therefore reduces the targeting efficiency (17, 18). Little is known how mesothelin is shed from cell surface.
In this study, we explored the mechanism of mesothelin shedding. We show that mesothelin shedding is mediated by the sheddase, TACE/ADAM17 a member of the MMP/ADAM family. In addition we show that by modulating mesothelin shedding, we can significantly improve the cytotoxic effect of SS1P. These findings could improve the clinical efficacy of mesothelin-targeted agents.
Materials and Methods
Reagents
Phosphatidylinositol-specific phospholipase C (PI-PLC), phosphatidylcholine-specific phospholipase C (PC-PLC), doxycyclin and cycloheximide were obtained from Sigma. Camostat, marimastat and batimastat were obtained from Tocris. EGF, TIMP-1, 2, 3 and 4 were from R&D. All other MMP/ADAM and phospholipase inhibitors including GM6001 were from EMD. GM6001, marimastat and batimastat are of the hydroxamate class of metallopeptidase inhibitors. They show potent but reversible inhibition of broad-spectrum MMPs/ADAMs by competitive binding of active site zinc. The small interference RNAs (siRNAs) against a tumor necrosis factor-α converting enzyme (TACE; Hs_ADAM17_1, Hs_ADAM17_7 and Hs_ADAM17_8) were purchased from Qiagen. The control siRNA against MMP-12 (Hs_ADAM12_1) is also from Qiagen. The siRNA against Luciferase (Luciferase GL2 Duplex) is from Dharmacon. The immunotoxin SS1P was prepared as previously described (7). The Alexa-labeling was done with a Alexa Fluor Protein Labeling Kit (Invitrogen).
To measure the amount of knock-down of TACE mRNA, an SYBR Green real-time PCR was performed with the following primers (forward: 5′ – GGTTTGACGAGCACAAAGAA – 3′; reverse 5′ – GGATCATGTTCTGCTCCAAA – 3′).
Cell culture
A431/H9 is a human mesothelin-transfected A431 cell line (epidermoid cancer) that highly expresses and sheds mesothelin. KB (cervical cancer) and A431/H9 cells were grown in DMEM with 10% FBS. Mesothelioma cell lines HAY and M30 were maintained in RPMI 1640 (10% FBS). Ascites and primary cells were isolated from mesothelioma patients before they received treatment. The M02 cell was from pleural fluid, and other primary cells were from the ascites of peritoneal mesothelioma. The MET-5A cell line is from ATCC. It is derived from non-cancerous mesothelial cells which expresses SV40 T antigen.
Shed mesothelin preparation and C-terminal sequencing
An SS1P affinity column was prepared with HiTrap NHS-activated HP (1 mL; GE Healthcare) according to the manufacturer’s instruction. Briefly, SS1P (2 mg) was incubated with the resin for 30 min at room temperature. The column was deactivated with buffer A (0.5 M ethanolamine, 0.5 M NaCl, pH 8.3) and washed with buffer B (0.1 M acetate, 0.5 M NaCl, pH 4.0) followed by PBS.
The supernatant from A431/H9 cell culture (600 mL) filtered through 0.22 μm membrane was loaded onto the SS1P affinity column at 0.5 mL/min. The column was washed with 10 mL each of PBS and citrate/phosphate buffer (pH 5.0), and eluted with 10 mM glycine-HCl (pH 2.4). Fractions were collected and neutralized with 1 M Tris-HCl, pH 8.0. For purifying shed mesothelin from human samples, 10 mL of ascites was diluted into 200 mL of PBS and filtered before loading.
Purified mesothelin was digested with trypsin (50 ng/μL; Roche Diagnostics) for 2 h at 37°C. An aliquot of the digest was purified by C18 ZipTip (Millipore). An in-gel digestion was done with mesothelin purified from patient ascites. The eluted peptides from ZipTip were analyzed by matrix-assisted laser desorption ionization time-of flight mass spectrometry (MALDI-TOF MS) using an Ultraflex III MALDI-TOF/TOF mass spectrometer (Bruker Daltonics). Spectra were collected in positive ion reflector mode using delayed extraction, with 1000–2000 laser shots acquired per spectrum. For Peptide Mass Fingerprint protein identification, mass spectrometry data were used to search the NCBI non-redundant human protein database using the Mascot search engine. Mass spectrometric sequencing of particular peptides was performed by MALDI-TOF MS/MS analysis of the same spots on the target plate. Fragment ion spectra were used for NCBI protein database search by the Mascot MS/MS ion search program.
SS1P internalization assay
A431/H9 cells were pretreated with GM6001 (20 μM) or DMSO (0.2%) for 48 h. After harvest by trypsin, cells were incubated with SS1P-Alexa488 (5 μg/mL) at 37°C for 10, 30, 60 and 120 min. DMSO or GM6001 were included during incubation. The cells were then stripped with glycine buffer (0.2 M Glycine HCl and 1 mg/mL BSA; pH 2.5) to remove surface-bound SS1P and analyzed with FACSCalibur. The signal from cells saturated by SS1P-Alexa488 was used as a standard. The amount of internalized SS1P was presented as the percentage of the standard.
Western blotting
To detect TACE expression, cells in 6-well plate were directly lysed with Laemmli buffer. The lysate was separated by SDS-PAGE (4–20%) under reducing conditions and transferred to a polyvinylidene difluoride membrane. The blocked membrane was incubated with rabbit anti-human TACE polyclonal antibody (1:5000; EMD) followed by horseradish peroxidase-conjugated goat anti-rabbit antibody (1:5000; Abcam). Proteins were visualized by enhanced chemiluminescence.
Cytotoxicity assay
Cells, pre-treated with GM6001 (20 μM) for 48 h, were seeded into 96-well plates at 5,000 per well and incubated at 37°C overnight. Serial dilutions of SS1P were added and incubated for another 72 h. GM6001 or the vehicle control was included during the incubation. Inhibition of cell growth was determined by WST-8 assays (Dojindo). Viability was expressed as the percentage of the absorbance value of untreated controls.
Statistics
All data are presented as mean ± SD. The error bars represent the standard deviation of triplicate wells. The experiments were repeated at least 3 times. Statistical differences between groups were measured by Student’s t test with two-tailed distribution. A P value of less than 0.05 was considered significant.
Results
A431/H9 cells share the same mechanism of mesothelin shedding as tumor cells obtained from mesothelioma patients
A431/H9 cells express very high levels of mesothelin on the cell surface and actively shed mesothelin (17). We chose these cells as a model to study mesothelin shedding after comparing the C-terminal sequence of mesothelin fragments shed from this cell line with clinical samples. Using an SS1P affinity column, we purified shed mesothelin from the ascites of two mesothelioma patients and the supernatant of A431/H9 cells grown in culture. Unprocessed mesothelin has a molecular weight (MW) of 75 kDa. It is processed by a furin-like enzyme to generate megakaryocyte-potentiating factor leaving behind mature mesothelin, which is attached to the cell surface and recognized by SS1P. Depending on its glycosylation status, the apparent MW of mature mesothelin is about 40 kDa on SDS-PAGE gel. Shed mesothelin from human ascites after isolation on an SS1P column is the major product and runs as a broad band located between 37 and 50 kDa (Fig. 1A). Several other bands are also present in the preparation. Sequencing results indicated they were derived from different types of antibodies. These antibodies are contaminants non-specifically binding to the SS1P affinity column.
Figure 1.

The determination of shed mesothelin C-terminal sequence. A. SDS-PAGE profile of shed mesothelin preparations from A431/H9 supernatant and ascites of a patient with mesothelioma. Bands in frame are shed mesothelin and subjected to the sequencing. B. The C-terminal cleavage sites of mesothelin.
To determine the C-terminal sequence of shed mesothelin from A431/H9 cells, the protein sample was digested with trypsin and the peptides were analyzed by MALDI-TOF MS. Two peaks not corresponding to the expected tryptic peptides, were C-terminal peptides of shed mesothelin and subjected to mass spectrometric sequencing by MALDI-TOF/TOF MS/MS (supplementary Fig. S1). In the Mascot search, the parameters were changed to reflect a potential cyclization of N-terminal glutamine residue into pyro-glutamic acid and the search resulted in confident identification of the peptides. Two sequences were identified, a longer one (QRQDDLDTLGLGLQGGIPNGYLVLDL) and a shorter one (QRQDDLDTLGLGLQGGIPNGY) (supplementary Fig. S2A and S2B). The MW of shed mesothelin from the ascites appeared smaller by SDS-PAGE compared with that from A431/H9 cell supernatant, but they have the same N-terminus confirmed by sequencing.
We used a similar method to determine the C-terminal sequences of shed mesothelin from ascites. Two sequences were identified and are the same as those from A431/H9 cells (supplementary Fig. S3A and S3B). The C-terminal cleavage sites of mesothelin are shown in Fig. 1B. The sequences are identical to those reported from ovarian cancer patients (19). The discrepancy in apparent MW could be due to the variation of glycosylation. The presence of two C-terminal peptides of mesothelin ending in Leu or Tyr most likely indicates simultaneous (not step-wise) cleavage of glycosylphosphatidylinositol (GPI)-anchored mesothelin. These results indicate that A431/H9 cells and patient’s cells share the same shedding mechanism. Therefore, A431/H9 appeared to be a good model to study mesothelin shedding.
Mesothelin sheddase belongs to MMP/ADAM family
Mesothelin was identified as a GPI-anchored protein. Such a protein has a glycolipid attached to its c-terminus during posttranslational modification. This structure anchors the protein to the cell membrane. Since the glypiation is the sole means of membrane attachment, cleavage of the group by phospholipases results in the release of the protein from the cell membrane (20). Phospholipase C (PLC) is an enzyme that is known to cleave the phospho-glycerol bond found in GPI-anchored proteins. We incubated A431/H9 cells with either PI-PLC or PC-PLC in HBSS buffer and examined mesothelin levels on the cell surface by Alexa488-labeled SS1P staining. SS1P is a mesothelin-targeting immunotoxin, which is composed of an anti-mesothelin Fv fused to a truncated PE38. After PLC treatment, cell-surface mesothelin was significantly decreased, indicating the presence of a GPI anchor structure (Fig. 2A). But PLC inhibitors (U73122, ET18-OCH3 and neomycin) failed to inhibit mesothelin shedding (Fig. 2B). Instead, sPLA2-IIA inhibitor I and D609 (PLC/phospholipase D [PLD] inhibitor) significantly increased mesothelin shedding by an unknown mechanism. These results suggest that endogenous PLCs do not significantly contribute to the mesothelin cleavage.
Figure 2.
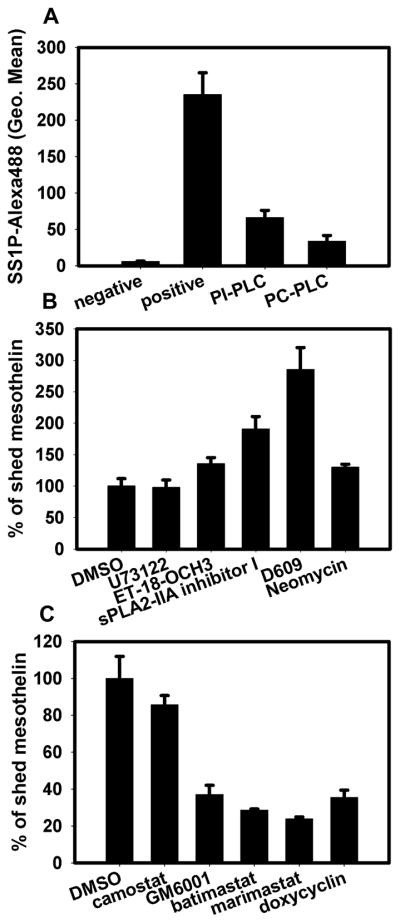
Mesothelin shedding is inhibited by broad-spectrum MMP/ADAM inhibitors. A. A431/H9 cells harvested by trypsinization were incubated with PI-PLC (1 U/mL) or PC-PLC (10 U/mL) for 1 h at 37°C. After wash with FACS buffer, cells were stained by Alexa488-labeled SS1P (2 μg/mL). The fluorescence signal was measured by flow cytometer. Cells without SS1P-Alexa488 staining were used as the negative control. Cells incubated with Hank’s buffer and stained by SS1P-Alexa488 were used as positive control. B and C. A431/H9 cells were seeded into 96-well plate and incubated with phospholipase inhibitor (B) or pan-MMP/ADAM inhibitors (C), including U73122 (10 μM), ET-18-OCH3 (10 μM), sPLA2-IIA inhibitor I (50 μg/mL), D609 (250 μg/mL), neomycin (1 mg/mL), GM6001 (20 μM), batimastat (20 μM), marimastat (20 μM) and camostat (20 μM). After 48 h, mesothelin in cell medium was measured by ELISA. DMSO-treated cell was negative control. The results were normalized with cell number in each well.
We then tried several broad-spectrum MMP/ADAM inhibitors (GM6001, batimastat and marimastat), and all of them showed an inhibitory effect on mesothelin shedding. The serine protease inhibitor camostat did not inhibit mesothelin shedding (Fig. 2C). Consistent with our results, GM6001 was previously shown to inhibit mesothelin shedding of primary mesothelioma cells (21). In addition, TAPI-1 and MMPI-III at 10 μM inhibited the shedding but the broad-spectrum cathepsin inhibitor I (EMD) did not (data not shown). The selective inhibition of mesothelin shedding by MMP/ADAM inhibitors suggests that members of the MMP/ADAM family are involved in or at least partly responsible for mesothelin shedding.
TACE is a potential mesothelin sheddase
To sort out the mesothelin sheddase from other members in MMP/ADAM family is challenging. Inhibitors highly restricted to a single protease are rare. We used endogenous MMP/ADAM inhibitors – TIMPs to narrow down the candidate list for the mesothelin sheddase, because their inhibition spectrums are well studied. TIMP-1, 2 and 4 did not show an inhibitory effect on mesothelin shedding, whereas TIMP-3 caused >50% inhibition comparable to the effect of GM6001 (Fig. 3A). TIMP-3 inhibits all MMPs, ADAM10, 12, 17, 33, ADAMTS-1, 2, 4 and 5. TIMP-1, 2 and 4 inhibit all MMPs, ADAM10, 12, and 33 (22). ADAM17, ADAMTS-1, 2, 4 and 5 appeared to be possible mesothelin sheddases. Of these, ADAM17/TACE is most frequently found as a sheddase for many cell-surface antigens, including TNF-α, EGFR ligands, etc. (23). The shedding activity of TACE seems to be sequestered in lipid rafts (24), which is a potential location for GPI-anchored proteins like mesothelin. We hypothesized TACE was responsible for mesothelin shedding. We found that shedding of mesothelin was increased by phorbol 12-myristate 13-acetate (PMA) and EGF, and that the EGF-induced mesothelin shedding could be inhibited by treatment with Iressa, a specific tyrosine kinase inhibitor of EGFR (Fig. 3B). This is also a feature of TACE-mediated shedding (25). TIMP-3 inhibited PMA-induced mesothelin shedding but TIMP-1, 2 and 4 did not (data not shown), indicating that constitutional and induced mesothelin shedding may share the same mechanism.
Figure 3.

TACE is a candidate as mesothelin sheddase. A and B. A431/H9 cells with fresh medium (1×105 cells/well in a 96-well plate) were incubated with TIMPs (5 μg/mL), EGF (50 ng/mL), PMA (10 μM) or EGF plus Iressa (1 μM) for 5 h. Shed mesothelin in the cell supernatant was measured by ELISA. Negative and positive controls are PBS and GM6001 (20 μM). C. Tumor cells isolated from the ascites of five mesothelioma patients were examined for TACE expression with western blot. Cells from patients NCI-M-02, 05, 10, 11 and 13 were included.
Most mesothelioma patients have elevated serum mesothelin levels in their blood. So TACE, as a mesothelin sheddase would be expected to be present in most of these mesotheliomas. We examined TACE expression in tumor cells isolated from the ascites of several mesothelioma patients. All samples expressed TACE protein, showing two bands in the western blot, pro-TACE and active TACE (Fig. 3C). We searched Gene Expression Omnibus (GEO) repository to check TACE expression profiles in mesothelioma samples. In a dataset (GDS1220) including 40 mesothelioma samples, 90% of them are TACE-positive. These data support our hypothesis that TACE is a mesothelin sheddase candidate.
TACE is involved in mesothelin shedding
Next we did a knock-down experiment with siRNA against TACE (Hs_ADAM17_7). The transfection of TACE siRNA into A431/H9 cells caused more than a 90% decrease of TACE protein expression (Fig. 4A) compared to mock and luciferase siRNA transfection. Cells with TACE knock-down demonstrated a 50% decrease in mesothelin shedding (Fig. 4B). We then measured cell-surface mesothelin expression with SS1P-Alexa488. We found a 50% increase, which we attribute to reduced mesothelin shedding (Fig. 4C). The cells were also two-fold more sensitive to SS1P’s cytotoxic effect as compared to controls (Fig. 4D). We also tried another two siRNAs against TACE (Hs_ADAM17_1 and 8), which have a comparable knock-down effect as Hs_ADAM17_7 measured by Real-Time PCR. They inhibited the shedding of mesothelin by 41 and 45% and increased surface mesothelin levels by 51 to 86% (data not shown). The other negative control (siRNA against MMP12) did not show any significant effect. These studies indicate the expression level of TACE has an effect on mesothelin shedding, the cell-surface mesothelin level and the cell’s sensitivity to SS1P.
Figure 4.

TACE is involved in mesothelin shedding. A431/H9 cells were transfected with TACE siRNA (Hs_ADAM17_7; 50 nM). After 48 h, the expression of TACE was examined by western blot (A). The mesothelin shedding was checked by incubating transfected cells in fresh medium for 5 h (B). The cells were stained by Alexa488-labeled SS1P (2.5 μg/mL) to measure mesothelin expression level on cell surface (C). D. The SS1P cytotoxicity on transfected cells. Two days after siRNA transfection, SS1P with serial dilutions was added for a short incubation period (2 h). After another 48 h, WST-8 reagent was added to determine IC50s. Mock and luciferase siRNA-transfected cells were negative controls. The error bars represent the standard deviation of triplicate wells. The experiments were repeated at least 3 times. A typical result is shown.
Modulating mesothelin shedding causes cells to be more sensitive to SS1P
Based on the TACE knock-down results, we evaluated whether mesothelin shedding can be modulated by other reagents to increase cell sensitivity to SS1P with the prospect of having potential clinical significance. We tested GM6001, the pan-MMP/ADAM inhibitor, because it had been shown to inhibit mesothelin shedding. GM6001 treatment increased cell-surface mesothelin level by about 2.6 folds (Fig. 5A). Because SS1P must be internalized into cells to be active, we examined the SS1P internalization process following treatment with GM6001 (Fig. 5B). Even at a very early time point (5 min), there was increased SS1P internalization in the GM6001-treated group. At the plateau phase (from 1–6 hours), internalized SS1P was increased 6-fold by GM6001 treatment. This increase could be attributed to the reduced dissociation of SS1P from the cell-surface. As expected, GM6001 made cells 5-fold more sensitive to SS1P (Fig. 5D). However, GM6001 did not change the cell’s sensitivity to cycloheximide (CHX) (Fig. 5C). Both Cycloheximide and SS1P kill cells by inhibiting protein synthesis but CHX diffuses through cell membrane instead of binding to mesothelin and being internalized by endocytosis. Therefore, the increased sensitivity to SS1P following pre-treatment with GM6001 is likely due to the modulation of the mesothelin shedding process.
Figure 5.

MMP/ADAM inhibitor regulates mesothelin-targeting process. A431/H9 cells were treated with GM6001 (20 μM) for 48 h. They were then stained with SS1P-Alexa488 (2.5 μg/mL) to determine cell-surface expression of mesothelin (A). Filled area, cells without staining; dash line, DMSO treatment; solid line, GM6001 treatment. B. The measurement of SS1P internalization rates on DMSO and GM6001-treated cells. C and D. Cytotoxicity of cycloheximide and SS1P on treated cells. Control is DMSO-treated cells.
To show that the effect of GM6001 is not limited to the A431/H9 cell line, we examined several other mesothelin-expressing cell lines (KB, HAY, M30). The results are summarized in Table 1. GM6001 inhibited mesothelin shedding by these cell lines. The cell-surface level of mesothelin was increased by the treatment, ranging from 50% to 260%. When treated with GM6001, the sensitivity of cells to SS1P increased 1.2–5 fold. We then examined the effect of GM6001 on primary mesothelioma cells isolated from two mesothelioma patients and found it increased cytotxicity by 2–2.5 fold. We also examined the MET-5A cell line, which is derived from normal mesothelium and expresses low level of mesothelin on the cell surface. MET-5A is not sensitive to SS1P even at the concentration of 1000 ng/mL. GM6001 treatment did not sensitize cells to this reagent. These results indicate that the modulation of mesothelin shedding is a potential approach to improve mesothelin-targeted therapies.
Table 1.
Modulation of mesothelin shedding and SS1P cytotoxicity by GM6001 on several cell lines
| Cell Line | Mesothelin Shedding (% change) | Mesothelin Surface Level (% change) | SS1P Cytotoxicity (fold change) | Cell Type |
|---|---|---|---|---|
| A431/H9 | −62 | 180 | 5 | Epidermoid Cancer |
| KB | −52 | 140 | 3.5 | Cervical Cancer |
| M30 | −44 | 50 | 2 | Mesothelioma |
| HAY | −62 | 40 | 1.2 | Mesothelioma |
| M16 | N/D | N/D | 2.5 | Primary Mesothelioma |
| M19 | N/D | N/D | 2 | Primary Mesothelioma |
| MET-5A | N/D | N/D | not sensitive | Normal Mesothelium |
M16 and M19 are primary mesothelioma cells isolated from patients
NCI-M-16 and NCI-M-19
N/D, not determined and data is not available.
Discussion
Mesothelin is a valuable biomarker and an attractive target for cancer therapy. Shedding of mesothelin could potentially have an effect on targeted therapies directed to mesothelin. In this study, we used the A431/H9 cell line as a model to study the mesothelin shedding process. Mesothelin sheddase was defined as a protease belonging to TACE, a member of the MMP/ADAM family and TIMP-3 was identified as an endogenous inhibitor of mesothelin shedding. Interestingly, modulating the shedding process had a significant impact on mesothelin-targeted therapy using the anti-mesothelin immunotoxin SS1P.
Mesothelin has been reported to be a GPI-anchored cell-surface protein. In this and previously reported studies, phospholipases (PI-PLC and PC-PLC) can release mesothelin from the cell surface. But several other lines of evidence suggest that TACE plays a more significant role in mesothelin shedding.
Several phospholipase inhibitors failed to reduce mesothelin shedding from A431/H9 cells. This indicates that phospholipases are not likely to play a significant role in mesothelin shedding. In addition, a GPI-anchored protein has a hydrophobic end which during posttranslational modification is cleaved off and replaced by the GPI-anchor. The GPI anchor attachment site is called the ω-site. We analyzed the mesothelin sequence for a ω-site with the prediction tool - big-PI Predictor 3.0 (26). Our sequencing data showed that the ω-site is positioned at serine, 25 amino acids away from the C-terminus of mesothelin that we determined. These results exclude phospholipase as the mesothelin sheddase.
MMP/ADAM inhibitors showed inhibitory effects on mesothelin shedding. These inhibitors included synthetic reagents (GM6001, batimastat and marimastat) and an endogenous factor (TIMP-3). GM6001, batimastat and marimastat are members of the hydroxamic acid class of reversible metallopeptidase inhibitors. The anionic state of the hydroxamic acid group binds to the active site zinc and inhibits enzymatic activity. TIMP-3 binds to the active site of mature metalloproteases via a 1:1 non-covalent interaction, blocking access of substrates to the catalytic site. The mesothelin sheddase is likely to belong to the MMP/ADAM family.
We identified TACE as the mesothelin sheddase through inhibition experiments with TIMPs. The specific knock-down of TACE expression by siRNA significantly decreased mesothelin shedding and increased membrane-bound mesothelin. This is the direct evidence that TACE is involved in the shedding. In addition, mesothelin shedding is PMA and EGF inducible, and EGF-induced shedding was blocked by gefatinib (Iressa). This is consistent with the profile of TACE-mediated shedding. GPI-anchored protein is preferentially localized in the cell membrane microdomain – lipid raft. This microdomain is enriched in cholesterol and sphingolipids and resistance to non-ionic detergents, such as Triton X-100 or Brij-98 at low temperatures (20). TACE, especially its mature form has been reported to be localized in lipid rafts, although this may be cell type-dependent (24). In our study, we found the active form of TACE in A431/H9 cells is insoluble in 2 × RIPA buffer at room temperature, supporting its presence in lipid rafts. The co-localization of TACE and mesothelin further supports the role of TACE in the shedding process. TACE has been shown to cleave some other lipid raft proteins (prion protein, CA4.4, CD30 and ErbB-4) (27–30).
It is not unusual for several endogenous proteases to be responsible for the shedding of a surface protein. This redundancy in enzymatic activity may have important biological significance. In this study, we chose TACE for mesothelin shedding from the inhibition spectrum of TIMPs. According to the literature, several other proteases fit the pattern and belong to MMP/ADAM family, including ADAMTS-1, 2, -4 and -5. They are also possible candidates for mesothelin sheddase. In this study TACE knock-down caused about a 50% reduction in mesothelin shedding, demonstrating the prominent role that TACE plays in mesothelin shedding.
Mesothelin expression, tumor burden and glomerular filtration rate have a big influence on mesothelin levels in the serum. Our study shows mesothelin shedding can also be regulated by other endogenous factors like TACE, EGF and TIMP-3. It is not clear whether there is correlation between their expression levels and the mesothelin levels in the blood. TACE, besides its direct role on mesothelin shedding, also participates in the shedding of EGFR ligands and is involved in the activation and EGFR signalling pathway (31). It may have an indirect effect on mesothelin shedding. In addition, some drugs may regulate mesothelin shedding. We showed that the EGFR tyrosine kinase inhibitor Irresa inhibited EGF induced mesothelin shedding. As a potentiator of TIMP-3 inhibitory action, the anti-arthritic agent, calcium pentosan polysulfate, enhances the binding affinity between TIMP-3 and its substrates more than 100-fold and also blocks the endocytosis of TIMP-3 by chondrocytes (32).
In this study we showed that mesothelin shedding decreased the cytotoxicity of SS1P by increasing its loss from the cell surface. Previously we demonstrated in vivo that shed mesothelin in tumor interstitial fluid was present at a high concentration and acts as a decoy receptor to block the SS1P targeting (18). Similar detrimental effects on shedding may apply to other mesothelin-targeted therapies. A TACE inhibitor would have double benefits in reducing the shed mesothelin barrier and increasing the drug association with tumor cells. A practical question is whether this effect would justify the design of a clinical trial. However, there is a lack of useful reagents for this purpose. Several broad-spectrum MMP/ADAM inhibitors have been tested in clinical trials and failed because of dose-limited toxicity and the lack of efficacy. This reflects our limited understanding of the diversity of the biological functions played by MMP/ADAM family members. Novel agents with more defined specificities should be the direction of drug development in this area (33). In fact, specific inhibitors of ADAM10 and 17 have been tested in combination with drugs targeting the EGFRs to achieve a more robust inhibition of the EGFR pathway in clinical settings (34). A similar approach could also be possibly used for mesothelin-targeted therapies. Alternatively, drugs which can modulate TACE activity could also be considered as a reagent for combination therapy.
Several approaches have been developed to target mesothelin-expressing tumors. In some clinical studies, mesothelin mediated toxicity has been shown to be dose limiting and manageable. A deeper understanding of the biology of mesothelin could be helpful in improving the efficacy of these treatments. Our study demonstrates the role of TACE in mesothelin shedding, contributing to the further development of mesothelin-based diagnosis and therapy.
Supplementary Material
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References
- 1.Chang K, Pastan I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci USA. 1996;93:136–40. doi: 10.1073/pnas.93.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang K, Pastan I. Molecular cloning and expression of a cDNA encoding a protein detected by the K1 antibody from an ovarian carcinoma (OVCAR-3) cell line. Int J Cancer. 1994;57:90–7. doi: 10.1002/ijc.2910570117. [DOI] [PubMed] [Google Scholar]
- 3.Ordonez NG. Application of mesothelin immunostaining in tumor diagnosis. Am J Surg Pathol. 2003;27:1418–28. doi: 10.1097/00000478-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Ordonez NG. Value of mesothelin immunostaining in the diagnosis of mesothelioma. Mod Pathol. 2003;16:192–7. doi: 10.1097/01.MP.0000056981.16578.C3. [DOI] [PubMed] [Google Scholar]
- 5.Hassan R, Bullock S, Premkumar A, et al. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. 2007;13:5144–9. doi: 10.1158/1078-0432.CCR-07-0869. [DOI] [PubMed] [Google Scholar]
- 6.Kreitman RJ, Hassan R, FitzGerald DJ, Pastan I. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin Cancer Res. 2009;15:5274–9. doi: 10.1158/1078-0432.CCR-09-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Xiang L, Hassan R, et al. Synergistic antitumor activity of taxol and immunotoxin SS1P in tumor-bearing mice. Clin Cancer Res. 2006;12:4695–701. doi: 10.1158/1078-0432.CCR-06-0346. [DOI] [PubMed] [Google Scholar]
- 8.Hassan R, Broaddus VC, Wilson S, Liewehr DJ, Zhang J. Anti-mesothelin immunotoxin SS1P in combination with gemcitabine results in increased activity against mesothelin-expressing tumor xenografts. Clin Cancer Res. 2007;13:7166–71. doi: 10.1158/1078-0432.CCR-07-1592. [DOI] [PubMed] [Google Scholar]
- 9.Hassan R, Cohen SJ, Phillips M, et al. Phase I clinical trial of the chimeric anti-mesothelin monoclonal antibody MORAb-009 in patients with mesothelin expressing cancers. Clin Cancer Res. 2010;16:6132–8. doi: 10.1158/1078-0432.CCR-10-2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Y, Moon E, Carpenito C, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70:9053–61. doi: 10.1158/0008-5472.CAN-10-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carpenito C, Milone MC, Hassan R, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. 2009;106:3360–5. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leao IC, Ganesan P, Armstrong TD, Jaffee EM. Effective depletion of regulatory T cells allows the recruitment of mesothelin-specific CD8 T cells to the antitumor immune response against a mesothelin-expressing mouse pancreatic adenocarcinoma. Clin Transl Sci. 2008;1:228–39. doi: 10.1111/j.1752-8062.2008.00070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fridlender ZG, Buchlis G, Kapoor V, et al. CCL2 blockade augments cancer immunotherapy. Cancer Res. 2010;70:109–18. doi: 10.1158/0008-5472.CAN-09-2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hung CF, Tsai YC, He L, Wu TC. Control of mesothelin-expressing ovarian cancer using adoptive transfer of mesothelin peptide-specific CD8+ T cells. Gene Ther. 2007;14:921–9. doi: 10.1038/sj.gt.3302913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson BW, Creaney J, Lake R, et al. Mesothelin-family proteins and diagnosis of mesothelioma. Lancet. 2003;362:1612–6. doi: 10.1016/S0140-6736(03)14794-0. [DOI] [PubMed] [Google Scholar]
- 16.Scholler N, Fu N, Yang Y, et al. Soluble member(s) of the mesothelin/megakaryocyte potentiating factor family are detectable in sera from patients with ovarian carcinoma. Proc Natl Acad Sci USA. 1999;96:11531–6. doi: 10.1073/pnas.96.20.11531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Hansen JK, Xiang L, et al. A flow cytometry method to quantitate internalized immunotoxins shows that taxol synergistically increases cellular immunotoxins uptake. Cancer Res. 2010;70:1082–9. doi: 10.1158/0008-5472.CAN-09-2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Xiang L, Hassan R, Pastan I. Immunotoxin and Taxol synergy results from a decrease in shed mesothelin levels in the extracellular space of tumors. Proc Natl Acad Sci USA. 2007;104:17099–104. doi: 10.1073/pnas.0708101104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hellstrom I, Raycraft J, Kanan S, et al. Mesothelin variant 1 is released from tumor cells as a diagnostic marker. Cancer Epidemiol Biomarkers Prev. 2006;15:1014–20. doi: 10.1158/1055-9965.EPI-05-0334. [DOI] [PubMed] [Google Scholar]
- 20.Chatterjee S, Mayor S. The GPI-anchor and protein sorting. Cell Mol Life Sci. 2001;58:1969–87. doi: 10.1007/PL00000831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sapede C, Gauvrit A, Barbieux I, et al. Aberrant splicing and protease involvement in mesothelin release from epithelioid mesothelioma cells. Cancer Sci. 2008;99:590–4. doi: 10.1111/j.1349-7006.2007.00715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. 2010;1803:55–71. doi: 10.1016/j.bbamcr.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arribas J, Esselens C. ADAM17 as a therapeutic target in multiple diseases. Curr Pharm Des. 2009;15:2319–35. doi: 10.2174/138161209788682398. [DOI] [PubMed] [Google Scholar]
- 24.Tellier E, Canault M, Rebsomen L, et al. The shedding activity of ADAM17 is sequestered in lipid rafts. Exp Cell Res. 2006;312:3969–80. doi: 10.1016/j.yexcr.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 25.Santiago-Josefat B, Esselens C, Bech-Serra JJ, Arribas J. Post-transcriptional up-regulation of ADAM17 upon epidermal growth factor receptor activation and in breast tumors. J Biol Chem. 2007;282:8325–31. doi: 10.1074/jbc.M608826200. [DOI] [PubMed] [Google Scholar]
- 26.http://mendel.imp.ac.at/gpi/gpi_server.html
- 27.Vincent B, Paitel E, Saftig P, et al. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J Biol Chem. 2001;276:37743–6. doi: 10.1074/jbc.M105677200. [DOI] [PubMed] [Google Scholar]
- 28.Esselens CW, Malapeira J, Colomé N, Moss M, Canals F, Arribas J. Metastasis-associated C4. 4A, a GPI-anchored protein cleaved by ADAM10 and ADAM17. Biol Chem. 2008;389:1075–84. doi: 10.1515/bc.2008.121. [DOI] [PubMed] [Google Scholar]
- 29.von Tresckow B, Kallen KJ, von Strandmann EP, et al. Depletion of cellular cholesterol and lipid rafts increases shedding of CD30. J Immunol. 2004;172:4324–31. doi: 10.4049/jimmunol.172.7.4324. [DOI] [PubMed] [Google Scholar]
- 30.Thiel KW, Carpenter G. ErbB-4 and TNF-alpha converting enzyme localization to membrane microdomains. Biochem Biophys Res Commun. 2006;350:629–33. doi: 10.1016/j.bbrc.2006.09.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borrell-Pages M, Rojo F, Albanell J, Baselga J, Arribas J. TACE is required for the activation of the EGFR by TGF-alpha in tumors. EMBO J. 2003;22:1114–24. doi: 10.1093/emboj/cdg111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Troeberg L, Fushimi K, Khokha R, Emonard H, Ghosh P, Nagase H. Calcium pentosan polysulfate is a multifaceted exosite inhibitor of aggrecanases. FASEB J. 2008;22:3515–24. doi: 10.1096/fj.08-112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nuti E, Tuccinardi T, Rossello A. Matrix metalloproteinase inhibitors: new challenges in the era of post broad-spectrum inhibitors. Curr Pharm Des. 2007;13:2087–100. doi: 10.2174/138161207781039706. [DOI] [PubMed] [Google Scholar]
- 34.Zhou BB, Peyton M, He B, et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell. 2006;10:39–50. doi: 10.1016/j.ccr.2006.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.