

Abstract
The c-jun proto-oncogene encodes a component of the mitogen-inducible immediate–early transcription factor AP-1 and has been implicated as a positive regulator of cell proliferation and G1-to-S-phase progression. Here we report that fibroblasts derived from c-jun−/− mouse fetuses exhibit a severe proliferation defect and undergo a prolonged crisis before spontaneous immortalization. The cyclin D1- and cyclin E-dependent kinases (CDKs) and transcription factor E2F are poorly activated, resulting in inefficient G1-to-S-phase progression. Furthermore, the absence of c-Jun results in elevated expression of the tumor suppressor gene p53 and its target gene, the CDK inhibitor p21, whereas overexpression of c-Jun represses p53 and p21 expression and accelerates cell proliferation. Surprisingly, protein stabilization, the common mechanism of p53 regulation, is not involved in up-regulation of p53 in c-jun−/− fibroblasts. Rather, c-Jun regulates transcription of p53 negatively by direct binding to a variant AP-1 site in the p53 promoter. Importantly, deletion of p53 abrogates all defects of cells lacking c-Jun in cell cycle progression, proliferation, immortalization, and activation of G1 CDKs and E2F. These results demonstrate that an essential, rate-limiting function of c-Jun in fibroblast proliferation is negative regulation of p53 expression, and establish a mechanistic link between c-Jun-dependent mitogenic signaling and cell-cycle regulation.
Keywords: c-Jun, cell cycle regulation, cell proliferation, mouse knockout fibroblasts, p53
The control of mammalian cell proliferation by mitogens occurs largely during the G1 phase of the cell cycle. During this period, extracellular signals are transduced by cytoplasmic signaling cascades to the nuclear cell cycle clock, where a decision is made between cell cycle progression and quiescence (Sherr 1994, 1996). Among the earliest responses to mitogenic signaling is activation of transcription factors such as c-Jun, a subunit of activator protein 1 (AP-1) (Angel and Karin 1991). Transcriptional activation of yet-to-be-identified target genes by c-Jun and other immediate early transcription factors is thought to be essential for mitogen-induced progression through the cell cycle. A causal role of c-Jun in promoting cell division was indeed suggested by studies using microinjection of neutralizing antibodies or antisense RNA, which cause a partial G0 arrest and block entry into S phase (Kovary and Bravo 1991; Riabowol et al. 1992; Smith and Prochownik 1992). Conversely, cell cycle distribution in cells overexpressing c-Jun is shifted toward S phase (Pfarr et al. 1994). c-Jun can act as an oncogene when mutated or expressed in a deregulated way, an ability that is shared by several cell cycle proteins promoting cell division such as cylin D1, cyclin-dependent kinase-4 (CDK4), c-Myc, and possibly E2F (Angel and Karin 1991; Sherr 1996; Weinberg 1996). In addition, c-Jun is involved in the control of apoptosis (Ham et al. 1995; Verheij et al. 1996; Bossy-Wetzel et al. 1997) and differentiation (Lord et al. 1993; Bohmann et al. 1994; Szabo et al. 1994; Patel and Sytkowski 1995). Although the mitogenic signaling pathways acting upstream of c-Jun are reasonably well understood, as is the basic cell cycle machinery, the connections between the two are still unclear (Cano and Mahadevan 1995; Karin 1995; Karin and Hunter 1995).
The core cell cycle clock itself operates by sequential activation and inactivation of a number of protein kinase complexes known as CDKs (Harper and Elledge 1996; Sherr 1996). In G1, these kinases consist of cyclin D1, D2, or D3 associated with CDK4 or CDK6 and cyclin E–CDK2 and phosphorylate, among others, the retinoblastoma protein (pRb) (Weinberg 1995). pRb phosphorylation leads to release of transcription factor E2F, which, once liberated, triggers expression of genes that enable cells to advance into late G1 and S phases (Grana and Reddy 1995; Zetterberg et al. 1995). Cyclin/CDK activity is regulated negatively by the association with CDK inhibitors (CKIs), such as p21, p27, p57, and the p16/INK4 family (Sherr and Roberts 1995; Elledge et al. 1996). In quiescent cells, CKIs are present in excess of cyclin/CDK complexes, whereas mitogen stimulation leads to increased expression of D-type cyclins (the first G1 cyclins). Consequently, CDK activity appears as soon as the putative CKI inhibitory threshold is exceeded (Harper and Elledge 1996). Conversely, expression of D-type cyclins declines when mitogens are withdrawn or antiproliferative agents are added, correlating with the failure of growth factor-deprived cells to progress into S phase (Sherr 1994, 1996).
Another important level of cell cycle control is executed by the tumor supressor gene p53, which is inactivated by mutations in >50% of human cancers (Levine 1997). p53 is up-regulated and activated by diverse cellular stresses and in turn it controls two major responses: cell cycle arrest and apoptosis (Bates and Vousden 1996; Jacks and Weinberg 1996). The role of p53 in G1 arrest caused by DNA damage is well established, and is largely dependent on transcriptional activation of one of its target genes, the CKI p21 (Levine 1997). A function of p53 in normal growth control is suggested by the fact that p53-deficient cells exhibit accelerated proliferation rates, and that overexpression of p53 inhibits cell cycle progression even in the absence of stress stimuli (Harvey et al. 1993; Agarwal et al. 1995; Bates and Vousden 1996).
We analyzed to what extent and by which mechanism the selective loss of c-Jun affects cell proliferation and cell cycle progression. Primary and spontaneously immortalized 3T3 fibroblasts lacking c-Jun were derived from previously generated knock-out mice (Hilberg et al. 1993), which proliferate slowly and exhibit several molecular defects in G1-to-S-phase progression. Furthermore, c-Jun null fibroblasts express elevated levels of p53 and p21, and their cell cycle defects can be reverted fully by simultaneous deletion of p53. These genetic and biochemical data identify a critical interaction between the proto-oncogene c-jun and the tumor suppressor gene p53, linking c-Jun-dependent mitogenic signaling to cell cycle regulation.
Results
Proliferation defects of fibroblasts lacking c-Jun
To determine the function of c-Jun in cell proliferation and cell cycle progression, mouse embryo fibroblasts (MEFs) were isolated from fetuses with a targeted null mutation in the c-jun gene (Hilberg et al. 1993). The proliferation rates of c-jun−/− MEFa (passage 1) were drastically reduced and saturation densities were only 30% of those attained by wild-type cells (Fig. 1A). Cells lacking a single wild-type c-jun allele (c-jun+/−) were also affected and exhibited proliferation rates and saturation densities intermediate between wild-type and c-jun−/− cells (Fig. 1A). Following the 3T3 protocol (Todaro and Green 1963), immortalized cell lines were established from wild-type and mutant MEFs, the latter undergoing a prolonged crisis before spontaneous immortalization (see below). Five independently derived and randomly chosen 3T3 fibroblast lines lacking c-Jun (passages 25–30) exhibited markedly reduced proliferation rates and doubling times of 75 ± 3.7 hr, whereas corresponding wild-type cells double every 23 ± 1 hr (Fig. 1B). Similar to primary MEFs, the saturation densities were considerably lower for c-jun−/− 3T3 fibroblasts at all serum concentrations ranging from 0.5% to 30% (e.g., only about one-third of the density achieved by wild-type cells at 30% serum). Nevertheless, a significant, almost linear increase in saturation density was observed for cells of both genotypes when the serum concentration was increased from 5% to 30%, indicating that mutant cells also respond to serum growth factors to some extent (Fig. 1C).
Figure 1.


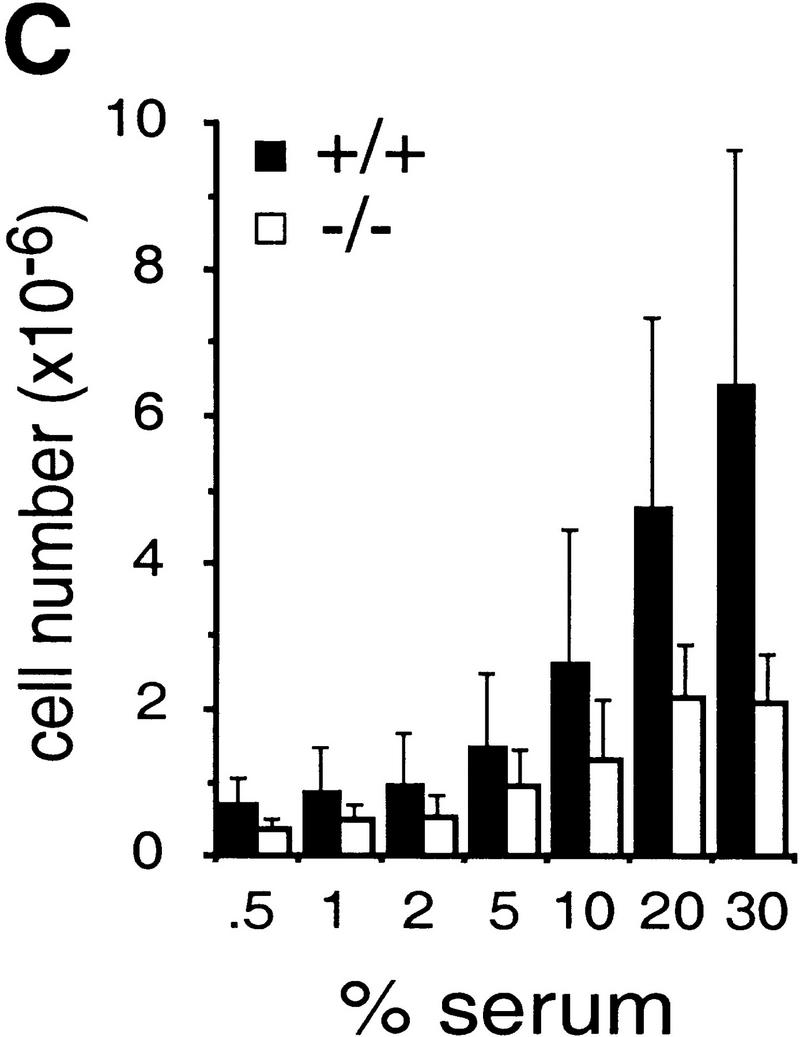



Growth properties of primary and immortalized fibroblasts lacking c-Jun. (A) Proliferation curves of wild-type (+/+), c-jun+/−, and c-jun−/− primary fibroblasts (MEFs; passage 1). The average ±s.d. numbers of cells isolated from three individual embryos of each genotype are shown, each determined in triplicate. (B) Proliferation curves of five independently established 3T3 fibroblast lines lacking c-Jun (−/−) and three corresponding wild-type 3T3 fibroblast lines (+/+). Cells were counted and passaged at 3-day intervals and cumulative cell numbers determined. (█) Standard NIH-3T3 fibroblasts obtained from ATCC. (C) Saturation densities of wild-type (+/+) and mutant (−/−) 3T3 fibroblasts as a function of serum concentration. Cells were counted after 13 days of culture in the presence of the indicated concentrations of fetal calf serum. Each point represents the average ±s.d. of two wild-type and three c-jun−/− cell lines, respectively. (D) Determination of the duration of the cell cycle of wild-type (+/+) and c-jun−/− 3T3 fibroblasts (−/−). [3H]thymidine was added to exponentially growing, asynchronous cultures, and cells were fixed and analyzed at various times after continuous thymidine incorporation (cumulative labeling). Each point represents the average ±s.d. of two independent cell lines of each genotype. (E, F) Parallel quantitative analysis of G1-to-S-phase progression (E) and cell growth (F) in wild-type (+/+) and c-jun−/− 3T3 fibroblasts (−/−) at the indicated times after serum stimulation of G0-synchronized cells (see Materials and Methods). Percentage of cells exiting G1 (i.e., the percentage of cells in S, G2, or M) was quantified by propidium iodide staining and FACS analysis. Cell volumes and cell numbers were determined in parallel with a CASY1 cell counter. The average of two independent cell lines of each genotype is shown.
When exponentially growing 3T3 fibroblasts were continuously labeled with [3H]thymidine for 1–96 hr (cumulative labeling), all cells in wild-type and c-jun−/− populations were actively cycling, as all nuclei eventually incorporated [3H]thymidine (Fig. 1D). Labeling of >90% of wild-type cells was achieved after ∼15–18 hr, whereas labeling of the same fraction of c-jun−/− cells was achieved only after ∼50–60 hr, indicating a dramatically expanded cell cycle. Furthermore, the fraction of the cell cycle occupied by S phase was much smaller in c-jun−/− cells: 13.8 ± 4.5% versus 56.6 ± 5% in wild-type cells, as judged by the fraction of cells labeled after 1 hr of incubation with [3H]thymidine (Fig. 1D). The length of S phase of c-jun−/− cells was calculated to be ∼12 hr, and the total cell cycle ∼72 hr. FACS analysis revealed that the decrease in the S phase population is accompanied by a similar decrease of G2/M cells and a concomitant increase in the G0/G1 population (data not shown). Using wild-type and c-jun−/− 3T3 lines synchronized in G0, we found that wild-type cells with a DNA content characteristic of S phase and later G2/M appeared within 21 hr of serum stimulation (Fig. 1E). This G1 exit was delayed and remarkably inefficient in mutant cells, as <40% of c-jun−/− cells, but >65% of wild-type cells were in S phase or in G2 at 24 hr after release from the G0 arrest (40% reduction in c-jun−/− cells; Fig. 1E). Parallel measurements of cell volumes showed that cell growth was not affected in c-jun−/− cells, as they increased in size at approximately the same rate as wild-type cells (Fig. 1F).
Impaired activation of G1 cyclin-dependent kinases
The inability of c-jun−/− cells to traverse G1 with normal efficiency suggested a defect in activation of essential G1 CDKs. Accordingly, activities of cyclin D1-CDK4/CDK6 and cyclin E–CDK2 complexes were measured, using immunoprecipitates from wild-type and c-jun−/− 3T3 fibroblasts that were synchronized in G0 and restimulated to enter the cell cycle. A significant reduction of cyclin D1-associated kinase activities was observed in c-jun−/− cells at all time points from 0 to 24 hr after restimulation (Fig. 2A). Likewise, cyclin E-associated kinase activity was potently induced in wild-type cells but only modestly increased in c-jun−/− cells at 18–24 hr after stimulation (Fig. 2A). On the basis of parallel FACS analysis, these time points correspond to late G1 and early S phase (Fig. 1E; data not shown). In spite of reduced CDK activities, the levels of all D-type cyclins (D1, D2, and D3) and of cyclin E were not reduced, and were even slightly elevated in both serum-starved and restimulated (Fig. 2B) as well as in exponentially growing c-jun−/− 3T3 fibroblasts (Fig. 2C). In contrast, the levels of cyclin A were slightly reduced in c-jun−/− cells, which may, however, be a secondary effect because of their inefficient cell cycle progression (Fig. 2B,C). No significant differences were observed in expression of CDK2, CDK4, and CDK6 (Fig. 2B,C). Thus, mitogen induction of cyclin D1- and cyclin E- dependent kinase activities is greatly reduced in c-jun−/− cells, although all cyclin and kinase subunits of these G1 CDKs are expressed at least at wild-type levels.
Figure 2.

Defective activation of G1 CDKs in fibroblasts lacking c-Jun. (A) Induction of cyclin D1 associated pRb kinase activity and cyclin E-associated histone H1 kinase activity in 3T3 fibroblasts. Quiescent wild-type (+/+) and c-jun−/− 3T3 fibroblasts (−/−) were stimulated to reenter the cell cycle (see Materials and Methods). At the indicated time points after serum stimulation, whole-cell extracts were prepared, anti-cyclin D1 and anti-cyclin E immunoprecipitates were isolated, and their GST–Rb and histone H1 phosphorylating activities were determined. The levels of coimmunoprecipitated CDK2 in the cyclin E/CDK2 immune complexes were determined by Western blotting. (B) Serum induction of expression of the indicated G1 cyclins and CDKs in G0 synchronized and restimulated wild-type (+/+) and c-jun−/− 3T3 fibroblasts (−/−) was determined by Western blotting. (C) Expression levels of G1 cyclins and CDKs in two independent wild-type (+/+, lanes 1,2) and two independent c-jun−/− (−/−, lanes 3,4) exponentially growing 3T3 fibroblast lines. Only the 33-kD splice variant of CDK2 is shown in B and C.
c-Jun negatively regulates p21 and p53 expression
After excluding that low CDK activities in c-jun−/− fibroblasts are due to reduced expression of their components, we determined the levels of p21, p27, and p16, three important CKIs that bind to CDKs and inhibit their activity (Sherr and Roberts 1995). Whereas expression of p27 and p16 was normal in all cell lines tested, the levels of p21 were significantly elevated in c-jun−/− 3T3 fibroblasts (Fig. 3A). p21 plays a key role in G1 checkpoint control (Harper and Elledge 1996), and its presence at high levels is likely to be causal to the low CDK activity and inefficient G1-to-S-phase progression of c-jun−/− cells. In agreement with its stimulatory function on p21 expression (Levine 1997), the tumor suppressor protein p53 also accumulates to significantly elevated levels in c-jun−/− 3T3 fibroblasts in the absence of exogenous stimuli (Fig. 3A). p53 and p21 levels are also elevated in early passage primary fibroblasts (MEFs) lacking c-Jun (Fig. 3C). However, the differences between c-jun−/− and wild-type MEFs are less pronounced than in 3T3 fibroblasts, which may be due to the fact that MEFs express higher basal levels of p53 than 3T3 fibroblasts (data not shown). High p53 expression is often associated with missense mutations, which result in loss of its transcriptional activator function and increase in its protein half-life (Midgley and Lane 1997). However, none of the mutant or wild-type 3T3 cell lines analyzed contained any mutations in the p53 open reading frame, and a p53 responsive luciferase reporter construct was transactivated efficiently in wild-type and c-jun−/− cells (data not shown). Furthermore, as in wild-type cells, the already higher basal levels of p53 in c-jun−/− cells can be further increased through protein stabilization in response to γ (Fig. 3B) or UV irradiation (Fig. 3E) and a number of other stress stimuli (not shown). Thus, the lack of c-Jun may not affect p53 expression through stabilization of the protein. To demonstrate this point more directly, we used pulse/chase experiments to determine the half-life of p53. The kinetics of p53 degradation were similar in untreated wild-type and mutant cells, with 41% and 37% of the initial amount of labeled protein detected in wild-type and c-jun−/− cells, respectively, after 2 hr of chase (Fig. 3D). UV irradiation leads to considerable stabilization of p53 in both cell types, and only 36% (wild-type cells) and 30% (c-jun−/− cells) of p53 protein were degraded after a 3-hr chase (Fig. 3D). The calculated half-lives of p53 in wild-type cells were 91 min before and 353 min after UV irradiation, and those in c-jun−/− cells were 84 min before and 382 min after UV irradiation.
Figure 3.
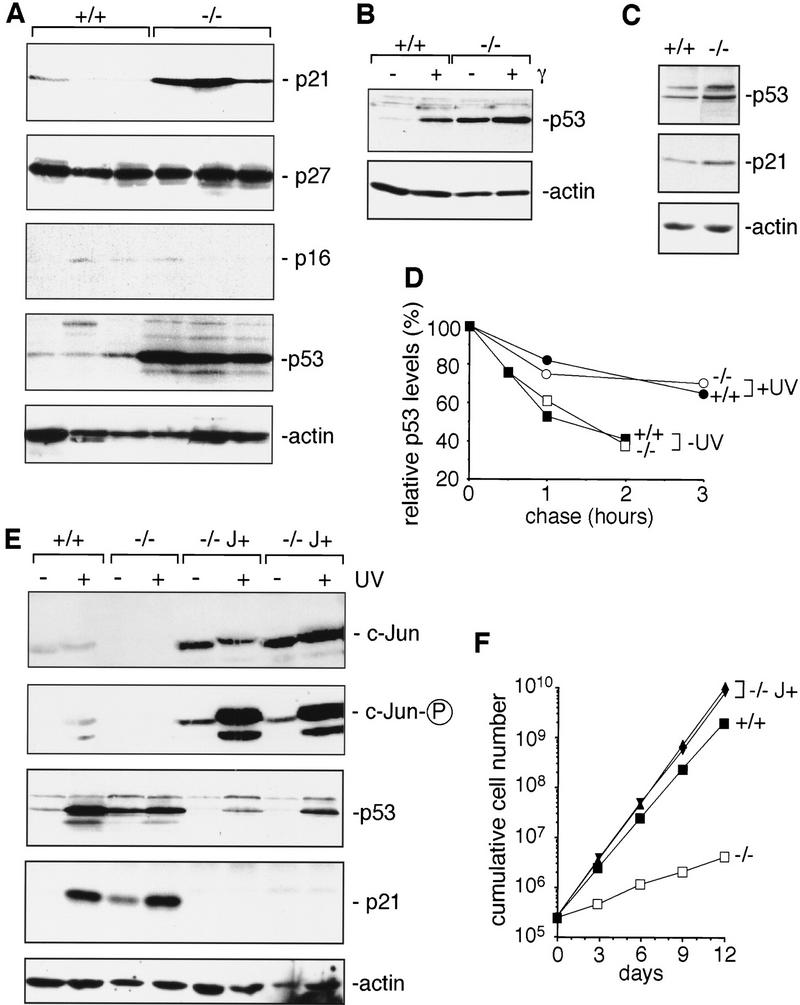
Increased expression of p21 and p53 in fibroblasts lacking c-Jun. (A) Western blot analysis of p21, p27, p16, p53, and actin (loading control) in three independent, exponentially growing wild-type (+/+) and c-jun−/− 3T3 fibroblast lines (−/−). The levels of p16 were very low in all wild-type and mutant 3T3 lines compared to primary fibroblasts (not shown). (B) Western blot analysis of p53 expression in wild-type (+/+) and c-jun−/− 3T3 fibroblasts (−/−) after γ-irradiation (20 Gy, 2 hr) or mock treatment (−). (C) Western blot analysis of p53, p21, and actin (loading control) in exponentially growing wild-type (+/+) and c-jun−/− (−/−) primary embryonic fibroblasts (MEFs, passage 1). (D) Turnover of p53 in wild-type (+/+) and c-jun−/− 3T3 fibroblasts, before and after UVC irradiation (+UV, 40 J/m2), was determined by pulse/chase experiments. The amount of labeled p53 protein at each time point was quantified with a PhosphorImager and normalized to the amount of labeled p53 detected after 0 min of chase. (E) Western blot analysis of c-Jun, phosphorylated c-Jun (c-Jun-P), p53, and p21 in wild-type (+/+), c-jun−/− (−/−), and c-Jun overexpressing 3T3 fibroblasts (−/− J+). Two independent derivatives of c-jun−/− 3T3 fibroblasts stably cotransfected with a human c-Jun expression vector and RSV–hygro (−/− J+), the parental c-jun−/− line (−/−), and one wild-type line (+/+) were irradiated (+) or not (−) with UVC (40 J/m2), and extracts were prepared 30 min later for the analysis of c-Jun, or after 24 hr for the analysis of p53 and p21. (Actin) Loading control for the p53 and p21 Western blot. (F) Reintroduction of functional c-Jun into c-jun−/− 3T3 fibroblasts rescues their proliferation defect. Proliferation rates of two independent stable lines expressing human c-Jun (−/− J+) were compared to the parental c-jun−/− and one wild-type (+/+) 3T3 fibroblast line.
To further analyze whether and how c-Jun regulates p53 expression, functional human c-Jun was introduced into c-jun−/− 3T3 fibroblasts by stable transfection. Two independent lines (−/− J+; C4 and C6) chosen randomly were analyzed. These cells express higher basal levels of c-Jun than normal wild-type fibroblasts (Fig. 3E). Both cell lines express significantly less p53 and p21 than untransfected c-jun−/− cells or even wild-type cells, before or after UV irradiation (Fig. 3E). Not only is the proliferation defect of these cells with a disrupted endogenous c-jun gene fully rescued, C4 and C6 cells proliferate even faster than wild-type cells (Fig. 3F). Thus, the proliferation defect and the elevated p53 and p21 levels of c-jun−/− fibroblasts are direct results of the specific loss of c-Jun, and any secondary mutations that may have occurred during immortalization seem to be irrelevant for these defects. Besides, it is unlikely that the same secondary mutations have occurred in all of the cell lines analyzed.
c-Jun binds to and represses the p53 promoter
Importantly, the effect of c-Jun on p53 protein levels is mirrored by the effect of c-Jun on p53 mRNA expression: Lack of c-Jun results in elevated p53 mRNA levels, whereas overexpression of c-Jun represses p53 mRNA well below the wild-type levels (Fig. 4A). Transient transfection assays were performed to test whether c-Jun represses p53 expression by way of a negative effect on the p53 promoter. The p53 promoter contains a conserved AP-1-like element that differs from a consensus AP-1 site by a single base-pair exchange (termed PF-1 site, Ginsberg et al. 1990; Fig. 4B). Luciferase expression directed by a 700-bp fragment of the murine p53 promoter containing this AP-1 site is increased about two- to threefold in c-jun−/− fibroblasts compared to wild-type cells, but reduced by more than twofold in c-Jun overexpressing cells (Fig. 4C). Mutation of the PF-1 site abolishes this repression by c-Jun, resulting in an approximately fourfold higher luciferase activity in wild-type cells and more than fivefold higher activity in c-Jun-overexpressing fibroblasts (Fig. 4C). In contrast, only a modest relief of repression was observed in c-jun−/− cells, indicating that c-Jun is the major AP-1 subunit mediating negative regulation of the p53 promoter through the PF-1 site (Fig. 4C). Parallel transfections were performed with the unrelated control reporter plasmid pGL2–Luc, which contains neither AP-1-binding sites nor sequences of the p53 promoter region. Luciferase activity driven by pGL2–Luc was neither increased in c-jun−/− cells nor reduced in Jun-overexpressing cells, demonstrating that the effects of c-Jun deletion or overexpression are specific for the p53 promoter (Fig. 4C). Cotransfection of a c-Jun expression plasmid reduced luciferase activity directed by the wild-type p53 promoter fragment by ∼70% (Fig. 4D). In contrast, cotransfection of c-Jun had only a minor effect (<10% reduction) on the reporter construct containing the mutated PF-1 element (Fig. 4D). Thus, repression of the p53 promoter by c-Jun is mediated primarily by the PF-1 element.
Figure 4.
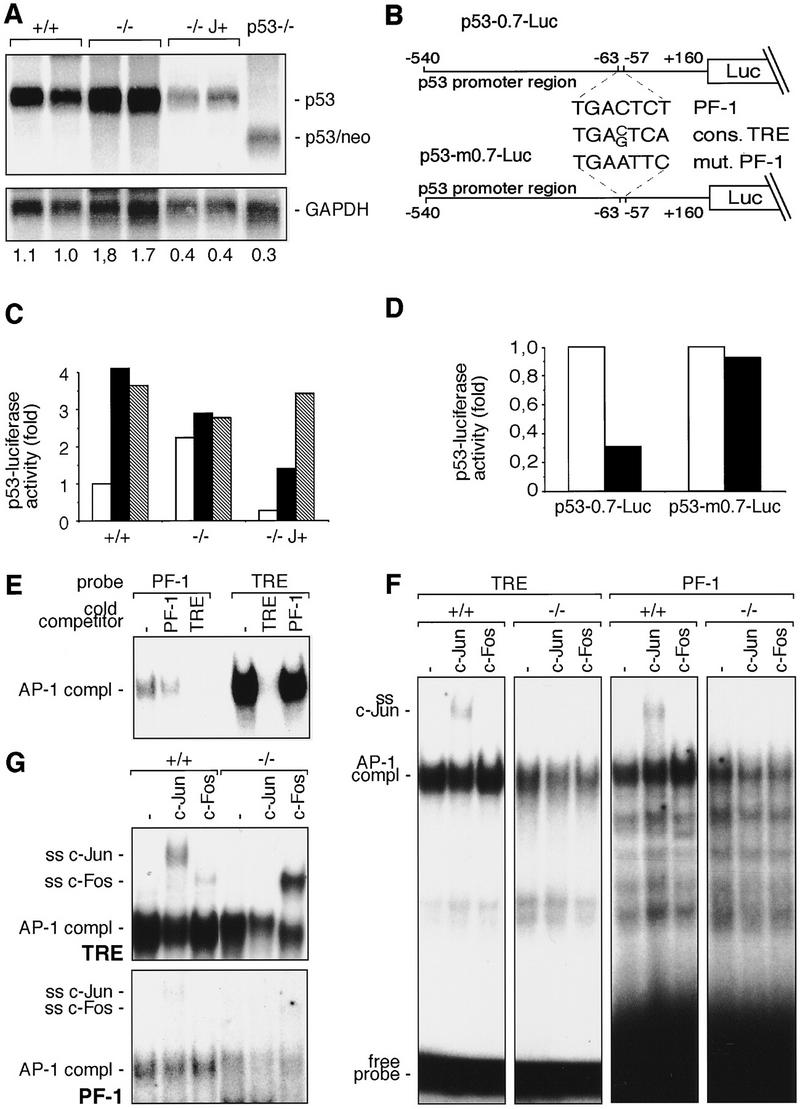
c-Jun binds to and represses the p53 promoter. (A) Northern blot analysis of p53 mRNA in two wild-type (+/+), two c-jun−/− (−/−), two c-Jun-overexpressing (−/− J+), and one p53−/− 3T3 fibroblast lines. Poly(A)+ RNA from untreated cells was separated on an agarose gel, transferred to Hybond N+ (Amersham) membrane, and probed with p53 and GAPDH cDNA probes. The intensities of bands were quantified with a PhosphorImager, and relative levels of p53 mRNA, normalized to GAPDH, are indicated at the bottom of each lane. (B) Schematic representation of the p53 promoter–luciferase reporter constructs used. p53-0.7–Luc contains a 700-bp fragment of the mouse p53 promoter region, which contains the PF-1 element (Ginsberg et al. 1990). p53-m0.7–Luc contains a mutated version of PF-1 (mut. PF-1). The consensus AP-1-binding site (cons. TRE) is shown for comparison. (C) p53-0.7–Luc (open bars), p53-m0.7–Luc (solid bars), or pGL2–Luc (shaded bars) (an unrelated control luciferase construct) was transfected into wild-type (+/+), c-jun null (−/−), and c-Jun-overexpressing (−/− J+) 3T3 fibroblasts, and luciferase activity was measured. (D) p53-0.7–Luc or p53-m0.7–Luc was cotransfected into wild-type 3T3 fibroblasts together with a c-Jun expression vector (solid bars) or empty expression vector (open bars), and luciferase activity was measured. Relative luciferase activities from one representative experiment performed in quadruplicate are shown in C and D. (E–G) Protein–DNA complexes formed with the consensus AP-1-binding site (TRE of the collagenase promoter) and the PF-1 element of the p53 promoter. Nuclear extracts were prepared from exponentially growing (E,F) or TPA treated (2 hr; 100 ng/ml; G) wild-type (+/+) and c-jun−/− (−/−) fibroblasts. Nuclear extracts were incubated with the labeled probes indicated on the top and complexes were resolved by PAGE. The positions of the DNA-bound AP-1 complexes are indicated on the left. For cross-competition analysis (E) an 80-fold molar excess of non-labeled competitor DNA (indicated at top) was used. For supershift analysis (F,G), extracts were preincubated with specific antiserum (indicated at top) for c-Jun (ss c-Jun) and c-Fos (ss c-Fos), and the positions of the resulting supershifts are indicated at left. Note that in F and G the time of exposure of the PF-1 analysis was sevenfold longer compared to the result obtained with the TRE.
Because the PF-1 site is highly homologous to the consensus AP-1-binding site, c-Jun may achieve this inhibitory effect by directly binding to this site. Accordingly, electrophoretic mobility shift assays were performed with an oligonucleotide containing the PF-1 site plus adjacent sequences from the mouse p53 promoter. The protein complex binding to this oligonucleotide exhibited an electrophoretic mobility indistinguishable from that of the protein–DNA complex formed on a high affinity consensus AP-1 site [i.e., TPA responsive element (TRE) of the collagenase gene; Schorpp et al. 1995; Fig. 4E]. This complex was specific, as it was efficiently competed by an excess of unlabeled wild-type PF-1 oligonucleotide, but not by a mutant version of PF-1 (Fig. 4E; data not shown). The relative binding affinity of the PF-1 site was significantly weaker than that of the TRE as evident from the lower intensity of the retarded bands obtained with the PF-1 probe (Fig. 4E). Moreover, cross-competition experiments showed that an excess of unlabeled PF-1 is a rather inefficient competitor for protein binding to the consensus AP-1 site, whereas addition of unlabeled consensus TRE sequence efficiently competes for binding to labeled PF-1 under conditions where competition by the homologous unlabeled PF-1 was not complete (Fig. 4E). These findings strongly suggest that the protein that binds to the PF-1 oligonucleotide is a bona fide AP-1 complex. Like the complex formed with the TRE, the complex binding to the PF-1 site contains c-Jun, as demonstrated by supershift analysis using a c-Jun specific antibody (Fig. 4F,G). As expected, no c-Jun-containing supershifted complexes were detected in c-jun−/− cells (Fig. 4F,G). Furthermore, the bands obtained in c-jun−/− cells were somewhat weaker than those in wild-type cells, confirming that c-Jun is an important component of the AP-1 complexes binding to both the PF-1 site and the TRE (Fig. 4F,G). No supershifted bands were obtained with a c-Fos specific antibody in exponentially growing, unstimulated cells with either the TRE or the PF-1 site (Fig. 4F). However, when TPA stimulation was used to induce expression of Fos proteins, substantial amounts of c-Fos were detected in the TRE- binding complexes, particularly in the mutant cells (Fig. 4G). In contrast, the complexes binding to the PF-1 site did not contain appreciable amounts of c-Fos even in TPA-stimulated cells, suggesting that only a subset of possible AP-1 dimers binds to this site (Fig. 4G). Similar amounts of Fra-2 bound to both the TRE and the PF-1 site (data not shown). Thus, the PF-1 site of the p53 promoter represents a variant AP-1-binding site to which c-Jun and some, but not all, other AP-1 subunits bind and that mediates overexpression of p53 in c-jun−/− cells and repression of p53 promoter activity in c-Jun overexpressing cells.
p53-dependent functions of c-Jun
Accumulation of p53 appears to be the most upstream event responsible for altered cell cycle regulation in c-jun−/− fibroblasts. Accordingly, fetuses derived from c-jun+/−p53+/− double heterozygous intercrosses were used to isolate c-jun−/−p53−/− double mutant fibroblasts along with wild-type, c-jun−/−p53+/+, and c-jun+/+p53−/− controls. To examine the role of c-Jun in the spontaneous immortalization process, two to five independent primary cultures of each of these four genotypes were subjected to the 3T3 protocol (Todaro and Green 1963). Wild-type fibroblasts were readily immortalized after a crisis period of ∼20–30 days (Fig. 5A). As described previously, p53−/− cells maintained a high proliferation rate for >30 passages tested and never entered a senescent phase (Harvey et al. 1993). In contrast, the crisis period was extremely prolonged in all five c-jun−/−p53+/+ cell cultures tested and lasted ∼6 months (Fig. 5A). Furthermore, these cells entered crisis prematurely, but the efficiency of immortalization was not reduced in the absence of c-Jun, and all primary cultures subjected to the 3T3 protocol eventually gave rise to established cell lines. Importantly, two independent c-jun−/−p53−/− cell populations behaved just like c-jun+/+p53−/− cells, but strikingly different from c-jun−/−p53+/+ cells, as they were rapidly immortalized without a prior crisis (Fig. 5A).
Figure 5.

Loss of p53 suppresses the cell cycle and immortalization defects of fibroblasts lacking c-Jun. (A) Immortalization of 3T3 fibroblast lines lacking functional c-Jun, p53, or both proteins. Primary fibroblasts isolated from embryonic day 11.5 (E11.5) mouse fetuses with the indicated genotypes were immortalized according to the standard 3T3 protocol (Todaro and Green 1963). An ‘immortalization curve’ of cumulative cell numbers for one representative example of each genotype is shown (day 0 in graph = passage 3). Wild-type (█) and c-jun−/− (□) primary fibroblasts were immortalized after ∼40 and 200 days in culture, respectively, whereas p53−/− (●) and c-jun−/−p53−/− (○) cells became immortalized without undergoing any detectable crisis. (B) Proliferation curves of primary fibroblasts (passage 1) lacking c-Jun, p53, or both proteins. Average ±s.d. numbers of cells isolated from two individual embryos of each genotype, determined in triplicates, is shown. (C) Proliferation curves of one wild-type, one c-jun−/−, two p53−/−, and two c-jun−/−p53−/− 3T3 fibroblast lines, each separately immortalized from primary cells isolated from individual E11.5 embryos. (D) Fraction of S-phase cells in asynchronous populations of 3T3 fibroblasts of the indicated genotypes, determined by 1-hr pulse-labeling with [3H]thymidine of exponentially growing cultures. Averages ±s.d. for two independent cell lines per genotype are shown. (E) Induction of cyclin D1-associated Rb kinase activity and cyclin E-associated histone H1 kinase activity in c-jun−/−p53−/− 3T3 fibroblasts. A representative experiment with immune complexes prepared at the indicated times after serum stimulation of G0 synchronized cells is shown (see Materials and Methods). (F) Western blot analysis of p21 in one c-jun−/−, one wild-type, two c-jun−/−p53−/−, and two p53−/− 3T3 fibroblast lines. The blot was reprobed for actin as a loading control.
Like the immortalization defects, the proliferation defects of primary and immortalized fibroblasts lacking c-jun are p53-dependent. c-jun+/+p53−/− primary fibroblasts proliferated about twice as fast and achieved higher saturation densities than corresponding wild-type cells (Fig. 5B). MEFs lacking both p53 and c-jun proliferated as fast as p53 single mutant cells, but about four to six times faster than c-jun single mutant cells (Fig. 5B). A very similar result was obtained with the 3T3 derivatives (Fig. 5C). Consistent with a longer doubling time, the cell volume of c-jun−/−p53+/+ primary fibroblasts was ∼20% larger than wild-type, whereas c-jun+/+p53−/− and c-jun−/−p53−/− cells were slightly smaller than wild-type cells (data not shown). Interestingly, smaller cell size has been observed in several genetically altered cells that, in contrast to c-jun−/− fibroblasts, exhibit an accelerated progression through G1, such as pRb−/− MEFs and cyclin D1 or cyclin E-overexpressing cells (Herrera et al. 1996 and references therein).
The fraction of [3H]thymidine-incorporating cells in exponentially growing c-jun−/− 3T3 fibroblast populations is reduced dramatically (Fig. 5D; see also Fig. 1D). In contrast, c-jun−/−p53−/− double mutant cell lines, like p53 single mutant cells, have at least as many S-phase cells as wild-type controls (Fig. 5D). Cyclin D1–CDK4/CDK6 and cyclin E–CDK2 kinase activities were mitogen-induced in c-jun−/−p53−/− 3T3 fibroblasts as efficiently as in wild-type cells, in sharp contrast to c-jun−/−p53+/+ cells (Fig. 5E; cf. Fig. 2A). The dominant reversion of the c-jun−/− phenotype by simultaneous deletion of p53 is also reflected at the level of p21 expression. In four different 3T3 lines lacking p53 (two c-jun+/+p53−/− and two c-jun−/−p53−/−), p21 was below detection, whereas in wild-type cells p21 was readily detectable, but lower than in c-jun−/−p53+/+ cells (Fig. 5F). These data demonstrate that the elevated levels of p21 in c-jun−/− fibroblasts are largely attributable to accumulation of p53. In summary, all proliferation-related defects observed in c-jun−/− cells were completely ameliorated by simultaneous deletion of p53.
Next, c-Jun- and p53-dependent activation of E2F was analyzed by electrophoretic mobility-shift assays using a consensus E2F oligonucleotide. G0-synchronized cells were stimulated with serum and analyzed as they progressed through G1 (Fig. 6A). In wild-type cells, the abundance of unbound E2F increased gradually early in G1 (0–11 hr), but rather abruptly late in G1 (14–20 hr), whereas the amount of E2F complexed to pRb family proteins remained relatively constant, consistent with earlier reports (e.g., Beijersbergen et al. 1995). In c-jun−/−p53+/+ cells, there was only a rather modest increase in DNA binding of uncomplexed E2F over time after serum stimulation, resulting in severalfold less free E2F in late G1 than in wild-type cells (Fig. 6A). Furthermore, expression of the E2F target gene proliferating cell nuclear antigen (PCNA; Lee et al. 1995) was significantly reduced in c-jun−/− cells, confirming the reduced transcriptional activity of E2F (Fig. 6B). Both the defects in E2F activation and PCNA expression were neutralized in c-jun−/−p53−/− double mutant cells (Fig. 6A; data not shown).
Figure 6.

Deletion of c-jun leads to poor activation of E2F, which can be rescued by simultaneous deletion of p53. (A) DNA-bound E2F complexes in synchronized wild-type (WT), c-jun−/−, and c-jun−/−p53−/− 3T3 fibroblasts were analyzed by electrophoretic mobility-shift assays (EMSA) using an oligonucleotide containing the consensus E2F-binding site. Whole-cell extracts were prepared at the indicated times after restimulation of quiescent cells. All complexes detected are specific as they were competed by an excess of unlabeled wild-type competitor but not by an oligonucleotide in which the E2F site was mutated (not shown). The amounts of extract used per lane were normalized by measuring protein concentrations and by quantitation of retarded bands formed with an SP-1 probe. The positions of free (active) E2F and of E2F complexed to pRb family proteins (compl. E2F) are indicated. (B) Western blot analysis of the E2F target gene PCNA in G0 synchronized wild-type and c-jun−/− 3T3 fibroblasts after serum stimulation for the indicated times.
Discussion
Previous analyses have suggested an important role of transcription factor AP-1 in proliferation and cell cycle progression. Here we examined to what extent c-Jun affects cell proliferation and immortalization of fibroblasts, and have analyzed the molecular mechanism by which c-Jun positively regulates cell cycle progression. Fibroblasts with a targeted null mutation in c-jun proliferate more slowly and exhibit pronounced defects in activation of G1 CDKs and E2F as well as in G1-to-S-phase progression. Furthermore, the tumor suppressor gene p53 and its target gene, the CDK inhibitor p21, are expressed at elevated levels, which is likely to be responsible for the low CDK activity and inefficient G1-to-S-phase progression in c-jun−/− cells, as p21 is a universal inhibitor of cyclin/CDK complexes and may also block DNA replication directly by inhibiting PCNA (Li et al. 1994; Sherr and Roberts 1995). Indeed, the absence of c-Jun has no negative effect on CDK activation, cell cycle progression, and proliferation of fibroblasts lacking also p53, in which p21 is expressed at low levels. This dual requirement for mitogenic activation of CDK4/CDK6 (accumulation of cyclin D1 and repression of p21) may be a safety device in the tight regulation of the mammalian cell cycle clock, ensuring that unscheduled activation of cyclin D1 will not trigger cell division (Fig. 7). Rather, a confirmatory signal mediated by c-Jun is needed. This signal promotes activation of CDK4/CDK6 and CDK2 by attenuating the accumulation of p53 and p21, both of which are inducible by mitogens (Macleod et al. 1995).
Figure 7.

Model for the dual functions of c-Jun in connecting mitogenic signaling to the cell cycle clock. Stimulation of cells by growth factors leads to increased expression of c-Jun, cyclin D1, p53, and p21. Cyclin D1 positively and p21 negatively influences activation of CDK4/CDK6, the first cyclin-dependent kinase to be activated as cells progress through G1. Once CDK4/CDK6 is activated and phosphorylates pRb and its related proteins, cells are committed to complete progression through the entire cell cycle in the absence of further mitogen stimulation (Harper and Elledge 1996; Sherr 1996). c-Jun contributes to mitogen-dependent activation of cyclin D1–CDK4/CDK6 in two ways. (1) It transduces mitogenic signals to induce cyclin D1 expression, although cyclin D1 can still be induced by mitogens in the absence of c-Jun (indicated by dashed arrow); (2) it attenuates accumulation of p21 through repression of p53 expression. In cells lacking p53, low levels of p21 allow efficient activation of cyclin D1–CDK4/CDK6, cyclin E–CDK2, E2F, and G1-to-S-phase progression whether or not c-Jun is present.
Importantly, accumulation of p53 in c-jun−/− fibroblasts appears to be attributable to a specific, physiological function of c-Jun in p53 regulation, and not to unspecific, constitutive activation of cellular stress response pathways. c-Jun represses p53 expression by negative regulation of its promoter activity, whereas protein stabilization does not contribute appreciably to accumulation of p53 in c-jun−/− cells. Interestingly, accumulation of p53 in response to serum growth factors is also achieved primarily at the mRNA level, but the basis for this induction response remains to be determined (Ginsberg et al. 1990). In contrast, it has been well established that accumulation of p53 in response to various stressful conditions, such as DNA damage, is achieved through a post-transcriptional mechanism, primarily metabolic stabilization of the p53 protein, whereas p53 mRNA levels are not altered (Maltzman and Czyzyk 1984; Levine 1997). Thus, the mechanism responsible for p53 accumulation in response to deletion of c-Jun is fundamentally different from the mechanism leading to p53 induction in response to stress signals. Furthermore, a stress–response model would fail to explain how overexpression of c-Jun represses p53 expression well below wild-type levels. We also found no evidence that stress–response pathways are constitutively active in c-jun−/− cells. The basal activities of the stress-activated protein kinases JNK1, JNK2, and p38, basal levels of DNA damage repair, and apoptosis rates are as low as in wild-type cells (data not shown). Finally, overexpression of c-Jun does not inhibit accumulation of p53 in response to DNA damage, and the higher basal levels of p53 in c-jun−/− cells can be further increased by stress stimuli, indicating that these two mechanisms of p53 regulation operate independently of each other.
Conversely to the deletion of c-jun, its overexpression considerably reduces p53 promoter activity, mRNA, and protein levels. Intriguingly, overexpression of c-Jun also recapitulates most of the phenotypic effects of p53 loss. Like fibroblasts lacking p53, fibroblasts overexpressing c-Jun express very low levels of p21, proliferate faster than wild-type cells, and fail to undergo G1 arrest upon UV irradiation (data not shown). In fact, the doubling times of two p53−/− cell lines, 21.2 and 18.7 hr, are remarkably similar to the 19.0- and 19.4-hr doubling times of the two c-Jun-overexpressing cell lines that were analyzed. Therefore, we conclude that a causal, previously unsuspected, physiological role of c-Jun is to attenuate the expression of the negative growth regulators p53 and p21. In fact, this functional interaction with p53 appears to be a rate-limiting function of c-Jun in fibroblast proliferation and cell cycle progression, as additional deletion of p53 abrogates the consequences of c-jun deletion. Similarly, the tumor suppressor protein pRb may be a rate-limiting substrate of cyclin D-CDK4/CDK6, and lack of cyclin D, or overexpression of the CDK4/CDK6 inhibitor p16 is without effect in cells lacking Rb (Weinberg 1995). Consistent with our findings, inactivation of p53 and pRb by SV40 large T also reverts the slower proliferation and facilitates efficient immortalization of c-jun−/− fibroblasts (Johnson et al. 1996). In analogy to the effect on p53 expression, c-Jun may further stimulate cell proliferation by antagonizing other transcription factors that slow down cell division, as has been shown for several nuclear hormone receptors and myogenic transcription factors (Angel and Karin 1991; Bengal et al. 1992; Li et al. 1992).
Because little is known about the regulation of p53 promoter activity, it is particularly interesting that c-Jun represses p53 expression at the transcriptional level. The p53 promoter contains a conserved element that differs from the consensus AP-1-binding site (TRE, Angel et al. 1987) by a single base-pair exchange and is involved in its regulation by serum growth factors (Ginsberg et al. 1990). Importantly, this site (termed PF-1 site) confers transcriptional repression onto a heterologous promoter, and may also down-regulate the endogenous p53 promoter (Ginsberg et al. 1990). Accordingly, we analyzed whether repression of p53 transcription by c-Jun is mediated by this element. Using electrophoretic mobility-shift assays we found that c-Jun indeed binds to the PF-1 site directly. Transient transfections of reporter constructs demonstrated that the PF-1 site is a negative regulatory element, as its mutation resulted in a severalfold increased activity of the p53 promoter. Furthermore, this site mediates the inhibitory effect of c-Jun on the p53 promoter, as cotransfection of c-Jun efficiently repressed the wild-type p53 promoter, but not a mutant in which the PF-1 site had been destroyed. The latter finding was confirmed in stable cell lines that overexpress c-Jun or in which the c-jun gene was deleted. Thus, c-Jun directly binds to and acts as a transcriptional repressor of the p53 promoter. Although c-Jun contains a transactivation domain (Angel and Karin 1991), a number of other examples where c-Jun inhibits expression of target genes by direct promoter binding were described. In several cases, such an inhibitory effect is mediated by variant c-Jun binding elements, such as two serum response elements (SREs) in the human α-actin promoter, or a c-Jun/ATF-2-binding site in the human urokinase enhancer (Bushel et al. 1995; De Cesare et al. 1995). On the other hand, a consensus AP-1 site plays a key role in the repression of MHC class I genes in neuronal cells (Murphy et al. 1996). The PF-1 site could also be considered as a variant AP-1-binding site, as it differs at one residue from the consensus TRE, exhibits weaker binding affinity than the TRE, and appears to bind selectively only a subset of Jun and Fos proteins. Most notably, the strong transactivator c-Fos was not detected to bind to the PF-1 site, whereas the PF-1 site binds Fra-2 efficiently, which lacks a transactivation domain and was shown to repress AP-1 activity (Suzuki et al. 1991; Yoshioka et al. 1995). These differences in binding specificity between the TRE and the PF-1 element might contribute to the negative regulatory properties of the latter sequence.
Activation of c-Jun and the Ras pathway in general were reported to induce cyclin D1 expression (Herber et al. 1994). Thus, elevated cyclin D1 expression in c-jun−/− cells is unexpected, and demonstrates that c-Jun may be sufficient, but is not required for mitogen induction of cyclin D1 expression. The fact that both cyclin D1 and cyclin E, which are regulated quite differently, are high in mutant cells, indicates that an indirect mechanism secondary to the partial cell cycle block causes this overexpression. Presumably cells respond to the higher inhibitory threshold imposed by overexpression of p21 with compensatory overexpression of these cyclins so that the corresponding CDKs can be activated at least to some extent. This cyclin accumulation is expected to require more time, which may explain the delay in progression into S phase.
How general is the p53-dependent role of c-Jun in proliferation? Unlike primary and immortalized fibroblasts, embryonic stem (ES) cells proliferate rapidly despite expression of high endogenous levels of p53 (Sabapathy et al. 1997). Thus, if c-Jun’s sole rate-limiting function is repression of p53 transcription, deletion of c-jun is expected to have no effect on ES cell proliferation. Indeed, this was found to be the case (Hilberg and Wagner 1992). In contrast, primary liver cell cultures from c-jun−/− embryos exhibit pronounced growth defects and reduced thymidine incorporation in vitro. In vivo, the contribution of c-jun−/− cells is progressively reduced in the livers of wild-type ↔ c-jun−/− chimeras during a postnatal stage that coincides with increased liver cell proliferation (K. Zatloukal, R. Eferl, and E.F. Wagner, unpubl.). However, unlike the in vitro proliferation defect, the embryonic lethality attributable to lack of c-Jun (Hilberg et al. 1993; Johnson et al. 1993) cannot be rescued by simultaneous deletion of p53, as no viable double mutant mice could be obtained (data not shown). In this context, it is important to note that c-Jun also plays a key role in biological processes other than proliferation. Most importantly, it promotes differentiation of many different lineages such as myeloid, erythroid, epithelial, and neuronal cells (Lord et al. 1993; Szabo et al. 1994; Patel and Sytkowski 1995). In some systems, such as Drosophila photoreceptor development, this function of c-Jun is exerted in postmitotic cells, demonstrating that it is independent of the function of c-Jun in proliferation (Bohmann et al. 1994). Although the cause of embryonic lethality of c-jun−/− mice has not been fully resolved, it is most likely not attributable to a defect in cell proliferation. Instead, defects in the differentiation of stromal and/or erythroid cells of the fetal liver and morphogenetic defects in the development of the heart outflow tract are strongly implicated in this lethality (Hilberg et al. 1993; K. Zatloukal, R. Eferl, and E.F. Wagner, unpubl.). We conclude that although lack of c-Jun negatively affects cell proliferation in vivo, at least in liver cells, this proliferation defect is not the cause of embryonic lethality. Rather, the embryonic lethality is most likely attributable to defects in differentiation and development, which presumably are p53 independent functions of c-Jun.
c-Jun is one of the key end points activated by mitogenic signaling cascades, most importantly the Ras/MAP kinase pathway (Cano and Mahadevan 1995; Karin 1995; Karin and Hunter 1995). Although the molecular mechanisms and growth-promoting functions of MAP kinase cascades were documented, it is not clear how they connect to the cell cycle clock (Downward 1997). Furthermore, the ‘classic’ target for the Ras/MAP kinase pathway, the c-fos gene, does not appear to be instrumental for stimulation of cell proliferation (Brüsselbach et al. 1995). Even a double deletion of c-fos and fosB has no effect on fibroblast proliferation (Gruda et al. 1996; R. Bravo, pers. comm.). The expression of cyclin D1 is determined by the extracellular mitogen environment, which reveals one possible connection between mitogenic signaling and cell cycle progression. However, the regulation of cyclin D1 expression is complex and not well understood (Grana and Reddy 1995; Sherr 1996). In contrast, the signaling pathways responsible for activation of c-jun transcription and potentiation of the activity of c-Jun are among the best examples of a clearly delineated pathway connecting growth factor receptors to the transcriptional machinery (Cano and Mahadevan 1995; Karin 1995; Karin and Hunter 1995). The present work thus identifies the connection between c-Jun and p53/p21 (Fig. 7) as a missing link between a cascade of events initiated by growth factor receptors at the cell surface and the nuclear cell cycle machinery for which most of the individual steps are reasonably understood at the molecular level.
The negative regulation of the tumor suppressor gene p53 could also contribute to the ability of c-Jun to promote oncogenic transformation, the mechanism of which is still unclear. If that is true, p53 mutations should not be required in tumors caused by deregulation of AP-1. Indeed, in the mouse skin carcinogenesis model, spontaneous malignant conversion of papillomas caused by epidermal expression of v-Fos does not involve p53 mutations, although such mutations are frequent events in malignant conversion of chemically induced papillomas (Greenhalgh et al. 1995). Furthermore, overexpression of c-Jun and mutation of p53 cooperate with activated Ras in oncogenic transformation, suggesting that these two events are functionally interchangeable (Hicks et al. 1991; Smeal et al. 1991). Similarly, osteosarcoma formation in transgenic mice overexpressing c-Fos is accelerated both by overexpression of c-Jun and deletion of p53 (Wang et al. 1995; W. Jochum and E.F. Wagner, unpubl.). However, whereas the presence of functional c-Jun is essential for transformation by Ras, mutation of p53 is not, suggesting that additional p53-independent functions of c-Jun are involved in this process (Johnson et al. 1996; M. Schreiber and E.F. Wagner, unpubl.). Nevertheless, it will be interesting to determine whether repression of p53 transcription contributes to the activating role of c-Jun not only in proliferation, but also in oncogenic transformation. Intriguingly, like cells lacking p53, the cell lines overexpressing c-Jun do not arrest efficiently in G1 after UV irradiation (M. Karin, unpubl.), and this defect in DNA damage-induced checkpoint arrest is thought to play a key role in the cancerogenic effect of p53 loss.
Materials and methods
Cell lines and culture conditions
Primary MEFs were isolated and immortalized as described (Todaro and Green 1963). Each primary fibroblast culture was isolated from a single E11.5–E12.5 mouse embryo of 129/Sv genetic background, and each 3T3 fibroblast line was immortalized from an individual primary culture. Wild-type, c-jun−/−, p53−/−, and c-jun−/−p53−/− embryos were derived from c-jun+/−p53+/− double heterozygous intercrosses, and additional wild-type, c-jun+/−, and c-jun−/− embryos were derived from heterozygous intercrosses of c-jun+/− mice. Mice carrying a mutated allele of c-jun or p53 used to initiate these intercrosses were generated by Hilberg et al. (1993) and Jacks et al. (1994), respectively. Genotyping of mutant embryos was performed by PCR, and the presence or absence of c-Jun or p53 protein was confirmed by Western blot analysis. Human c-Jun was introduced into c-jun−/− 3T3 fibroblasts by cotransfection with SRa HA–c-Jun (Deng and Karin 1993) and an RSV-hygro plasmid encoding for hygromycin B resistance. Several stable clones expressing higher levels of c-Jun than untransfected wild-type cells were identified by Western blotting, and two of them, designated −/−J+ C4 and −/−J+ C6, were randomly chosen for further analysis. Fibroblasts were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum (FCS). Cell numbers and cell size were determined with a CASY1 cell counter (Schärfe System, Reutlingen, Germany). To obtain synchronized cells for various experiments, 3T3 fibroblasts were arrested in G0 by contact inhibition followed by culturing in medium containing 0.5% FCS for 48 hr and were released into the cell cycle by replating at standardized cell density in medium containing 10% FCS. Cell cycle progression was monitored by propidium iodide staining and fluorescence-activated cell sorter (FACS) analysis using a Becton Dickinson FACScan system. [3H]thymidine labeling was performed as described (Riabowol et al. 1992) and at least 500 single cells per time point were scored.
Protein analysis
Whole cell extracts were prepared as described (Matsushime et al. 1994) and 100 μg of protein extract were used for immunoprecipitation of cyclin D1 (DCS11 antibody, gift from J. Bartek, Danish Cancer Society, Copenhagen, Denmark) or cyclin E (M20, Santa Cruz). Immune complexes were incubated with Gst-Rb (C-pocket, 728-928) or histone H1 (Boehringer Mannheim) as described (Matsushime et al. 1994). For Western blot analysis, 30 μg of protein samples were separated by SDS-PAGE, blotted onto nitrocellulose and immunodetection was performed with an enhanced chemoluminescence system (Amersham). The following primary antibodies were used (1:1000 unless otherwise indicated): CM1 (1:2000) and CM5 (Medac) for p53; M20 (Santa Cruz, sc-481) for cyclin E; HD11 (Santa Cruz, sc-246) for cyclin D1; M20 (Santa Cruz, sc-593) for cyclin D2; C16 (Santa Cruz, sc-182) for cyclin D3; M2 (Santa Cruz, sc-163) for CDK2; C22 (Santa Cruz, sc-260) for CDK4; C21 (Santa Cruz, sc-177) for CDK6; KM-1 (Santa Cruz, sc-822) for amino-terminally phosphorylated c-Jun; C19 (Santa Cruz, sc-528) for p27; C19 (Santa Cruz, sc-397) for p21; M-156 (Santa Cruz, sc-1207) for p16; PC10 (Santa Cruz, sc-056) for PCNA; A-2066 (Sigma) for actin (1:100). The antibody for cyclin A was a generous gift from M. Pagano (New York University Medical Center, New York, NY), and the antibody for c-Jun was described previously (Deng and Karin 1993). To determine the half-life of p53 protein, pulse/chase experiments were performed. Cells were UV irradiated (40 J/m2) or mock treated 2 hr before labeling, pulse-labeled with [35S]methionine (250 μCi/ml medium) for 45 min, washed twice with PBS and once with medium, and chased by culturing in medium containing an excess of cold methionine for the times indicated in Figure 3D. Whole cell extracts were prepared and 500 μg of protein extract were used for immunoprecipitation of p53 with Ab-1 and Ab-4 (Oncogene Science, OP03 and OP32). Immunopreciptitates were separated by SDS-PAGE, and intensities of p53 bands quantified by PhosphorImager analysis. An exponential function of amount of labeled p53 protein over time was derived and protein half-life values calculated.
Transient transfections
3T3 fibroblasts were transfected using Superfect (Qiagen) according to the manufacturer’s recommendations. Luciferase reporter construct (0.1 μg), CMV–β-gal (0.1 μg), and 1.5 μl of Superfect per 0.26 cm2 well were used. In cotransfection experiments, 0.1 μg of SRa c-Jun (Jun expression vector, Deng and Karin 1993) or 0.1 μg of SRa (empty control plasmid) was added to the above transfection mixture. p53-0.7–Luc was a gift from Moshe Oren and contains a 0.7-kb EcoRI–HindIII fragment (−540/+160) of part of the promoter and the first noncoding exon of the mouse p53 gene (Ginsberg et al. 1990) linked to the firefly luciferase gene. p53-m0.7–Luc was generated from p53-0.7–Luc using the QuickChange site-directed mutagenesis kit (Stratagene) according to the manufacturer’s recommendations. The putative AP-1-binding site at position −63/−57 was changed from TGACTCT to TGAATTC, thereby generating an EcoR1 site allowing an easy screening of mutant clones. Successful mutagenesis was confirmed by sequence analysis; thus, p53-m0.7–Luc differs from p53-0.7–Luc only at positions −60, −58, and −57 of the p53 promoter (see Fig. 4B). pGL2–Luc was obtained from Stratagene. Luciferase and β-galactosidase activities were measured using standard chemiluminescence procedures. Each transfection was performed in quadruplicate, and transfection efficiency was normalized by determination of β-galactosidase activity of the cotransfected cytomegalovirus (CMV)-β-gal expression plasmid.
Electrophoretic mobility-shift assays
Nuclear extracts from wild-type and c-jun −/− fibroblasts were prepared as described (Schreiber et al. 1995). Gel retardation assays (EMSAs) were performed as described previously (Schreiber et al. 1995). Briefly, 2 μg of nuclear extract was incubated in 4% glycerol, 12 mm HEPES-KOH (pH 7.9), 5 mm MgCl2, 4 mm Tris-HCl (pH 7.9), 0.6 mm EDTA, 10 mm dithiothreitol, 1 μg poly[d(I-C)], 2 μg BSA, and 20 fmoles of radioactively labeled probe in a final volume of 20 μl for 30 min at room temperature, followed by separation of the DNA–protein complexes on 4% polyacrylamide gel in 0.25× TBE (1× TBE is 89 mm Tris-base, 2.5 mm EDTA, 89 mm boric acid). For supershift assays, 1 μl of antiserum was added to the nuclear extracts and incubated for 1 hr on ice before addition of labeled probe. The following double-stranded oligonucleotides were used as probes: 5′-AGCTAAAGTGGTGACTCATCACTAT-3′ and 5′-AGCTATAGTGATGAGTCACCACTTT-3′ (TRE of the murine collagenase-3 gene; Schorpp et al. 1995); 5′-TCGACAATCCTGACTCTGCAAG-3′ and 5′-TCGACTTGCAGAGTCAGGATTG-3′ (PF-1 site of the murine p53 promoter; Ginsberg et al. 1990); 5′-TCGACAATCCTGAATTCGCAAG-3′ and 5′-TCGACTTGCGAATTCAGGATTG-3′ (mutant version of PF-1; Fig.4 B). c-Jun-specific antiserum was provided by Curt Pfarr and Moshe Yaniv (both at Institut Pasteur, Paris, France), c-Fos-specific antiserum was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). For the analyses of E2F, whole cell extracts were prepared and E2F binding activity was analyzed using 0.5 ng of [32P]-labeled oligonucleotide containing the consensus E2F-binding site (Santa Cruz) as described by Beijersbergen et al. (1995).
Acknowledgments
We thank M. Oren for the p53-0.7–Luc reporter construct, M. Pagano, J. Bartek, C. Pfarr, and M. Yaniv for antibodies, and E. Harlow for the GST–Rb bacterial expression vector. We are grateful to G. Ammerer, M. Eilers, K. Nasmyth, and K. Zatloukal for critical reading of the manuscript, and members of our groups for valuable discussions. This work was supported by the Austrian Federal Ministry of Science, Transport and the Arts, by grants from the Deutsche Forschungsgemeinschaft (He 581/8-2), the Max Planck Research Prize to E.F.W. and U. Rüther, the Training and Mobility of Researchers Programme of the European Community (ERB4061PL95-078), and by grants from the National Institutes of Health (ES 04151, ES 06376) and the Department of Energy (DE-FG03-86ER60429). F.P. and J.T. were supported by postdoctoral fellowships from the Human Frontiers Science program.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL wagner@ht.imp.univie.ac.at; FAX 43-1798 71 53.
References
- Agarwal ML, Agarwal A, Taylor WR, Stark GR. p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc Natl Acad Sci. 1995;92:8493–8497. doi: 10.1073/pnas.92.18.8493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- Angel P, Imagawa M, Chiu R, Stein B, Imbra RJ, Rahmsdorf HJ, Jonat C, Herrlich P, Karin M. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell. 1987;49:729–739. doi: 10.1016/0092-8674(87)90611-8. [DOI] [PubMed] [Google Scholar]
- Bates S, Vousden KH. p53 in signaling checkpoint arrest or apoptosis. Curr Opin Genet Dev. 1996;6:12–19. doi: 10.1016/s0959-437x(96)90004-0. [DOI] [PubMed] [Google Scholar]
- Beijersbergen RL, Carlee L, Kerkhoven RM, Bernards R. Regulation of the retinoblastoma protein-related p107 by G1 cyclin complexes. Genes & Dev. 1995;9:1340–1353. doi: 10.1101/gad.9.11.1340. [DOI] [PubMed] [Google Scholar]
- Bengal E, Ransone L, Scharfmann R, Dwarki VJ, Tapscott SJ, Weintraub H, Verma IM. Functional antagonism between c-Jun and MyoD proteins: Direct physical association. Cell. 1992;68:507–519. doi: 10.1016/0092-8674(92)90187-h. [DOI] [PubMed] [Google Scholar]
- Bohmann D, Ellis MC, Staszewski LM, Mlodzik M. Drosophila Jun mediates Ras-dependent photoreceptor determination. Cell. 1994;78:973–986. doi: 10.1016/0092-8674(94)90273-9. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Bakiri L, Yaniv M. Induction of apoptosis by the transcription factor c-Jun. EMBO J. 1997;16:1695–1709. doi: 10.1093/emboj/16.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüsselbach S, Möhle-Steinlein U, Wang Z-Q, Schreiber M, Lucibello FC, Müller R, Wagner EF. Cell proliferation and cell cycle progression are not impaired in fibroblasts and ES cells lacking c-Fos. Oncogene. 1995;10:79–86. [PubMed] [Google Scholar]
- Bushel P, Kim JH, Chang W, Catino JJ, Ruley HE, Kumar CC. Two serum response elements mediate transcriptional repression of human smooth muscle alpha-actin promoter in ras-transformed cells. Oncogene. 1995;10:1361–1370. [PubMed] [Google Scholar]
- Cano E, Mahadevan LC. Parallel signal processing among mammalian MAPKs. Trends Biochem Sci. 1995;20:117–122. doi: 10.1016/s0968-0004(00)88978-1. [DOI] [PubMed] [Google Scholar]
- De Cesare D, Vallone D, Caracciolo A, Sassone-Corsi P, Nerlov C, Verde P. Heterodimerization of c-Jun with ATF-2 and c-Fos is required for positive and negative regulation of the human urokinase enhancer. Oncogene. 1995;11:365–376. [PubMed] [Google Scholar]
- Deng T, Karin M. JunB differs from c-Jun in its DNA binding and dimerization domains, and represses c-Jun by formation of inactive heterodimers. Genes & Dev. 1993;7:479–490. doi: 10.1101/gad.7.3.479. [DOI] [PubMed] [Google Scholar]
- Downward J. Cell cycle: Routine role for Ras. Curr Biol. 1997;7:R258–260. doi: 10.1016/s0960-9822(06)00116-3. [DOI] [PubMed] [Google Scholar]
- Elledge SJ, Winston J, Harper JW. A question of balance: The role of cyclin-kinase inhibitors in development and tumorigenesis. Trends Cell Biol. 1996;6:388–392. doi: 10.1016/0962-8924(96)10030-1. [DOI] [PubMed] [Google Scholar]
- Ginsberg D, Oren M, Yaniv M, Piette J. Protein-binding elements in the promoter region of the mouse p53 gene. Oncogene. 1990;5:1285–1290. [PubMed] [Google Scholar]
- Grana X, Reddy EP. Cell cycle control in mammalian cells: Role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs) Oncogene. 1995;11:211–219. [PubMed] [Google Scholar]
- Greenhalgh DA, Wang XJ, Eckardt JN, Roop DR. 12-O-tetradecanoylphorbol-13-acetate promotion of transgenic mice expressing epidermal-targeted v-fos induces rasHA-activated papillomas and carcinomas without p53 mutation: Association of v-fos expression with promotion and tumor autonomy. Cell Growth Differ. 1995;6:579–586. [PubMed] [Google Scholar]
- Gruda MC, van Amsterdam J, Rizzo CA, Durham SK, Lira S, Bravo R. Expression of FosB during mouse development: Normal development of FosB knockout mice. Oncogene. 1996;12:2177–2185. [PubMed] [Google Scholar]
- Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ. Cdk inhibitors in development and cancer. Curr Opin Genet Dev. 1996;6:56–64. doi: 10.1016/s0959-437x(96)90011-8. [DOI] [PubMed] [Google Scholar]
- Harvey M, Sands AT, Weiss RS, Hegi ME, Wiseman RW, Pantazis P, Giovanella BC, Tainsky MA, Bradley A, Donehower LA. In vitro growth characteristics of embryo fibroblasts isolated from p53-deficient mice. Oncogene. 1993;8:2457–2467. [PubMed] [Google Scholar]
- Herber B, Truss M, Beato M, Müller R. Inducible regulatory elements in the human cyclin D1 promoter. Oncogene. 1994;9:2105–2107. [PubMed] [Google Scholar]
- Herrera RE, Sah V, Williams BO, Makela TP, Weinberg RA, Jacks T. Altered cell cycle kinetics, gene expression, and G1 restriction point regulation in Rb-deficient fibroblasts. Mol Cell Biol. 1996;16:2402–2407. doi: 10.1128/mcb.16.5.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks GG, Egan SE, Greenberg AH, Mowat M. Mutant p53 tumor suppressor alleles release ras-induced cell cycle growth arest. Mol Cell Biol. 1991;11:1344–1352. doi: 10.1128/mcb.11.3.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilberg F, Wagner EF. Embryonic stem (ES) cells lacking functional c-jun: Consequences for growth and differentiation, AP-1 activity and tumorigenicity. Oncogene. 1992;7:2371–2380. [PubMed] [Google Scholar]
- Hilberg F, Aguzzi A, Howells N, Wagner EF. c-jun is essential for normal mouse development and hepatogenesis. Nature. 1993;365:179–181. doi: 10.1038/365179a0. [DOI] [PubMed] [Google Scholar]
- Jacks T, Weinberg RA. Cell-cycle control and its watchman. Nature. 1996;381:643–644. doi: 10.1038/381643a0. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Johnson RS, van Lingen B, Papaioannou VE, Spiegelman BM. A null mutation at the c-jun locus causes embryonic lethality and retarded cell growth in culture. Genes & Dev. 1993;7:1309–1317. doi: 10.1101/gad.7.7b.1309. [DOI] [PubMed] [Google Scholar]
- Johnson R, Spiegelman B, Hanahan D, Wisdom R. Cellular transformation and malignancy induced by ras require c-jun. Mol Cell Biol. 1996;16:4504–4511. doi: 10.1128/mcb.16.8.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Karin M, Hunter T. Transcriptional control by protein phosphorylation: Signal transmission from the cell surface to the nucleus. Curr Biol. 1995;5:747–757. doi: 10.1016/s0960-9822(95)00151-5. [DOI] [PubMed] [Google Scholar]
- Kovary K, Bravo R. The jun and fos protein families are both required for cell cycle progression in fibroblasts. Mol Cell Biol. 1991;11:4466–4472. doi: 10.1128/mcb.11.9.4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Chiang WH, Chiang SH, Liu YC, Hwang J, Ng SY. Regulation of cyclin D1, DNA topoisomerase I, and proliferating cell nuclear antigen promoters during the cell cycle. Gene Expr. 1995;4:95–109. [PMC free article] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Li L, Chambard JC, Karin M, Olson EN. Fos and Jun repress transcriptional activation by myogenin and MyoD: The amino terminus of Jun can mediate repression. Genes & Dev. 1992;6:676–689. doi: 10.1101/gad.6.4.676. [DOI] [PubMed] [Google Scholar]
- Li R, Waga S, Hannon GJ, Beach D, Stillman B. Differential effects by the p21 CDK inhibitor on PCNA-dependent DNA replication and repair. Nature. 1994;371:534–537. doi: 10.1038/371534a0. [DOI] [PubMed] [Google Scholar]
- Lord KA, Abdollahi A, Hoffman-Lieberman B, Liebermann DA. Proto-oncogenes of the fos/jun family of transcription factors are positive regulators of myeloid differentiation. Mol Cell Biol. 1993;13:841–851. doi: 10.1128/mcb.13.2.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod KF, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K, Vogelstein B, Jacks T. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes & Dev. 1995;9:935–944. doi: 10.1101/gad.9.8.935. [DOI] [PubMed] [Google Scholar]
- Maltzman W, Czyzyk L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol Cell Biol. 1984;4:1689–1694. doi: 10.1128/mcb.4.9.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato JY. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol. 1994;14:2066–2076. doi: 10.1128/mcb.14.3.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgley CA, Lane D. p53 protein stability in tumour cells is not determined by mutation but is dependent on Mdm2 binding. Oncogene. 1997;15:1179–1189. doi: 10.1038/sj.onc.1201459. [DOI] [PubMed] [Google Scholar]
- Murphy C, Nikodem D, Howcroft K, Weissman JD, Singer DS. Active repression of major histocompatibility complex class I genes in a human neuroblastoma cell line. J Biol Chem. 1996;271:30992–30999. doi: 10.1074/jbc.271.48.30992. [DOI] [PubMed] [Google Scholar]
- Patel HR, Sytkowski AJ. Erythropoietin activation of AP1 (Fos/Jun) Exp Hematol. 1995;23:619–625. [PubMed] [Google Scholar]
- Pfarr CM, Mechta F, Spyrou G, Lallemand D, Carillo S, Yaniv M. Mouse JunD negatively regulates fibroblast growth and antagonizes transformation by ras. Cell. 1994;76:747–760. doi: 10.1016/0092-8674(94)90513-4. [DOI] [PubMed] [Google Scholar]
- Riabowol K, Schiff J, Gilman MZ. Transcription factor AP-1 is required for initiation of DNA synthesis and is lost during cellular aging. Proc Natl Acad Sci. 1992;89:157–161. doi: 10.1073/pnas.89.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabapathy K, Klemm M, Jaenisch R, Wagner EF. Regulation of ES cell differentiation by functional and conformational modulation of p53. EMBO J. 1997;16:6217–6229. doi: 10.1093/emboj/16.20.6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorpp M, Mattei A-G, Herr I, Gack S, Schaper J, Angel P. Structural organization and chromosomal localization of the mouse collagenase type I gene. Biochem J. 1995;308:211–217. doi: 10.1042/bj3080211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber M, Baumann B, Cotten M, Angel P, Wagner EF. Fos is an essential component of the mammalian UV response. EMBO J. 1995;14:5338–5349. doi: 10.1002/j.1460-2075.1995.tb00218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. G1 phase progression: Cycling on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- ————— Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes & Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- Smeal T, Binetruy B, Mercola DA, Birrer M, Karin M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature. 1991;354:494–496. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- Smith MJ, Prochownik EV. Inhibition of c-jun causes reversible proliferative arrest and withdrawal from the cell cycle. Blood. 1992;79:2107–2115. [PubMed] [Google Scholar]
- Suzuki T, Okuno H, Yoshida T, Endo T, Nishina H, Iba H. Difference in transcriptional regulatory function between c-Fos and Fra-2. Nucl Acids Res. 1991;25:5537–5542. doi: 10.1093/nar/19.20.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo E, Preis LH, Birrer MJ. Constitutive c-Jun expression induces partial macrophage differentiation in U-937 cells. Cell Growth Differ. 1994;5:439–446. [PubMed] [Google Scholar]
- Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol. 1963;17:299–313. doi: 10.1083/jcb.17.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, Haimovitz-Friedmann A, Fuks Z, Kolesnick RN. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature. 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- Wang Z-Q, Liang J, Schellander K, Wagner EF, Grigoriadis AE. c-Fos induced osteosarcoma formation in transgenic mice: Cooperativity with c-Jun and the role of endogenous c-Fos. Cancer Res. 1995;55:6244–6251. [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- ————— E2F and cell proliferation: A world turned upside down. Cell. 1996;85:457–459. doi: 10.1016/s0092-8674(00)81244-1. [DOI] [PubMed] [Google Scholar]
- Yoshioka K, Deng T, Cavigelli M, Karin M. Antitumor promotion by phenolic antioxidants: Inhibition of AP-1 activity through induction of Fra expression. Proc Natl Acad Sci. 1995;92:4972–4976. doi: 10.1073/pnas.92.11.4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterberg A, Larsson O, Wiman KG. What is the restriction point? Curr Opin Cell Biol. 1995;7:835–842. doi: 10.1016/0955-0674(95)80067-0. [DOI] [PubMed] [Google Scholar]