

Abstract
Chromosome segregation depends on kinetochores, the structures that mediate chromosome attachment to the mitotic spindle. We isolated mutants in IPL1, which encodes a protein kinase, in a screen for budding yeast mutants that have defects in sister chromatid separation and segregation. Cytological tests show that ipl1 mutants can separate sister chromatids but are defective in chromosome segregation. Kinetochores assembled in extracts from ipl1 mutants show altered binding to microtubules. Ipl1p phosphorylates the kinetochore component Ndc10p in vitro and we propose that Ipl1p regulates kinetochore function via Ndc10p phosphorylation. Ipl1p localizes to the mitotic spindle and its levels are regulated during the cell cycle. This pattern of localization and regulation is similar to that of Ipl1p homologs in higher eukaryotes, such as the human aurora2 protein. Because aurora2 has been implicated in oncogenesis, defects in kinetochore function may contribute to genetic instability in human tumors.
Keywords: sister chromatid separation, chromosome segregation, Ipl1p/aurora kinase, kinetochores, microtubules, yeast
Accurate chromosome segregation depends on the precise coordination of events in the chromosome cycle. Chromosomes replicate and establish linkage between the sister chromatids during S phase. The linkage must be maintained until all of the chromosomes have become aligned on the mitotic spindle, with the opposing sister kinetochores attached to opposite poles of the spindle. At the onset of anaphase, the linkage between the sisters must be destroyed promptly (sister chromatid separation) to allow the kinetochore-dependent movement of the separated sisters along microtubules to opposite poles of the spindle (chromosome segregation).
Several proteins involved in sister chromatid linkage have been identified (for review, see Biggins and Murray 1998). In budding yeast, the Mcd1/Scc1 cohesin protein is required for sister chromatid cohesion (Guacci et al. 1997; Michaelis et al. 1997). This protein associates with chromosomes during replication and dissociates from them at anaphase, making it an excellent candidate for a physical link between sister chromatids (Michaelis et al. 1997). In budding yeast, sister chromatid separation at the metaphase-to-anaphase transition requires ubiquitin-mediated proteolysis of Pds1p (Cohen-Fix et al. 1996; Ciosk et al. 1998) that is catalyzed by a multiprotein ubiquitin ligase known as the cyclosome or anaphase-promoting complex (APC) (for review, see Cohen-Fix and Koshland 1997). The destruction of Pds1p allows an interacting protein, Esp1p, to promote sister chromatid separation by inducing the dissociation of Mcd1p/Scc1p from chromosomes by an unknown mechanism (Ciosk et al. 1998).
Accurate chromosome segregation depends on proper spindle assembly and regulating the onset of anaphase. In particular, the sisters must be attached to opposite poles before the linkage between them is destroyed and the separated chromosomes must be able to move along the microtubules to the poles of the spindle. Chromosomes attach to the spindle through kinetochores, multiprotein complexes that assemble onto centromeric DNA (for review, see Hyman and Sorger 1995). The CBF3 complex, which contains four components (Ndc10p, Cep3p, Ctf13p, and Skp1p; Lechner and Carbon 1991; Stemmann and Lechner 1996; Kaplan et al. 1997), binds to centromeric DNA and this interaction is essential for kinetochore function (Lechner and Carbon 1991; Sorger et al. 1995). The CBF3 complex does not bind microtubules, but beads coated with centromeric DNA–CBF3 complexes can bind to microtubules after they have been incubated in yeast extracts (Sorger et al. 1994), suggesting that CBF3 can recruit unknown microtubule-binding proteins to the kinetochore. The kinetochore attachment to microtubules is monitored by the spindle assembly checkpoint that arrests cells at metaphase until defects in chromosome attachment are corrected (for review, see Rudner and Murray 1996).
The IPL1 gene encodes a protein kinase that was identified originally in a screen for yeast mutants that increase in ploidy (Chan and Botstein 1993; Francisco et al. 1994). It has been suggested that Ipl1p opposes the protein phosphatase I activity of Glc7p in budding yeast (Francisco and Chan 1994; Francisco et al. 1994), and protein phosphatase I activity is required for proper chromosome segregation in several organisms (Doonan and Morris 1989; Ohkura et al. 1989; Axton et al. 1990; Hisamoto et al. 1994). Ipl1p homologs have been identified in many eukaryotic organisms and one of the human homologs, aurora2, is amplified in colorectal and breast cancer cell lines (Sen et al. 1997; Bischoff et al. 1998). In addition, overexpression of aurora2 can transform rat cell lines (Bischoff et al. 1998), suggesting that regulating this protein is important for maintaining genomic stability in higher eukaryotes.
We identified ipl1 mutants in a screen for mutants that exhibit defective chromosome behavior during mitosis. Several lines of evidence suggest that ipl1 mutants are defective in chromosome segregation, not sister chromatid separation. In vivo and in vitro assays suggest that the segregation defect in ipl1 mutants is caused by defective kinetochore function. The ipl1 kinase phosphorylates Ndc10p in vitro, suggesting that Ipl1p regulates the kinetochore by phosphorylating Ndc10p.
Results
Analysis of sister chromatids in ipl1 mutants
We used chromosomes marked with green fluorescent protein (GFP; Straight et al. 1996) to isolate mutants defective in chromosome behavior during mitosis. Chromosome IV, one of the largest budding yeast chromosomes, was marked by integrating a tandem repeat of lactose operators (LacO) near the centromere and visualized by expressing a GFP-lactose repressor fusion (GFP–LacI). We isolated 2000 temperature-sensitive mutants in this strain, screened them by fluorescence microscopy for defects in mitotic chromosome behavior, and isolated nine complementation groups that appeared to be defective in sister chromatid separation (N. Bhalla, S. Biggins, and A.W. Murray, unpubl.). We cloned one of these genes by complementing the temperature-sensitive defect and determined that it was the previously isolated IPL1 gene (Chan and Botstein 1993; Francisco et al. 1994).
We used the GFP-marked chromosome to analyze chromosome behavior during the cell cycle of ipl1 mutants. Wild-type and ipl1-321 mutant cells were arrested in G1 with α-factor and released to the nonpermissive temperature (37 °C) in the absence of α-factor. An hour after the release, we added α-factor back to prevent cells from entering the next cell cycle and monitored sister chromatid separation by microscopy. Figure 1A shows that in wild-type cells, sister chromatid separation began 80 min after the release from G1 and was complete by 120 min. In the ipl1-321 mutant, sister separation only occurred in half the cells. We obtained similar results when the Lac operators were integrated midway along the long arm or close to the telomere of chromosome IV, or at the centromere of chromosome III, the second smallest chromosome in Saccharomyces cerevisiae (data not shown).
Figure 1.
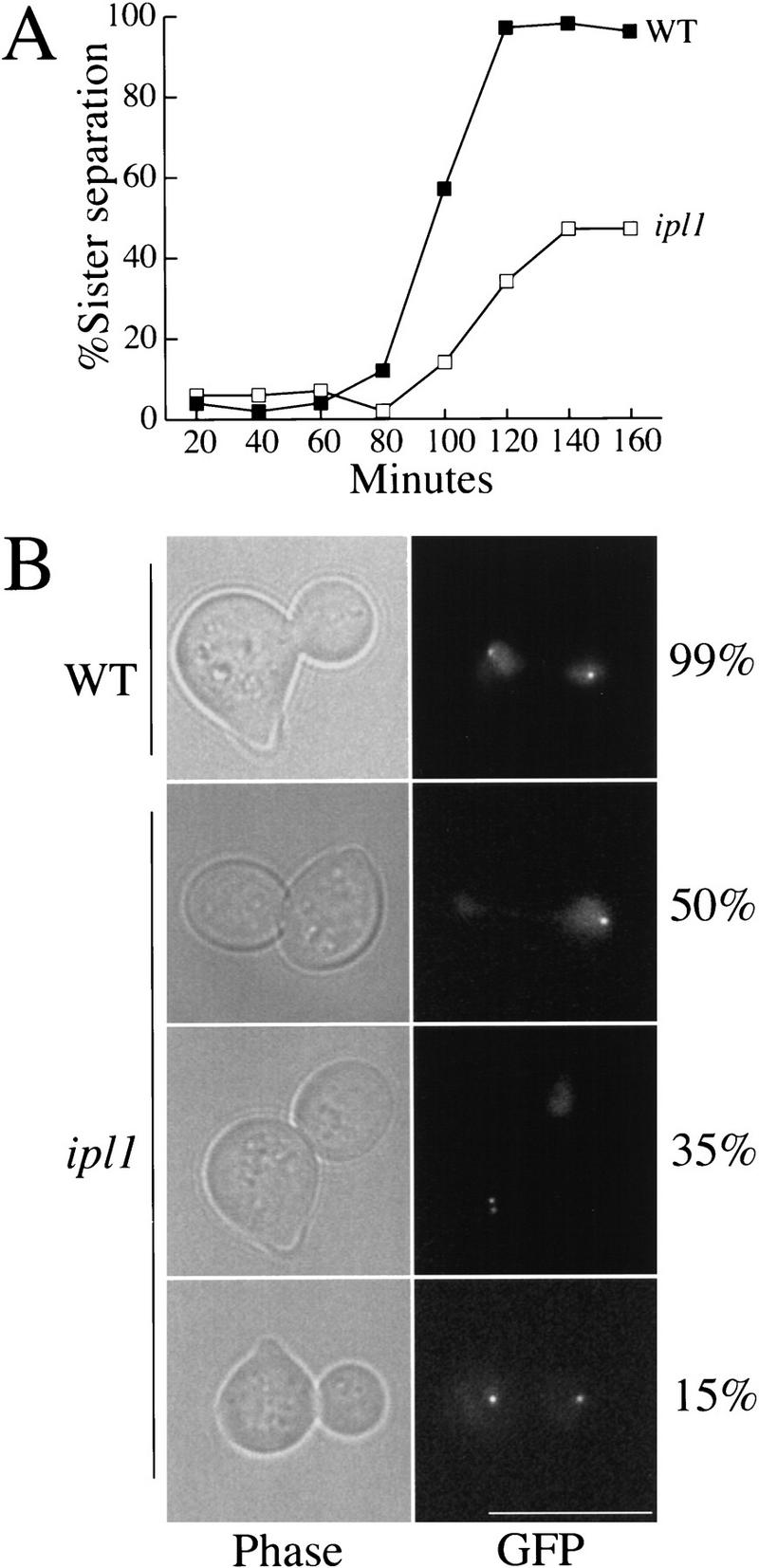
(A) Sister chromatids in ipl1 mutants. Wild-type and ipl1-321 cells released from α-factor arrest (T = 0) into the nonpermissive temperature (37°C) were scored for sister chromatid separation by microscopy. These strains contained integrated lactose operators at the centromere of chromosome IV. Although sister chromatids completely separate in wild-type cells (SBY214; █), they appear to separate only 50% of the time in ipl1-321 (SBY322; □ ). (B) The phenotypes of cells at 140 min after α-factor release are shown. Sister chromatids were visualized by GFP–LacI fluorescence and the nuclear DNA was visualized by the background GFP fluorescence. Three ipl1 mutant phenotypes are observed: Sisters appear to be held together 50% of the time, both sisters separate at one pole 35% of the time, and sisters separate to opposite poles 15% of the time. Phase contrast is shown in the left panels; GFP fluorescence is shown in the right panels. Bar, 10 μm.
Detailed analysis of the GFP-marked chromosomes suggested the ipl1 cells had additional defects in chromosome segregation (Fig. 1B). At 140 min, wild-type large-budded cells had segregated their sister chromatids to opposite poles (Fig. 1B). At the same time, the large-budded ipl1 cells exhibited three phenotypes: 50% of the cells had a single GFP dot at one spindle pole, 35% had two closely separated GFP dots both at one spindle pole, and 15% had two GFP spots at opposite spindle poles. In all three types of cells, the bulk chromosomal DNA was segregated unequally, as reported previously for other ipl1 alleles (Francisco et al. 1994). When the two GFP-marked sister chromatids segregated to the same pole of the spindle, this pole was located in the bud in 60% of the mitoses (data not shown).
Sister chromatids separate in ipl1 mutants
Our initial observations could not distinguish defects in sister chromatid separation from those in chromosome segregation. Because yeast chromosomes are confined in a nucleus ∼1.5 μm in diameter and the practical resolution limit of standard optical microscopes is 0.3 μm, it is hard to distinguish between two possible defects: the failure of sister separation or normal sister separation followed by abnormal segregation. If both sisters are pulled to the same spindle pole, they will often be so close to each other that the LacO arrays on the two sisters cannot be resolved as separate objects. To distinguish between these possibilities, we analyzed sister chromatid separation in the absence of a spindle; in this situation, ordered, microtubule-dependent segregation cannot occur and separated sisters diffuse slowly apart from each other (Marshall et al. 1997). Normally, the absence of microtubules arrests the cell cycle by activating the spindle checkpoint (Hoyt et al. 1991; Li and Murray 1991), but mad and bub mutants that inactivate the checkpoint bypass this arrest and reveal that sister chromatid separation does not require microtubules (Straight et al. 1996, 1997).
To determine whether sister chromatids separated in ipl1 mutants, we analyzed sister chromatid separation in the absence of microtubules in wild-type, ipl1-321, and mad2Δ strains, and in an ipl1-321 mad2Δ double mutant. Cells were arrested in G1 with α-factor, then released into medium containing the microtubule-depolymerizing drugs nocodazole and benomyl at 37°C. Figure 2 shows that sister chromatids do not separate in wild-type cells because the checkpoint is activated by the absence of microtubules and arrests cells at metaphase. We found that ipl1 mutants also activate the spindle assembly checkpoint and arrest at metaphase in the presence of nocodazole and benomyl. In mad2 mutants, the cell cycle continues because the checkpoint cannot be activated, resulting in sister chromatid separation. Because there is no segregation machinery, the two separated sister chromatids cannot always be resolved from each other and the level of separation never reaches 100%. In the ipl1 mad2 double mutant, sister chromatids separated at a similar rate and to a similar extent as they do in the mad2 mutant. We performed the same experiment with ipl1-321 mad3Δ and ipl1-321 bub2Δ double mutants and obtained similar results (data not shown). Unlike ipl1 mad2 double mutants, esp1-1 mad2Δ double mutants released into nocodazole and benomyl do not separate sister chromatids (data not shown; and similar experiments in Ciosk et al. 1998). These data show that ipl1 mutants are not defective in sister chromatid separation and thus imply that their primary defect is in chromosome segregation.
Figure 2.

Sister chromatids separate in ipl1 mutants. Wild-type (SBY214; █), ipl1-321 (SBY322; ○), mad2Δ (SBY99; ▴), and mad2Δ ipl1-321 (SBY100; ●) strains were arrested in G1 with α-factor (T = 0), released to the nonpermissive temperature (37°C) in nocodazole and benomyl, and scored for the percentage sister chromatid separation over time. In the absence of a spindle, ipl1 mutants are competent to separate sister chromatids.
We performed a second, independent test for sister chromatid separation in ipl1 mutants by examining the localization of the sister chromatid cohesion protein Mcd1/Scc1 (Guacci et al. 1997; Michaelis et al. 1997). In the esp1 mutant, Mcd1p/Scc1p does not dissociate from chromosomes at anaphase and sister chromatids do not separate, suggesting Mcd1p/Scc1p dissociation is critical for sister chromatid separation (Ciosk et al. 1998). If ipl1 mutants were defective in sister chromatid separation, Mcd1p/Scc1p should not dissociate from chromosomes at anaphase. We performed chromosome spreads to localize Mcd1p/Scc1p during the cell cycle in wild-type and ipl1-321 strains that contained an integrated Mcd1/Scc1 protein tagged with three copies of the hemagglutinin (HA) epitope. Cells were arrested in G1 with α-factor and released to fresh media at the nonpermissive temperature and chromosome spreads and indirect immunofluorescence microscopy were performed on samples throughout the time course. The spreads were stained with DAPI to visualize DNA, anti-HA antibodies to visualize Mcd1p/Scc1p, and anti-GFP antibodies to visualize the marked sister chromatids. Figure 3 shows that Mcd1p/Scc1p associates and dissociates from the chromosomes identically in wild-type and ipl1-321 cells: Mcd1p/Scc1p is not bound to the chromosomes during G1, associates with the chromosomes during S phase, and dissociates from them at anaphase. In wild-type cells, anaphase can be identified by the stretched DNA and the separated GFP staining of the sister chromatids (Fig. 3A). In ipl1 mutants, Mcd1p/Scc1p is not associated with chromosomes at anaphase even though the sister chromatids do not appear to have separated (Fig. 3A). Figure 3B quantifies the fraction of chromosome spreads that stain for Mcd1p/Scc1p during the cell cycle of wild-type, ipl1-321 and esp1-1 cells. There are no detectable differences in Mcd1p/Scc1p association with chromosomes in wild-type and ipl1 strains throughout the cell cycle, whereas mutants in esp1 prevent the dissociation of Mcd1p/Scc1p as reported previously (Ciosk et al. 1998). This experiment provided additional evidence that sister chromatid separation is normal in ipl1 mutants and that the defect in chromosome behavior is an error in chromosome segregation that results in both sister chromatids being pulled to a single spindle pole.
Figure 3.
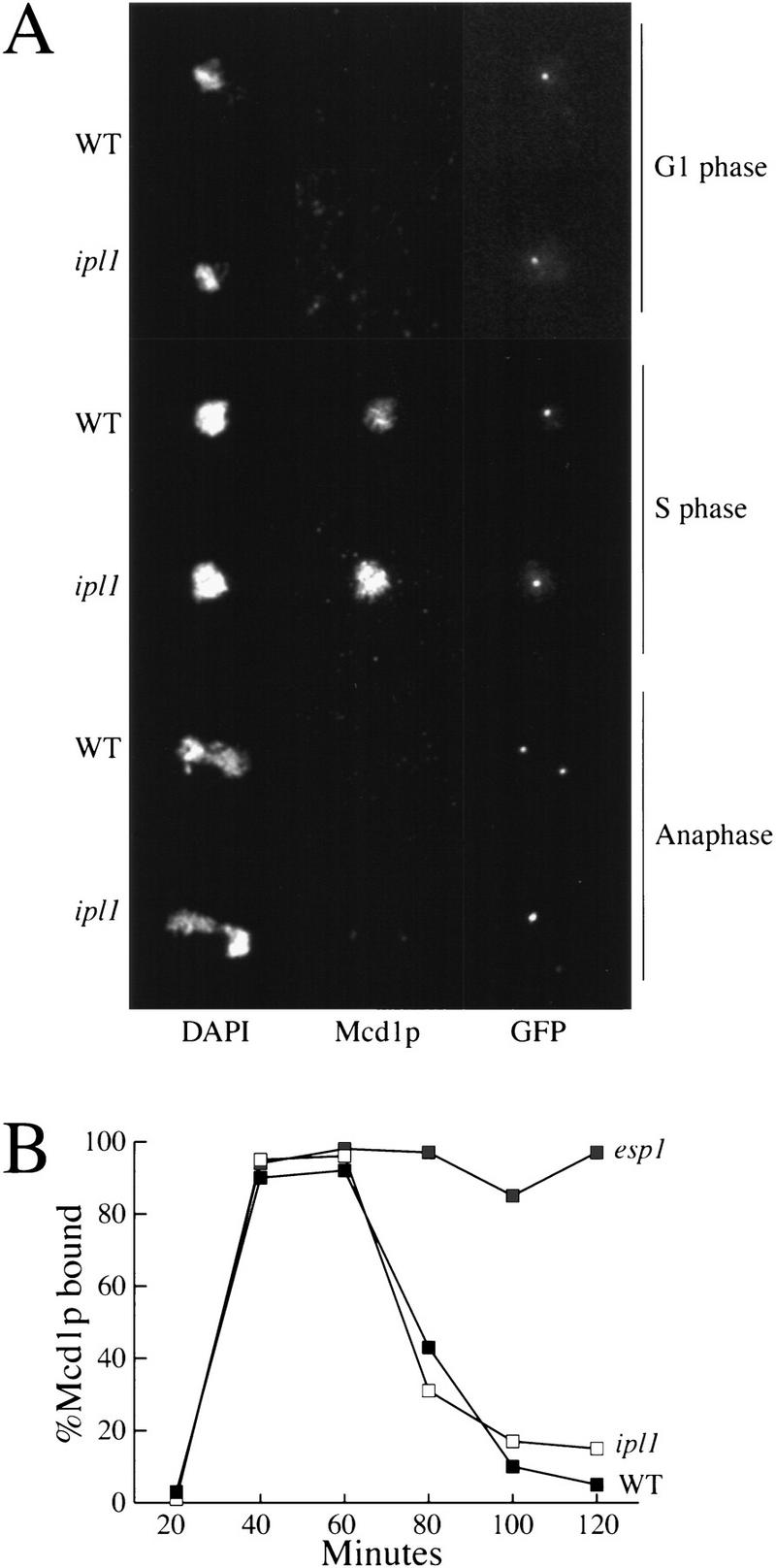
Mcd1p localization is normal in ipl1 mutants. (A) Mcd1p was localized to chromosomes by indirect immunofluorescence on chromosome spreads in wild-type (SBY376) and ipl1-321 (SBY378) strains containing HA3-epitope-tagged Mcd1p throughout the cell cycle. DAPI staining is shown in the left panels, anti-HA antibody staining is shown in the middle panels, and anti-GFP antibody staining is shown in the right panels. G1-phase, S-phase, and anaphase cells are indicated. Mcd1p is localized to chromosomes during S phase but not during G1 or anaphase in both wild-type and ipl1 cells. Bar, 10 μm. (B) The results of Mcd1p localization to chromosomes were quantified and the percentage of chromosome spreads with Mcd1p localized to chromosomes is graphed vs. time during the cell cycle for wild-type (SBY214; WT), ipl1-321 (SBY322; ipl1), and esp1-1 strains (SBY102; esp1).
Ipl1 is a cell-cycle-regulated protein associated with the mitotic spindle
If Ipl1p regulates chromosome segregation during mitosis, its protein levels or localization might be cell-cycle regulated. To test this, Ipl1p was epitope-tagged with 12 copies of the Myc epitope at its carboxyl terminus and Ipl1 protein levels were analyzed at various points in the cell cycle. Cells were arrested in G1 with α-factor, S phase with hydroxyurea, and mitosis with nocodazole, and protein extracts were prepared and Western blotted with anti-Myc antibodies. Ipl1p levels are low during G1 and peak during S phase and mitosis (Fig. 4A). These results are not caused by the cell-cycle arrests because similar results were obtained when Ipl1p levels were analyzed in a synchronous cell-cycle experiment (data not shown).
Figure 4.
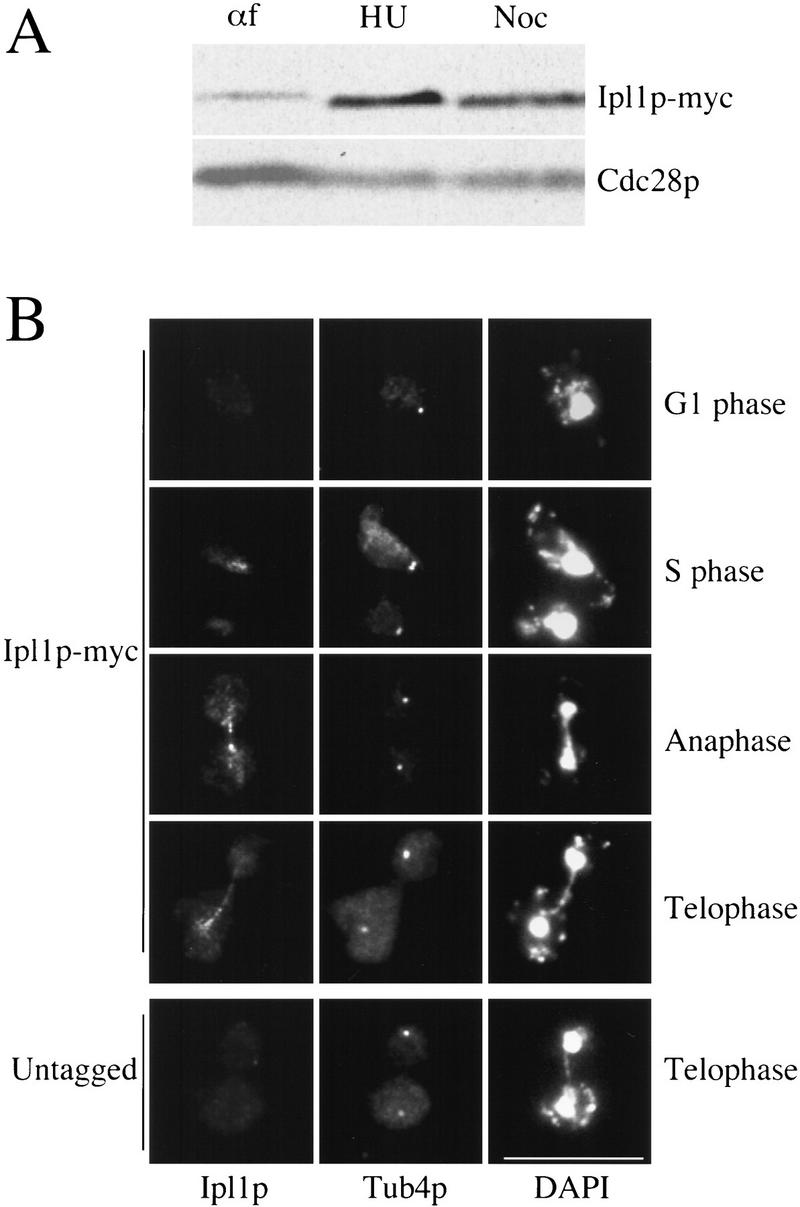
Ipl1p is a cell-cycle-regulated protein associated with the spindle. (A) The levels of Ipl1p–Myc12 (SBY388) in α-factor-arrested cells (αf, left lane), hydroxyurea-arrested cells (HU, middle lane), and nocodazole-arrested cells (Noc, right lane) were compared by Western blotting protein extracts with anti-Myc antibodies. The levels of Cdc28p are shown as a control. (B) Indirect immunofluorescence microscopy was performed on Ipl1p–Myc12 (SBY146) and an untagged control (SBY3) strain. Anti-Myc antibodies that recognize Ipl1p are shown in the left panels, anti-Tub4 antibodies are in the middle panels, and DAPI staining is in the right panels. The experiment was performed on cells throughout the cell cycle as indicated. Ipl1p localizes to the mitotic spindle. Bar, 10 μm.
To localize Ipl1p, we examined strains containing either the Ipl1p–Myc12 fusion or no epitope tag throughout the cell cycle. Cells were synchronized in G1 with α-factor, then released into medium free of α-factor. Indirect immunofluorescence microscopy was performed using antibodies to the Myc epitope to visualize Ipl1p, antibodies to Tub4p, the budding yeast γ-tubulin to visualize the spindle pole body (Marschall et al. 1996), and DAPI to visualize DNA (Fig. 4B). We did not detect the Ipl1 protein in G1-arrested cells (Fig. 4B), possibly because the protein levels are very low (see Figure 4A). During S phase, Ipl1p accumulated in the nucleus (Fig. 4B). At the onset of mitosis, the nuclear fluorescence disappeared and Ipl1p was localized to the mitotic spindle, as a series of dots stretching between the two spindle pole bodies (Fig. 4B). Although we do not know whether the punctate spindle staining is significant or reflects an accessibility problem of the antibody caused by the 12 myc tags, we see this pattern under a variety of fixation conditions. In addition, the staining is specific to Ipl1p because strains lacking an epitope tag do not exhibit any nuclear or spindle staining. The Ipl1p localization patterns are consistent with Ipl1p having a role in chromosome segregation.
The spindles and spindle poles appear normal in ipl1 mutants
The localization of Ipl1p to the mitotic spindle and the ipl1 mutant chromosome missegregation phenotypes suggested that Ipl1p could be involved in the function of the spindle, the spindle pole bodies, or the kinetochore. We therefore analyzed the spindle in wild-type and ipl1-321 strains and detected no major differences between wild-type and ipl1 mutants in the timing of spindle formation and breakdown (data not shown). In addition, we did not detect any gross morphological defects in spindle appearance in ipl1 mutants (data not shown), consistent with data published previously on additional ipl1 alleles (Francisco and Chan 1994).
We next examined spindle pole body duplication and separation in wild-type and ipl1 strains. Cells were arrested in G1 with α-factor and released to fresh media at 37°C. We performed indirect immunofluorescence microscopy on cells harvested throughout the time course using DAPI to stain DNA and antibodies against Tub4p to stain the spindle pole bodies (Fig. 5). At the beginning of the cell cycle, there was a single spindle pole body in both wild-type and ipl1-321 strains. During S phase, the spindle pole body duplicated and began to separate. This can be seen as two adjacent spindle pole bodies in both wild-type and ipl1 strains. At anaphase, when the DNA separated in wild-type cells, the spindle pole bodies were found at opposite poles in each bud. At anaphase in the ipl1 mutant, the spindle pole bodies still separated to opposite poles and stained equally brightly with anti-Tub4 antibodies, even though the DNA was segregated asymmetrically. In addition, we quantified the percentage of cells that had two spindle poles bodies throughout the cell cycle and found no differences between wild-type and ipl1 cells (data not shown). Therefore, spindle pole bodies duplicate and separate normally in the ipl1 mutant, and there is no mislocalization of an integral spindle pole body component.
Figure 5.

Spindle pole body duplication is normal in ipl mutants. Indirect immunofluorescence microscopy was performed to detect spindle pole bodies in wild-type (SBY214) and ipl1-321 (SBY322) strains throughout the cell cycle at the nonpermissive temperature. Strains were harvested to analyze spindle pole bodies in G1, S phase, and mitosis as indicated in wild-type (left panels) and ipl1-321 cells (right panels). DAPI staining and anti-Tub4p staining are indicated. Bar, 10 μm.
The kinetochore is defective in ipl1 mutants
The combination of a defect in chromosome segregation with apparently normal spindle pole bodies and spindle formation suggested that ipl1 mutants have a kinetochore defect. Kinetochore function was assayed in vivo by examining spindle length in an ipl1 mutant during a metaphase arrest. In wild-type cells the metaphase spindle is short because forces that separate the spindle poles are opposed by forces on the kinetochore that pull the kinetochores toward the spindle poles. Because the two sister kinetochores are linked to each other and face opposite poles, the forces on them pull the poles towards each other and keep the spindle short (Waters et al. 1996). If there is no linkage between the sisters, the metaphase spindle is long and the chromosomes are located close to the poles giving an appearance that is very similar to wild-type cells in anaphase (Piatti et al. 1995; Michaelis et al. 1997). The same anaphase-like prometaphase occurs if the microtubules do not properly attach to the kinetochores, even if the sisters remain linked to each other (F. Severin and A.A. Hyman, unpubl.). We therefore examined spindle length in ipl1 and wild-type cells at the metaphase arrest produced by the cdc23-1 mutant, which is defective in anaphase promoting complex-mediated proteolysis. We shifted asynchronous cdc23-1 and cdc23-1 ipl1-321 mutants to 37°C for 4 hr and then performed indirect immunofluorescence microscopy using anti-tubulin antibodies to visualize the spindle, anti-GFP antibodies to visualize the marked sister chromatids, and DAPI to visualize the DNA (Fig. 6A). In cdc23-1, the spindle remains short because sister chromatids remain linked and the DNA does not segregate. In cdc23-1 ipl1-321 mutants, however, the spindle partially elongates and begins to break down even though the marked pair of sister chromatids remained unseparated (Fig. 6A) and the level of the mitotic cyclin Clb2p remains high during the arrest (data not shown), indicating tht the metaphase arrest is intact in the cdc23-1 ipl1-321 double mutant. We quantified the defect by measuring the spindle length in cdc23-1 versus cdc23-1 ipl1-321 mutants and found that the spindle was significantly lengthened in the cdc23-1 ipl1 mutant (Fig. 6B). The spindle elongation in cdc23-1 ipl1-321 mutants was similar to a control cdc23-1 pds1-296 mutant where the spindle elongates because of the separation of sister chromatids during the metaphase arrest (Fig. 6B).
Figure 6.
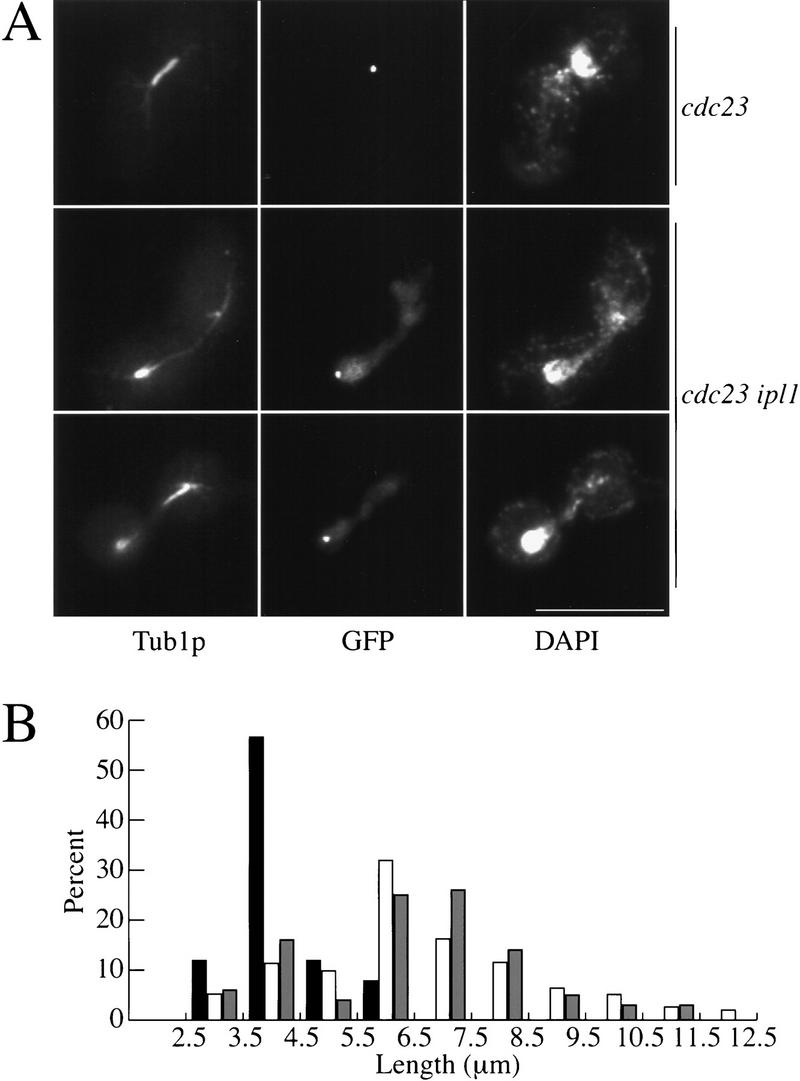
The spindle elongates in ipl1 mutants during a metaphase arrest. (A) Indirect immunofluorescence was performed on cdc23-1 (SBY134) and cdc23-1 ipl1-321 (SBY133) mutants shifted to the nonpermissive temperature (37°C) for 4 hr as indicated. Anti-tubulin staining is shown in the left panels, anti-GFP staining (to recognize the marked sister chromatid) is in the middle panels, and DAPI staining is shown in the right panels. Bar, 10 μm. (B) The spindle length (μm) in cdc23-1 (solid bars), cdc23-1 ipl1-321 (open bars), and cdc23-1 pds1-296 (SBY340; shaded bars) strains was measured in 100 cells, and the percentage of cells containing each spindle length graphed.
The elongation of the spindle suggested a defect in the interactions between microtubules and the kinetochore in ipl1 mutants. To test for genetic interactions between kinetochore components and IPL1, we made double mutants between kinetochore component mutants and the ipl1-321 mutant. We tested mutants in CBF3 components (ndc10-1, ndc10-2, cep3-1, ctf13-1) as well as additional kinetochore mutants (cse4-323, mif2-1). Double mutants between ipl1-321 and ndc10-1, ndc10-2 and cep3-1 all exhibited lower permissive temperatures than either single mutant but the other double mutants did not (data not shown). In addition, ndc10-1 and ndc10-2 mutants are sensitive to increased dosage of IPL1. When IPL1 was overexpressed from the inducible galactose promoter (pGAL–IPL1) in these mutants, it severely inhibited their growth at the permissive temperature even though wild-type cells were not affected by overexpression of IPL1 (Fig. 7A; data not shown). These data suggest a specific interaction between Ipl1p and CBF3 proteins.
Figure 7.

Ipl1p interacts with Ndc10p. (A) Serial dilutions of wild-type (WT; SBY214) and ndc10-1 (SBY164) cells in the presence or absence of pGAL–IPL1 (pSB169) at 23°C on glucose and galactose media are shown. Although pGAL–IPL1 does not affect wild-type cells (SBY394), it severely inhibits the growth of ndc10-1 cells (SBY196), suggesting a specific interaction between NDC10 and IPL1. (B) Ipl1p phosphorylates Ndc10p. Kinase assays were performed using the GST–Ipl1p kinase (lanes 1, 3–7) or GST as a control (lane 2). The substrates tested were GST–Ipl1, GST–Ndc10p, GST, histone HI, myelin basic protein (MBP), and casein, as indicated. The GST–Ndc10p is a carboxy-terminal fragment of Ndc10p that contains codons 679–894 of the 956-amino-acid protein. Two micrograms of substrate was used in each reaction except GST–Ipl1p autophosphorylation. The autophosphorylation of GST–Ipl1p and the phosphorylation of GST–Ndc10p and MBP by GST–Ipl1p is indicated at right. There are GST–Ndc10p breakdown products in addition to the GST–Ndc10 fusion protein in lane 3.
To test directly for kinetochore defects, we used two in vitro assays for kinetochore function. Wild-type and ipl1-321 mutant extracts prepared from cells grown at the permissive or nonpermissive temperatures were incubated with centromere DNA to allow kinetochores to assemble in vitro. Kinetochore assembly was assayed by monitoring the mobility shift of centromere DNA on a nondenaturing gel that results from binding CBF3 proteins (Fig. 8A). Kinetochore assembly was normal in wild-type and ipl1 mutants but did not occur in the ndc10-1 mutant as reported previously (Sorger et al. 1995).
Figure 8.
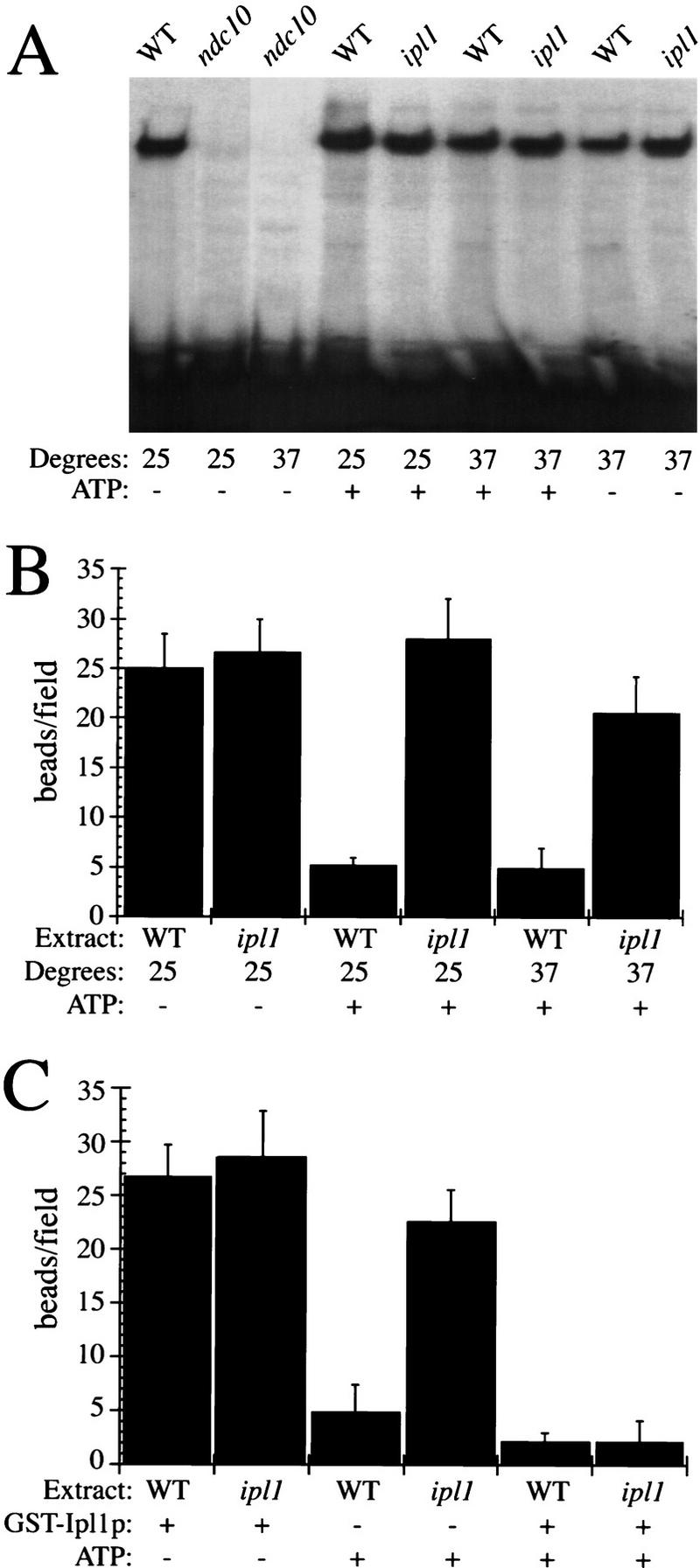
ipl1 mutants affect kinetochore function. (A) Nondenaturing gel analysis of kinetochore assembly was performed in the presence or absence of ATP using radiolabeled centromere DNA and extracts prepared from wild-type (SBY214), ndc10-1 (SBY164), or ipl1-321 (SBY322) cells grown at either 23°C or 37°C. The ndc10-1 kinetochore mutant is defective in assembling kinetochores but ip11 mutants and wild-type cells assemble normal kinetochores. (B) The microtubule-binding activity (no. of beads bound per field) in the presence or absence of ATP of wild-type amd ipl1-321 extracts made from cells grown at 25°C or 37°C is graphed. The ipl1-321 mutant is not sensitive to the addition of ATP at either temperature. (C) The microtubule-binding activity (no. of beads bound per field) in the presence or absence of ATP, and recombinant GST–Ipl1p from cells grown at 25°C is graphed. The addition of GST–Ipl1p to ipl1-321 extracts restores the microtubule inhibition by ATP.
We next tested whether microtubule binding to in vitro assembled kinetochores is altered in the ipl1 mutant. Although this assay measures microtubule binding, it does not fully reconstitute kinetochore function because the beads do not move along microtubules. Beads carrying centromeric DNA were added to wild-type and ipl1-321 mutant extracts and assayed for their ability to bind taxol-stabilized microtubules. This assay is sensitive to the addition of ATP, with high levels of ATP inhibiting microtubule binding. This effect is caused by the regulation of Ndc10p phosphorylation by the balance between the protein phosphatase I Glc7p and at least one unknown kinase (Sassoon et al. 1999). Ndc10p is hyperphosphorylated in glc7-10 extracts, and addition of recombinant Ndc10p reconstitutes the microtubule binding activity of glc7-10 extracts. Because genetic data suggest that Ipl1p opposes Glc7p activity (Francisco and Chan 1994; Francisco et al. 1994), it is possible that the Ipl1p kinase opposes Glc7p in the kinetochore assays.
We assayed the microtubule-binding activity of kinetochore beads assembled in wild-type and ipl1 extracts. In the absence of ATP, kinetochore beads assembled in wild-type and ipl1 mutant extracts both bound microtubules (Fig. 8B). When kinetochore beads assembled in wild-type extracts that had been supplemented with ATP, binding was reduced strongly, consistent with the idea that Ndc10 phosphorylation reduces binding in this assay. However, when ATP was added to ipl1-312 mutant extracts (prepared from cells were grown at either the permissive or nonpermissive temperatures), there was little reduction in the microtubule-binding activity of kinetochore beads. To determine whether this effect was specific to Ipl1p, we tested whether recombinant Ipl1p could complement the ipl1-321 mutant extracts in vitro. We fused Ipl1p to glutathione-S-transferase (GST) and bacterially expressed and purified GST–Ipl1p to add to the in vitro assays (Fig. 8C). When GST–Ipl1p was added to extracts in the absence of ATP, there was no affect on microtubule binding. However, when GST–Ipl1p was added to extracts in the presence of ATP, microtubule binding was inhibited to wild-type levels.
We eliminated the possibility that the reduced ATP sensitivity of kinetochore activity in ipl1 mutants is caused by increased protein phosphatase 1 activity by adding ATP together with microcystin, an inhibitor of protein phosphatase I. These conditions did not reduce microtubule binding of ipl1 extracts below the level seen with ATP alone, suggesting that the reduced ATP sensitivity of ipl1 extracts is caused by decreased kinase activity rather than increased phosphatase activity (data not shown). In addition, when the assays were performed using extracts prepared from nocodazole-arrested cells, the results were similar to the log-phase extracts, eliminating the possibility that there is a cell-cycle effect on the assays (data not shown). Taken together, these results suggest that Ipl1p opposes directly or indirectly the activity of Glc7p in regulating microtubule binding.
Ipl1p phosphorylates Ndc10p in vitro
The effects of ipl1 mutants on kinetochore activity prompted us to ask whether Ipl1p phosphorylates Ndc10p directly. GST and GST–Ipl1p fusions were purified from bacteria and tested for kinase activity towards recombinant GST, a GST fusion to a carboxy-terminal fragment of Ndc10p, and a variety of standard kinase substrates (Fig. 7B). Purified GST–Ipl1p phosphorylates GST–Ndc10p but not GST. GST–Ipl1p fails to phosphorylate casein or histone H1 and phosphorylates Ndc10p more strongly than myelin basic protein, suggesting that Ipl1 has specific kinase activity for Ndc10p. Like many protein kinases, GST–Ipl1p phosphorylates itself, producing a labeled product that migrates at a slightly higher molecular weight than the GST–Ndc10p protein fusion. To determine whether the mutant Ipl1-321 protein could phosphorylate Ndc10p, we recovered the ipl1-321 mutant gene and purified a GST–ipl1-321 fusion protein in bacteria. This protein does not exhibit any measurable kinase activity towards Ndc10p or MBP (data not shown). This result is consistent with the ipl1-321 mutation changing the conserved aspartic acid 283 in subdomain IX of the catalytic domain to an asparagine (data not shown).
Discussion
We isolated ipl1 mutants in a screen for mutants with defects in sister chromatid separation and segregation. In ipl1 mutants, the sister chromatids segregate to one spindle pole instead of opposite poles in 85% of cells. Two independent tests show that the ipl1 defect affects chromosome segregation rather than sister chromatid separation. The cytological behavior of ipl1 mutants is consistent with defects in kinetochore function and in vitro assays for kinetochore–microtubule interactions show altered kinetochore behavior in ipl1 extracts. Finally, Ipl1p can phosphorylate the kinetochore protein Ndc10 in vitro, whose activity is regulated by phosphorylation. Together these results suggest that an important function of Ipl1p is to regulate kinetochore behavior by phosphorylating Ndc10p.
ipl1 mutants are defective in chromosome segregation
In a normal mitosis, sister chromatid separation and chromosome segregation are tightly coupled events. Separation will induce segregation as long as the sister kinetochores are attached to microtubules from opposite spindle poles and are being pulled towards the poles. Measurements of kinetochore tension in insect and tissue culture cells show that both of these conditions are fulfilled in a normal metaphase (Nicklas 1983; Waters et al. 1996). In mutants that affect spindle or kinetochore function, sister separation may fail to induce chromosome segregation. We have used two assays to distinguish separation from segregation. The first is to monitor chromosome separation in the absence of microtubules. Under these conditions there is no difference between the extent of chromosome separation in wild-type and ipl1 cells, showing that ipl1 mutants are not defective in sister chromatid separation. The second is to follow the dissociation of the cohesion protein Mcd1/Scc1 from chromosomes. Although the loss of this protein has not been shown to induce sister separation directly, it is correlated tightly with separation and occurs normally in ipl1 cells. These data suggest that sister chromatid separation occurs normally in ipl1 mutants but chromosome segregation is defective.
IPL1 is required for kinetochore function
We examined kinetochore function in the ipl1 mutant. The significant elongation of ipl1 spindles during a metaphase arrest is consistent with the failure of linked sister kinetochores to counteract forces that oppose spindle elongation. We cannot exclude the possibility that this phenotype is caused by a spindle abnormality, but like a previous report (Francisco et al. 1994), we did not detect any defects in spindle pole body or spindle morphology. A specific kinetochore defect was revealed by in vitro tests. Although the ability to assemble the CBF3 complex on centromeric DNA was normal, the microtubule-binding of kinetochore beads assembled in ipl1 extracts could not be inhibited by ATP. This result suggests that the kinase activity of Ipl1 plays a role in regulating kinetochore activity. The exact nature of this role is unclear because we do not understand how the phosphorylation-induced inhibition of microtubule binding in vitro corresponds to kinetochore function in vivo. We speculate that this reaction may be involved in the ability of kinetochores to move along microtubules. Consistent with this idea, ipl1 mutants exhibit synthetic lethality with a deletion of CIN8 (Geiser et al. 1997), one of the microtubule motor proteins in budding yeast (Hoyt et al. 1992; Roof et al. 1992).
We propose that Ipl1p regulates kinetochore function via phosphorylation of the CBF3 component Ndc10p based on a variety of data: recombinant Ipl1p phosphorylated a carboxy-terminal fragment of Ndc10p in vitro, the ipl1 mutant phenotype is similar to the partial loss of ndc10 activity in an Ndc10p shut-off experiment as described previously (Goh and Kilmartin 1993) and there are two genetic interactions between the two genes, ipl1 ndc10 double mutants are more temperature-sensitive than either single mutant and overexpression of IPL1 strongly impairs the growth of ndc10 mutants. Finally, the Glc7p phosphatase appears to oppose Ipl1p activity in vivo (Francisco et al. 1994), and Glc7p regulates Ndc10p dephosphorylation and kinetochore function (Sassoon et al. 1999). Taken together, these data suggest that Ipl1p phosphorylates Ndc10p to regulate microtubule binding. To directly test this, we analyzed Ndc10p phosphorylation in vivo. Although we can detect Ndc10p phosphorylation in vivo when Ndc10p is overexpressed, we have yet to detect a significant difference in Ndc10p phosphorylation between ipl1-321 and Ipl1p overexpressing strains (data not shown). Future work will be needed to determine whether Ipl1p directly phosphorylates specific sites on Ndc10p in vivo.
Models for IPL1 function
How do ipl1 mutants affect kinetochore activity? The segregation of both sister chromatids to one pole in the majority of ipl1 cells as well as the elongation of the spindle during a metaphase arrest in ipl1 cells suggests that kinetochore function has been affected. There is clearly some kinetochore activity in ipl1 mutants, however, as the marked centromeres move to the spindle poles in anaphase and there is microtubule-binding activity in vitro.
One explanation is that one of the two sister kinetochores is defective. For example, if kinetochore replication was conservative, the old kinetochore could be fully functional, whereas the new one was defective. As a result both sister chromatids would segregate to the pole that was attached to the old kinetochore. Another explanation for the ipl1 mutant phenotype is that the two sister kinetochores attach preferentially to microtubules from the same pole instead of those from opposite poles as they do in wild-type cells. Finally, it is possible that both sister kinetochores are equally active and attach to opposite poles, but that the details of their interaction with microtubules is altered in a way that prevents normal chromosome segregation. For example, the interactions between kinetochores and microtubules in ipl1 cells might be too transient to allow accurate chromosome segregation. Kinetochores bind to the plus end of microtubules, and must therefore maintain attachment to a microtubule end that is alternately growing and shrinking. During anaphase, movement of the chromosomes to the poles is largely dependent on the ability of kinetochores to follow the shrinking plus end of microtubules. In vertebrate cells, there is evidence that kinetochore behavior can be regulated in at least three ways: Kinetochores can actively move either away or towards the spindle pole to which they attach (Skibbens et al. 1993), tension at the kinetochore can affect the direction of chromosome movement by determining whether microtubules that terminate at the kinetochore grow or shrink (Skibbens et al. 1993), and the interactions by which a kinetochore first captures the microtubule differ from those that occur after the kinetochore has bound to microtubules (Rieder and Alexander 1990). Any of these steps could be regulated by changes in kinetochore phosphorylation, and alterations in kinetochore phosphorylation could impair chromosome segregation by altering the dynamics of kinetochore–microtubule interactions or the kinetochore’s ability to generate force. Although we cannot distinguish amongst the different models for how kinetochore function is altered in ipl1 cells, the observation that ipl1 and glc7 mutants both affect kinetochore function argues phosphorylation–dephosphorylation balances are likely to play an important role in kinetochore function.
Ipl1p and homologs in higher eukaryotes
Ipl1p has homologs in several organisms, including humans (Kimura et al. 1997; Sen et al. 1997; Bischoff et al. 1998), frogs (Roghi et al. 1998), mice (Gopalan et al. 1997), and flies (Glover et al. 1995). In all of these organisms, the homologs associate with mitotic structures such as the spindle, the centrosome, and the spindle midzone and the protein levels are cell-cycle regulated. We showed that Ipl1p in budding yeast is not detected by immunofluorescence in G1 cells, appears in the nucleus during S phase and is seen as a series of dots on the spindle of mitotic cells. It is unclear whether Ipl1p associates with the spindle pole body. Unlike Tub4p, which exhibits a strong signal at the spindle pole body, there is little colocalization between Ipl1p and Tub4p and Ipl1p does not exhibit a strong signal at the spindle pole body.
Like its homologs, Ipl1p protein levels are low in G1-arrested cells and high in S phase and mitotic cells and Ipl1 mRNA shows a similar pattern of abundance (Cho et al. 1988). The IPL1 promoter contains a promoter element (the MluI cell-cycle box) which is associated with transcripts that peak at the G1/S boundary. As has been suggested for the Ipl1p homologs Aik1 (Kimura et al. 1997), Eg2 (Roghi et al. 1998), and AIM-1 (Terada et al. 1998), we speculate that Ipl1p is degraded by ubiquitin-mediated proteolysis as cells enter G1 via destruction boxes that could be recognized by the APC.
A variety of functions for Ipl1p homologs in higher eukaryotes have been reported. The Drosophila aurora protein is required for centrosome separation (Glover et al. 1995). The rat AIM-1 protein is implicated in cytokinesis because overexpression of a kinase-inactive protein does not affect nuclear division but disrupts cleavage furrow formation (Terada et al. 1998). In Xenopus, the addition of kinase-inactive Eg2 to egg extracts prevents the assembly of a bipolar spindle in vitro (Roghi et al. 1998). Therefore, Ipl1p homologs appear to have functions in centrosome separation, spindle formation, and cytokinesis.
We find that Ipl1p regulates the interaction between microtubules and kinetochores in budding yeast. Although a kinetochore defect has not been reported for mutants in Ipl1p homologs, identifying this function in yeast required cytological and in vitro assays that are not available for other eukaryotes. In addition, it is possible that the centrosome separation and spindle formation phenotypes caused by perturbing aurora and Eg2 are caused by defects in kinetochore–microtubule interactions. Because multiple Ipl1p homologs exist in higher eukaryotes, such as humans, worms, and frogs (Bischoff et al. 1998), different homologs may perform different functions in mitosis. The human aurora2 gene partially complements an ipl1 mutant in yeast but the aurora1 gene does not (Bischoff et al. 1998).
Although our studies have shown that Ipl1p regulates kinetochore function, it remains possible that the protein has additional functions. There are no apparent defects in cytokinesis in ipl1 mutants, suggesting that Ipl1p in budding yeast does not regulate cytokinesis like AIM-1 in mammalian cells (Terada et al. 1998). Although we did not detect a defect in spindle pole body function in ipl1 mutants, the spindle pole body-associated protein Nuf2p is asymmetrically localized in ipl1 mutants (Chan, pers. comm.) as opposed to wild-type cells in which it stains the spindle poles equally (Osborne et al. 1994). As our examination of the integral spindle pole body component, Tub4p, revealed no defect in spindle pole body localization or appearance, it remains an open question whether Ipl1p has a role in spindle pole body function. In addition, although we have not detected a defect in the mitotic spindle, the localization of Ipl1p to the spindle may reflect an unidentified role in spindle function.
Aurora2, a human Ipl1p homolog, is amplified in some colorectal and breast cancers and overexpression of the protein transforms tissue culture cells (Sen et al. 1997; Bischoff et al. 1998). This observation shows that overexpression of Ipl1p homologs induce genomic instability, and it is tempting to speculate that this is caused by altered kinetochore phosphorylation that leads to defects in chromosome segregation. The combination of our observations and the effect of ipl1 overexpression in tissue-culture cells suggest that kinetochore defects could give rise to genetic instability in human tumors. A majority of cell lines from human colorectal tumors show a high frequency of chromosome loss and gain. In some cell lines this instability appears to be caused by defects in the spindle checkpoint (Cahill et al. 1998), but it is possible that in others it is caused by defects in kinetochore behavior.
Materials and methods
Microbial techniques and yeast strain constructions
Media and genetic and microbial techniques were essentially as described (Sherman et al. 1974; Rose et al. 1990). All cytological experiments were carried out by arresting cells in 1 μg/ml α-factor at the permissive temperature (23°C) for 4 hr, washing cells twice in α-factor-free media and resuspending them in prewarmed media at 37°C. After 1 hr, α-factor was added back to prevent cells from entering the next cell cycle. All experiments reported were repeated a minimum of two times with similar results. In all experiments, at least 100 cells were counted. Stock solutions of inhibitors were: 60 mg/ml benomyl (DuPont, Boston, MA) in DMSO, 10 mg/ml nocodazole (Sigma, St. Louis, MO) in DMSO, 10 mg/ml α-factor (Biosynthesis, Lewisville, TX) in DMSO. All stocks were stored at −20°C. Hydroxyurea (Sigma) was added directly to media at a final concentration of 0.2 m. For benomyl/nocodazole experiments, cells were released into 30 μg/ml benomyl and 15 μg/ml nocodazole at 37°C.
Yeast strains are listed in Table 1. Yeast strains were constructed by standard genetic techniques. Diploids were isolated on selective media at 23°C and subsequently sporulated at 23°C. The pGAL–Δ176–CLB2 fusion that is contained in some strains is not expressed in glucose media. The strains XL1-Blue and DH5α were used for all bacterial manipulations.
Table 1.
Yeast strains used in this study
| SBY3 | MATa ura3-1 leu2-3,112 his3-11 trp1-1 ade2-1 can1-100 bar1Δ |
| SBY99 | MATa ura3-1 leu2-3,112 his3-11:pCUP1–GFP12–lacI12:HIS3 trp1-1:lacO:TRP1 ade2-1 can1-100 bar1Δ mad2::URA3 |
| SBY100 | MATa ura3-1 leu2-3,112 his3-11:pCUP1–GFP12–lacI12:HIS3 trp1-1:lacO:TRP1 ade2-1 can1-100 bar1Δ mad2::URA3 ipl1-321 |
| SBY102 | MATa ura3-1 leu2-3,112 his3-11:pCUP1–GFP12–lacI12:HIS3 trp1-1:lacO:TRP1 ade2-1 can1-100 MCD1:HA3:URA3: HA3 lys2Δ:pGAL–Δ176–Clb2:LYS2 esp1-1 |
| SBY133 | MATa ura3-1 leu2-3,112 his3-11:pCUP1–GFP12–lacI12:HIS3 ade2-1 bar1Δ ipl1-321 cdc23-1 |
| SBY134 | MATa ura3-1 leu2-3,112 his3-11:pCUP1–GFP12–lacI12:HIS3 ade2-1 bar1Δ cdc23-1 |
| SBY146 | MATa ura3-1 leu2-3,112 his3-11 trp1-1 ade2-1 can1-100 bar1Δ IPL1:IPL1-myc12 |
| SBY164 | MATa ura3-1 leu2-3,112 his3-11 trp1-1 ade2-1 ndc10-1 |
| SBY196 | MATa ura3-1 leu2-3,112 his3-11 trp1-1 ade2-1 ndc10-1 IPL1:pGAL–IPL1–myc12:URA3 |
| SBY214 | MATa ura3-1 leu2-3,112 his3-11:pCUP1–GFP12–lacI12:HIS3 trp1-1:lacO:TRP1 ade2-1 can1-100 bar1Δ lys2Δ |
| SBY215 | MATa ura3-1 leu2-3,112 his3-11:pCUP1–GFP12–lacI12:HIS3 trp1-1:lacO:TRP1 ade2-1 can1-100 bar1Δ lys2Δ:pGAL– Δ176–CLB2:LYS2 |
| SBY254 | MATα ura3-1 leu2-3,112 his3-11 trp1-1 ade2-1 can1-100 bar1Δ lys2Δ [pRS315] |
| SBY320 | MATa ura3-1 leu2-3,112 his3-11:pCUP1–GFP12–lacI12:HIS3 trp1-1:lacO:TRP1 ade2-1 can1-100 bar1Δ lys2Δ:pGAL– Δ176–CLB2:LYS2 ipl1-321 |
| SBY321 | MATα ura3-1 leu2-3,112 his3-11:pCUP1–GFP12–lacI12:HIS3 trp1-1:lacO:TRP1 ade2-1 can1-100 bar1Δ lys2Δ:pGAL– Δ176–CLB2:LYS2 ipl1-321 |
| SBY322 | MAYaura3-1 leu2-3,112 his3-11:pCUP1–GFP12–lacI12:HIS3 trp1-1:lacO:TRP1 ade2-1 can1-100 bar1Δ lys2Δ ipl1-321 |
| SBY340 | MATα ura3-1 leu2,3-112 his3-11:pCUP1–GFP12–lacI12:HIS3 trp1-1:lacO:TRP1 ade2-1 can1-100 cdc23-1 pds1-296 |
| SBY373 | MATα ura3-1 leu2-3,112 his3-11 trp1-1 ade2-1 can1-100 bar1Δ lys2Δ IPL1:URA3 [pRS315] |
| SBY376 | MATa ura3-1 leu2-3,112 his3-11:pCUP1–GFP12–lacI12:HIS3 trp1-1:LacO:TRP1 ade2-1 can1-100 bar1Δ lys2Δ:pGAL– Δ176-CLB2:LYS2 MCD1:HA3:URA3:HA3 |
| SBY378 | MATa ura3-1 leu2-3,112 his3-11:pCUP1–GFP12–lacI12:HIS3 trp1-1:lacO:TRP1 ade2-1 can1-100 bar1Δ lys2Δ:pGAL– Δ176–CLB2:LYS2 MCD1:HA3:URA3:HA3 ipl1–321 |
| SBY388 | MAYa ura3-1 leu2-3,112 his3-11:pCUP1–GFP12–lacI12:HIS3 trp1-1:lacO:TRP1 ade2-1 can1-100 bar1Δ lys2Δ IPL1:IPL1– myc12 |
| SBY394 | MATa ura3-1 leu2-3,112 his3-11:pCUP1–GFP12–lacI12:HIS3 trp1-1:lacO:TRP1 ade2-1 can1-100 bar1Δ lys2Δ IPL1: pGAL–IPL1–myc12:URA3 |
All strains are isogenic with the W303 strain background. Plasmids are indicated in brackets.
Plasmid constructions
IPL1 subclones to test complementation and linkage were constructed by digesting the isolated genomic clone with KpnI–SacI and ligating the 2788-bp fragment into the KpnI–SacI sites of pRS316 and pRS306 (Sikorski and Hieter 1989) to create pSB148 and pSB149, respectively. pSB149 was digested with HpaI to integrate at the IPL1 locus. To create pGAL–IPL1, IPL1 was PCR amplified from pSB148 using primers IPL1.BAM5 (5′-GAT/GGC/ATC/GGA/TCC/ATG/CAA/CGC/AAT/AGT/TTA/G-3′) and IPL1.XBA3 (5′-GAT/GCC/ATC/TCT/AGA/CTA/TAA/CCG/CTT/ATT/TCC-3′). These primers engineer BamHI and XbaI sites at the 5′ and 3′ ends of the IPL1 gene, respectively, and the BamHI site is adjacent to the ATG codon at the start site of IPL1. The resulting PCR product was digested with BamHI–XbaI and ligated into the BamHI–XbaI sites of pDK20 (gift of Doug Kellogg, University of California, Santa Cruz) to create pSB164. To integrate into yeast, it was digested with StuI.
A clone that encoded Ipl1p tagged with 12 copies of the Myc epitope was constructed in three steps. First, a 450-bp BamHI–SacI fragment from pLH71 (L. Hwang, unpubl.) that encodes Myc12 was ligated into the BamHI–SacI sites of pRS306 to create pSB162. This plasmid was then digested with SpeI and filled in with the Klenow fragment of DNA polymerase I to insert a stop codon after the Myc12 tag, creating pSB167. Second, the IPL1 gene was PCR amplified using primers IPL1.XHO5 (5′-CAT/GCG/ATC/CTC/GAG/GCA/TTG/CTT/ATT/GAA/CAG-3′) and IPL1.BAM3 (5′-GCA/TGG/ATC/GGA/TCC/TAA/CCG/CTT/ATT/TTC/CC-3′), cleaved with XhoI and BamHI and ligated into pGEM-2T (Promega, Madison, WI) to create pSB165. Finally, pSB165 was digested with XhoI–BamHI and the IPL1 gene was ligated into pSB167 digested with XhoI–BamHI to create pSB169, an Ipl1p–Myc12 integrating plasmid.
To create a pGAL–IPL1–MYC12 clone, pSB169 was digested with HpaI–SacI and this fragment was ligated into pSB164 cut with HpaI–SacI to create pSB171. A GST–Ipl1p fusion was made by PCR amplifying ipl1 from pSB148 with primers IPL1.BAM5 and IPL1.BAM3 and digesting the PCR product with BamHI. This was ligated into the pGEX–2T (Qiagen, Chatsworth, CA) BamHI site to create pSB181, an in-frame GST–Ipl1p fusion expressed in bacteria. A pCUP–GFP12–lacI12 fusion protein, pSB116, was constructed by isolating the 435-bp BamHI–EcoRI CUP1 promoter fragment from pPW66T (Dohmen et al. 1994) filling it in with the Klenow fragment of DNA polymerase I and ligating it into pAFS135 (Straight et al. 1998 ), which encodes a GFP–Lac repressor fusion that had been digested with XhoI and filled in with Klenow.
Isolation and analysis of IPL1
The LOC screen was performed on the mutagenized parent strain SBY215 and the details of the screen will be published elsewhere (N. Bhalla, S. Biggins, and A.W. Murray, unpubl.). To confirm that IPL1 is linked to the loc2 mutation, pSB149 was digested with HpaI to linearize the plasmid in the IPL1 gene and subsequently integrated into SBY254 to create SBY373. Integration at IPL1 was confirmed by Southern analysis, and then SBY373 was crossed to SBY320. The resulting diploid was dissected and all temperature-senstive spores (loc2 mutants) segregated away from the marked IPL1:URA3 gene in 22 tetrads dissected, confirming that the IPL1 gene is tightly linked to the loc2 mutation. In addition, plasmids (ex:pSB164) containing solely PCR-amplified IPL1 complement the loc2 temperature sensitive mutation, confirming that the IPL1 gene corresponds to LOC2.
MCD1–HA3 strains were made by transformation using PCR amplified HA epitopes as described (Schneider et al. 1995). The triple HA epitope was PCR amplified from plasmidpMPY–3×HA using primers MCD–HA–up (5′-GGA/AAT/ATT/AAA/ATA/GAC/GCC/AAA/CCT/GCA/CTA/TTT/GAA/AGG/TTT/ATC/ATA/GCT/AGG/GAA/CAA/AAG/CTG/G-3′) and MCD–HA–down (5′-CAT/CAG/CTT/ATT/GGG/TCC/ACC/AAG/AAA/TCC/CCT/CGG/CGT/AAC/TAG/GTT/TTA/CTA/TAG/GGC/GAA/TTG/G-3′) that are designed to integrate after the last codon in the MCD1 gene. The PCR product was transformed into SBY215 and SBY322 to create strains SBY376 and SBY378, respectively.
IPL1 cloning
The loc2 mutant was cloned by complementation of the temperature-sensitive phenotype of SBY321 using a centromere-based yeast genomic library as described by Hardwick and Murray 1995. Plasmid DNA from colonies that grew at 37°C was isolated (Schneider et al. 1995) and transformed into bacteria. The DNA was retransformed into SBY321 and plasmids that conferred temperature resistance were sequenced with the Sequenase version 2.0 kit supplied by United States Biochemical Corp. (Cleveland, OH) according to the manufacturer’s instructions. To identify the complementing region of the loc2 suppressing clones, a 2788-bp fragment containing the IPL1 gene and flanking genomic DNA was subcloned and found to complement the temperature-sensitive phenotype.
Protein and immunological techniques
Protein extracts for Western blotting were made as described (Minshull et al. 1996). Lysates were separated on denaturing polyacrylamide gels and transferred to nitrocellulose (Schleicher & Schuell, Keene, NH) and blotted as described (Minshull et al. 1996). 12CA5 antibodies that recognize the HA tag were from BabCO (Berkeley, CA) and 9E10 antibodies that recognize the Myc tag were a gift of Sue Jaspersen (UCSF). Both antibodies were used at a 1:1000 dilution. Cdc28 antibodies were used at a 1:1000 dilution (Charles et al. 1998). Anti-Tub4p antibodies were a gift from L. Marschall and Tim Stearns (Stanford, CA) and used at a 1:500 dilution. Anti-tubulin antibodies, yol 1/34, were obtained from Accurate Chemical and Scientific Corp. (Westbury, NY) and used at a 1:100 dilution. Anti-GFP antibodies were a gift of A. Straight (UCSF) and used at a 1:1000 dilution. GST and GST–ipl1 proteins were purified from bacterial strain DH5α as described by (Kellogg et al. 1995).
Microscopy
To perform microscopy, 1 ml of cells were harvested, pelleted, and resuspended in 100 μl paraformaldehyde at room temperature for 15 min. Cells were pelleted, washed once in 1 ml of 0.1 m potassium phosphate, 1.2 m Sorbitol buffer, and resuspended in the same buffer. Cells were sonicated prior to microscopy. Immunofluorescence was carried out as described (Rose et al. 1990). DAPI was obtained from Molecular Probes (Eugene, OR) and used at 1 μg/ml final concentration. Chromosome spreads were performed as described (Michaelis et al. 1997; Loidl et al. 1998). Lipsol was obtained from Lip Ltd. (Shipley, England).
Kinase assays and kinetochore assays
Purified GST and GST–Ipl1p (0.8 μg) were added to 25-μl kinase reactions containing 50 mm Tris-Cl (pH 7.4), 25 mm β-glycerophosphate, 1 mm DTT, 10 μm ATP, 5 mm magnesium chloride, and 2.5 μCi of [γ-32P]ATP (Amersham, Arlington Heights, IL) for 15 min at 30°C. Two micrograms of each of the following substrates were added to the reaction: myelin basic protein (Sigma), histone HI (Sigma), dephosphorylated casein (Sigma), and carboxy-terminal GST–Ndc10p. Reactions were stopped with 25 μl of 2× sample buffer (Hardwick and Murray 1995) and loaded on 15% polyacrylamide gels. Gels were stained with Coomassie blue, dried, and exposed to film. Microtubule binding assays were performed as described (Sassoon et al. 1999). Gel-shift assays were performed as described (Sorger et al. 1995).
Acknowledgments
We are grateful to Laura Marschall and Tim Stearns for Tub4p antibodies, the Morgan lab for Cdc28p and anti-myc antibodies and Aaron Straight for GFP antibodies. We thank members of the Murray and Morgan labs for critical reading of the manuscript and many insightful discussions and suggestions and Clarence Chan for sharing unpublished information. In addition, we thank the Sorger laboratory for contributions to studies on Ndc10p. This work was supported by a Jane Coffin Childs postdoctoral fellowship to S.B., a National Science Foundation predoctoral fellowship to N.B., and grants from the National Institutes of Health and the Human Frontier Science Program to A.W. Murray.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL sbiggins@cgl.ucsf.edu; FAX (415) 476-4929.
References
- Axton JM, Dombradi V, Cohen PT, Glover DM. One of the protein phosphatase 1 isoenzymes in Drosophila is for mitosis. Cell. 1990;63:33–46. doi: 10.1016/0092-8674(90)90286-n. [DOI] [PubMed] [Google Scholar]
- Biggins S, Murray A. Sister chromatid cohesion in mitosis. Curr Opin Cell Biol. 1998;10:769–775. doi: 10.1016/s0955-0674(98)80120-8. [DOI] [PubMed] [Google Scholar]
- Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, Chan CS, Novotny M, Slamon DJ, Plowman GD. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998;17:3052–3065. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- Chan CS, Botstein D. Isolation and characterization of chromosome-gain and ploidy mutants in yeast. Genetics. 1993;135:677–691. doi: 10.1093/genetics/135.3.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles J, Jaspersen SL, Tinker-Kulberg RL, Hwang L, Szidon A, Morgan DO. The Polo-related kinase Cdc5 activates and is destroyed by the mitotic cyclin destruction machinery in S. cerevisiae. Curr Biol. 1998;9:497–507. doi: 10.1016/s0960-9822(98)70201-5. [DOI] [PubMed] [Google Scholar]
- Cho R, Campbell M, Winzeler E, Steinmetz L, Conway A, Wodicka L, Wolfsberg T, Gabrielian A, Landsman D, Lockhart D, Davis R. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell. 1988;2:65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- Ciosk R, Zachariae W, Michaelis C, Shevchenko A, Mann M, Nasmyth K. An ESP1/PDS1 complex regulates loss of sister chromatid metaphase to anaphase transition in yeast. Cell. 1998;93:1067–1076. doi: 10.1016/s0092-8674(00)81211-8. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O, Koshland D. The metaphase-to-anaphase transition: Avoiding a mid-life crisis. Curr Opin Cell Biol. 1997;9:800–806. doi: 10.1016/s0955-0674(97)80080-4. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O, Peters JM, Kirschner MW, Koshland D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes & Dev. 1996;10:3081–3093. doi: 10.1101/gad.10.24.3081. [DOI] [PubMed] [Google Scholar]
- Dohmen RJ, Wu P, Varshavsky A. Heat-inducible degron: A method for constructing temperature-sensitive mutants. Science. 1994;263:1273–1276. doi: 10.1126/science.8122109. [DOI] [PubMed] [Google Scholar]
- Doonan JH, Morris NR. The bimG gene of Aspergillus nidulans, required for completion of anaphase, encodes a homolog of mammalian phosphoprotein phosphatase 1. Cell. 1989;57:987–996. doi: 10.1016/0092-8674(89)90337-1. [DOI] [PubMed] [Google Scholar]
- Francisco L, Chan CS. Regulation of yeast chromosome segregation by Ipl1 protein type 1 protein phosphatase. Cell Mol Biol Res. 1994;40:207–213. [PubMed] [Google Scholar]
- Francisco L, Wang W, Chan CS. Type 1 protein phosphatase acts in opposition to Ipl1 protein kinase in regulating yeast chromosome segregation. Mol Cell Biol. 1994;14:4731–4740. doi: 10.1128/mcb.14.7.4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser JR, Schott EJ, Kingsbury TJ, Cole NB, Totis LJ, Bhattacharyya G, He L, Hoyt MA. Saccharomyces cerevisiae genes required in the absence of the CIN8-encoded spindle motor act in functionally diverse mitotic pathways. Mol Biol Cell. 1997;8:1035–1050. doi: 10.1091/mbc.8.6.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover DM, Leibowitz MH, McLean DA, Parry H. Mutations in aurora prevent centrosome separation leading to formation of monopolar spindles. Cell. 1995;81:95–105. doi: 10.1016/0092-8674(95)90374-7. [DOI] [PubMed] [Google Scholar]
- Goh PY, Kilmartin JV. NDC10: A gene involved in chromosome segregation in Saccharomyces cerevisiae. J Cell Biol. 1993;121:503–512. doi: 10.1083/jcb.121.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalan G, Chan CS, Donovan PJ. A novel mammalian, mitotic spindle-associated kinase is related yeast and fly chromosome segregation regulators. J Cell Biol. 1997;138:643–656. doi: 10.1083/jcb.138.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guacci V, Koshland D, Strunnikov A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell. 1997;91:47–57. doi: 10.1016/s0092-8674(01)80008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick K, Murray AW. Mad1p, a phosphoprotein component of the spindle assembly checkpoint in budding yeast. J Cell Biol. 1995;131:709–720. doi: 10.1083/jcb.131.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisamoto N, Sugimoto K, Matsumoto K. The Glc7 type 1 protein phosphatase of Saccharomyces is required for cell cycle progression in G2/M. Mol Cell Biol. 1994;14:3158–3165. doi: 10.1128/mcb.14.5.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt MA, Trotis L, Roberts BT. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 1991;66:507–517. doi: 10.1016/0092-8674(81)90014-3. [DOI] [PubMed] [Google Scholar]
- Hoyt MA, He L, Loo KK, Saunders WS. Two Saccharomyces cerevisiae kinesin-related gene products required for mitotic spindle assembly. J Cell Biol. 1992;118:109–120. doi: 10.1083/jcb.118.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA, Sorger PK. Structure and function of kinetochores in budding yeast. Annu Rev Cell Dev Biol. 1995;11:471–495. doi: 10.1146/annurev.cb.11.110195.002351. [DOI] [PubMed] [Google Scholar]
- Kaplan KB, Hyman AA, Sorger PK. Regulating the yeast kinetochore by ubiquitin-dependent degradation and Skp1p-mediated phosphorylation. Cell. 1997;91:491–500. doi: 10.1016/s0092-8674(00)80435-3. [DOI] [PubMed] [Google Scholar]
- Kellogg DR, Kikuchi A, Fujii-Nakata T, Turck CW, Murray AW. Members of the NAP/SET family of proteins interact specifically with B-type cyclins. J Cell Biol. 1995;130:661–673. doi: 10.1083/jcb.130.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Kotani S, Hattori T, Sumi N, Yoshioka T, Todokoro K, Okano Y. Cell cycle-dependent expression and spindle pole localization of novel human protein kinase, Aik, related to Aurora of Drosophila and yeast Ipl1. J Biol Chem. 1997;272:13766–13771. doi: 10.1074/jbc.272.21.13766. [DOI] [PubMed] [Google Scholar]
- Lechner J, Carbon J. A 240 Kd multisubunit complex, CBF3, is a major component of the budding yeast centromere. Cell. 1991;64:717–725. doi: 10.1016/0092-8674(91)90501-o. [DOI] [PubMed] [Google Scholar]
- Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell. 1991;66:519–531. doi: 10.1016/0092-8674(81)90015-5. [DOI] [PubMed] [Google Scholar]
- Loidl J, Klein F, Engebrech J. Genetic and morphological approaches for the analysis of meiotic chromosomes in yeast. Meth Cell Biol. 1998;53:257–285. doi: 10.1016/s0091-679x(08)60882-1. [DOI] [PubMed] [Google Scholar]
- Marschall LG, Jeng RL, Mulholland J, Stearns T. Analysis of Tub4p, a yeast gamma-tubulin-like protein: Implications for microtubule-organizing center function. J Cell Biol. 1996;134:443–454. doi: 10.1083/jcb.134.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall WF, Straight A, Marko JF, Swedlow J, Dernburg A, Belmont A, Murray AW, Agard DA, Sedat JW. Interphase chromosomes undergo constrained diffusional motion in living cells. Curr Biol. 1997;7:930–939. doi: 10.1016/s0960-9822(06)00412-x. [DOI] [PubMed] [Google Scholar]
- Michaelis C, Ciosk R, Nasmyth K. Cohesins: Chromosomal proteins that prevent premature separation of sister chromatids. Cell. 1997;91:35–45. doi: 10.1016/s0092-8674(01)80007-6. [DOI] [PubMed] [Google Scholar]
- Minshull J, Straight A, Rudner A, Dernburg A, Belmont A, Murray AW. Protein phosphatase 2A regulates MPF activity and sister chromatid cohesion in budding yeast. Curr Biol. 1996;6:1609–1620. doi: 10.1016/s0960-9822(02)70784-7. [DOI] [PubMed] [Google Scholar]
- Nicklas RB. Measurements of the force produced by the mitotic spindle in anaphase. J Cell Biol. 1983;97:542–548. doi: 10.1083/jcb.97.2.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkura H, Noriyuki K, Miyatani S, Toda T, Yanagida M. The fission yeast dis2+ gene required for chromosome disjoining encodes one of two putative type 1 protein phosphatases. Cell. 1989;57:997–1007. doi: 10.1016/0092-8674(89)90338-3. [DOI] [PubMed] [Google Scholar]
- Osborne M, Schlendstedt G, Jinks T, Silver P. Nuf2, a spindle pole body-associated protein required for nuclear division in yeast. J Cell Biol. 1994;125:853–866. doi: 10.1083/jcb.125.4.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatti S, Lengauer C, Nasmyth K. Cdc6 is an unstable protein whose de novo synthesis in G1 is important for the onset of S phase and for preventing a ’reductional’ anaphase in the budding yeast Saccharomyces cerevisiae. EMBO J. 1995;14:3788–3799. doi: 10.1002/j.1460-2075.1995.tb00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL, Alexander SP. Kinetochores are transported poleward along a single astral microtubule during chromosome attachment to the spindle in newt lung cells. J Cell Biol. 1990;110:81–95. doi: 10.1083/jcb.110.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roghi C, Giet R, Uzbekov R, Morin N, Chartrain I, Le Guellec R, Couturier A, Doree M, Philippe M, Prigent C. The Xenopus protein kinase pEg2 associates with the cell cycle-dependent manner, binds to the spindle microtubules involved in bipolar mitotic spindle assembly. J Cell Sci. 1998;111:557–572. doi: 10.1242/jcs.111.5.557. [DOI] [PubMed] [Google Scholar]
- Roof DM, Meluh PB, Rose MD. Kinesin-related proteins required for assembly of the mitotic spindle. J Cell Biol. 1992;118:95–108. doi: 10.1083/jcb.118.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose M, Winston F, Heiter P. Methods in yeast genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1990. p. 198. [Google Scholar]
- Rudner AD, Murray AW. The spindle assembly checkpoint. Curr Opin Cell Biol. 1996;8:773–780. doi: 10.1016/s0955-0674(96)80077-9. [DOI] [PubMed] [Google Scholar]
- Sassoon, I., F.F. Severin, P.D. Andrews, M.-R. Taba, K.B. Kaplan, A.J. Ashford, M.J.R. Stark, P.K. Sorger, and A.A. Hyman. Regulation of Saccharomyces cerevisiae kinetochores by the type I phosphatase Glc7p. Genes & Dev. (this issue). [DOI] [PMC free article] [PubMed]
- Schneider BL, Seufert W, Steiner B, Yang QH, Futcher AB. Use of polymerase chain reaction epitope tagging for protein Saccharomyces cerevisiae. Yeast. 1995;11:1265–1274. doi: 10.1002/yea.320111306. [DOI] [PubMed] [Google Scholar]
- Sen S, Zhou H, White R. A putative serine/threonine kinase encoding gene BTAK on chromosome 20q13 is amplified and overexpressed in human breast cancer cell lines. Oncogene. 1997;14:2195–2200. doi: 10.1038/sj.onc.1201065. [DOI] [PubMed] [Google Scholar]
- Sherman F, Fink G, Lawrence C, editors. Methods in yeast genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1974. [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibbens RV, Skeen VP, Salmon ED. Directional instability of kinetochore motility during chromosome congression and segregation in mitotic newt lung cells: A push-pull mechanism. J Cell Biol. 1993;122:859–875. doi: 10.1083/jcb.122.4.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorger PK, Severin FF, Hyman AA. Factors required for the binding of reassembled yeast kinetochores to microtubules in vitro. J Cell Biol. 1994;127:995–1008. doi: 10.1083/jcb.127.4.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorger PK, Doheny KF, Hieter P, Kopski KM, Huffaker TC, Hyman AA. Two genes required for the binding of an essential cerevisiae kinetochore complex to DNA. Proc Natl Acad Sci. 1995;92:12026–12030. doi: 10.1073/pnas.92.26.12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmann O, Lechner J. The Saccharomyces cerevisiae kinetochore contains a cyclin-CDK complexing homologue, as identified by in vitro reconstitution. EMBO J. 1996;15:3611–3620. [PMC free article] [PubMed] [Google Scholar]
- Straight AF, Belmont AS, Robinett CC, Murray AW. GFP tagging of budding yeast chromosomes reveals that protein–protein interactions can mediate sister chromatid cohesion. Curr Biol. 1996;6:1599–1608. doi: 10.1016/s0960-9822(02)70783-5. [DOI] [PubMed] [Google Scholar]
- Straight AF, Marshall WF, Sedat JW, Murray AW. Mitosis in living budding yeast: Anaphase A but no metaphase plate. Science. 1997;277:574–578. doi: 10.1126/science.277.5325.574. [DOI] [PubMed] [Google Scholar]
- Straight A, Sedat J, Murray A. Time lapse microscopy reveals unique roles for kinesins during anaphase in budding yeast. J Cell Biol. 1998;143:687–694. doi: 10.1083/jcb.143.3.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada Y, Tatsuka M, Suzuki F, Yasuda Y, Fujita S, Otsu M. AIM-1: a mammalian midbody-associated protein required for cytokinesis. EMBO J. 1998;17:667–676. doi: 10.1093/emboj/17.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters JC, Skibbens RV, Salmon ED. Oscillating mitotic newt lung cell kinetochores are, on average, under tension and rarely push. J Cell Sci. 1996;109:2823–2831. doi: 10.1242/jcs.109.12.2823. [DOI] [PubMed] [Google Scholar]