

Abstract
We have investigated the role of protein phosphorylation in regulation of Saccharomyces cerevisiae kinetochores. By use of phosphatase inhibitors and a type 1 protein phosphatase mutant (glc7-10), we show that the microtubule binding activity, but not the centromeric DNA-binding activity, of the kinetochore complex is regulated by a balance between a protein kinase and the type 1 protein phosphatase (PP1) encoded by the GLC7 gene. glc7-10 mutant cells exhibit low kinetochore-microtubule binding activity in vitro and a high frequency of chromosome loss in vivo. Specifically, the Ndc10p component of the centromere DNA-binding CBF3 complex is altered by the glc7-10 mutation; Ndc10p is hyperphosphorylated in glc7-10 extracts. Furthermore, addition of recombinant Ndc10p reconstitutes the microtubule-binding activity of a glc7-10 extract to wild-type levels. Finally, the glc7-10-induced mitotic arrest is abolished in spindle checkpoint mutants, suggesting that defects in kinetochore–microtubule interactions caused by hyperphosphorylation of kinetochore proteins activate the spindle checkpoint.
Keywords: Checkpoint, kinetochore, mitosis, type 1 protein phosphatase
One of the most important events in eukaryotic cell division is the faithful segregation of chromosomes to the two daughter cells. Chromosome segregation is mediated in large part by kinetochores, DNA–protein complexes that assemble on centromeric DNA and bind to microtubules of the mitotic spindle. Sister chromatid pairs make bipolar attachments to the spindle, with one chromatid bound to microtubules from one pole and the other chromatid bound to microtubules from the opposite pole. Once both kinetochores of each chromosome have established proper microtubule attachment, anaphase is triggered: Sister chromatids disjoin and move toward opposite spindle poles. Ensuring the faithful execution of this process requires tight regulation of the assembly, microtubule-binding properties, and motility of kinetochores. As yet, little is known about the mechanisms that control these important processes.
Evidence from several organisms suggests that type 1 protein phosphatases (PP1) are required for chromosome segregation. In Saccharomyces cerevisiae, the catalytic subunit of PP1 is encoded by a single essential gene GLC7. Glc7p mutants arrest before anaphase onset with replicated DNA, short spindles, and high levels of Cdc28p kinase activity (Hisamoto et al. 1994; Black et al. 1995; MacKelvie et al. 1995). In the distantly related fission yeast Schizosaccharomyces pombe, sds21 dis2 double mutant cells carrying loss-of-function mutations in both of the genes encoding catalytic PP1 subunits, also arrest in mitosis at the restrictive temperature, and have short metaphase spindles and condensed and unseparated chromosomes (Ohkura et al. 1988, 1989; Ishii et al. 1996). Similarly, in Aspergillus nidulans, BimG (PP1) mutants at the restrictive temperature are blocked in mitosis with condensed chromosomes and short metaphase spindles (Doonan and Morris 1989). In Drosophila melanogaster, mutations in one of the four genes encoding PP1 isoenzymes result in overcondensed chromosomes, and no elongated anaphase spindles (Axton et al. 1990). Finally, experiments in mammalian cells have shown that the microinjection of anti-PP1 antibodies causes cells to arrest their division at metaphase (Fernandez et al. 1992). These data show that PP1 function is essential for the onset of anaphase and for correct chromosome segregation in a wide variety of eukaryotes. The reasons for the mitotic arrest are unknown. One possibility is that in the absence of PP1, kinetochores are incorrectly assembled or regulated, thereby triggering the spindle checkpoint that monitors the attachment of kinetochores to microtubules and prevents cell cycle progression to anaphase when attachment is defective (for review, see Wells 1996; Hardwick 1998).
In S. cerevisiae, centromeric sequences are well conserved among all 16 chromosomes and are composed of three elements, CDEI, CDEII, and CDEIII (Clarke and Carbon 1980; Fitzgerald et al. 1982; Hegemann and Fleig 1993). Point mutations in the highly conserved central bases of CDEIII inactivate the centromere in vivo, showing that CDEIII plays a critical role in kinetochore function (McGrew et al. 1986; Ng and Carbon 1987). CDEIII is bound by CBF3, a complex consisting of four proteins, Skp1p (23 kD), Ctf13p (58 kD), Cep3p (64 kD), and Ndc10p (110 kD) all of which are necessary for DNA-binding activity and for cell viability (for review, see Hyman and Sorger 1995; Clarke 1998). Previously, we described a method to partially reassemble yeast kinetochores on centromeric DNA in vitro and measure the binding of these centromere complexes to microtubules (Sorger et al. 1994). This assay established that CBF3 plays an obligate, though not sufficient role in the microtubule-binding activity of kinetochores. However, the molecular nature of this microtubule-binding activity and how it is regulated is unclear to date.
Here, we report that PP1 regulates the attachment of kinetochores to microtubules in S. cerevisiae. Budding yeast contain a single PP1 catalytic subunit, Glc7p. Kinetochores reconstituted in vitro from Glc7p mutant cell extracts have reduced microtubule binding relative to those reconstituted from wild-type extracts. CEN DNA binding by the CBF3 protein complex is identical in Glc7p mutant and wild-type extracts, suggesting that Glc7p specifically regulates the microtubule-binding activity of kinetochores. Kinetochore microtubule binding in Glc7p mutant extracts is restored to wild-type levels by addition of recombinant Ndc10p but not Cep3p or Ctf13p–Skp1p, suggesting that Ndc10p is the target of the Glc7p pathway in budding yeast. These results suggest that the mitotic arrest of Glc7p mutants is triggered by defective kinetochore–microtubules interactions.
Results
Microtubule binding by kinetochore complexes is regulated by phosphorylation
To investigate the role of phosphorylation and dephosphorylation in the regulation of kinetochore activity, we added ATP to the in vitro microtubule binding assay for reconstituted yeast kinetochores (Sorger et al. 1994). This assay consists of reassembling kinetochore complexes on centromeric DNA linked to fluorescent latex beads by incubation in a yeast extract. Then, the beads are incubated with taxol-stabilized microtubules and bead–microtubule interactions are monitored by fluorescence microscopy. In extracts from wild-type cells, we observed high levels of bead binding, corresponding to reconstitution of ∼10%–20% of the kinetochores present in the starting culture. Using a luciferase assay, we determined that the concentration of ATP in these reactions was <0.1 μm, a concentration insufficient for kinases to phosphorylate their substrates. When ATP was added to extracts to a final concentration of 1 mm, the number of beads bound to microtubules decreased approximately threefold (Fig. 1A). When apyrase was added after having incubated the extract in the presence of ATP, microtubule-binding activity returned to wild-type levels. Thus, kinetochore–microtubule binding activity was reduced by the addition of ATP and restored by depletion of the added ATP by apyrase. This may reflect the presence of both a kinase activity that is inhibitory for microtubule binding in the presence of ATP, as well as an opposing phosphatase activity that activates kinetochore–microtubule attachment, as suggested by the restoration of microtubule-binding activity after apyrase addition.
Figure 1.


(A) Microtubule-binding activity (number of beads bound per field) of extracts coming from wild-type exponential cultures grown at 30°C. ATP, and/or phosphatase inhibitors were added to the extract as described. Apyrase was added 40 min after ATP and/or phosphatase inhibitors and microtubule-binding activity was measured soon thereafter. The background level of binding typically observed with beads carrying nonfunctional CEN-DNA was around two per field. Microtubule binding was normalized to 106 beads/μl and a field size of 25,000 μm2. (Solid bars) No apyrase added; (shaded bars) apyrase added. (B) DNA-binding activity of extracts coming from a wild-type exponential culture grown at 30°C, in the presence of ATP and/or phosphatase inhibitors. The ATP concentration in both microtubule-binding and band-shift reactions was 1 mm, the okadaic acid concentration was 0.1 μm, the microcystin-LR concentration was 1 μm, and the apyrase concentration was 1 U/μl. (OA) Okadaic acid; (MC) microcystin; (Ap) Apyrase; (C) control (no additions).
To determine the type of phosphatase involved in the ATP-dependent regulation of microtubule-binding activity, we added two different phosphatase inhibitors. Microcystin-LR (Runnegar et al. 1995) is a potent inhibitor of PP1 and PP2A, whereas okadaic acid at low concentration is a selective inhibitor of PP2A (Zhang et al. 1994). When microcystin-LR and ATP were added simultaneously to reactions, microtubule-binding activity was reduced 15-fold relative to untreated reactions. The subsequent addition of apyrase to the microcystin and ATP-treated reactions did not restore activity (Fig. 1A). These data suggest that microcystin is inhibiting the function of a phosphatase that normally stimulates microtubule binding. To determine whether this is a type 1 or type 2A phosphatase, we repeated the experiment using okadaic acid at the PP2A-selective concentration of 0.1 μm. In ATP and okadaic acid-treated reactions, microtubule binding was sixfold lower than in untreated reactions but, significantly, binding could be restored by the addition of apyrase (Fig. 1A). As a control, when microcystin-LR or okadaic acid alone was added to extracts, no decrease in kinetochore–microtubule binding activity was observed. This suggests that without ATP, endogenous kinase activity is negligible and substrates are not phosphorylated. These findings strongly indicate that PP1 activates microtubule binding. In microcystin-treated reactions, the PP1 is inactivated, and thus, following the addition of ATP, inhibitory kinases are able to down-regulate microtubule binding.
Next, we asked whether ATP was regulating the DNA-binding activity of CBF3 or some other aspect of kinetochore formation. The binding of CBF3 to DNA was measured by incubation of extracts with radiolabeled CDEIII–DNA (Sorger et al. 1995) and then testing of CBF3–CDEIII complex formation with a bandshift assay. We observed that the addition of ATP to CDEIII-binding reactions, with or without either microcystin or okadaic acid, did not alter the amount of DNA-bound CBF3 (Fig. 1B). Thus, ATP did not appear to be altering the ability of CBF3 to bind to DNA. Instead, it must regulate another step in the formation of a microtubule-attachment site.
Reduced microtubule-binding activity in a PP1 mutant
To test directly whether PP1 regulates microtubule binding by kinetochore complexes, we assayed extracts from cells carrying a mutation in the unique PP1 catalytic subunit GLC7. One temperature-sensitive allele of GLC7, glc7-10, exhibits a G2/M arrest at the restrictive temperature (P.D. Andrews and M.J.R. Stark, in prep.). Following the shift of glc7-10 to 37°C, the majority of cells exhibit a large budded phenotype with an elongated bud, show a short mitotic spindle with poor tubulin staining, and have their nucleus positioned at the bud neck. These phenotypes are consistent with a defect in kinetochore function. We prepared extracts from glc7-10 mutant cells grown at the permissive temperature and at the nonpermissive temperature, and observed three- to sixfold lower microtubule-binding activity in both extracts compared with congenic wild-type extracts (Fig. 2A). We could not observe a significant difference between glc7-10 extracts from cells grown at 26°C and 37°C, due to the difficulty of making accurate quantification of low numbers in the in vitro assay. Interestingly, the three- to sixfold decrease in microtubule-binding activity of glc7-10 relative to wild type is similar to that observed by treatment of extracts from wild-type cells with ATP (Fig. 1A). We then assayed the effect of ATP addition to glc7-10 and wild-type extracts prepared from either cells grown at the permissive temperature (Fig. 2B, glc7-10 26°C) or shifted to 37°C for 3 hr (Fig. 2B, glc7-10 37°C). When ATP was added to these extracts, microtubule-binding activity decreased, as observed previously (Fig. 1A). After ATP depletion by apyrase, the microtubule-binding activity was restored in wild-type extracts (Fig. 2C). In contrast, microtubule-binding activity was not restored by apyrase addition in glc7-10 37°C extracts and marginally restored in glc7-10 26°C extracts (Fig. 2B). This result suggests that at the restrictive temperature, Glc7-10p protein function is necessary for re-establishing kinetochore–microtubule attachment on ATP depletion.
Figure 2.
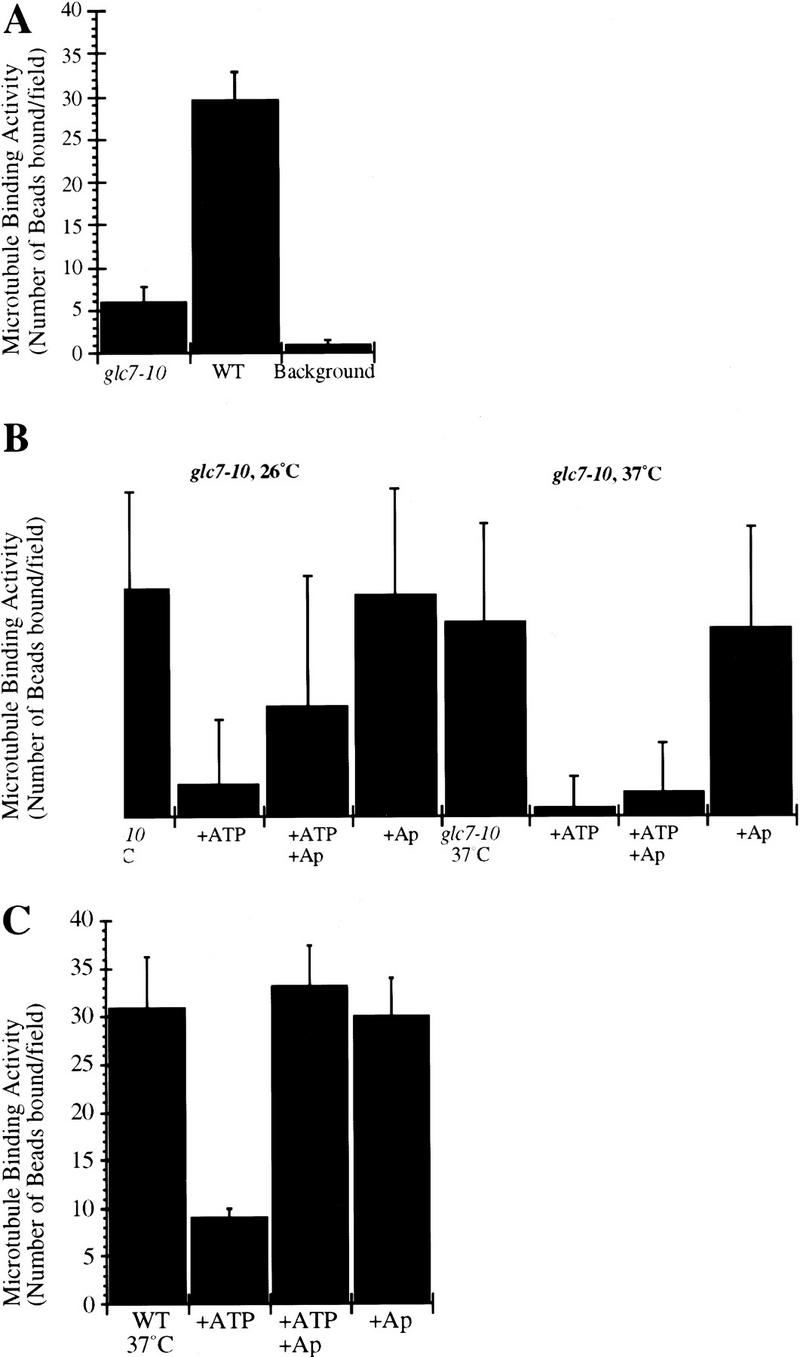
(A) Microtubule-binding activity (number of beads bound per field) of glc7-10 mutant and wild-type extracts prepared from cells grown at the permissive temperature. The background level of binding typically observed with beads carrying nonfunctional CEN–DNA was around two per field. Microtubule binding was normalized to 106 beads/μl and a field size of 25,000 μm2. (B) Microtubule-binding activity (number of beads bound per field) of glc7-10 mutant extracts from cells grown at the permissive temperature (glc7-10 26°C) or shifted for 3 hr to the nonpermissive temperature (glc7-10 37°C) after incubation of the extracts in the presence of ATP or after depletion of ATP by apyrase. ATP was added at 1 mm, apyrase at 1 U/μl of the reaction volume after a 40-min incubation with ATP. For better visualization of the differences in microtubule binding activity, note the difference in the y-axis compared with those in graphs A and C. (C) Microtubule-binding activity (number of beads bound per field) of wild-type extracts from cells shifted for 3 hr to the nonpermissive temperature. ATP was added at 1 mm, apyrase at 1 U/μl of the reaction volume after incubation with ATP for 40 min.
To know whether the decrease in microtubule-binding activity was specific for glc7-10 allele, we assayed the kinetochore–microtubule binding activity of a glc7-1 mutant extract. glc7-1 cells are defective in glycogen synthesis (Cannon et al. 1994). This extract does not show any defect in our in vitro assay (data not shown), suggesting that the lack of microtubule-binding activity in glc7-10 mutant is likely to be specific for mitotic alleles of GLC7.
To determine whether microtubule binding was also decreased in PP2A mutants, we examined cells carrying temperature-sensitive mutations in PP2A genes (Evans and Stark 1997). S. cerevisiae contains two PP2A catalytic subunits encoded by the PPH21 and PPH22 genes. Mutations in either pph21 or pph22 block cells in mitosis, although pph21 mutants eventually proceed through a round of aberrant chromosome segregation (Evans and Stark 1997). No significant differences in microtubule-binding activity between wild-type and PP2A mutant cells were observed (D.R.H. Evans, I. Sassoon, P.D. Andrews, A.A. Hyman, and M.J.R. Stark, in prep.). Taken together, these results suggest that Glc7p is part of a kinase–phosphatase balance that regulates the microtubule binding activity of kinetochores.
In vivo evidence for Glc7p function in chromosome segregation
We wanted to relate the observation that glc7-10 cells have reduced microtubule-binding activity in vitro with their in vivo mitotic phenotype. To test this relationship, we determined whether glc7-10 mutants had an increased frequency of chromosome loss. We visualized the mitotic stability of an artificial chromosome containing the SUP11 marker, using the red/white sectoring assay (see Materials and Methods) at permissive and semipermissive temperature (Fig. 3A,B). Red sectored colonies were already present at 26°C and greatly increased in the glc7-10 mutant at 35.5°C. Thus, the dramatic increase in chromosome loss in vivo in the glc7-10 mutant indicates that Glc7p is required for the fidelity of mitotic chromosome segregation and is consistent with the reduction of microtubule–kinetochore interactions in glc7-10 cell extracts in our in vitro assay at the permissive and nonpermissive temperatures.
Figure 3.


(A) Chromosome loss phenotype of glc7-10 cells at the semipermissive temperature. Chromosome loss was monitored visually by red sectored colonies after growth of strains on YPD medium at 35.5°C. (B) Chromosome loss quantitation: Fraction of total colonies that were red or sectored at 26°C, 32°C, 34°C, and 35.5°C. (The number of total colonies counted are in parentheses).
A defect in kinetochore–microtubule interaction has been proposed to activate the spindle checkpoint and delay the onset of anaphase (for review, see Wells 1996). Thus, the G2/M arrest in a glc7-10 mutant could result from kinetochore defects, triggering the spindle checkpoint. To test this possibility, we generated double mutants between glc7-10 and mutants preventing checkpoint activation. First, we constructed a double mutant containing glc7-10 and either ndc10-1 (Goh and Kilmartin 1993) or ndc10-42 (Doheny et al. 1993). As mentioned above, glc7-10 cells arrest in mid-mitosis. In contrast, ndc10-1 cells do not arrest but exhibit polyploidy (Goh and Kilmartin 1993). This later finding is surprising, as one might expect that a cell with defective kinetochores would arrest as a consequence of engaging the mitotic checkpoint (Spencer and Hieter 1992; Pangilinan and Spencer 1996; Wells and Murray 1996; Wang and Burke 1997). An attractive explanation is that in ndc10-1 mutants, kinetochores are sufficiently impaired at the restrictive temperature that they can neither bind microtubules nor activate the checkpoint. We generated synchronous cultures of the glc7-10 ndc10-1 double mutants by releasing cells arrested at G1/G0 by nutritional deprivation into fresh medium at 37°C and analyzed the DNA content of cells at different time points using flow cytometry (FACS). As expected, glc7-10 cells primarily contained 2C DNA, indicative of a G2/M arrest (Fig. 4A,b). ndc10-1 cells (Fig. 4A,c) did not arrest but went through an aberrant mitosis, resulting in the generation of polyploid cells (2C and 4C DNA at 4–7 hr) consistent with earlier findings (Goh and Kilmartin 1993). Double mutant glc7-10 ndc10-1 cells were similar in their phenotype to the ndc10-1 single mutant (Fig. 4A,d), a result that was also obtained with glc7-10 ndc10-42 cells (data not shown). These data show that ndc10-1 is epistatic to glc7-10 and suggest that the mitotic arrest in the glc7-10 mutant phenotype is derived from a kinetochore defect.
Figure 4.
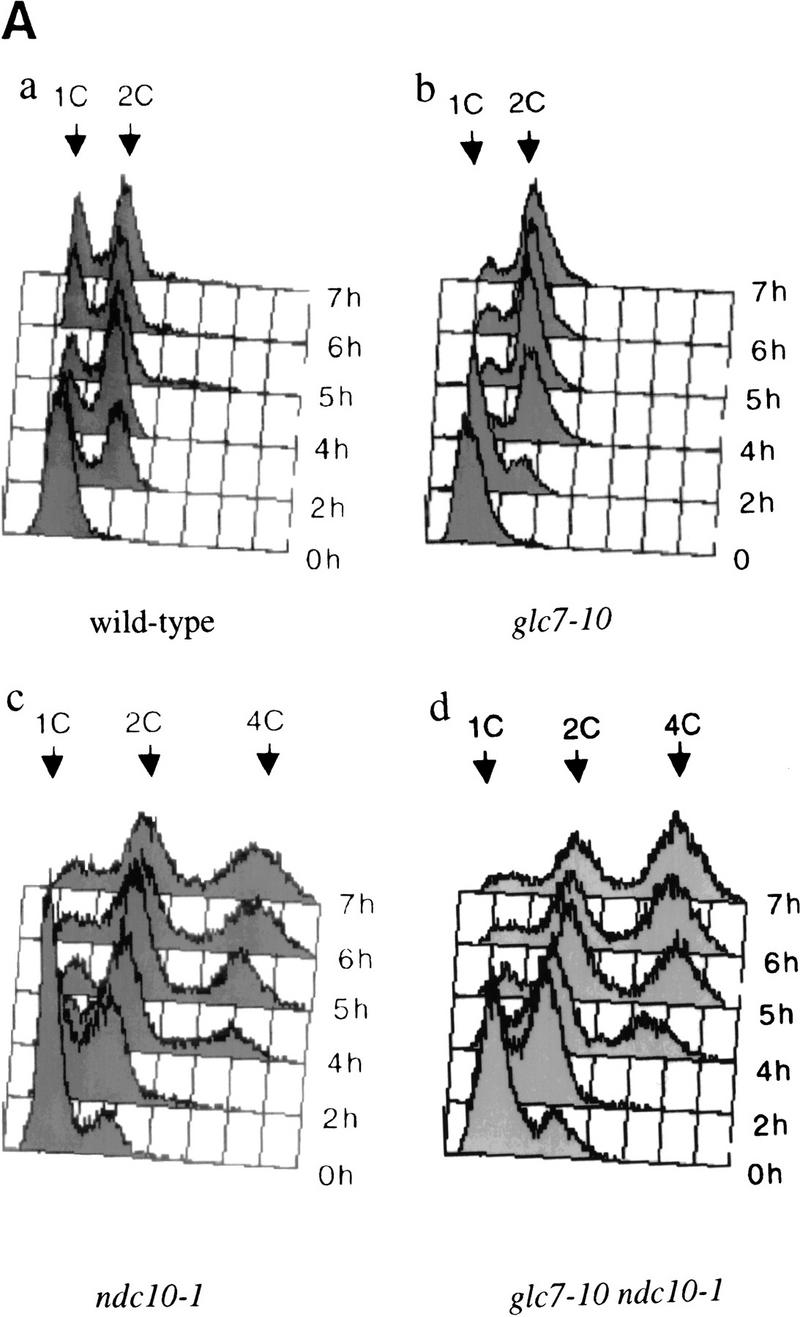

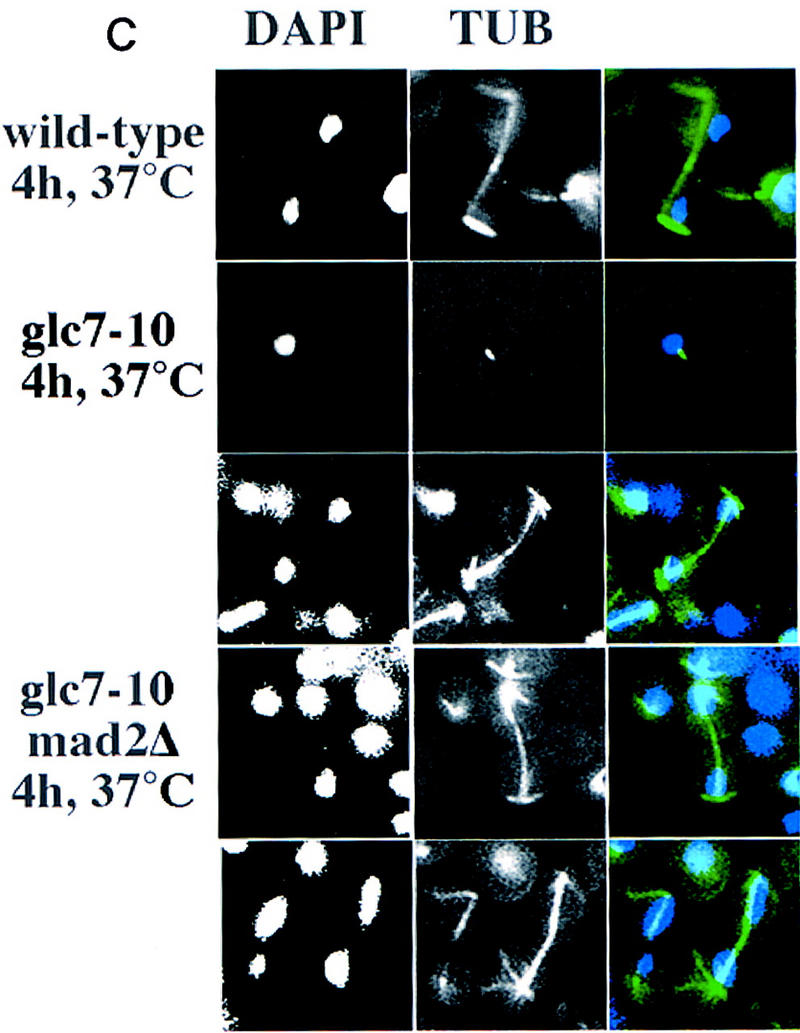
(A) DNA flow cytometry of wild-type (a), glc7-10 (b), ndc10-1 (c), and glc7-10 ndc10-1 (d) cultures synchronized by starvation on plates for 2–3 days at the permissive temperature. Samples were taken at different time points of the incubation at the restrictive temperature and processed for DNA content measurement (FACS). The arrows show whether cells have 1C, 2C, or 4C of DNA mass, corresponding to an unreplicated, a replicated, or a re-replicated genome, respectively. (B) DNA flow cytometry of mad2Δ (a) and glc7-10 mad2Δ (b) cultures synchronized by centrifugal elutriation and released in complete medium at 37°C. Samples were taken at different time points and processed for DNA content measurements. (C) Tubulin immunofluorescence was performed on wild-type, glc7-10, and glc7-10 mad2Δ double mutants shifted to the nonpermissive temperature for 4 hr. Three examples of glc7-10 mad2Δ anaphase elongated spindles are shown. (Left) DAPI staining; (middle) anti-tubulin staining; (right) merge of both stainings.
One of the genes required for the spindle checkpoint in yeast is MAD2 (Li and Murray 1991). In the absence of MAD2, the kinetochore-dependent arrest of cells containing unattached chromosomes is abolished (Wang and Burke 1997). Therefore, we tested whether deletions in MAD2 would also bypass the glc7-10-dependent mitotic arrest. We isolated glc7-10 mad2Δ double mutant G1 cells by centrifugal elutriation, released them at restrictive temperature, and analyzed their DNA content by flow cytometry (Fig. 4B). The double mutant glc7-10 mad2Δ cells grown at the restrictive temperature (Fig. 4B,b) show a wild-type profile for several generations, suggesting that they bypass the glc7-10 arrest. Tubulin immunofluorescence analyses of glc7-10 mad2Δ cells at the nonpermissive temperature confirmed that the spindle was elongating like in wild-type cells at anaphase and that DNA segregated between the mother and the daughter cell (Fig. 4C).
To confirm this result, we plated single G0 cells coming from stationary phase and observed division patterns of single cells at 37°C. In the glc7-10 mutant, 89% of the cells arrested at the first mitosis and the other 11% arrested at the next mitosis (Fig. 5). In mad2Δ mutant, cells continued dividing although they showed a small percentage of cell death after each division. In the glc7-10 mad2Δ mutant, cells continued to divide at the nonpermissive temperature, although some fraction (14%) arrested at each division. Similar results were obtained with glc7-10 bub2Δ (data not shown), suggesting that spindle checkpoint proteins such as Mad2p and Bub2p are required for glc7-10 cells to arrest at the restrictive temperature.
Figure 5.
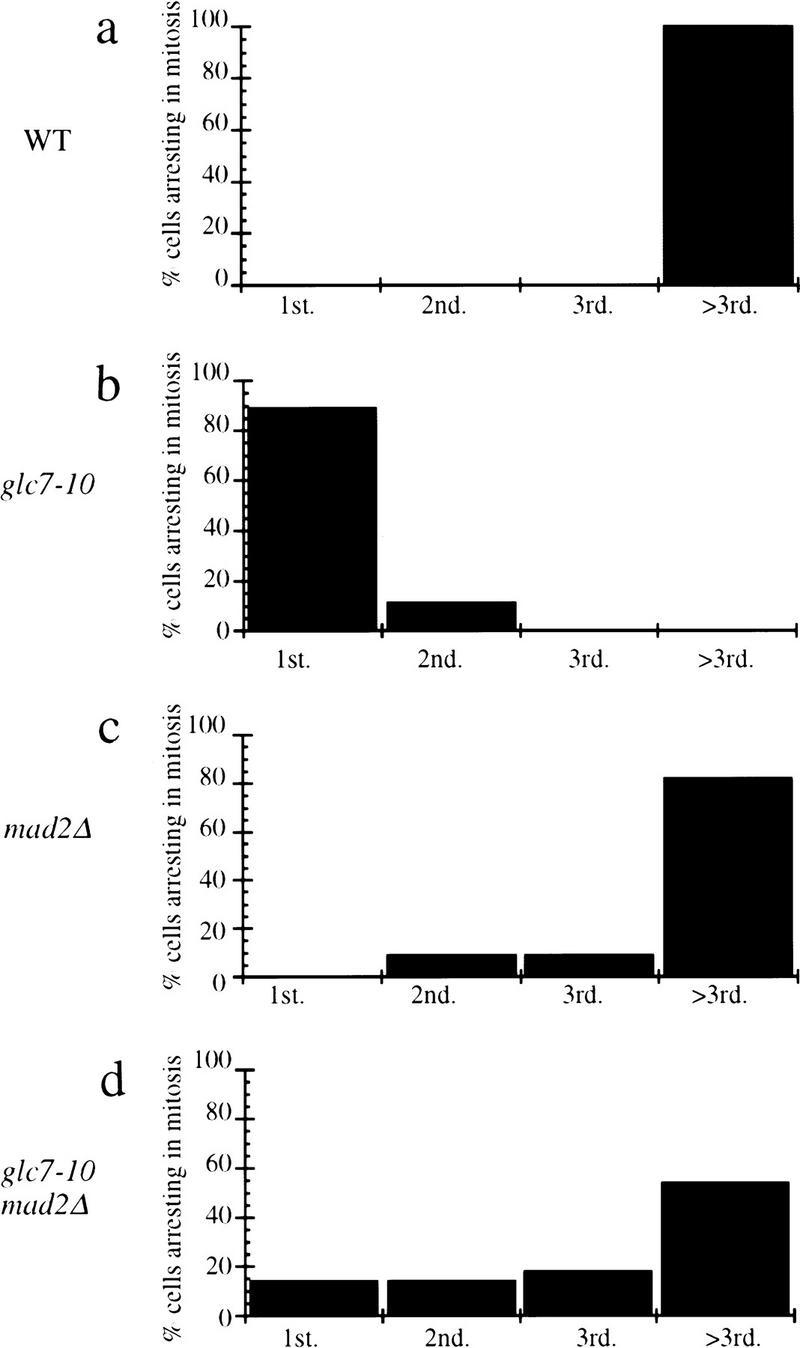
Single cell analyses. Single G0 cells coming from stationary phase at the permissive temperature were isolated by micromanipulation and grown at the nonpermissive temperature on complete solid medium. Single cells coming from wild-type (a), glc7-10 (b), mad2Δ (c), or glc7-10 mad2Δ (d) cultures were examined. We recorded the percentage of cells arresting at the first division (1st), the second division (2nd), and the third division (3rd), and after the third division (>3rd). Fifty individual cells per cell type were analyzed.
Ndc10p is hyperphosphorylated in glc7-10 extracts
Next, we asked why glc7-10 extracts had low microtubule binding activity compared with wild-type extracts. One possibility was that the levels of CBF3 complex proteins may be lower in glc7-10 cells than in wild-type cells. Therefore, we compared the amounts of Ndc10p, Skp1p, and Cep3p by immunoblotting in wild-type and glc7-10 extracts (Fig. 6A). Currently available probes for Ctf13p are not sensitive enough to detect this protein in our extracts. Bands corresponding to Cep3p and Skp1p were equal in intensity in wild-type and glc7-10 cells, but the Ndc10p band appeared to be two- to threefold weaker (Fig. 6B). The lower intensity of the Ndc10p signal might reflect lower levels of the Ndc10 protein, or it might be a consequence of reduced blotting efficiency. To distinguish between these possibilities, we inserted a myc tag at the 3′ end of the NDC10 ORF and repeated the immunoblotting using anti-myc antibodies. In this case, we observed equally intense Ndc10p bands in wild-type and glc7-10 extracts. The rabbit anti-Ndc10p antisera were raised against a 215-residue carboxy-terminal fragment of Ndc10p that had been expressed in Escherichia coli, and that was therefore unphosphorylated. To explain the lower blotting efficiency of Ndc10p in glc7-10 extracts, we hypothesized that the antisera were less efficient at recognizing phosphorylated Ndc10p than dephosphorylated Ndc10p. To test this hypothesis, we treated wild-type and glc7-10 extracts with alkaline phosphatase and then immunoblotted samples using the rabbit anti-Ndc10p antisera (Fig. 6B). In this case, we observed equal staining for Ndc10p in samples from wild-type and mutant extracts (Fig. 6B). We conclude from these experiments that rabbit antibodies that are sensitive to the phosphorylation state of Ndc10p have fortuitously been raised. Thus, although Ndc10p is present at equal levels in wild-type and glc7-10 cells, it cannot be recognized efficiently when Glc7p activity is reduced. We conclude that at least the carboxyl terminus of Ndc10p is hyperphosphorylated in cells in which PP1 has been mutated.
Figure 6.
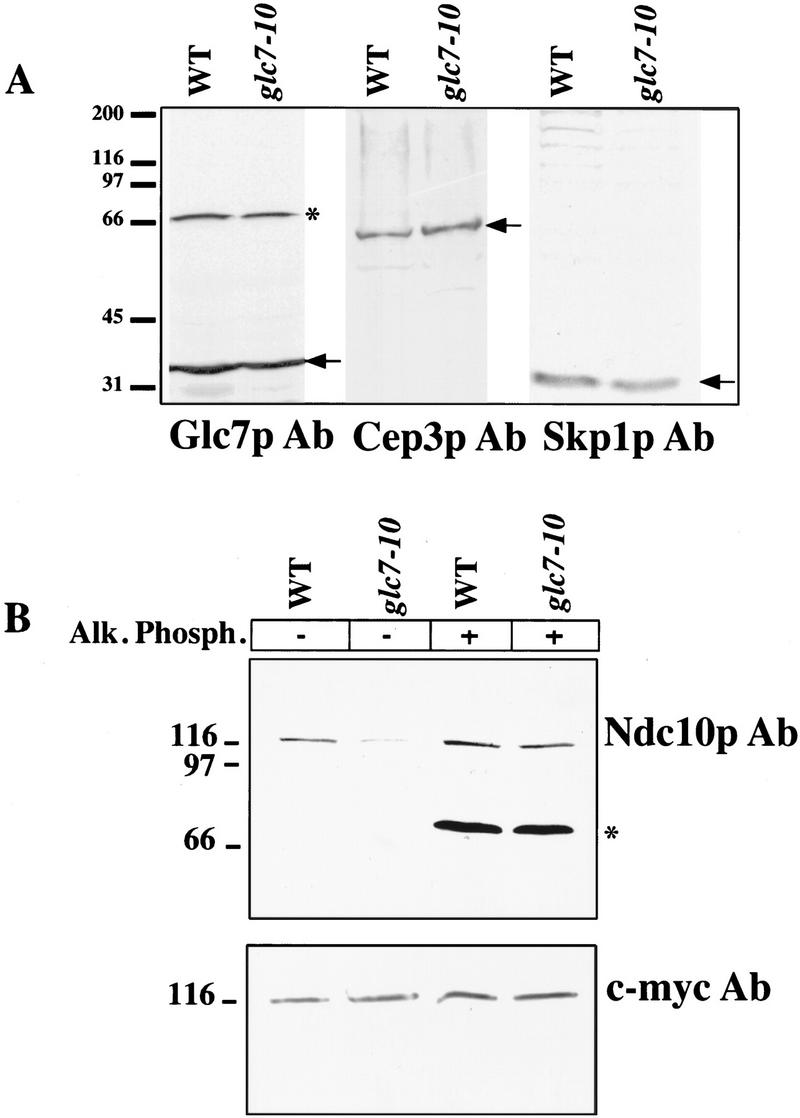
(A) Protein level comparison in glc7-10 and wild-type extracts. Extracts from wild-type and glc7-10 cultures shifted to 37°C for 3 hr were prepared and run on a 10% SDS–polyacrylamide gel. Western blots were performed with Skp1p and Cep3p purified antisera. (Arrows) Expected band: Cep3p (64 kD), Skp1p (24 kD). The same blot performed with a purified antiserum against Glc7p (37 kD) is shown as a loading control. Molecular masses (kD) of protein standards are indicated. (B) Alkaline phosphatase treatment of glc7-10 and wild-type crude extracts. Both wild-type and glc7-10 strains used contained a c-myc-tagged integrated version of NDC10. Both extracts were made after shifting of the cultures to the restrictive temperature for 3 hr. Subsequently, they were treated with alkaline phosphatase and blotted with Ndc10p and c-myc antisera. (*) Cross-reacting bands unrelated to our study. Molecular masses (kD) of protein standards are indicated.
Does Ndc10p hyperphosphorylation impair the binding of CBF3 to CEN-DNA? To address this question, we measured the CDEIII-binding activity of CBF3 in wild-type and glc7-10 extracts from cells grown at both permissive and restrictive temperatures and found that the band shift intensities were similar (Fig. 7A). Thus, as surmised previously from our analysis of ATP-treated extracts (Fig. 1), increased phosphorylation of CBF3, specifically Ndc10p, does not impair its DNA-binding activity.
Figure 7.
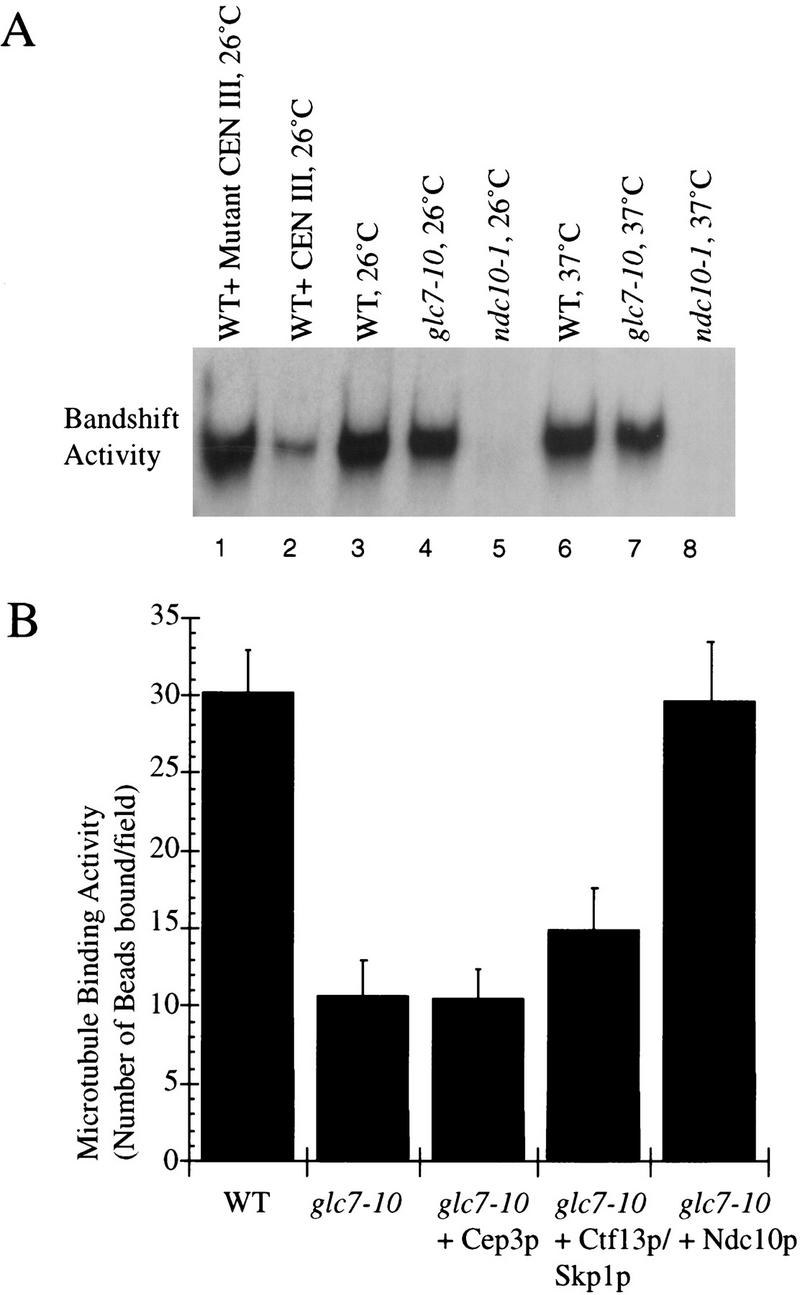
(A) Analysis of CDE III-binding activity in extracts from glc7-10 (lanes 4,7) and wild-type cells (lanes 3,6) grown at permissive and restrictive temperatures, respectively (Sorger et al. 1995). The band-shift activity of a wild-type extract in the presence of an excess of wild-type (lane 2) or mutant (lane 1) CEN III DNA cold competitor is shown as a control, as well as the band-shift activity of a ndc10-1 extract from cells grown at 26°C (lane 5) or 37°C (lane 8). (B) Reconstitution of the microtubule-binding activity (number of beads bound per field) and the DNA-binding activity of a glc7-10 extract from cells grown at the permissive temperature by addition of baculovirus-produced Ndc10p, Cep3p, and Ctf13p–Skp1p. The reason for addition of Ctf13p–Skp1p as a complex rather than as separated proteins is explained elsewhere (Kaplan et al. 1997). The microtubule-binding activity has been normalized as described previously.
The addition of Ndc10p to glc7-10 extracts reconstitutes its microtubule-binding activity
To test directly whether Ndc10p hyperphosphorylation is responsible for the low microtubule-binding activity of glc7-10 extracts, we added fractionated lysates from insect cells expressing recombinant Ndc10p, Cep3p, or Ctf13p–Skp1p as described (Kaplan et al. 1997). It has been shown previously that these four proteins are necessary and sufficient to assemble functional CBF3 (Lechner and Carbon 1991; Kaplan et al. 1997). The association among CBF3 subunits is weak in the absence of centromeric DNA: When recombinant CBF3 subunits are added to yeast cell extracts, mixed complexes can form that contain both recombinant and native subunits (Grancell and Sorger, unpubl.). When recombinant CBF3 subunits were added to glc7-10 extracts, we observed that Ndc10p, but not Cep3p or Ctf13p–Skp1p, restored microtubule-binding activity to wild-type levels (Fig. 7B). To demonstrate that our preparations of Ndc10p, Cep3p, and Ctf13p–Skp1p were active, we added them to ndc10-1, cep3-2, or ctf13-30 extracts and showed that microtubule binding was restored to wild-type levels (data not shown). The amount of Ndc10p required to reconstitute microtubule-binding activity in glc7-10 extracts was the same as that required in extracts from ndc10-1 cells grown at the permissive temperature, in which Ndc10p is completely inactive (Sorger et al. 1995). From these data, we conclude that the low microtubule-binding activity of glc7-10 extracts is a consequence of Ndc10p hyperphosphorylation, which results in reduced activity of CBF3 toward microtubule attachment.
Discussion
In this report we examine the role of protein phosphorylation in regulating the activity of S. cerevisiae kinetochores. Using an in vitro system in which kinetochore–microtubule binding can be reconstituted in cell extracts, we show that this association is stimulated by a type 1 protein phosphatase and inhibited by an opposing kinase activity. We found that phosphorylation alters the ability of the essential centromere-binding complex CBF3 to mediate microtubule attachment, but that it does not alter its ability to bind to DNA. The component of CBF3 subject to regulation by the Glc7p phosphatase pathway seems to be Ndc10p, because this protein is hyperphosphorylated in the glc7-10 mutant. As glc7-10 arrest is mediated by the spindle checkpoint, it is possible that a change in Ndc10p phosphorylation is able to generate a checkpoint-dependent arrest.
Regulation of centromere–microtubule attachment by Glc7p
We used ATP and apyrase additions to show that phosphorylation regulates the microtubule-binding activity of yeast kinetochores in vitro. The use of microcystin at a concentration sufficient to inhibit PP1 and PP2A, and the use of okadaic acid at a concentration sufficient to inhibit only PP2A allowed us to conclude that PP1 promotes microtubule-binding activity of kinetochores in our in vitro assay. Furthermore, PP1 mutant extracts but not PP2A mutant extracts show low microtubule-binding activity in the same assay, supporting the hypothesis that PP1 activates kinetochore–microtubule attachment. Altogether, these findings can be explained by postulating that microtubule-binding activity is regulated by phosphorylation and that the ratio of phosphorylated and nonphosphorylated kinetochore proteins is controlled by the competing actions of a protein kinase and a type 1 protein phosphatase. The decrease in microtubule-binding activity on ATP addition is in agreement with previous experiments analyzing binding of minichromosomes to taxol-stabilized microtubules (Kingsbury and Koshland 1993).
Ndc10p is a downstream substrate of the Glc7p phosphatase pathway
What substrate of Glc7p regulates kinetochore activity? Our data show that the kinetochore protein Ndc10p is hyperphosphorylated in the glc7-10 mutant extracts. This hyperphosphorylation appears to be correlated with the low microtubule-binding activity of kinetochore complexes observed in glc7-10 extracts because the addition of recombinant Ndc10p, but not other CBF3 subunits, restores microtubule-binding activity to wild-type levels. Although these data do not show that Ndc10p is a direct target of Glc7p, they define a regulatory pathway acting on kinetochores via Ndc10p. This pathway includes Glc7p and a kinase, both upstream from Ndc10p, which regulate kinetochore–microtubule attachment by phosphorylation. A potential kinase is the essential Ipl1 Ser/Thr protein kinase (Chan and Botstein 1993), which genetically counteracts Glc7p (Francisco et al. 1994) and has been shown to phosphorylate Ndc10p in vitro. Consistent with Ipl1p counteracting the Glc7p pathway on kinetochore activity, ipl1 mutant extracts are not sensitive to ATP addition in our in vitro kinetochore–microtubule-binding assay (Biggins et al. 1999).
Interestingly, Ndc10p hyperphosphorylation is deleterious for the microtubule-binding activity of kinetochores but does not impair the binding of CBF3 to centromeric DNA. Thus, Ndc10p has two independently regulated functions in kinetochores: The first function is to bind to centromeric DNA, the second function is to participate in mediating the assembly of a microtubule attachment site. Only the second function is regulated by Glc7p. We know little about how CBF3 mediates microtubule attachment. CBF3 alone does not bind to microtubules (Sorger et al. 1994) but does associate with additional cellular factors, at least one of which can be purified from extracts (K.B. Kaplan, F.F. Severin, A.A. Hyman, and P.K. Sorger, in prep.), to link centromeric DNA to microtubules in vitro. Thus, it seems likely that Glc7p acts to remove the phosphorylated epitopes of Ndc10p that are inhibitory for its association with microtubule-binding proteins.
What is the role of phosphorylation of Ndc10p in the wild-type cell? Previous experiments have shown that the binding of CBF3 to CDEIII does not vary through the vegetative cell cycle, but that microtubule-binding activity is low in G1, higher in S phase and reaches maximal levels at mitosis (I. Sassoon and A.A. Hyman, unpubl.). One explanation is that these changes in microtubule binding are derived from cell-cycle dependent changes in Ndc10p phosphorylation mediated, in part, by Glc7p.
Ndc10p, Glc7p, and the spindle checkpoint
We found that glc7-10 arrest at the G2/M transition is mediated by the spindle checkpoint, because glc7-10 ndc10-1, glc7-10 mad2Δ, and glc7-10 bub2Δ do not arrest at mitosis. Why do glc7-10 cells trigger the spindle assembly checkpoint? One possibility is that Glc7p has a role in control of spindle assembly, for instance by controlling microtubule organization. Defects in microtubule organization can trigger a spindle checkpoint-dependent arrest. Another possibility is that the spindle checkpoint proteins can sense the hyperphosphorylation of Ndc10p and activate a signaling cascade to stop the cell cycle at the metaphase–anaphase transition. Although we understand some of the downstream pathways required for the spindle checkpoint, we have no idea as to how kinetochores actually sense microtubule attachment. Perhaps the Glc7p-dependent phosphorylation state of Ndc10p plays a role in sensing microtubule attachment. We cannot yet distinguish between these possibilities, which will require the mapping of the Ndc10p phosphorylation sites, but it is interesting to note that in a glc7-10 mad2Δ double mutant, the cells can divide for several generations, suggesting that there are no gross defects in spindle organization in glc7-10 mutants.
A general role for PP1 in mitosis
In all eukaryotes in which genetic analysis has been performed, mutations in type 1 protein phosphatase generate a mitotic arrest, and as shown in this study and others (Hisamoto et al. 1994), these mutations decrease the fidelity of mitotic chromosome segregation at permissive temperatures. However, the critical mitotic substrates for PP1 have not been identified. In S. pombe, a genetic interaction between PP1 and components of the APC has been detected, suggesting that APC could be activated by PP1 (Yamada et al. 1997). However, results in Xenopus egg extracts do not support a role of PP1 in the direct activation of APC (Vorlaufer and Peters 1998). Our results suggest that PP1 is required for correct kinetochore assembly in S. cerevisiae (see model in Fig. 8). Impairment of kinetochore–microtubule interactions may be contributing to the G2/M arrest mediated by the spindle checkpoint proteins Mad2p and Bub2p. It is possible that the G2/M arrest seen in other organisms could also be due to kinetochore defects.
Figure 8.

Glc7p regulates the attachment of kinetochores to microtubules in S. cerevisiae. The CEN–DNA binding activity of the kinetochore complex is not regulated by the Glc7p type 1 phosphatase pathway. On the contrary, the Glc7p pathway specifically regulates the microtubule-binding activity of kinetochores, probably through phosphorylation of specific kinetochore proteins.
Materials and methods
Yeast strains
All strains used in this study are summarized in Table 1. In all cases, the background is W303A.
Table 1.
Yeast strains used in this study
| Strains
|
Relevant genotypes
|
Sources/Reference
|
|---|---|---|
| TH3 | MATa, ctf13-30 | Doheny et al. (1993) |
| TH269 | MATa, cep3-2 | Strunnikov et al. (1995) |
| TH113 | MATa, ndc10-42 | Doheny et al. (1993) |
| TH271 | MATa, ndc10-1 | Goh and Kilmartin (1993) |
| PAY700-4 | MATa, glc7::LEU2, trpl::glc7-10::TRP1 | P.D. Andrews and M.J.R. Stark (in prep.) |
| TH397 | MATa, mad2::URA3 | this study |
| TH548 | MATa, bub2::HIS3 | this study |
| TH523 | MATa, glc7::LEU2, trp1::glc7-10::TRP1, mad2::URA3 | this study |
| TH545 | MATa, glc7::LEU2, trp1::glc7-10::TRP1, bub2::HIS3 | this study |
| TH539 | MATa, glc7::LEU2, trp1::glc7-10::TRP1, ndc10-1 | this study |
| TH496 | MATa, glc7:LEU2, trp1::glc7-10::TRP1, NDC10 myc6 Cterm::TRP1 | this study |
| TH411 | NDC10 myc6 Cterm::TRP1 | S. Piatti (unpubl.) |
| TH549 | MATa, glc7::LEU2, trp1::glc7-10::TRP1, ndc10-42 | this study |
| PAY704-1 | MATa, glc7::LEU2, trp1::GLC7::TRP1 | P.D. Andrews and M.J.R. Stark (in prep.) |
| K5043 | MATα, ura3-52 lys2-801 ade2-101, his3Δ200, leu2Δ1, TRP1, CFIII (CEN3.L.YPH278) URA3 SUP11 | K. Nasmyth (IMP, Vienna, Austria) |
Reagents
BSA, nocodazole, α factor, propidium iodide, okadaic acid, microcystin, taxol, apyrase, and cytohelicase were purchased from Sigma. PMSF, high concentration alkaline phosphatase, and ATP were purchased from Boehringer Mannheim. Restriction enzymes and buffers were purchased from New England Biolabs. Bovine brain tubulin was prepared in house (Ashford et al. 1998). ECL+ Kit and secondary antibody goat anti-rabbit or goat anti-mouse IgGs conjugated with horse radish peroxidase were purchased from Amersham.
General yeast methods and DNA manipulations
All yeast growth medium (rich medium, minimal medium, and sporulation medium) and general yeast methods are described in Rose et al. (1990). Yeast strains were transformed by the lithium acetate procedure as described by Ito et al. (1983). General molecular biology methods were carried out as described by Sambrook et al. (1989). One-step gene disruption was performed by the method of Langle-Rouault (Langle and Jacobs 1995). Verification of the disruption was confirmed by PCR with appropriate primers.
Chromosome loss assay
Yeast cells carrying ade2 ochre mutations accumulate a read pigment, the appearance of which is suppressed by a single copy of the ochre suppressor gene SUP11. The ade2 strain K5043 contains SUP11 on a nonessential chromosome fragment and produces white colonies, allowing chromosome loss to be scored by the appearance of red sectors during colony development (Spencer et al. 1990). The SUP11 chromosome fragment was transferred into the mutant glc7-10 and control GLC7 backgrounds by crossing K5043 with PAY700-4 and PAY700-1, respectively, identifying spores that germinated to give Trp+ Leu+ Ura+ colonies following sporulation and tetrad dissection. Loss of the SUP11 chromosome fragment was monitored visually after 7 days by the production of red sectors following growth of the strains on YPD plates lacking supplementary adenine at 26°C and 35.5°C.
Yeast cultures and extracts preparation
Yeast temperature-sensitive cells were grown at 26°C (permissive temperature) to a density of 2–5 × 107 cells/ml. Whole-cell extracts were prepared by centrifugation of the culture 4 min at 4000 rpm and at 4°C in a J6-B centrifuge (Beckman) and washing once in water at 4°C [in a 50-ml Falcon tube in a megafuge 10R (Heraeus)]. The pellet was then weighed and resuspended in the same weight of breakage buffer 2× [200 mm β-glycerophosphate, 100 mm bis-Tris-propane (pH 7.0), 400 mm KCl, 10 mm EDTA, 10 mm EGTA, 20% glycerol, 4°C] with protease inhibitors (final concentrations: 1 mm PMSF and 10 μg/ml each pepstatin, leupeptin, and chymostatin). The cell paste obtained was then dripped in a 50-ml Falcon tube filled with liquid nitrogen. The frozen paste was pulverized with a porcelain mortar (Coors 60322, 13.5 cm O.D.) and pestle cooled in liquid nitrogen (∼300 strokes are required to obtain a very fine powder and to fragment >50% of the cells). The efficiency of breakage was monitored by examination of the cells by phase-contrast microscopy. The powder was then thawed on ice and cell debris was removed by centrifugation at 12,000 rpm for 10 min at 4°C. Typical protein concentrations in the extracts were 10–40 mg/ml. In extracts prepared in this manner, the ATP concentration (determined by luciferase luciferin) was <1 μm for an extract prepared at 20 mg/ml. The extracts were frozen in liquid nitrogen and stored at −70°C for several months. Alkaline phosphatase treatment of extracts was performed with Boehringer Mannheim high concentration alkaline phosphatase (1 U/μl) and incubation at 37°C for 15 min.
Band-shift assay
The DNA probe used for this experiment consisted of 89 bp derived from the centromeric sequence of chromosome III of S. cerevisiae (Espelin et al. 1997). The probe was radiolabeled (Sorger et al. 1995), gels were prepared (Lechner and Carbon 1991), and band-shift reactions were performed (Sorger et al. 1995) essentially as described.
Microtubule-binding assay
The microtubule-binding assay has been described previously (Sorger et al. 1994). Typically, we use a 30-μl reaction containing 50–80 μg of extract with 3 × 107 beads and 10 μg of sonicated salmon sperm DNA in kinetochore buffer [10 mm HEPES (pH 8), 6 mm MgCl2, 10% glycerol] and adjusted to a final KCl concentration of 140 mm. For the experiment involving inhibitors of phosphatases, ATP was added at 1 mm, microcystin was added at 1 μm, okadaic acid was added at 0.1 μm (parallel experiments involving okadaic acid titration confirmed that 0.1 μm was sufficient to inhibit only type 2A protein phosphatases), and apyrase was added for 5 min at 1 U/μl after the 40-min incubation. Microtubule-bound beads were counted manually, 10–20 fields were averaged, and standard deviations and the mean calculated.
Flow cytometry analysis
Small unbudded G1 cells were isolated by centrifugal elutriation, performed as described previously (Schwob and Nasmyth 1993). Cells were grown overnight on YPRaffinose and small G1 cells were released into YPD at 37°C. Samples were processed for flow cytometry analysis as described previously (Schwob and Nasmyth 1993).
Production of yeast proteins in baculovirus
CBF3 subunits were produced in baculovirus insect cells as described previously (Kaplan et al. 1997). Each protein was purified and was able to complement its corresponding extract for DNA binding and microtubule-binding activity (Espelin et al. 1997; Kaplan et al. 1997). Ndc10p produced in insect cells is extensively phosphorylated as measured by in vivo 32P labeling. Bacterially produced Ndc10p, or alkaline phosphatase-treated baculovirus-produced Ndc10p are incapable of CBF3 complex formation (K.B. Kaplan, unpubl.).
Antibody production
The plasmid pGEX–GST–NDC10 was kindly provided by J. Kilmartin (Medical Research Council, Cambridge, UK). It contains the carboxy-terminal part of the NDC10 gene (amino-acids 679–894). It was transformed into BL21 bacteria and the GST–Ndc10p was renatured, purified over a GST column, and injected into rabbits. The sera obtained were subsequently purified over a GST–Ndc10p column and stored in 50% glycerol at −20°C. Small peptides suitable for raising antisera were chosen by computer analysis of the sequences of the Cep3p, Skp1p, and Glc7p proteins, synthesized, conjugated to carrier proteins (Imject Activated Immunogen Conjugation kits, Pierce, 77107-8) and injected into rabbits. The sera obtained were purified with the peptides coupled to a column (Sulfolink coupling gel, Pierce 20401-2). The purified antibodies were stored at −20°C in 50% glycerol.
Acknowledgments
We thank J. Kilmartin for kindly providing the Ndc10p antibodies and the GST–NDC10 plasmid, S. Piatti for providing the c-myc-tagged NDC10 strain, A. Tóth for the BUB2 disruption oligonucleotides, A. Atzberger for technical assistance with the FACS Scan, M. Glotzer, A. Murray, and K. Nasmyth for helpful discussions, and A. Deshai, C. Echeverri, A. Murray, S. Schuyler, and R. Tournebize for critical reading of the manuscript. This work was carried out with support from the Wellcome Trust to M.J.R.S. (project grants 046956 and 049778). This work was supported by a Leukemia Society Special Fellowship to K.B.K and by the Searle Scholars Program, the North Atlantic Treaty Organization (travel grant CRG.960163) and by National Institutes of Health grant R01-GM51464.
Footnotes
Corresponding author.
E-MAIL hyman@embl-heidelberg.de; FAX 49 6221 387 512.
References
- Ashford JA, Andersen SSL, Hyman AA. Preparation of tubulin from bovine brain. In: Celis J E, editor. Cell biology: A laboratory handbook. San Diego, CA: Academic Press; 1998. pp. 205–212. [Google Scholar]
- Axton JM, Dombradi V, Cohen PT, Glover DM. One of the protein phosphatase 1 isoenzymes in Drosophila is essential for mitosis. Cell. 1990;63:33–46. doi: 10.1016/0092-8674(90)90286-n. [DOI] [PubMed] [Google Scholar]
- Biggins, S., F. Severin, N. Bhalla, I. Sassoon, A. Hyman, and A. Murray. 1999. The conserved protein kinase Ip11 regulates microtubule binding to kinetochores in budding yeast. Genes & Dev. (this issue). [DOI] [PMC free article] [PubMed]
- Black S, Andrews PD, Sneddon AA, Stark MJ. A regulated MET3-GLC7 gene fusion provides evidence of a mitotic role for Saccharomyces cerevisiae protein phosphatase 1. Yeast. 1995;11:747–759. doi: 10.1002/yea.320110806. [DOI] [PubMed] [Google Scholar]
- Cannon JF, Pringle JR, Fiechter A, Khalil M. Characterization of glycogen deficient glc mutants of Saccharomyces cerevisiae. Genetics. 1994;136:485–503. doi: 10.1093/genetics/136.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Botstein D. Isolation and characterization of chromosome-gain and increase-in-ploidy mutants in yeast. Genetics. 1993;135:677–691. doi: 10.1093/genetics/135.3.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke L. Centromeres: Proteins, protein complexes, and repeated domains at centromeres of simple eukaryotes. Curr Opin Genet Dev. 1998;8:212–218. doi: 10.1016/s0959-437x(98)80143-3. [DOI] [PubMed] [Google Scholar]
- Clarke L, Carbon J. Isolation of a yeast centromere and construction of functional small circular chromosomes. Nature. 1980;287:504–509. doi: 10.1038/287504a0. [DOI] [PubMed] [Google Scholar]
- Doheny KF, Sorger PK, Hyman AA, Tugendreich S, Spencer F, Hieter P. Identification of essential components of the S. cerevisiae kinetochore. Cell. 1993;73:761–774. doi: 10.1016/0092-8674(93)90255-O. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doonan JH, Morris NR. The bimG gene of Aspergillus nidulans, required for completion of anaphase, encodes a homolog of mammalian phosphoprotein phosphatase 1. Cell. 1989;57:987–996. doi: 10.1016/0092-8674(89)90337-1. [DOI] [PubMed] [Google Scholar]
- Espelin CW, Kaplan KB, Sorger PK. Probing the architecture of a simple kinetochore using DNA-protein crosslinking. J Cell Biol. 1997;139:1383–1396. doi: 10.1083/jcb.139.6.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DR, Stark MJ. Mutations in the Saccharomyces cerevisiae type 2A protein phosphatase catalytic subunit reveal roles in cell wall integrity, actin cytoskeleton organization and mitosis. Genetics. 1997;145:227–241. doi: 10.1093/genetics/145.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez A, Brautigan DL, Lamb NJ. Protein phosphatase type 1 in mammalian cell mitosis: Chromosomal localization and involvement in mitotic exit. J Cell Biol. 1992;116:1421–1430. doi: 10.1083/jcb.116.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald HM, Clarke L, Carbon J. Nucleotide sequence comparisons and functional analysis of yeast centromere DNAs. Cell. 1982;29:235–244. doi: 10.1016/0092-8674(82)90108-8. [DOI] [PubMed] [Google Scholar]
- Francisco L, Wang W, Chan CS. Type 1 protein phosphatase acts in opposition to Ipl1 protein kinase in regulating yeast chromosome segregation. Mol Cell Biol. 1994;14:4731–4740. doi: 10.1128/mcb.14.7.4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh PY, Kilmartin JV. NDC10: A gene involved in chromosome segregation in Saccharomyces cerevisiae. J Cell Biol. 1993;121:503–512. doi: 10.1083/jcb.121.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick KG. The spindle checkpoint. Trends Genet. 1998;14:1–4. doi: 10.1016/S0168-9525(97)01340-1. [DOI] [PubMed] [Google Scholar]
- Hegemann JH, Fleig UN. The centromere of budding yeast. BioEssays. 1993;15:451–460. doi: 10.1002/bies.950150704. [DOI] [PubMed] [Google Scholar]
- Hisamoto N, Sugimoto K, Matsumoto K. The Glc7 type 1 protein phosphatase of Saccharomyces cerevisiae is required for cell cycle progression in G2/M. Mol Cell Biol. 1994;14:3158–3165. doi: 10.1128/mcb.14.5.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA, Sorger PK. Structure and function of kinetochores in budding yeast. Annu Rev Cell Dev Biol. 1995;11:471–495. doi: 10.1146/annurev.cb.11.110195.002351. [DOI] [PubMed] [Google Scholar]
- Ishii K, Kumada K, Toda T, Yanagida M. Requirement for PP1 phosphatase and 20S cyclosome/APC for the onset of anaphase is lessened by the dosage increase of a novel gene sds23+ EMBO J. 1996;15:6629–6640. [PMC free article] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan KB, Hyman AA, Sorger PK. Regulating the yeast kinetochore by ubiquitin-dependent degradation and Skp1p-mediated phosphorylation. Cell. 1997;91:491–500. doi: 10.1016/s0092-8674(00)80435-3. [DOI] [PubMed] [Google Scholar]
- Kingsbury J, Koshland D. Centromere function on minichromosomes isolated from budding yeast. Mol Biol Cell. 1993;4:859–870. doi: 10.1091/mbc.4.8.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langle RF, Jacobs E. A method for performing precise alterations in the yeast genome using a recycable selectable marker. Nucleic Acids Res. 1995;23:3079–3081. doi: 10.1093/nar/23.15.3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner J, Carbon J. A 240 kd multisubunit protein complex, CBF3, is a major component of the budding yeast centromere. Cell. 1991;64:717–725. doi: 10.1016/0092-8674(91)90501-o. [DOI] [PubMed] [Google Scholar]
- Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell. 1991;66:519–531. doi: 10.1016/0092-8674(81)90015-5. [DOI] [PubMed] [Google Scholar]
- MacKelvie SH, Andrews PD, Stark MJ. The Saccharomyces cerevisiae gene SDS22 encodes a potential regulator of the mitotic function of yeast type 1 protein phosphatase. Mol Cell Biol. 1995;15:3777–3785. doi: 10.1128/mcb.15.7.3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrew J, Diehl B, Fitzgerald HM. Single base-pair mutations in centromere element III cause aberrant chromosome segregation in Saccharomyces cerevisiae. Mol Cell Biol. 1986;6:530–538. doi: 10.1128/mcb.6.2.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng R, Carbon J. Mutational and in vitro protein-binding studies on centromere DNA from Saccharomyces cerevisiae. Mol Cell Biol. 1987;7:4522–4534. doi: 10.1128/mcb.7.12.4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkura H, Adachi Y, Kinoshita N, Niwa O, Toda T, Yanagida M. Cold-sensitive and caffeine-supersensitive mutants of the Schizosaccharomyces pombe dis genes implicated in sister chromatid separation during mitosis. EMBO J. 1988;7:1465–1473. doi: 10.1002/j.1460-2075.1988.tb02964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkura H, Kinoshita N, Miyatani S, Toda T, Yanagida M. The fission yeast dis2+ gene required for chromosome disjoining encodes one of two putative type 1 protein phosphatases. Cell. 1989;57:997–1007. doi: 10.1016/0092-8674(89)90338-3. [DOI] [PubMed] [Google Scholar]
- Pangilinan F, Spencer F. Abnormal kinetochore structure activates the spindle assembly checkpoint in budding yeast. Mol Biol Cell. 1996;7:1195–1208. doi: 10.1091/mbc.7.8.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose MD, Winston F, Hieter P. Methods in yeast genetics: A laboratory course manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1990. [Google Scholar]
- Runnegar M, Berndt N, Kong S-M, Lee EY-C, Zhang L. In vivo and in vitro binding of microcystin to protein phosphatases 1 and 2A. Biochem Biophys Res Commun. 1995;216:162–169. doi: 10.1006/bbrc.1995.2605. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schwob E, Nasmyth K. CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae. Genes & Dev. 1993;7:1160–1175. doi: 10.1101/gad.7.7a.1160. [DOI] [PubMed] [Google Scholar]
- Sorger PK, Severin FF, Hyman AA. Factors required for the binding of reassembled yeast kinetochores to microtubules in vitro. J Cell Biol. 1994;127:995–1008. doi: 10.1083/jcb.127.4.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorger PK, Doheny KF, Hieter P, Kopski KM, Huffaker TC, Hyman AA. Two genes required for the binding of an essential Saccharomyces cerevisiae kinetochore complex to DNA. Proc Natl Acad Sci. 1995;92:12026–12030. doi: 10.1073/pnas.92.26.12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer F, Gerring SL, Connelly C, Hieter P. Mitotic chromosome transmission fidelity mutants in Saccharomyces cerevisiae. Genetics. 1990;124:237–249. doi: 10.1093/genetics/124.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer F, Hieter P. Centromere DNA mutations induce a mitotic delay in Saccharomyces cerevisiae. Proc Natl Acad Sci. 1992;89:8908–8912. doi: 10.1073/pnas.89.19.8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strunnikov AV, Kingsbury J, Koshland D. CEP3 encodes a centromere protein of Saccharomyces cerevisiae. J Cell Biol. 1995;128:749–760. doi: 10.1083/jcb.128.5.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorlaufer E, Peters JM. Regulation of the cyclin B degradation system by an inhibitor of mitotic proteolysis. Mol Biol Cell. 1998;9:1817–1831. doi: 10.1091/mbc.9.7.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Burke DJ. Cdc55p, the B-type regulatory subunit of protein phosphatase 2A, has multiple functions in mitosis and is required for the kinetochore/spindle checkpoint in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:620–626. doi: 10.1128/mcb.17.2.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells AE. The spindle-assembly checkpoint: Aiming for a perfect mitosis, every time. Trends Cell Biol. 1996;6:228–234. doi: 10.1016/0962-8924(96)10018-0. [DOI] [PubMed] [Google Scholar]
- Wells WA, Murray AW. Aberrantly segregating centromeres activate the spindle assembly checkpoint in budding yeast. J Cell Biol. 1996;133:75–84. doi: 10.1083/jcb.133.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Kumada K, Yanagida M. Distinct subunit functions and cell cycle regulated phosphorylation of 20S APC/cyclosome required for anaphase in fission yeast. J Cell Sci. 1997;110:1793–1804. doi: 10.1242/jcs.110.15.1793. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zhao S, Long F, Zhang L, Bai G, Shima H, Nagao M, Lee EY-C. J Biol Chem. 1994;269:16997–17000. [PubMed] [Google Scholar]