

Abstract
We have isolated and characterized two suppressor genes, SUI4 and SUI5, that can initiate translation in the absence of an AUG start codon at the HIS4 locus in Saccharomyces cerevisiae. Both suppressor genes are dominant in diploid cells and lethal in haploid cells. The SUI4 suppressor gene is identical to the GCD11 gene, which encodes the γ subunit of the eIF-2 complex and contains a mutation in the G2 motif, one of the four signature motifs that characterizes this subunit to be a G-protein. The SUI5 suppressor gene is identical to the TIF5 gene that encodes eIF-5, a translation initiation factor known to stimulate the hydrolysis of GTP bound to eIF-2 as part of the 43S preinitiation complex. Purified mutant eIF-5 is more active in stimulating GTP hydrolysis in vitro than wild-type eIF-5, suggesting that an alteration of the hydrolysis rate of GTP bound to the 43S preinitiation complex during ribosomal scanning allows translation initiation at a non-AUG codon. Purified mutant eIF-2γ complex is defective in ternary complex formation and this defect correlates with a higher rate of dissociation from charged initiator-tRNA in the absence of GTP hydrolysis. Biochemical characterization of SUI3 suppressor alleles that encode mutant forms of the β subunit of eIF-2 revealed that these mutant eIF-2 complexes have a higher intrinsic rate of GTP hydrolysis, which is eIF-5 independent. All of these biochemical defects result in initiation at a UUG codon at the his4 gene in yeast. These studies in light of other analyses indicate that GTP hydrolysis that leads to dissociation of eIF-2 ⋅ GDP from the initiator-tRNA in the 43S preinitiation complex serves as a checkpoint for a 3-bp codon/anticodon interaction between the AUG start codon and the initiator-tRNA during the ribosomal scanning process.
Keywords: GTP hydrolysis, translation initiation, ribosomal scanning, AUG selection, eIF-2, eIF-5
The ribosomal scanning model is a well-accepted account for the mechanism to initiate translation initiation at the majority of eukaryotic mRNAs (Kozak 1978, 1989a). During this process, the 43S preinitiation complex traverses the leader region until it finds the start site for translation. For the vast majority of mRNAs, the first AUG codon serves as the start site for translation initiation. Previous biochemical studies have led to the following general model for how translation initiates in eukaryotic cells. eIF-2 (eukaryotic initiation factor) binds to the charged initiator-tRNA and forms a ternary complex which, in conjunction with eIF-3 and the 40S ribosome subunit, forms the 43S preinitiation complex. eIF-4F and eIF-4B, two additional initiation factors, melt secondary structure in the 5′-untranslated region (UTR) and facilitate the binding of the preinitiation complex to the 5′ end of mRNA. Following binding, this complex then scans for the first downstream AUG start codon. Once found, eIF-5 stimulates the hydrolysis of GTP bound to the 43S preinitiation complex. After GTP hydrolysis, the initiation factors are released, which leaves the initiator-tRNA at the P site of the 40S ribosomal subunit. The 60S ribosomal subunit can now join the 40S subunit and elongation of the peptide chain begins (for review, see Hershey 1991; Merrick 1992).
One fundamental difference between eukaryotic and prokaryotic translation initiation is that the former is quite stringent in only selecting an AUG codon as the start site for translation initiation. For example, none of the nine possible point mutations of the AUG start site at the HIS4 locus in yeast can serve as a signal for translation initiation (Donahue and Cigan 1988). In contrast, prokaryotic translation initiation at some genes use alternative codons, such as UUG and GUG (Gualerzi and Pon 1990). Nevertheless, there is nothing fundamentally important about the AUG codon per se for eukaryotic translation initiation. We have shown previously that an AGG codon can serve as a translation initiation site at the HIS4 gene provided a compensatory change in the anticodon of an initiator-tRNA gene (5′-CCU-3′) was present in the cell (Cigan et al. 1988a). Therefore, what is fundamentally important to the mechanism of eukaryotic translation initiation is that a 3-bp codon/anticodon interaction needs to be established between the start site and the initiator-tRNA.
We have used a genetic approach to gain insight into the mechanism of ribosomal recognition of the start site. Point mutations in the start codon of HIS4 gene were generated and extragenic suppressors that could restore a His+ phenotype were isolated. Through this reversion analysis, three suppressor genes were found that can initiate translation via a mismatched codon/anticodon between a UUG codon and the initiator-tRNA. The sui1 gene copurifies in part with the yeast translation initiation factor, eIF-3 (Naranda et al. 1996), and a mammalian homolog of Sui1 has been reported to correspond to the translation initiation factor eIF-1 (Kasperaitis et al. 1995). At present the function of Sui1 is unknown. The other two suppressors, sui2 and SUI3 encode the α and β subunits, respectively, of the eIF-2 complex. eIF-2 is a three-subunit complex (α, β, and γ) that is characterized biochemically to bind tRNAiMet in a GTP-dependent fashion (Hershey 1991; Merrick 1992). Our ability to isolate mutations in these two subunits of eIF-2 that confer the ability to initiate at a UUG codon implicated eIF-2 to have an additional function in ribosomal recognition of the start codon.
In this paper, we describe the isolation and characterization of two additional suppressor genes, SUI4 and SUI5. Both suppressor genes were identified as dominant suppressors in diploid cells and have a lethal phenotype in haploid cells. SUI4 is identical to the GCD11 gene in yeast that encodes the γ subunit of the eIF-2 complex (Hannig et al. 1993). The γ subunit of eIF-2 is classified as a GTP-binding protein based on sequence homology to conserved motifs in the GTPase superfamily (Bourne et al. 1991). SUI5 is identical to the TIF5 that encodes the translation initiation factor, eIF-5 (Chakravarti and Maitra 1993). eIF-5 has been shown biochemically to be involved in stimulating the hydrolysis of GTP bound to eIF-2, which is required for eIF-2 dissociation from the 43S pre-initiation complex and subsequent 60S ribosome junction (Chakrabarti and Maitra 1991). Biochemical characterizations of the mutant eIF-2 complex from a SUI4 strain and mutant eIF-5 from a SUI5 strain suggest that the ability to initiate at a UUG codon in vivo results from increased dissociation of eIF-2 from the initiator-tRNA in the absence of GTP hydrolysis, and enhanced stimulation of eIF-2 GTP hydrolysis, respectively. Furthermore, we characterized biochemically eIF-2 from SUI3 suppressor mutants and show that mutant complexes have an increased intrinsic rate of GTP hydrolysis in the absence of eIF-5. Our in vivo and in vitro analyses indicate that the GTP hydrolysis step that leads to dissociation of eIF-2 from initiator-tRNA serves as a checkpoint in ensuring a 3-bp codon/anticodon interaction during the ribosomal scanning process and preventing initiation at non-AUG codons.
Results
Genetic and molecular characterization of the SUI4 and SUI5 alleles
We reported previously the isolation of a group of suppressor mutants from diploid cells that were capable of initiating translation at the HIS4 gene in the absence of an AUG start codon (Castilho-Valavicius et al. 1992). Among these dominant His+ suppressors were a subgroup, which when sporulated, yielded only two viable His− spores indicating that the dominant suppressor allele conferred a lethal phenotype in a haploid cell. The recessive lethal phenotype of some of these suppressor mutants could be rescued when a CEN plasmid containing the wild-type SUI3 gene was present in the dipolid cells before sporulation. However, a subgroup of these mutants were not rescued by SUI3+ suggesting that they contained mutations in different translation initiation components, possibly one of two suppressor genes isolated previously sui1 and sui2 (Cigan et al. 1989; Yoon and Donahue 1992) or the GCD11 gene (Hannig et al. 1993) that encodes the γ subunit of eIF-2, the only subunit of eIF-2 not yet isolated as a suppressor mutant through our reversion studies.
To ascertain whether these genes could rescue the recessive lethal phenotype we transformed Ura− derivatives of the diploid suppressor mutants AR171, AR172, AR173, AEC6, AEC7, AEC8, AEC10, and AR168 with CEN plasmids containing either the SUI1, SUI2, SUI3, or GCD11 genes and subjected them to tetrad analysis. As shown in Table 1, the recessive lethal phenotype associated with the suppressor strains AR171, AR172, AR173, and AEC8 was capable of being complemented by the plasmid containing the wild-type GCD11 gene as indicated by consistently observing four- and three-spore tetrads. This indicated that these strains contained a suppressor mutation in the gene encoding the γ subunit of eIF-2. We refer to this suppressor allele as SUI4 (GCD11). In contrast, AEC6, AEC7, AEC10, and AR168 were not rescued by either of the four plasmids (data not shown). To be certain that the suppressor gene in these latter four strains did not correspond to any of these four genes we integrated a copy of either SUI1, SUI2, SUI3, or GCD11 as part of the URA3+, YIp5 plasmid into each diploid strain and analyzed them by tetrad analysis following the segregation of the Ura3+ phenotype relative to the recessive lethal phenotype. Table 2 shows the results for the AR168 strain. The Ura3+ phenotype associated with each of these plasmids in the different strains segregated independently of the recessive lethal phenotype, indicating that the suppressor mutation in AR168 is not an altered allele of SUI1, SUI2, SUI3, or GCD11. We refer to the suppressor allele in AR168 as the SUI5 gene.
Table 1.
GCD11 rescues the lethal phenotype of SUI4
| Segregation of His phenotypes in tetrads
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diploid/Plasmid
|
4 spores
|
3 spores
|
2 spores
|
1 spore
|
||||||||||
| 4+:0−
|
3+:1−
|
2+:2−
|
1+:3−
|
0+:4−
|
3+:0−
|
2+:1−
|
1+:2−
|
0+:3−
|
2+:0−
|
1+:1−
|
0+:2−
|
1+:0−
|
0+:1−
|
|
| AR171/YCp50 | 8 | 2 | ||||||||||||
| p1200 (SUI1) | 9 | 1 | ||||||||||||
| p591 (SUI2) | 8 | 2 | ||||||||||||
| pBE30 (SUI3) | 5 | 5 | ||||||||||||
| Ep293 (GCD11) | 1 | 3 | 3 | 1 | 1 | 1 | ||||||||
| AR172/YCp50 | 6 | 3 | ||||||||||||
| p1200 (SUI1) | 9 | 1 | ||||||||||||
| p591 (SUI2) | 9 | |||||||||||||
| pBE30 (SUI3) | 5 | 5 | ||||||||||||
| Ep293 (GCD11) | 4 | 1 | 1 | 3 | 1 | |||||||||
| Ar173/YCp50 | 10 | |||||||||||||
| p1200 (SUI1) | 10 | |||||||||||||
| p591 (SUI2) | 8 | 2 | ||||||||||||
| pBE30 (SUI3) | 1 | 3 | 6 | |||||||||||
| Ep293 (GCD11) | 1 | 2 | 4 | 1 | 2 | |||||||||
| AEC8/YCp50 | 8 | 1 | ||||||||||||
| p1200 (SUI1) | 9 | |||||||||||||
| p591 (SUI2) | 9 | 1 | ||||||||||||
| pBE30 (SUI3) | 8 | 2 | ||||||||||||
| Ep293 (GCD11) | 3 | 3 | 1 | 2 | 1 | |||||||||
Table 2.
The dominant, recessive lethal suppressor mutation in the AR168 strain is not linked to SUI1, SUI2, SUI3, or SUI4 (GCD11)
| Tetrad analysis | ||||||
|---|---|---|---|---|---|---|
| (segregation of Ura3 phenotype among viable ascospores)
|
||||||
| Diploid strains | 2+:0− | 1+:1− | 0+:2− | 1+:0− | 0+:1− | Ura3+:Ura3− |
| AR168, YIp5–SUI1 | 1 | 2 | 1 | 3 | 3 | 1.0 |
| AR168, YIp5–SUI2 | 0 | 6 | 0 | 5 | 6 | 0.9 |
| AR168, YIp5–SUI3 | 0 | 2 | 0 | 2 | 2 | 1.0 |
| AR 168, YIp5–SUI4 (GCD11) | 1 | 1 | 0 | 2 | 2 | 1.6 |
Part of the amino-acid sequence of the eIF-2γ subunit matches the four highly conserved motifs in the GTPase superfamily, suggesting that it might function as a GTPase in translation initiation, such as EF-Tu functions as a GTPase during elongation (Hannig et al. 1993). The SUI4 mutant alleles from strains AR171, AR172, AR173, and AEC8 were isolated by the integration-excision method (Roeder and Fink 1980). DNA sequence analysis of these alleles revealed that they all contained the same point mutation changing Asn-135 to Lys in the G2 motif (eIF-2γN135K). This amino acid residue is conserved in the G2 motif of yeast and human eIF-2γ (Gaspar et al. 1994). These observations suggest that an alteration in the function of this putative G-protein might allow the preinitiation complex to initiate at a non-AUG codon at his4.
The dominant SUI5 suppressor allele was cloned directly from a DNA library constructed from yeast strain AR168 and screened for the dominant His+ suppressor phenotype indicative of the SUI5 suppressor gene being associated with a plasmid. Subsequent subcloning/deletion analysis narrowed the suppressor gene to an ∼2.8-kb DNA fragment. Preliminary DNA sequence analysis identified this DNA fragment to contain only one complete open reading frame (ORF) that corresponded to the TIF5 gene, which encodes the eukaryotic translation initiation factor 5, eIF-5 (Chakravarti and Maitra 1993). eIF-5 is analogous to a GTPase-activating protein in that it stimulates GTP hydrolysis on eIF-2 at the time of translation initiation (Hershey 1991; Merrick 1992). The complete DNA sequence of the coding region of the SUI5 suppressor gene revealed a single-base mutation that altered Gly-31 to Arg (eIF-5G31R). Gly-31 is not only conserved in relative position but is located within a region that is conserved most highly between yeast (Chakravarti and Maitra 1993) and mammalian (Das et al. 1993) eIF-5 (Fig. 1). Additional studies also identified the suppressor mutants AEC6, AEC7, and AEC10 to contain the same SUI5 allele. Therefore, we conclude that the SUI5 suppressor gene represents a mutated form of eIF-5. Because eIF-5 has been shown to stimulate the hydrolysis of GTP bound to eIF-2 as part of the 43S preinitiation complex, the identification of SUI5 indicates that the ability to initiate translation at his4 in the absence of an AUG codon can occur through an alteration in the mechanism of stimulation of eIF-2 GTPase activity.
Figure 1.
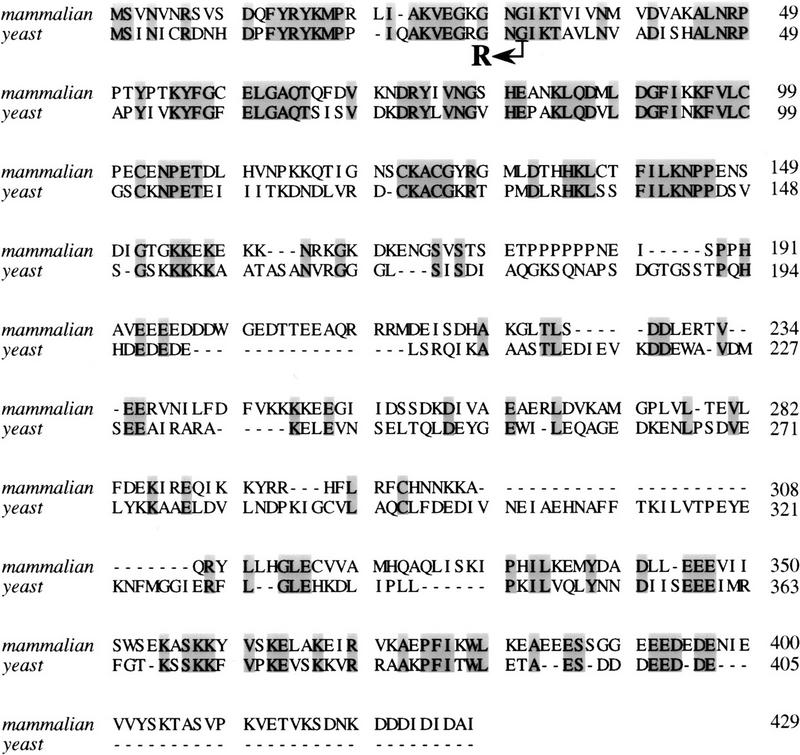
Protein sequence alignment of mammalian (top) and yeast (bottom) eIF-5. The amino acid sequences of mammalian and yeast eIF-5 are compared for maximal alignment. The shaded regions indicate identical amino acid residues among the two proteins. The SUI5 suppressor allele contained a point mutation (GGT → CGT) that altered Gly-31 to Arg as indicated by the arrow.
SUI4 (eIF-2γN135K) and SUI5 (eIF-5G31R) allow initiation of translation at a UUG codon
Using direct protein sequence analysis, we demonstrated previously that the sui1, sui2 (eIF-2α), and SUI3 (eIF-2β) suppressor mutants initiate translation at his4 in the absence of an AUG start codon by allowing a mismatched codon/anticodon interaction between a UUG codon and the initiator-tRNA (Yoon and Donahue 1992; H. Yoon and T.F. Donahue, unpubl.). This UUG codon is located two codons downstream (+3 amino acid position) from the AUG start site at HIS4 (Donahue et al. 1982). To discern which codon the SUI4 and SUI5 suppressor mutants use for translation initiation, his4–lacZ fusion chimerae were generated that lacked an AUG start codon and either had the normal UUG codon at the +3 codon position or a GUG, CUG, or UUA codon in place of the +3 UUG. The β-galactosidase specific activity of each chimera was measured in SUI4 and SUI5 strains as an indication of specificity and efficiency of translation initiation. As shown in Table 3 (lines 1–4), SUI4 and SUI5 strains use the UUG codon at the +3 position to initiate translation similar to that observed with the sui1, sui2, and SUI3 suppressor strains as substitution of UUA, CUG, or GUG for UUG greatly reduces β-galactosidase activity. The level of activity seen with these strains, despite mutation of the UUG, is either a result of some residual translation initiation either at the +3 amino acid position or at some undefined position in the his4–lacZ region. In addition, a four- to fivefold induction of the his4 transcript levels has been shown to occur as a result of mutations in eIF-2, which alter the translational regulation of GCN4, a transcriptional regulator of HIS4 expression (Williams et al. 1989; Castilho-Valavacius et al. 1990). This transcriptional induction enhances the level of β-galactosidase activity in some of our suppressors. Nevertheless, these studies indicate that the mechansim of non-AUG initiation in the SUI4 and SUI5 strains is functionally related to the mechanism of non-AUG initiation in sui1, sui2, and SUI3 strains.
Table 3.
Efficiency and specificity of suppression
|
his4–lacZ fusiona
|
β-Galactosidase specific activityb (suppressor strains)
|
||||
|---|---|---|---|---|---|
|
sui1
|
sui2 (eIF-2α)
|
SUI3 (eIF-2β)
|
SUI4/+ (eIF-2γ)
|
SUI5/+ (eIF-5)
|
|
| 1. 5‘+1AUUGUUUUG | 239 (100%) | 57 (100%) | 403 (100%) | 115 (100%) | 440 (100%) |
| 2. 5‘+1AUUGUUUUA | 17 (7%) | 6 (11%) | 58 (14%) | 18 (16%) | 5 (1%) |
| 3. 5‘+1AUUGUUCUG | 25 (10%) | 8 (14%) | 67 (17%) | 20 (17%) | 4 (1%) |
| 4. 5‘+1AUUGUUGUG | 27 (11%) | 10 (18%) | 73 (18%) | 19 (17%) | 4 (1%) |
| 5. 5‘−44GUG+1AUUGUUUUG | 209 (100%) | 16 (100%) | 355 (100%) | 71 (100%) | 365 (100%) |
| 6. 5‘−44GUU+1GUGGUUGUG | 61 (29%) | 2 (13%) | 132 (37%) | 37 (52%) | 3 (1%) |
| 7. 5‘−44GUU+1GUGGUAUUA | 69 (33%) | 2 (13%) | 108 (30%) | 28 (39%) | 5 (1%) |
Boldface letters indicate the codons change(s) compared to the wild-type HIS4 sequence.
Boldface numbers indicate the β-galactosidase specific activity (units). Numbers in parentheses indicate the percentage of activity compared to UUG control in line 1 or 5, respectively.
The inability to see comparable β-galactosidase activity with CUG and GUG codons at the +3 amino acid position in the his4–lacZ fusion strains could suggest that our suppressor mutants prefer to use UUG as oppose to CUG or GUG as a start site for suppression. Alternatively, naturally occurring upstream and out-of-frame CUG and GUG codons that are present in the HIS4 leader region (Donahue et al. 1982) might preclude the ability to detect efficient suppression at these codons, similar to the effect of an upstream AUG start codon precluding initiation at a downstream AUG during the scanning process (Donahue and Cigan 1988). Therefore, to gain insight into whether our suppressor strains could use a codon other than UUG for suppression we mutated an out-of frame GUG codon at position −44 in the HIS4 leader region and introduced a GUG codon either at the +1 amino acid position or at both the +1 and +3 amino acid positions. We focused on the GUG codon as only one base change was required to eliminate an upstream GUG codon at the −44 position whereas multiple CUG codons exist in the HIS4 leader region and would have needed to be mutated (Donahue et al. 1982). In addition, GUG is used as an alternative start codon at some genes in Escherichia coli (Gualerzi and Pon 1990). As shown in Table 3 (lines 6,7), the level of β-galactosidase activity increases in sui1, SUI3, and SUI4 suppressor strains that contain these his4–lacZ fusion constructs relative to the level of activity in the UUG his4–lacZ control (line 5). However, the level of β-gal activity obtained is still lower than strains that have a UUG codon at +3 as part of the his4–lacZ fusion. In contrast, the SUI5 strain does not use GUG to any appreciable level. It is currently unclear why the SUI5 mutant has such a strong preference for a UUG codon as the site for suppression. In addition, sui2 is a very poor suppressor (Castilho-Valavacius et al. 1990) even with the UUG his4–lacZ fusion, which obscures our ability to draw a sound conclusion about its ability to use GUG inefficiently as an alternative site for suppression. Nevertheless, our conclusion for most of our suppressor strains is that they prefer to use a UUG codon as the start site for suppression and some will use GUG albeit less efficiently. This suggests that the effects of these suppressor mutations is to breakdown the mechanism for achieving translation initiation fidelity.
Biochemical characterizations of the SUI4 (eIF-2γN135K) suppressor
Given that the mutation of eIF-2γN135K is located in the G2 motif (Bourne et al. 1991), it is conceivable that the SUI4 mutant might allow translation to initiate at the UUG codon by altering GTP binding or the rate of GTP hydrolysis. To investigate these possible suppression mechanisms, the mutant eIF-2γN135K complex was purified and characterized for basic biochemical properties related to eIF-2 function compared with wild-type eIF-2. For these and other experiments we purified mutant and wild-type eIF-2 using a two-step purification procedure, phosphocellulose cation exchange resin (P11) and nickel affinity resin. A histidine tag (His tag) was introduced at the amino terminus between the first and second codons of the eIF-2γ-coding regions and the wild-type or mutant recombinant protein was produced in yeast under the control of its own native promoter. A plasmid containing the His-tagged wild-type eIF-2γ allele complements the lethal effects of a GCD11 null allele and the level of tagged protein synthesized in vivo is not significantly different from native eIF-2γ levels (data not shown), suggesting that the His-tagged eIF-2γ subunit is functionally active in vivo. The His-tagged eIF-2γN135K was also considered active in vivo as it was capable of generating a dominant His+ suppressor phenotype in haploid yeast. This was not a result of the His tag at the amino end of the protein as the His-tagged version of the wild-type eIF-2γ subunit when expressed in yeast did not confer a His+ suppressor phenotype.
This purification scheme allowed reasonable yields of eIF-2 complex without contamination of eIF-5. As shown by Western blot analysis, eIF-5 was eluted from the P-11 column at low salt concentration (Fig. 2A, lane 3), whereas the His-tagged eIF-2 complex remained bound under these conditions and was eluted at higher salt concentration (Fig. 2A, lane 4). The stronger binding of the His-tagged complex to the P11 column served as a key step in separating mutant eIF-2 complex from wild-type complex for further characterization. The purified eIF-2 complex when resolved on a 10% SDS-PAGE gel and followed by Coomassie blue staining was not homogeneous, but highly purified with very few impurity proteins (Fig. 2B, lane 2). Furthermore, as shown by Coomassie blue staining (Fig. 2B) and Western blot analysis (Fig. 2C), the amount of eIF-2 in the wild-type preparation is very similar to that in the mutant preparation. The stoichiometry of the three different subunits of the mutant eIF-2 complex purified by the overexpression scheme is comparable with that of the wild-type eIF-2 complex (Fig. 2). Also, the mutant eIF-2 preparation did not contain any native γ subunit that migrates faster on gels as it lacks a His tag (Fig. 2C, lane 2).
Figure 2.

Purification of the His-tagged wild-type eIF-2 and the eIF-2γN135K complexes. (A) Crude extracts and fractions from P-11 and Ni chromatography columns were resolved by 10% SDS-PAGE, transferred to a nitrocellulose membrane, and analyzed by Western blot analysis using antisera directed against eIF2-α and eIF2-γ (top) or eIF2-β and eIF-5 (bottom). Purification of wild-type eIF-2: (Lane 1) 50 μg of crude extract; (lane 2) 50 μg of protein from the flowthrough fraction of the P-11 column; (lane 3) 20 μg of protein from the 450 mm KCl wash fraction of the P-11 column; (lane 4) 10 μg of protein from the 750 mm KCl elution fraction of the P-11 column (predialysis); (lane 5) 10 μg of protein from the 750 mm KCl elution fraction of the P-11 column (after dialysis); (lane 6) 10 μg of protein from the flowthrough fraction from the nickel affinitiy column; (lane 7) 5 μg of protein from the pooled elution fraction from the nickel affinity column; (lane 8) 5 μg of wild-type eIF-2 after concentration of the pooled elution fraction by centricon-30. (B) Coomassie blue staining of purified wild-type eIF-2 and eIF-2γN135K complexes. Fifteen micrograms of protein from the concentration step after Ni affinity chromatography were resolved on a 10% SDS–polyacrylamide gel. (Lane 1) Molecular mass markers with positions noted as (kD); (lane 2) wild-type eIF-2 complex; and (lane 3) mutant eIF-2γN135K complex. Arrows point to the position of each of the eIF-2 subunits as determined by Western blot analysis. (C) Western blot analysis of purified wild-type eIF-2 and eIF-2γN135K complexes. Five micrograms of protein was resolved by 10% SDS-polyacrylamide gel and probed for the presence of the α, β, and γ subunits of eIF-2 by Western blot analysis. The position of each is noted by an arrow. (Lane 1) Wild-type eIF-2, (lane 2) mutant eIF-2γN135K.
The first assay we employed was to measure the ability of eIF-2γN135K complex to bind charged initiator-tRNA (Met-tRNAiMet) in a GTP-dependent fashion, otherwise known as ternary complex formation (Hershey 1991; Merrick 1992). As shown in Figure 3A, mutant eIF-2 is only capable of forming ternary complex at ∼15%–20% of wild-type levels. Therefore, as observed previously with sui2 (α) and SUI3 (β) mutants (Donahue et al. 1988; Cigan et al. 1989), a suppressor mutation in eIF-2γ also leads to a defect in ternary complex formation. The inability to see significant binding of eIF-2γN135K complex to initiator-tRNA may be a result of a number of possibilities. One possibility is that the mutant complex does not bind GTP. Alternatively, it may bind GTP but the mutation in the γ subunit either alters the ability of eIF-2 to bind or stay bound to initiator-tRNA, or confers to eIF-2 the ability to autohydrolyze GTP in an eIF-5 independent fashion that leads to dissociation of eIF-2 from initiator-tRNA. Therefore, we tested these possibilities.
Figure 3.


Ternary complex formation by purified wild-type and mutant eIF-2 complexes. (A) Purified wild-type eIF-2 (□) and mutant eIF-2γN135K (⋄) were each assayed for the ability to promote GTP-dependent binding to the [3H] methionine charged initiator-tRNA [3H]Met–tRNAiMet, 60,000 cpm/pmole, 0.075 μm) as a function of protein concentration (μg). Identical reactions without GTP were performed as a control for nonspecific GTP-independent binding activity. The number of picomoles of [3H]Met–tRNAiMet bound in the absence of GTP was subtracted from the number of pimoles of [3H]Met–tRNAiMet] bound in the presence of GTP to determine the GTP-dependent-binding activity, for each respective eIF-2 complex. (B) Wild-type eIF-2 (□), the mutant eIF-2γN135K complex (⋄), and the mutant eIF-2βS264Y complex (○) were each assayed for their ability to dissociate from the [3H]Met–tRNAiMet in the presence of the nonhydrolyzable GTP analog, GppNp. For the wild-type eIF-2 and eIF-2βS264Y complexes, ternary complex formed after 5 min of incubation with GppNp and [3H]Met–tRNAiMet (0.075 μm) was competed with various concentrations of unlabeled, charged initiator-tRNA for an additional 5 min and the level of labeled ternary complex determined by the filter-binding assay. The same reaction conditions were used to assay the eIF-2γN135K complex with the exception that a 10-min incubation time was used in the initial step to enhance the level of labeled ternary complex. Ternary complex without addition of unlabeled charged initiator-tRNA was stable at 37°C for up to 15 min (data not shown). Identical reactions without GppNp were performed as a control for nonspecific binding of the labeled tRNA and subtracted from the respective assays as background. The amount of eIF-2 preparation in each reaction was adjusted to compensate for similar initial levels of ternary complex formation in the presence of GppNp. Total protein in each reaction was 1.25 μg for the wild-type eIF-2 and 5 μg for each of the mutant complexes. (C) The eIF-2γN135K complex (5 μg of total protein) was assayed for its ability to bind [3H]Met–tRNAiMet (60,000 cpm/pmole) in the presence of GppNp as compared with to the wild-type eIF-2 (5 μg of total protein). (Lanes 1–3) The amount of ternary complex formed by wild-type eIF-2 in the presence of GTP (25 μm), and 25 μm, and 1 mm GppNp, respectively, in a 5-min reaction. (Lanes 4–6) The amount of ternary complex formed by mutant eIF-2γN135K complex in the presence of GTP (25 μm); and 25 μm, and 1 mm GppNp, respectively, in a 10-min reaction. The 10-min time point used for the mutant complex analysis was to maximize the amount of initiator-tRNA binding. However, only a 10% increase in binding is observed by using a 10-min incubation vs. a 5-min incubation period. (D) Same as C except using purified mutant eIF-2βS264Y complex (5 μg of total protein) compared with wild-type eIF-2 complex (2.5 μg of total protein). The different levels of total protein added to each reaction adjusts for the lower yields of eIF-2 in the former preparation as determined by Western blot analysis using antibodies directed against the α subunit of eIF-2. (Lanes 1,2) The amount of ternary complex formed by wild-type eIF-2 in the presence of GTP (25 μm); and 25 μm GppNp, respectively, in a 5-min reaction. (Lanes 3–5) The amount of ternary complex formed by mutant eIF-2βS264Y complex in the presence of GTP (25 μm), and 25 μm, and 1 mm GppNp, respectively, in a 5-min reaction. (E) Same as C except using purified mutant eIF-2βL254P complex (5 μg of total protein) compared with wild-type eIF-2 complex (0.17 μg of total protein). The different levels of total protein added to each reaction adjusts for the lower yields of eIF-2 in the former preparation as determined by Western blot analysis using antibodies directed against the α subunit of eIF-2. (Lanes 1,2) The amount of ternary complex formed by wild-type eIF-2 in the presence of GTP (25 μm), and 25 μm GppNp, respectively in a 5-min reaction. (Lanes 3,4) The amount of ternary complex formed by mutant eIF-2βL254P complex in the presence of GTP (25 μm); and 25 μm GppNp, respectively, in a 5-min reaction. The data in panels B–E represent the average of two independent experiments with a standard deviation <15%.
To test for a GTP-binding defect we performed three different assays. The first two assays determined the ability of the eIF-2γN135K complex to bind either [3H]GTP or [γ-32P]GTP compared with wild-type eIF-2 complex. As shown in Figure 4, A and B, the eIF-2γN135K complex binds each of these labeled nucleotides in a similar fashion to that observed with the wild-type complex. The third assay we performed was a competition assay between [α-32P]GTP bound to eIF-2 and unlabeled GDP. The reason for using GDP in these competition assays is that eIF-2 has a 400-fold higher affinity for GDP than GTP and therefore GDP would act as a stronger competitor (Hershey 1991). As shown in Figure 4C, 2 min after addition of an equal concentration of GDP, the amount of [α-32P]GTP bound to eIF-2γN135K complex and wild-type eIF-2 complex achieves equilibrium. Further incubation does not change this equilibrium suggesting that the rate of dissociation of GTP from the eIF-2γN135K complex is not significantly different than the rate of dissociation of GTP from the wild-type eIF-2 complex. In addition, the dissociation rate of GTP from the eIF-2γN135K mutant complex is virtually identical to wild-type eIF-2 complex in the first minute (data not shown). GDP-binding assays and competition assays using labeled GDP and unlabeled GDP also did not show any significant differences in nucleotide binding/dissociation between eIF-2γN135K complex and wild-type eIF-2 (data not shown). These data suggest that the initiator-tRNA-binding defect observed with the eIF-2γN135K complex (Fig. 3) is not a result of a major change in its GTP- or nucleotide-binding activity. In agreement with this, increasing the concentration of GTP does not increase significantly the initiator-tRNA-binding activity of the eIF-2γN135K complex relative to wild-type eIF-2 activity (data not shown). In fact, the initiator-tRNA-binding activity seems to be more sensitive to higher GTP concentration, ∼50% reduced at 20-fold excess of GTP, whereas wild-type eIF-2-binding activity is unaffected by a 40-fold excess of GTP.
Figure 4.



GTP-binding activity of wild-type and mutant eIF-2 complexes. (A) Purified wild-type eIF-2 complex (□; 1.25 μg of total protein), mutant eIF-2γN135K complex (⋄; 1.25 μg of total protein) and mutant eIF-2βS264Y (○; 2.5 μg of total protein) were assayed for their ability to bind [3H]GTP (1 μm final concentration) using a filter-binding assay. The different levels of total protein added to each reaction adjusts for the lower yields of eIF-2 in the latter preparation as determined by Western blot analysis using antibodies directed against the α subunit of eIF-2. The number of picomoles of [3H]GTP bound in the absence of protein was subtracted from the number of picomoles of [3H]GTP bound in the presence of protein to determine the binding activity. The amount of [3H]GTP bound at the 6-min timepoint was arbitrarily chosen as the 100% level for comparative purposes. (B) Same as A except using [γ-32P]GTP. For this reaction the specific activity of [γ-32P]GTP was adjusted with [3H]GTP, using a [3H]GTP:[γ-32P]GTP ratio of 1000:1 (10 μm final concentration), as [3H]GTP is purer than unlabeled GTP (see Materials and Methods). The protein–[γ-32P]GTP complex was quantitated using a scintillation counter but without scintillation fluid to avoid interference with [3H]GTP counts. The data represent the average of two independent experiments with a standard deviation <7%. (C) Purified wild-type eIF-2 complex (□; 1.25 μg of total protein), mutant eIF-2γN135K complex (⋄; 1.25 μg of total protein), and mutant eIF-2βS264Y (○; 2.5 μg of total protein) were assayed for their ability to dissociate from [α-32P]GTP (1 μm final concentration) using a filter-binding assay. The different levels of total protein added to each reaction adjusts for the lower yields of eIF-2 in the latter preparations as determined by Western blot analysis using antibodies directed against the α subunit of eIF-2. For this reaction the specific activity of [α-32P]GTP was adjusted with [3H]GTP, using a [3H]GTP:[α-32P]GTP ratio of 100:1 (1 μm final concentration), as [3H]GTP is purer than unlabeled GTP (see Material and Methods). The protein–[α-32P]GTP complex was quantitated using a scintillation counter but without scintillation fluid to avoid interference with [3H]GTP counts.
To test whether mutant complex might hydrolyze GTP in the absence of eIF-5 we performed ternary complex formation assays in the presence of GppNp, a nonhydrolyzable GTP analog. Substitution of GppNp for GTP has a modest stimulatory effect on the ability of wild-type eIF-2 to bind initiator-tRNA, 30%–64%, based on independent eIF-2 preparations (Fig. 3C,D,E, cf. lanes 1 and 2). A stimulatory effect is also observed on the ability of eIF-2γN135K complex to bind initiator-tRNA in the presence of GppNp, approximately twofold. This represents ∼50% of wild-type eIF-2-binding levels in the presence of GTP (Fig. 3C, lanes 5 and 1, respectively). Increasing the concentration of GppNp 40-fold does not increase the ability of the wild-type eIF-2 to bind initiator-tRNA but further increases the ability of eIF-2γN135K to bind; ∼2.8-fold better than GTP (Fig. 3C, lane 4 vs. lane 6). This represents ∼63% of wild-type eIF-2-binding levels in the presence of GTP (Fig. 3C, lanes 1,6). The ability of a nonhydrolyzable analog of GTP to stabilize initiator-tRNA binding suggests that the mutation in the G2 motif of the γ subunit confers some eIF-5-independent autohydrolytic activity on the eIF-2γN135K complex.
Finally, we tested whether the mutation in the γ subunit of eIF-2 might cause a higher dissociation rate from the charged initiator-tRNA. For these experiments we first formed ternary complex with [3H]Met–tRNAiMet in the presence of GppNp and then assayed the ability of the complex to remain bound in the presence of increasing concentrations of unlabeled, charged initiator-tRNA. The reason for using GppNp is that this analog should block any autohydrolytic activity that might lead to dissociation of eIF-2γN135K from the labeled-tRNA. In addition, the amount of wild-type eIF-2 used for the control experiment was adjusted to account for the same level of [3H]Met–tRNAiMet bound by the mutant complex. As shown in Figure 3B, [3H]Met–tRNAiMet bound to eIF-2γN135K in the presence of GppNp is competed very efficiently with increasing amounts of unlabeled charged initiator-tRNA, whereas the majority of wild-type eIF-2 remains bound even at a 1:10 ratio of labeled to unlabeled tRNA. Furthermore, in the presence of sixfold excess of unlabeled charged initiator-tRNA, ∼95% of labeled charged initiator-tRNA remained bound to the wild-type eIF-2 complex after 1 min (data not shown). In contrast, only ∼40% of labeled charged initiator-tRNA remained bound to the mutant eIF-2γN135K complex after 1 min (data not shown). These data suggest that the mutation in the G2 motif of the γ subunit confers on the eIF-2γN135K complex an increased dissociation rate from the initiator-tRNA in the absence of GTP hydrolysis.
Biochemical characterizations of the SUI5 (eIF-5) suppressor mutant
eIF-5 mediates the hydrolysis of GTP bound to eIF-2 ternary complex as part of the 43S ribosomal pre-initiation complex (Hershey 1991; Merrick 1992). This GTP hydrolysis allows eIF-2 to dissociate from the preinitiation complex in the eIF-2 ⋅ GDP binary form. This dissociation leaves the initiator-tRNA in the P site of the ribosome and is required for subsequent ribosome junction (Chakrabarti and Maitra 1991). We therefore assumed that the mutation in SUI5 affects the ability of eIF-5 to hydrolyze GTP. To assay GTPase activity mediated by eIF-5, we reconstituted the 43S preinitiation complex in vitro in the presence of an AUG triplet using purified 40S ribosomes and eIF-2. The 43S complex was purified from free [γ-32P]GTP and purified wild-type eIF-5 or mutant eIF-5G31R was then added and GTP hydrolysis was measured by quantitation of radiolabeled 32Pi released. For purification of eIF-5, a His tag was introduced at the carboxyl terminus of the wild-type and SUI5 alleles and purified from yeast using P11 and nickel affinity chromatography. The introduction of the His tag did not affect the ability of the wild-type protein to complement a null mutation of the essential eIF-5 gene, nor the dominant suppressor phenotype of the SUI5 allele, indicating that both modified proteins retain function in vivo. Figure 5A shows the Coomassie blue-stained gel of purified wild-type and mutant eIF-5. Both proteins were highly purified with very few impurity proteins. However, in comparison with the amount of eIF-5 protein in wild-type preparation (Fig. 5A, lanes 2,3), we always observed lower yields of eIF-5G31R protein (Fig. 5A, lanes 4,5) and therefore we had to adjust for equal amounts of wild-type and mutant eIF-5 proteins added to comparative assays as quantitated by Western blot analysis (Fig. 5B). The quantitation of the Western blots agreed with scanning of Coomassie blue-stained gels (data not shown).
Figure 5.

Purification and quantitation of wild-type and mutant eIF-5. (A) Coomassie blue staining of purified wild-type and mutant eIF-5 protein. Protein was resolved by 10% SDS-acrylamide gel electrophoresis. (Lane 1) Molecular mass markers, the positions of which are noted as kD; (lanes 2,3) 15 and 30 μg, respectively, of concentrated wild-type eIF-5 from the Ni affinity column; (lanes 4,5) 15 and 30 μg, respectively, of concentrated mutant eIF-5G31R from the Ni affinity column. (B) Quantitation of the level of wild-type eIF-5 and mutant eIF-5G31R by Western blot analysis. Five different amounts of total protein in both the wild-type (⋄) and mutant eIF-5 (□) purified preparations were resolved on a 10% SDS-acrylamide gel and transferred to a nitrocellulose membrane and detected by Western blot analysis (bottom) using antiserum directed against eIF-5 protein (1:25,000). 125I-labeled protein A (Amersham, 30 mCi/mg) was used as the secondary probe (1:4000). The membrane was also exposed to a PhosphorImager screen (Molecular Dynamics) to determine levels of radioactivity. Data were plotted using a linear-regression program (CA-Cricket Graph III; Computer Associates) in a Macintosh computer (top). The ratio of the two slopes (WT:G31R = 2.67:1) was used as the difference in eIF-5 protein levels in the two preparations.
Figure 6 shows the results of the GTPase assays using two different amounts of protein. In both cases the mutant eIF-5G31R protein shows an initial rate of hydrolysis that is approximately twofold greater than the initial rate of hydrolysis of wild-type eIF-5. This eIF-5-dependent GTP hydrolysis requires formation of preinitiation complex and is not observed when eIF-5 is incubated with ternary complex alone (data not shown). In addition, this twofold difference is observed with two independent preparations of wild-type and mutant eIF-5 proteins (data not shown). This observation suggests that the mutant eIF-5 is more active in stimulating eIF-2-dependent GTP hydrolysis on the 43S preinitiation complex.
Figure 6.

Comparison of the rate of hydrolysis of GTP bound to eIF-2 by purified wild-type eIF-5 and mutant eIF-5G31R protein. Wild-type eIF-5 and mutant eIF-5G31R were each assayed for the ability to promote hydrolysis of GTP bound to eIF-2 when part of the preinitiation complex. Formation and purification of the 43S preinitiation complex is described in Material and Methods. The amount of total protein added to each reaction was adjusted to compensate for the lower yields of eIF-5G31R in final purified preparations (i.e., 2.67 times more mutant total protein than wild-type total protein as described in Fig. 5B). Identical results were determined with independent preparations of both wild-type and mutant eIF-5. (♦) Three micrograms of wild-type eIF-5; (•) 8 μg of mutant eIF-5G31R; (⋄) 6 μg of wild-type eIF-5; (○) 16 μg of mutant eIF-5G31R; (□) no eIF-5.
Biochemical characterizations of the SUI3 (eIF-2βS264Y) suppressor mutants
Previous studies in our lab identified dominant mutations in the SUI3 gene that encodes the β subunit of eIF-2 that allows initiation at a UUG codon (Donahue et al. 1988; Yoon and Donahue 1992). One of these suppressor alleles, SUI3-2 (eIF-2βS264Y), was identified in a haploid yeast strain and eIF-2 partially purified from this strain was shown to have an in vitro defect in ternary complex formation similar to that described above for the SUI4 suppressor gene (Donahue et al. 1988). Another SUI3 allele, SUI3-40 (eIF-2βL254P), was isolated as a dominant suppressor in a diploid yeast strain that showed a recessive lethal phenotype on sporulation of the diploid (Castlho-Valavicius et al. 1992). To determine whether these mutant eIF-2 complexes have defects related to GTP binding or GTP hydrolysis, we purified these mutant complexes and assayed their activities as performed above for the SUI4 mutant complex. The mutant eIF-2 complexes were purified as described for eIF-2γ with the exception that the mutant β subunit contained the His tag that was introduced at the amino terminus of the SUI3-coding region. Here again, the His tag did not interfere with the ability of the wild-type SUI3 allele to complement a SUI3 null allele nor the ability of the mutant SUI3 His-tagged alleles to confer a dominant suppressor phenotype (data not shown). Coomassie blue staining of SDS-polyacrylamide gels revealed that there was less purified mutant eIF-2 complex/total protein in final preparations than was observed with wild-type eIF-2 preparations (data not shown). Therefore, for comparative assays of these mutant complexes to wild-type eIF-2 we adjusted for the level of eIF-2 as determined by Western blot analysis using antisera directed against the α subunit of eIF-2. Typically, we found that the eIF-2βS264Y complex to be present in final preparations at ∼50% the level of wild-type eIF-2 per total protein and the eIF-2βL254P complex to constitute only ∼3% the level of wild-type eIF-2 per total protein in final preparations.
Figure 3, D and E (lanes 3), shows that in the presence of GTP, eIF-2βS264Y complex and eIF-2βL254P complex, each show a defect in initiator-tRNA binding compared with wild-type eIF-2. Interestingly, when the same assay is performed in the presence of GppNp, the nonhydrolyzable analog of GTP, both mutant complexes can now bind initiator-tRNA at a comparable level to that of wild-type eIF-2 binding in the presence of GTP (Fig. 3, D and E, cf. lanes 1 and 4). However, the binding activity was not restored completely by the GppNp analog, representing ∼70%–80% of wild-type activity stimulated by GppNp (Fig. 3, D and E, cf. lanes 2 and 4). In contrast to what was observed with the eIF-2γN135K, a 40-fold increase in GppNp concentration did not increase further the initiator-tRNA-binding activity of the eIF-2βS264Y complex (Fig. 3D, lane 5). Nevertheless, these data suggest that the lower initiator-tRNA-binding activity associated with these mutant complexes is caused by an aberrantly higher intrinsic GTPase activity as a result of a mutation in the β subunit of eIF-2.
To further address whether the mutant eIF-2βS264Y complex has a higher intrinsic GTP hydrolysis rate, this complex was assayed for its ability to bind [γ-32P]GTP and [3H]GTP. The rationale was that if the mutant had higher intrinsic hydrolysis activity the [γ-32P] moiety would be hydrolyzed and mutant eIF-2 complex would not be detected as binding GTP. In contrast, if the mutant complex can bind GTP, the complex would still be labeled after binding [3H]GTP as subsequent to hydrolysis GDP will remain bound stably to eIF-2. For these experiments we attempted to adjust the concentration of each complex to bind an equivalent number of pmoles of GTP. As shown in Figure 4A, the eIF-2βS264Y complex has a rate of [3H]GTP binding that is similar to the wild-type complex and is associated stably with the complex for 15 min. In contrast, Figure 4B indicates that the amount of [γ-32P]GTP label initially associated with eIF-2βS264Y is reduced to ∼60% after 60 sec, whereas the vast majority of label initially associated with the wild-type complex remains associated in this time frame. Therefore, loss of label is associated with using a γ-labeled phosphate but not when the guanine part of the GTP moiety contains the label. The simplest interpretation of these results in light of restoration of initiator-tRNA-binding activity in the presence of GppNp is that the mutant eIF-2βS264Y complex binds GTP, but the mutation in the β subunit results in the complex intrinsically hydrolyzing GTP in the absence of eIF-5.
We also assayed the eIF-2βS264Y complex to determine if it might have an initiator-tRNA dissociation defect as we observed for eIF-2γN135K. As shown in Figure 3B, eIF-2βS264Y complex shows a difference from the wild-type eIF-2 complex in its ability to be dissociated from initiator-tRNA in the presence of GppNp. However, this difference, when compared with the dissociation defect associated with the eIF-2γN135K complex, is less severe. Instead, the major difference between the eIF-2βS264Y complex and the eIF-2γN135K complex is that the mutation in the former complex confers a greater increase in intrinsic hydrolysis of GTP.
Discussion
Using a genetic reversion analysis we have now identified mutations in the α, β, and γ subunits of eIF-2 as well as eIF-5 that confer aberrant initiation properties on the 43S preinitiation complex in vivo. Specifically, mutations in these proteins allow a mismatched codon/anticodon interaction between a UUG codon and the initiator-tRNA. These data suggest that one of the in vivo functions of eIF-2 and eIF-5 is to assure that the 43S preinitiation complex only recognize an AUG codon and to prevent initiation at non-AUG codons, as suppressor mutations in these proteins can support a non-AUG initiation event. One unique aspect of our study is that it provides a connection between ribosomal recognition of the AUG start codon and biochemical steps that occur during the initiation process, namely GTP hydrolysis and dissociation of eIF-2 from initiator-tRNA. Based on our data, translation initiation can be viewed as a process that has parallel to molecular switches associated with other biological processes that incorporate G proteins (Thompson et al. 1986; Thompson 1988; von Mollard et al. 1994; Koepp and Silver 1996; Rybin et al. 1996; Balch 1990; Wu et al. 1996). eIF-2, as a G-protein complex, exists in either of two states. The active state of eIF-2 is the GTP-bound form that can bind initiator-tRNA and associate with the 40S ribosome to scan mRNA. The inactive form of eIF-2 is the GDP-bound form that cannot bind initiator-tRNA. Therefore, GTP hydroysis at the time of translation initiation serves as a switch to convert eIF-2 from its active to its inactive state that allows it to dissociate from initiator-tRNA. This results in leaving the initiator-tRNA in the P site of the ribosome, which signals the transition from the initiation phase to the elongation phase of protein synthesis. Based on previous data, we speculate that there is only one requirement for such a switch in yeast. This requirement is a 3-bp codon/anticodon interaction between the AUG start codon and the initiator-tRNA. Justification for this speculation is based on the observation that sequence context in yeast has an insignificant role compared with the AUG start codon in the overall process of translation initiation (Cigan et al. 1988b). In addition, we have shown that an AGG codon at HIS4 can be recognized for initiation by the scanning ribosome when one copy of the initiator-tRNA genes has been mutated to have a complementary UCC anticodon (Cigan et al. 1988a). Once the 3-bp codon/anticodon interaction is established, eIF-5 can stimulate GTP hydrolysis that leads to release of eIF-2 ⋅ GDP (and other associated initiation factors). eIF-2 ⋅ GDP is converted to eIF-2 ⋅ GTP by the guanine nucleotide exchange factor, eIF-2B (Bushman et al. 1993a,b; Dever et al. 1995), which rounds out the GTPase cycle during the initiation process.
For translation initiation to occur at a non-AUG codon, the main obstacle to overcome is to dissociate initiation factors and leave the initiator-tRNA in the P site of the ribosome mismatched base-paired with a non-AUG codon. All of the biochemical characterizations of mutant eIF-2 complexes and mutant eIF-5 we have presented are compatible with such an aberrant initiation event. Figure 7 presents our model to explain the in vivo events that lead to UUG initiation, in the context of our in vivo and in vitro data. Figure 7, A and B, depicts the situation in a wild-type cell whereby the ribosome scans mRNA and may encounter and pause at a UUG codon in the leader region. In the absence of a 3-bp codon/anticodon interaction, GTP is not hydrolyzed and the ribosome continues to scan the leader. Finally, a 3-bp codon/anticodon interaction is realized at an AUG start codon, GTP is hydrolyzed, which leads to release of the translation initiation factors, leaving the initiator-tRNA in the P site, therefore, 60S joining and elongation can begin. Figure 7C depicts the situation in our SUI3 and SUI5 suppressor strains. Here again the ribosome scans and pauses at a UUG codon. However, in the case of eIF-5G31R, GTP hydrolysis is stimulated too quickly, or in the case of eIF-2βS264Y and eIF-2βL254P, eIF-2 now has intrinsic GTP hydrolysis activity. Either event leads to premature hydrolysis of GTP during the pause period, eIF-2 ⋅ GDP dissociates, and the initiator-tRNA remains in the P site. As a result, the initiator-tRNA is mismatched base-paired with the UUG codon. Nevertheless, the ribosome has initiated translation and is now committed to the elongation phase of protein synthesis. Figure 7D explains how one biochemical defect associated with the eIF-2γN135K complex, that is, dissociation from the initiator-tRNA in the absence of GTP hydrolysis, might relate to initiation at the UUG codon. At the time of ribosomal pausing at a UUG codon, eIF-2γN135K might dissociate prematurely from the initiator-tRNA despite the fact that GTP has not been hydrolyzed. Again, this would leave the initiator-tRNA in the P site mismatched base-paired with the UUG codon. Therefore, as a result of altering the rate of GTP hydrolysis or dissociation of eIF-2, the ribosome will initiate aberrantly at a UUG codon and to a lesser extent, as seen with some of our mutants, at a GUG codon as well (Table 3). This represents a breakdown in translation initiation fidelity that is controlled by the GTP hydrolysis step. Why inefficient suppression at a GUG codon is not observed with the SUI5 suppressor is a curiosity.
Figure 7.
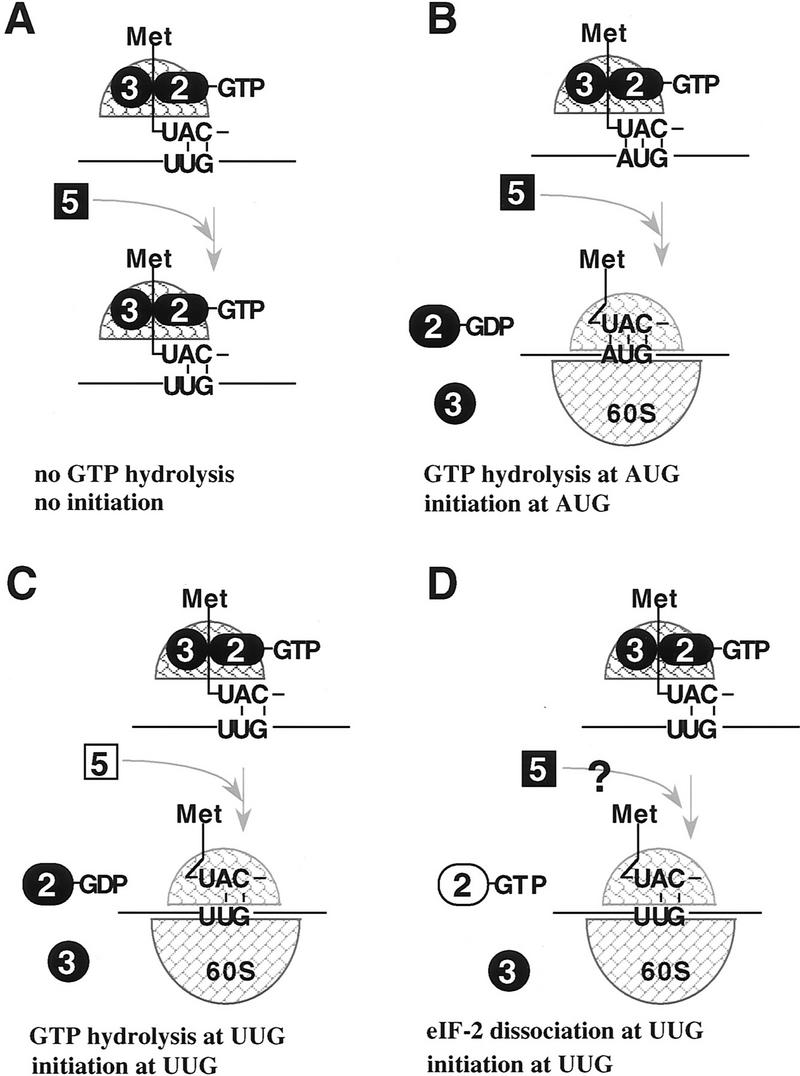
Schematic diagram of how GTP hydrolysis controls AUG selection during ribosomal scanning in wild-type cells and SUI suppressor mutants. (A,B) Translation initiation in wild-type cells. When the 43S preinitiation complex (including eIF-2 ⋅ GTP, eIF-3, 40S ribosomal subunit, and the initiator-tRNA) pauses at a non-AUG codon, such as UUG, GTP hydrolysis is inhibited or not induced attributable to the absence of the stringent 3-bp codon/anticodon signal and no translation initiation occurs. Therefore, the ribosome continues to scan the leader and encounters an AUG start codon (B). A 3-bp codon/anticodon interaction occurs, which signals the eIF-5-dependent hydrolysis of GTP bound to eIF-2. Translation initiation factors are released leaving the initiator-tRNA in the P site and the 60S ribosome joins such that elongation can begin. (C,D) Translation initiation at a non-AUG codon in SUI3, SUI4, and SUI5 strains. The 43S preinitiation complex pauses at a UUG codon, a mutation in eIF-5 or eIF-2β allows GTP to be hydrolyzed without a 3-bp codon/anticodon interaction between the UUG and the initiator-tRNA (C). Alternatively, when the 43S preinitiation complex pauses at UUG, the mutation in eIF-2γ results in dissociation of eIF-2 from the initiator-tRNA (D). Either event results in leaving the initiator-tRNA at the P site mismatched base-paired with the UUG codon. The 60S ribosome joins and the ribosome is committed to the elongation phase of translation. ( ) Initiator tRNA; (➋) eIF-2; (➌) eIF-3; (
) Initiator tRNA; (➋) eIF-2; (➌) eIF-3; ( ) eIF-5; (
) eIF-5; ( ) the 40S ribosomal subunit; (
) the 40S ribosomal subunit; ( ) the 60S ribosomal subunit.
) the 60S ribosomal subunit.
In contrast to prokaryotes, translation initiation at a non-AUG codon is extremely rare in eukaryotes. However, CUG has been reported to serve as a translational start site at, for example, the c-myc gene (Hann et al. 1988). The physiological significance of this initiation event at c-myc is not clear in light of a shorter protein being synthesized from a downstream AUG. However, Kozak (1989b) has proposed that an increased pause of the ribosome as a result of nearby and downstream secondary structure may contribute to rare non-AUG codon initiation events. Our data could suggest that by increasing the pause period at this CUG, it might increase the chance of GTP hydrolysis that would lead to an aberrant initiation event.
Our data suggest that the initiation factors eIF-2 and eIF-5 maintain the fidelity of translation initiation and that the GTP hydrolysis step is an important link in signalling or responding to ribosomal recognition of an AUG start codon. The observations that the γ subunit of eIF-2 and eIF-5 are involved in this process in vivo are consistent with the γ subunit being related in signature sequence motifs to other G-proteins and eIF-5 having been characterized previously biochemically to stimulate GTP hydrolysis in vitro (Charabarti and Maitra 1991). Characterization of the eIF-2γN135K complex reveals that it has a defect in premature dissociation from initiator-tRNA. A defect in intrinsic GTP hydrolysis was also detected at the level of the nonhydrolyzable analog of GTP, GppNp, being able to stimulate eIF-2γN135K complex to bind initiator-tRNA. At this time it is unclear whether these two defects are interrelated. However, the mutation in the G2 motif as we observed with eIF-2γN135K is compatible with conferring a hydrolysis and tRNA-binding defect based on co-crystal structures of EF-Tu (Nissen et al. 1995). Clearly, the initiator-tRNA-binding defect is a result of either or both of the two defects. Furthermore, both defects would be expected to add to the efficiency of initiation at a UUG codon (Fig. 7C,D). This latter point would also hold true for eIF-2βS264Y complex that appears to have a major defect in intrinsic GTP hydrolysis and a less severe defect in initiator-tRNA dissociation (Figs. 3B and 4B).
Another unique aspect of our analysis in terms of structure/function of eIF-2 is the observation that mutations in the β subunit have such a significant effect on the GTP hydrolysis reaction. Both point mutations in the β subunit (eIF-2βS264Y and eIF-2βL254P) are located at a putative zinc-finger motif in the carboxyl end of the SUI3-coding region and each confers intrinsic GTP hydrolysis activity on the eIF-2 complex in the absence of eIF-5. In fact, all SUI3 suppressor alleles we have characterized each contain a point mutation either within or nearby this motif (Castilho-Valavicius et al. 1992). This motif is conserved in the human β subunit and mutations that confer suppression reside at conserved amino acid positions between the human and yeast protein (Pathak et al. 1988). Taken together, our data suggest that this putative motif in the human and yeast β subunit has some essential function related to the GTP hydrolysis step, which is consistent with two-hybrid studies that the β subunit of eIF-2 interacts with eIF-5 (H. Yoon and T.F. Donahue, unpubl.). In fact, all of our mutational observations at the α, β (Donahue et al. 1988; Cigan et al. 1989; Castilho-Valavicius et al. 1992), and γ (this report) subunits of eIF-2 as well as eIF-5 (Fig. 1) are at residues conserved in the corresponding mammalian proteins, which points to the overall relevance of our studies to the general mechanism of ribosomal recognition of a start codon in all eukaryotes.
Materials and methods
Yeast strains and genetic methods
All strains in this study (Table 4) are related to TD28, an ascospore derivative of yeast strain S288C, which has been used extensively for studies of HIS4 translation initiation (Huang and Donahue 1997). Standard genetic techniques and media used for this analysis have been described (Guthrie and Fink 1991). The construction of yeast strains containing initiator codon mutations and the genetic selection scheme to identify suppressors of initiation have been described previously using haploid (Donahue et al. 1988) and diploid (Castilho-Valavicius et al. 1992) yeast cells.
Table 4.
Yeast strains
| 76-3D | MATα his4-303[ATT] ura3-52 leu2-3,112 |
| 304-1D | MATa sui2-1 his4-303[AUU] ura3-52 |
| 301-4D | MATa sui1-1 his4-303[AUU] lev2-3,112 ura3-52 |
| 301-5B | MATa sui1-1 his4-303[AUU] ura3-52 ino1-13 |
| 2119-11D | MATa SUI3-3 his4-303[AUU] ura3-52 leu2-3,112 ino1-13 |
| AEC6 | MATa/MATα ura3-52/ura3-52 LEU2/leu2-3,112 INO1/ino1-13 his4-302[CTG]/his4Δ401[ABC−] SUI5/SUI5G31R |
| AEC7 | MATa/MATα ura3-52/ura3-52 LEU2/leu2-3,112 INO1/ino1-13 his4-302[CTG]/his4Δ401[ABC−] SUI5/SUI5G31R |
| AEC8 | MATa/MATα ura3-52/ura3-52 LEU2/leu2-3,112 INO1/ino1-13 his4-300[ACC]/his4Δ401[ABC−] SUI4/SUI4N135K |
| AEC10 | MATa/MATα ura3-52/ura3-52 LEU2/leu2-3,112 INO1/ino1-13 his4-305[GTG]/his4Δ401[ABC−] SUI5/SUI5G31R |
| AR168 | MATa/MATα ura3-52/ura3-52 LEU2/leu2-3,112 INO1/ino1-13 his4-300[ACC]his4Δ401[ABC−] SUI5/SUI5G31R |
| AR171 | MATa/MATα ura3-52/ura3-52 LEU2/leu2-3,112 INO1/ino1-13 his4-302[CTG]/his4Δ401[ABC−] SUI4/SUI4N135K |
| AR172 | MATa/MATα ura3-52/ura3-52 LEU2/leu2-3,112 INO1/ino1-13 his4-300[ACC]/his4Δ401[ABC−] SUI4/SUI4N135K |
| AR173 | MATa/MATα ura3-52/ura3-52 LEU2/leu2-3,112 INO1/ino1-13 his4-302[CTG]/his4Δ401[ABC−] SUI4/SUI4N135K |
| BCV59 | MATa/MATα ura3-52/ura3-52 leu2-3,112/leu2-3,112his4-306/his4-306 sui3Δ::URA3/SUI3 |
| HH705 | MATα his4-303[ATT] ura3-52 leu2-3,112 pYEp24–URA3–SUI2–SUI3 pYO325–LEU2–SUI4N135K |
| HH725 | MATα his4-303[ATT] ura3-52 leu2-3,112 pYEp24–URA3–SUI2 pYO325–LEU2-SUI4–SUI3L254P |
| HH839 | MATa ura3-52 ino1-13 pYEp24–URA3–SUI5 |
| HH849 | MATa ura3-52 ino1-13 pYEp24–URA3–SUI4 |
| HH860 | MATa his4-301[ACG] ura3-52 leu2-3,112 pRS315–LEU2–SUI5G31R |
| HH868 | MATα ura3-52 leu2-3,112 his4-316 gcd11Δ::URA3 pRS315–LEU2–SUI4 |
| HH875 | MATα ura3-52 leu2-3,112 ino1-13 his4-306 tif5Δ::URA3 pRS315–LEU2–SUI5 |
| HKH873 | MATa/MATα ura3-52/ura3-52 leu2-3,112/leu2-3,112 his4-306/his4-306ino1-13/ino1-13tif5Δ::URA3/TIF5 |
| JAR10-1-2 | MATa/MATα ura3-52/ura3-52 leu2-3,112/leu2-3,112 his4-316/his4-316 gcd11Δ::URA3/GCD11 |
| JJ1 | MATa his4-300[ACC] ura3-52 ino1-13 |
| JJ5 | MATa his4-302[CTG] ura3-52 ino1-13 |
| JJ6 | MATa his4-303[ATT] ura3-52 ino1-13 |
| JJ7 | MATa his4-303[ATT] ura3-52 ino1-13 |
| JJ209 | MATa his4-306[TTG] ura3-52 ino1-13 |
| JJ210 | MATa his4-306[TTG] ura3-52 ino1-13 |
| JRC30-3B | MATa his4-301[ACG] ura3-52 leu2-3,112 |
| KT7 | MATa ura3-52 leu2-3,112 sui3Δ::URA3 pRS315–LEU2–SUI3S264Y |
| TD28 | MATa ura3-52 ino1-13 |
Complementation analysis of the dominant, recessive lethal suppressor mutation in yeast strains AR171, AR172, AR173, and AEC8 was performed by transformation of these strains with the CEN plasmids, p1200 (SUI1+), p591(SUI2+), pBE30 (SUI3+), and Ep293 (GCD11+), and subsequent tetrad analysis. The SUI4 (GCD11) mutant alleles from these strains were isolated by the integration-excision method (Roeder and Fink 1980) using plasmid Ep488 (Hannig et al. 1993). Genomic DNA was isolated from each strain, restricted with HindIII, ligated, and used to transform E. coli. Plasmids isolated from E. coli were used to transform haploid yeast cells containing an his4 initiator codon mutation. Nonselective acquisition of a His+ phenotype among the transformants was used as an indication that the corresponding plasmid contained the dominant SUI4 suppressor allele. DNA sequence analysis was performed as described previously for the GCD11 gene (Hannig et al. 1993)
The dominant, recessive lethal suppressor mutation in yeast strains AEC6, AEC7, AEC10, and AR168 was not rescued after transformation with plasmids p1200 (SUI1+), p591 (SUI2+), pBE30 (SUI3+), and Ep293 (GCD11+) and subsequent tetrad analysis, suggesting that these strains contained a suppressor mutation in a different gene. We confirmed this by genetic linkage analysis. The Ura+ integrating plasmids, p636 (SUI1+), p615 (SUI2+), p504 (SUI3+), and Ep488 (GCD11+), were each used to transform yeast strain AR168 and the segregation of the Ura+ phenotype was compared with the recessive lethal phenotype by tetrad analysis. None of these plasmids were detected to be linked to the SUI5 suppressor locus. To clone this suppressor gene, genomic DNA was isolated from the SUI5 mutant strain AR168 and digested with BamHI. DNA was ligated into the BamHI site of the centromere containing yeast vector YCp50 (Parent et al. 1985) and was used to transform the his4 initiator codon mutant strains JJ1, JJ5, JJ6, JJ7, JJ209, and JJ210 to Ura+. Transformants were replica-plated to synthetic dextrose minus histidine plate and analyzed for their ability to nonselectively restore a His+ phenotype, indicative of the dominant SUI5 phenotype. Total DNA was isolated from Ura+, His+ transformants and was used to transform the E. coli. Plasmids were isolated and subjected to restriction/deletion analysis. The wild-type SUI5 allele on a plasmid (p2005) was isolated by gap–duplex repair of the mutant SUI5 gene on a plasmid (p1972). The position of the SUI5 mutation was identified by DNA sequence analysis of the entire coding region and compared with the wild-type allele. The recessive lethal suppressor mutation in strains AEC6, AEC7, and AEC10 was shown to be rescued by the wild-type SUI5 gene on a plasmid (p2005). The SUI5 suppressor gene from each of these strains was isolated by the gap–duplex repair method (Orr-Weaver et al. 1983) using plasmid p2020 after restriction with PvuII and SacII to linearize the plasmid. Plasmids p2286, p2289, and p2287 from AEC6, AEC7, and AEC10, respectively, were isolated and subjected to DNA sequence analysis.
A His tag was introduced at the amino termini of the wild-type and mutant eIF-2γ alleles by standard PCR procedures (Sambrook et al. 1989). In the first PCR reaction, an XmaI restriction site was introduced ∼0.5 kb upstream of the AUG start codon of the GCD11 gene and a His10 tag, flanked by a BglII and a BamHI restriction site, respectively, was introduced immediately 3′ to the AUG start codon. This PCR fragment was subcloned into the XmaI and BamHI restriction sites of the LEU2+, CEN plasmid, pRS315 (Sikorski and Hieter 1989), to generate plasmid p2119. For the second PCR reaction, the forward primer introduced a BamHI site preceding the second codon and the reverse primer created an XbaI site 0.23 kb from the end of the GCD11 codon region end. This PCR fragment was inserted into the BamHI and XbaI sites of the pRS315, which contained the first PCR fragment. Therefore, the 10-histidine tag was introduced in-frame between the first and the second codons of either wild-type (p2115) or mutant eIF-2γ (p2117).
The diploid yeast strain JAR 10-1-2, containing one homolog of the GCD11 gene disrupted with the URA3+ gene, was generated by the one-step gene disruption method (Rothstein 1983). An HpaI fragment containing most of the GCD11 coding region (from ∼25 bp upstream from the start codon to ∼120 bp upstream from the stop codon) was deleted from plasmid p2138 that contained the intact GCD11 gene. The wild-type URA3 gene as part of a SmaI DNA fragment was ligated into the HpaI site to generate plasmid p2178. Tetrad analysis of JAR10-1-2 yielded two viable spores that were Ura− and two inviable spores that should have been URA3+. JAR10-1-2 was transformed with plasmid p2115, which contains the wild-type His-tagged eIF-2γ allele. Yeast strain HH868, which was used as the source of purification of the wild-type His tagged eIF-2γ complex, was identified as a haploid, Ura+ and Leu+ ascospore indicating that the His-tagged eIF-2γ gene as part of the LEU2 plasmid could functionally substitute for the gcd11::URA3+ allele.
Plasmid p2117 containing the His-tagged eIF-2γN135K allele was used to transform the his4 initiator strain, JRC30-3B to Leu+ and analyzed nonselectively for a His+ phenotype indicative of the dominant SUI4 suppressor phenotype. eIF-2γN135K complex was purified from yeast strain HH705, which overexpressed the wild-type α and β subunits in addition to eIF-2γN135K. This strain contained two high-copy-number plasmids. One plasmid, pTD1778, is derived from YEp24 and contains the SUI2 and SUI3 wild-type genes (kindly provided by Dr. Tom Dever, National Institutes of Health, Bethesda, MD). The second plasmid, pYO325, is derived from pRS305 and contains the LEU2 gene, a 2 μ ARS sequence (provided by Dr. Yosh Ohya, University of Tokyo, Japan). The His-tagged SUI4 (eIF-2γN135K) mutant allele from plasmid p2117 was subcloned as a SacI–XhoI DNA fragment into pYO325 to generate plasmid p2262. Overexpression of eIF-2γN135K in strain HH705 results in a His+ suppressor phenotype and confers a slow-growth phenotype on SD medium lacking uracil and leucine.
A His tag was introduced at the carboxyl termini of the wild-type and mutant eIF-5 alleles by PCR. In the first step, an His10 tag flanked by a BamHI restriction site and a BglII restriction site, respectively, was introduced immediately 5′ to the stop codon, and an XbaI restriction site was introduced at ∼0.84 kb downstream from the end of the SUI5-coding region. This DNA fragment was subcloned into the BamHI and XbaI sites of plasmid pRS315. In the second step, an XmaI restriction site was introduced ∼0.45 kb upstream of the AUG start codon of the SUI5 gene and a BamHI site was introduced immediately 3′ to the last codon in the SUI5-coding region. This DNA fragment was subcloned into the XmaI and BamHI sites of plasmid pJLC101 (kindly provided by Dr. Norman Pace, University of California, Berkely). Finally, a SalI–BamHI fragment from pJLC101 containing the second PCR fragment was subcloned into the SalI–BamHI sites of the pRS315 plasmid containing the BamHI and XbaI PCR fragment generated in the first step. Therefore, the His10 tag was introduced in-frame immediately 5′ to the stop codon in both the wild-type eIF-5 (p2185) and the eIF-5G31R (p2187) genes.
The diploid yeast strain HKH873, containing one homolog of the TIF5 gene disrupted with the URA3+ gene, was generated by the one-step gene disruption method (Rothstein 1983). Using PCR methods, the TIF5-coding region was deleted between amino acid position 23 and the translational stop codon and the URA3+ gene was inserted at the novel junction to generate plasmid p2206. Tetrad analysis of HKH873 yielded two viable spores that were Ura− and two inviable spores that are presumably URA3+. HKH873 was transformed with plasmid p2185, which contains the wild-type His-tagged eIF-5 allele. Yeast strain HH875, which was used as the source of purification of the wild-type His-tagged eIF-5 protein was identified as a haploid, Ura+ and Leu+ ascospore, indicating that the His-tagged eIF-5 gene as part of the LEU2 plasmid could functionally substitute for the tif5:URA3+ allele. Yeast strain HH860, producing His-tagged mutant SUI5 (eIF-5G31R), was generated by transforming the his4 initiator codon mutant strain JRC30-3B to Ura+ with the plasmid p2187 containing the His-tagged mutant eIF-5G31R allele. The presence of the His-tagged SUI5 (eIF-5G31R) allele in strain HH860 results in a dominant His+ suppressor phenotype.
A His tag was introduced at the amino termini of the wild-type and mutant eIF-2β by PCR. In the first PCR reaction, an XbaI restriction site was introduced ∼0.76 kb upstream from the SUI3 start codon and a His10 tag followed by a BamHI restriction site was introduced immediately 3′ to the start codon. This PCR fragment was subcloned into the XbaI and BamHI sites of plasmid pRS315 to generate plasmid p2120. In the second PCR reaction, a BamHI site was introduced immediately 5′ to the second codon of the SUI3-coding region, and a HindIII restriction site was introduced ∼0.24 kb downstream from the stop codon. This PCR fragment was subcloned into the BamHI and HindIII restriction sites of p2120 to generate plasmid p2122 containing the wild-type SUI3 gene with a His tag introduced in-frame between the AUG start codon and the second codon of the coding region. To generate His-tagged versions of the SUI3-2 (eIF-2βS264Y) and SUI3-40 (eIF-2βL254P) suppressor alleles, a BglII–HindIII DNA fragment from plasmids pBE66 (SUI3-2) and p2197 (SUI3-40) were subsituted for the BglII–HindIII fragment in plasmid p2122 to generate plasmids p2192 and p2199, respectively. The presence of each respective suppressor mutation was confirmed by DNA sequence analysis (Sanger et al. 1977).
The wild-type and mutant SUI3-2 His-tagged constructs were used to transform BCV59 strain to test their ability to rescue a sui3::URA3+ gene disruption (Donahue et al 1988). Both the SUI3+ and SUI3-2 His-tagged constructs complement the lethality associated with the sui3::URA3+ allele in haploid ascospores. Plasmid p2199 containing the His-tagged SUI3-40 (eIF-2βL254P) allele was used to transform the his4 initiator strain BCV50 to Leu+ and analyzed nonselectively for a His+ phenotype, indicative of the dominant SUI3-40 suppressor phenotype. Yeast strain HH725 used for purification of the eIF-2βL254P complex was generated by transformation of yeast strain 76-3D with the high-copy plasmids p2305 and p2309. Plasmid p2305 is a YEp24-based plasmid and contains the wild-type SUI2 (eIF-2α) gene, which was generated by deletion of the KpnI fragment containing the wild-type SUI3 gene in plasmid pTD1778, as described above. Plasmid p2309 is the high-copy vector pYO325 that contains the His-tagged mutant SUI3 -40 (eIF-2βL254P) allele and the wild-type GCD11 (eIF-2γ) allele. Plasmid p2309 was generated by first deleting the BamHI–BglII fragment containing the His-tagged portion of the GCD11 gene as part of plasmid p2260 and subsequently subcloning the XhoI–PvuII fragment from plasmid p2199 containing the His-tagged SUI3-40 (eIF-2βL254P) allele into the XhoI and SmaI restriction sites. Overexpression of eIF-2βL254P in strain HH725 results in a His+ suppressor phenotype and confers a slow-growth phenotype on SD medium lacking uracil and leucine.
Site-directed mutatgenesis was performed to introduce GUG, UUA, and CUG at the +3 amino acid position in the HIS4-coding region as described previously for other site-directed changes at HIS4 (Cigan et al. 1988b). For these experiments, the SalI DNA fragment from the proximal region of the HIS4 gene (Donahue et al. 1982) was subcloned into the SalI site of the single-stranded phage vector mp18. For construction of UUA and CUG changes, two oligonucleotides were designed, each changed the AUG start codon to AUU but changed the third amino acid codon from UUG to either CUG or UUA. Three different his4–lacZ constructions were made by site-directed mutagenesis to assess the ability of GUG to serve as a site for suppression. One construct was constructed in the identical fashion as UUA and CUG with the exception that a GUG codon was introduced at the +3 amino acid position. This construct then served as a templete to change a GUG codon at position −44 in the his4 leader region to GUU, which we refer to as the GUU template. The GUU template was then used to make two additional his4–lacZ constructs. One construct has an additional GUG codon at the +1 amino acid position in addition to the GUG at the +3 postion. The second construct only has a GUG codon at the the +1 position, a GUA codon at the +2 position amino acid position, and a UUA codon at the +3 position. As a result of these changes, no GUG or UUG codons were present upstream or nearby the GUG codon at the +1 position that might possibly interfere with the ability to see a maximal level of suppression. The presence of these mutations in all constructs were confirmed by DNA sequencing. A 763-bp Sau3A DNA fragment containing the HIS4 promoter/enhancer region, the HIS4 leader region, and approximately the first 11 amino acids of the coding region (Donahue 1982) from each construct was then subcloned into the BamHI site of plasmid p349 that only contains the lacZ-coding region (Cigan et al. 1988b). As a result of this subcloning the his4-coding region is fused in-frame with the lacZ-coding region, as described previously (Donahue and Cigan 1988). DNA sequence analysis was used to confirm the site-directed changes and Sau3A/BamHI junction being in-frame with the lacZ-coding region.
The his4–lacZ plasmids containing UUG (p440; Donahue and Cigan 1988), UUA (p2042), CUG (p2032), GUG (p2030), GUU/GUG/GUG (p2222), and GUU/GUG (p2223) site-directed changes were each used to transform the sui1-1 suppressor strain 301-4D (Castilho-Valavicius et al. 1990), the sui2-1 suppressor strain 304-1D (Castilho-Valavicius et al. 1990), the SUI3-2 suppressor strain 2119-11D (Castilho-Valavicius et al. 1990), the SUI4-1 suppressor strains AEC8, AR171, AR172, and AR173, and the SUI5-1 suppressor strains AEC6, AEC7, AEC10, and AR168. β-Galactosidase assays were performed as described previously (Donahue and Cigan 1988). Assays were performed in duplicate and the average value reported. For assays of SUI4 and SUI5 suppressor strains, the specific activity represents the average value derived from all related suppressor strains that are isogenic.
Western blot analysis
Polyclonal antiserum directed against eIF-2γ and eIF-5 were raised in New Zealand white rabbits as described previously for generating antibodies against eIF-2β and eIF-2α (Donahue et al. 1988; Cigan et al. 1989). For antibody production eIF-2γ and eIF-5 protein were purified from E. coli using the IPTG-inducible expression vector pET-19b (Novagen). Plasmids p2076 and p2072 expressing eIF-2γ and eIF-5, respectively, were constructed by PCR using a forward primer that introduced an NdeI restriction site immediately 5′ to the start codon and a reverse primer that introduced a BamHI restriction site immediately 3′ to the stop codon. Each of the entire coding regions were subcloned in-frame into the NdeI and BamHI sites of plasmid pET-19b and the in-frame fusion was confirmed by DNA sequence analysis. The recombinant proteins were overexpressed and purified from E. coli according to the manufacturer’s protocol.
The titer of both antisera was followed by Western blot analysis (Towbin et al. 1979) using yeast crude extracts (50 μg) prepared from the wild-type strain (TD28) and the strains HH849 and HH839, which overexpress eIF-2γ and eIF-5, respectively. To construct the eIF-2γ overexpression plasmid, the EcoRI–SalI fragment from plasmid Ep293 containing the wild-type GCD11 was first subcloned into the EcoRI and SalI restriction sites of polylinker containing plasmid pCT3, to generate plasmid p2096. A SalI–SpeI DNA fragment from p2096 was then subcloned into the SalI and NheI restriction sites of high-copy-number plasmid YEp24, to generate plasmid p2099. To construct the eIF-5 overexpression plasmid, the XhoI fragment from plasmid p2005 containing wild-type eIF-5 was subcloned into YEp24 at the SalI site to generate plasmid p2095. Yeast crude extracts were prepared as described previously (Donahue et al. 1988) and the protein concentration was determined by the Bradford method (Bio-Rad protein assay, microassay procedures). Extracts were resolved on 10% SDS-polyacrylamide gels, transferred to nitrocellulose membrane (1 A for 1 hr), and blocked with 2% nonfat dry milk in PBS containing 0.5% Triton X-100 (Sigma). After overnight incubation with the primary antiserum (1:25,000), peroxidase conjugated anti-rabbit IgG (1:25,000; Sigma) was used in a 2-hr incubation as the secondary antibody. The antibody–antigen complex was detected by the ECL system (Amersham) according to the manufacturer’s protocol.
Protein purification
A combination of P-11 cation exchanger (Whatman) and nickel affinity (Novagen) column chromatography was used to purify the wild-type eIF-2 (HH868), mutant eIF-2γN135K (HH705), wild-type eIF-5 (HH875), mutant eIF-5G31R (HH860), mutant eIF-2βS264Y (KT7), and mutant eIF-2βL254P (HH725) proteins. Briefly, 12 liters of yeast culture (30°C) were harvested at an OD600 of ∼1.0. Cell pellets were washed once with ice-cold ddH2O and resuspended in 25 ml lysis buffer [20 mm KPO4 (pH 7.5), 100 mm KCl, 1 mm DTT; 1 mm PMSF; 2 mg/ml of aprotinin, 1 mg/ml of pepstatin A; 2 mg/ml of bestatin; and 10% glycerol]. All remaining steps were performed at 4°C. Cells were lysed by French press (1,135 psi; Spectronic Instrument, Inc.), and the lysate was centrifuged for 30 min at 39,000g to remove insoluble debris. The cleared lysate was filtered through 0.45 mm filter membranes (Gelman).
Filtered lysate was loaded on a P-11 column (6.5 × 2.5 cm, ∼30 ml) that had been pre-equilibrated with 10 column volumes of P-100 buffer containing 20 mm KPO4 (pH 7.5), 100 mm KCl, 1 mm PMSF, and 10% glycerol. The column was washed with five column volumes of the P-100 buffer and subsequently with five column volumes of the P-450 buffer (same as P-100 buffer except for 450 mm KCl). Total protein was eluted with five column volumes of the P-750 buffer (same as P-100 buffer except for 750 mm KCl). Peak fractions were pooled (determined by UV monitor) and dialyzed twice against 4 liters of Ni-5 buffer [20 mm Tris-HCl (pH 7.5), 5 mm imidazole (pH 7.9), 500 mm NaCl, 1 mm PMSF, and 10% glycerol].
Dialysate was loaded on a nickel column (2 × 1.5 cm, ∼3 ml) that had been charged with 10 column volumes of 50 mm NiSO4 (Sigma) and pre-equilibrated with 10 column volumes of Ni-5 buffer. The column was washed with five column volumes of Ni-5 buffer and subsequently with five column volumes of Ni-30 buffer (same as Ni-5 buffer except that it contains 30 mm imidazole). Protein bound to the column was then eluted with five column volumes of Ni-200 buffer (same as Ni-5 buffer except for 200 mm imidazole). Peak fractions were pooled and concentrated using a Centricon-30. During the concentration steps, the buffer was changed to 20 mm Tris-HCl (pH 7.5), 500 mm KCl, 1 mm PMSF, 0.1% NP-40, 0.1 mm EDTA, 1 mm DTT, and 10% glycerol. Protein was aliquoted and saved at −70°C. Under these conditions, protein activity was stable (with no loss of activity detected) over the course of 1 year.
Charged initiator-tRNA and 40S ribosome subunits preparations
Total yeast tRNA (Boehringer Mannhein) was charged with [3H]methionine (70–85 Ci/mmole, Amersham) and purified by a protocol published previously (Donahue et al. 1988). The typical yield (from 400 μg total yeast tRNA) was 75–80 pmoles of charged initiator-tRNA with a specific activity of about 60,000 cpm/pmole. The unlabeled charged initiator-tRNA was charged by the same protocol except replacing the [3H]methionine with the same amount of unlabeled l-methionine (Sigma).
The 40S ribosomal subunits were purified from the wild-type strain TD28 by modification of two procedures reported previously (Toraño et al. 1974; Warner and Gorenstein 1978). Briefly, one-half liter of cells was grown at 30°C and harvested at an OD600 of ∼1.0. Cell pellets were washed once with ice-cold ddH2O and resuspended in 6 ml of TMN buffer [50 mm Tris-acetate (pH 7.0), 50 mm NH4Cl, 12 mm MgCl2, and 1 mm DTT]. The cells were lysed by French press and the lysate was centrifuged for 30 min at 39,000g to remove insoluble debris. Approximately 3.5 ml of cleared lysate was layered over 4 ml 10% sucrose in HKB buffer [20 mm Tris-HCl (pH 7.4), 5 mm MgCl2, 500 mm KCl, and 1 mm DTT] and centrifuged for 2 hr at 170,000g. The ribosomal pellet was resuspended in 1.5 ml of density gradient buffer containing 50 mm Tris-HCl (pH 7.5), 800 mm KCl, 12 mm Mg(OAc)2, and 20 mm β-mercaptoethanol. Approximately 75 OD260 units of the crude ribosome preparation were layered on a 35-ml linear sucrose density gradient (15%–30%) and centrifuged for 20 hr at 15°C (SW 27 rotor, 18,000 rpm) to separate the 40S and the 60S subunits. The 40S subunits were collected through an ISCO density gradient fractionator and then diluted with an equal volume of ribosome dilution buffer containing 20 mm Tris-HCl (pH 7.5), 30 mm Mg(OAc)2, and 20 mm β-mercaptoethanol. The diluted 40S subunits were centrifuged overnight at 4°C (Ti-60 rotor, 40,000 rpm). The 40S pellet was then resuspended in a small volume of ribosome suspension buffer (a maximum volume of 300 μl, depending on the size of the pellet) containing 20 mm Tris-HCl (pH 7.5), 5 mm MgCl2, 100 mm KCl, 1 mm DTT, and 50% glycerol and stored at −70°C. The 40S ribosome subunits purified by this procedure were determined not to be contaminated with eIF-5 and eIF-2γ proteins by Western blot analysis.
In vitro assays
Ternary complex formation was assayed by filter binding similar to published procedures (Donahue et al. 1988; Cigan et al. 1993). Reactions conditions were as follows: 20 mm Tris-HCl (pH 7.5), 5 mm MgCl2, 100 mm KCl, 1 mm DTT, 0.1% NP-40, 25 μm GTP (Sigma), 0.15 μm charged initiator-tRNA [3H]Met–tRNAiMet, 60,000 cpm/pmole) and purified eIF-2 in a total volume of 20 μl. The reaction mixture was incubated at 37°C for either 5 min (wild-type eIF-2 and mutant eIF-2βS264Y) or 10 min (mutant eIF-2γN135K) and then kept on ice. The reaction was stopped by addition of 1 ml of ice-cold wash buffer [20 mm Tris-HCl (pH 7.5), 5 mm MgCl2, 100 mm KCl, and 1 mm DTT]. Reactions were filtered through a prewetted nitrocellulose membrane (Schleicher and Schuell, BA85, 0.45 μm). The filter membrane was washed twice with 1 ml of ice-cold wash buffer and air-dried for 10 min. The membrane was soaked in 8 ml of scintillation fluid (Bio-Safe II, Research Products, Inc.) for 10 min and counted by a scintillation counter (Beckman LS-230). GTP-independent initiator-tRNA binding was determined by omitting GTP from the reactions.
Ternary complex assays in the presence of the nonhydrolyzable analog of GTP were performed identically with the exception that GppNp (Sigma) was substituted for GTP (Sigma). For assays of dissociation of charged initiator-tRNA from eIF-2 complexes, the reactions were allowed to reach equilibrium for either 5 or 10 min in the presence of 0.075 μm charged initiator-tRNA [3H]Met–tRNAiMet. Different amounts of unlabeled, charged initiator-tRNA were then added to individual reactions, incubated for 5 min at 37°C, and radioactivity bound to eIF-2 was assayed by filter binding as described above.
GTP-binding assays were performed as described previously (Panniers et al. 1988; Erickson and Hannig 1996) with minor modifications. The standard reaction conditions for the [3H]GTP (Amersham, 7.5 Ci/mmole)-binding assays contained 20 mm Tris-HCl (pH 7.5), 5 mm MgCl2, 100 mm KCl, 1 mm DTT, 0.1% NP-40, and 1 μm of [3H]GTP in a total volume of 55 μl. After incubation with eIF-2 at 37°C, 10-μl aliquots were taken and added to 0.5 ml of ice-cold wash buffer containing 20 mm Tris-HCl (pH 7.5), 5 mm MgCl2, 100 mm KCl, and 1 mm DTT, and counts bound to eIF-2 quantitated by the filter-binding assay as described above. The [γ-32P]GTP (Amersham, >5000 Ci/mmole)-binding assays, were performed in a similar manner with the exception that 10 μm of [3H]GTP and 0.01 μm of [γ-32P]GTP in a 1000:1 ratio, respectively, were present in a total initial reaction volume of 10 μl. Also, for the latter assays the dry membranes were counted without scintillation fluid to avoid [3H]GTP counts. For assaying the dissociation rate of [α-32P]GTP (Amersham, 3000 Ci/mmole), the reactions were performed in a total volume of 55 μl in the same buffer conditions as above except using 1 μm of [3H]GTP and 0.01 μm of [α-32P]GTP (100:1). After a 3-min incubation at 37°C, a 10-μl aliquot of the reaction mixture was measured for bound labeled nucleotide, which served as the zero timepoint. Unlabeled GDP was then added to the remaining reaction mixture to arrive at a final total concentration of 1 μm GDP. The reaction mixture were incubated at 37°C and 10-μl aliquots of reaction mixture were taken at different time points and [α-32P]GTP radioactivity that remained bound to eIF-2 was detected by filter-binding assay. Filters were air-dried for >30 min and then counted without scintillation fluid to avoid [3H]GTP counts.
eIF-5-dependent GTP hydrolysis assays were performed as described previously (Dubnoff and Maitra 1972; Peterson et al. 1979; Chaudhuri et al. 1981; Chakravarti et al. 1993) with minor modifications. 43S complex was formed by incubation of ternary complex, except using [γ-32P]GTP (>5000 Ci/mmole, Amersham) to label eIF-2 at a 100:1 ratio of cold GTP to [γ-32P]GTP=100:1] with 0.2 OD260 units of purified 40S ribosome subunits, and 2 nmoles of AUG triplet ribonucleotide (synthesized by NBI). After a 4-min incubation at 37°C, the reaction mixture (22 μl) was loaded on a Sepharose CL-6B (Sigma) column (3.7 × 0.7 cm, ∼1.5 ml) that had been pre-equilibrated with 43S buffer containing 20 mm Tris-HCl (pH 7.5), 5 mm MgCl2, 100 mm KCl, 1 mm DTT, 0.1% NP-40, and 10 μm GTP. The peak fractions (determined by a Geiger counter), which elute in the void volume were pooled, kept on ice, and used immediately. GTP hydrolysis assays were performed in a total reaction volume of 530 μl. Equal amounts of wild-type eIF-5 or mutant eIF-5G31R were added to each reaction and GTP hydrolysis was measured according to published procedure (Chakravarti et al. 1993). Equivalent amounts of eIF-5 were determined by Western blot analysis using anti-eIF-5 antibody.
Acknowledgments
We thank Dr. John Richardson and Dr. Cheng Kao for critical suggestions about biochemical assays, Pauline Donahue for assistance with the genetic analysis, Ana Raman and Amy Clewell for their assistance with cloning and subcloning of the SUI4 and SUI5 suppressor genes, and Sue Kim and Kristin Taylor-Lubell for their assistance with DNA sequencing of SUI5 alleles and protein purifications. This work was supported by a Public Health Service Grant from the National Institutes of Health, GM32263 (T.F.D.), and a National Science Foundation grant MCB-9631066 (E.M.H.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL donahue@sunflower.bio.indiana.edu; FAX (812) 855-6705.
References
- Balch WE. Small GTP-binding proteins in vesicular transport. Trends Biochem Sci. 1990;15:473–477. doi: 10.1016/0968-0004(90)90301-q. [DOI] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature. 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- Bushman JL, Asuru AI, Matts RL, Hinnebusch AG. Evidence that GCD6 and GCD7, translational regulator of GCN4, are subunits of guanine nucleotide exchange factor for eIF-2 in Saccharomyces cerevisiae. Mol Cell Biol. 1993a;13:1920–1932. doi: 10.1128/mcb.13.3.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman JL, Foiani M, Cigan AM, Paddon CJ, Hinnebusch AG. Guanine nucleotide exchange factor for eukaryotic translation initiation factor 2 in Saccharomyces cerevisiae: Interactions between the essential subunits GCD2, GCD6 and GCD7 and the regulatory subunit GCN3. Mol Cell Biol. 1993b;13:4618–4631. doi: 10.1128/mcb.13.8.4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilho-Valavacius B, Yoon H, Donahue TF. Genetic characterization of the Saccharomyces cerevisiae translation initiation suppressors sui1, sui2, and SUI3 and their effects on HIS4 expression. Genetics. 1990;24:483–495. doi: 10.1093/genetics/124.3.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilho-Valavacius B, Thompson GM, Donahue TF. Mutation analysis of the Cys-X2-Cys-X19-Cys-X2-Cys motif in the β subunit of eukaryotic translation initiation factor 2. Gene Exp. 1992;2:297–309. [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti A, Maitra U. Function of eukaryotic initiation factor 5 in the formation of 80S ribosome polypeptide chain initiation complex. J Biol Chem. 1991;266:14039–14045. [PubMed] [Google Scholar]
- Chakravarti D, Maitra U. Eukaryotic translation initiation factor 5 from Saccharomyces cerevisiae. J Biol Chem. 1993;268:10524–10533. [PubMed] [Google Scholar]
- Chakravarti D, Maiti T, Maitra U. Isolation and immunochemical characterization of eukaryotic translation initiation factor 5 from Saccharomyces cerevisiae. J Biol Chem. 1993;268:5754–5762. [PubMed] [Google Scholar]
- Chaudhuri A, Stringer EA, Valenzuela D, Maitra U. Characterization of eukaryotic initiation factor 2 containing two polypeptide chains of Mr=48,000 and 38,000. J Biol Chem. 1981;256:3988–3994. [PubMed] [Google Scholar]
- Cigan AM, Feng L, Donahue TF. tRNAiMet functions in directing the scanning ribosome to the start site of translation. Science. 1988a;242:93–97. doi: 10.1126/science.3051379. [DOI] [PubMed] [Google Scholar]
- Cigan AM, Pabich EK, Donahue TF. Mutational analysis of the HIS4 translational initiator region in Saccharomyces cerevisiae. Mol Cell Biol. 1988b;8:2964–2975. doi: 10.1128/mcb.8.7.2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cigan AM, Pabich EK, Feng L, Donahue TF. Yeast translation initiation suppressor sui2 encodes the α subunit of eukaryotic initiation factor 2 and shares identity with the human α subunit. Proc Natl Acad Sci. 1989;86:2784–2788. doi: 10.1073/pnas.86.8.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cigan AM, Bushman JL, Boal TR, Hinnebusch AG. A protein complex composed of translational regulators of GCN4 mRNA is the guanine nucleotide exchange factor for translation initiation factor 2 in yeast. Proc Natl Acad Sci. 1993;90:5350–5354. doi: 10.1073/pnas.90.11.5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das K, Chevesich J, Maitra U. Molecular cloning and expression of cDNA for mammalian translation initiation factor 5. Proc Natl Acad Sci. 1993;90:3058–3062. doi: 10.1073/pnas.90.7.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Yang W, Åström S, Byström AS, Hinnebusch AG. Modulation of tRNAiMet, eIF-2, and eIF-2B expression shows that GCN4 translation is inversely coupled to the level of eIF-2 ⋅ GTP ⋅Met-tRNAiMet ternary complexes. Mol Cell Biol. 1995;15:6351–6363. doi: 10.1128/mcb.15.11.6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue TF, Cigan AM. Genetic selection for mutations that reduce or abolish ribosomal recognition of the HIS4 translational initiator region. Mol Cell Biol. 1988;8:2955–2963. doi: 10.1128/mcb.8.7.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue TF, Farabaugh PJ, Fink GR. The nucleotide sequence of the HIS4 region of yeast. Gene. 1982;18:47–59. doi: 10.1016/0378-1119(82)90055-5. [DOI] [PubMed] [Google Scholar]
- Donahue TF, Cigan AM, Pabich EK, Castilho-Valavicius B. Mutations at a Zn(II) finger motif in the yeast eIF-2β gene alter ribosomal start-site selection during the scanning process. Cell. 1988;54:621–632. doi: 10.1016/s0092-8674(88)80006-0. [DOI] [PubMed] [Google Scholar]
- Dubnoff JS, Maitra U. Characterization of the ribosome-dependent guanosine triphosphatase activity of polypeptide chain initiation factor IF 2. J Biol Chem. 1972;247:2876–2883. [PubMed] [Google Scholar]
- Erickson FL, Hannig EM. Ligand interactions with eukaryotic translation initiation factor 2: Role of the γ-subunit. EMBO J. 1996;15:6311–6320. [PMC free article] [PubMed] [Google Scholar]
- Gaspar NJ, Kinzy TG, Scherer BJ, Hümbelin M, Hershey JWB, Merrick WC. Translation initiation factor eIF-2: Cloning and expression of the human cDNA encoding the γ-subunit. J Biol Chem. 1994;269:3415–3422. [PubMed] [Google Scholar]
- Gualerzi CO, Pon CL. Initiation of mRNA translation in prokaryotes. Biochemistry. 1990;29:5881–5889. doi: 10.1021/bi00477a001. [DOI] [PubMed] [Google Scholar]
- Guthrie, C. and G.R. Fink. 1991. Guide to yeast genetics and molecular biology. Methods Enzymol. 194: ..... [PubMed]
- Hann SR, King MW, Bentley DL, Anderson CW, Eisenman CW. A non-AUG translational initiation in c-myc exon 1 generates an N-terminally distinct protein whose synthesis is disrupted in Burkitt’s lymphomas. Cell. 1988;52:185–195. doi: 10.1016/0092-8674(88)90507-7. [DOI] [PubMed] [Google Scholar]
- Hannig EM, Cigan AM, Freeman BA, Kinzy TG. GCD11, a negative regulator of GCN4 expression, encodes the γ subunit of eIF-2 in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:506–520. doi: 10.1128/mcb.13.1.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershey JWB. Translational control in mammalian cells. Annu Rev Biochem. 1991;60:717–755. doi: 10.1146/annurev.bi.60.070191.003441. [DOI] [PubMed] [Google Scholar]
- Huang H-k, Donahue TF. The fundamentals of translation initiation. In: Harford JB, Morris DR, editors. mRNA metabolism and post-transcriptional gene regulation. Vol. 17. New York, NY: John Wiley & Sons, Inc.; 1997. pp. 147–164. [Google Scholar]
- Kasperaitis MAM, Voorma HO, Thomas AAM. The amino acid sequence of eukaryotic translation initiation factor 1 and its similarity to yeast initiation factor SUI1. FEBS Lett. 1995;365:47–50. doi: 10.1016/0014-5793(95)00427-b. [DOI] [PubMed] [Google Scholar]
- Koepp DM, Silver PA. A GTPase controlling nuclear trafficking: Running the right way or walking randomly? Cell. 1996;87:1–4. doi: 10.1016/s0092-8674(00)81315-x. [DOI] [PubMed] [Google Scholar]
- Kozak M. How do eukaryotic ribosomes select initiation regions in messenger RNA? Cell. 1978;15:1109–1123. doi: 10.1016/0092-8674(78)90039-9. [DOI] [PubMed] [Google Scholar]
- ————— The scanning model for translation: An update. J Cell Biol. 1989a;108:229–241. doi: 10.1083/jcb.108.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Context effects and inefficient initiation at non-AUG codons in eukaryotic cell-free translation system. Mol Cell Biol. 1989b;9:5073–5080. doi: 10.1128/mcb.9.11.5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick WC. Mechanism and regulation of eukaryotic protein synthesis. Microbiol Rev. 1992;56:291–315. doi: 10.1128/mr.56.2.291-315.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naranda T, MacMillan SE, Donahue TF, Hershey JWB. SUI1/p16 is required for the activity of eukaryotic translation initiation factor 3 in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2307–2313. doi: 10.1128/mcb.16.5.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen P, Kjeldgaard M, Thirup S, Polekhina G, Reshetnikova L, Clark BFC, Nyborg J. Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP analog. Science. 1995;270:1464–1472. doi: 10.1126/science.270.5241.1464. [DOI] [PubMed] [Google Scholar]
- Orr-Weaver TL, Szostak JW, Rothstein RJ. Genetic applications of yeast transformation with linear and gapped plasmids. Methods Enzymol. 1983;101:228–245. doi: 10.1016/0076-6879(83)01017-4. [DOI] [PubMed] [Google Scholar]
- Panniers R, Rowlands AG, Henshaw EC. The effect of Mg2+ and guanine nucleotide exchange factor on the binding of guanine nucleotides to eukaryotic initiation factor 2. J Biol Chem. 1988;263:5519–5525. [PubMed] [Google Scholar]
- Parent SA, Fenimore CM, Bostain KA. Vector systems for the expression, analysis and cloning of DNA sequences in S. cerevisiae. Yeast. 1985;1:83–138. doi: 10.1002/yea.320010202. [DOI] [PubMed] [Google Scholar]
- Pathak VK, Nielsen PJ, Trachsel H, Hersey JWB. Structure of the β subunit of translation initiation factor eIF-2. Cell. 1988;54:633–639. doi: 10.1016/s0092-8674(88)80007-2. [DOI] [PubMed] [Google Scholar]
- Peterson DT, Safer B, Merrick WC. Role of eukaryotic initiation factor 5 in the formation of 80S initiation complex. J Biol Chem. 1979;254:7730–7735. [PubMed] [Google Scholar]
- Rothstein RJ. One-step gene disruption in yeast. Methods Enzymol. 1983;101:202–210. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- Roeder GS, Fink GR. DNA rearrangements associated with a transposable element in yeast. Cell. 1980;21:239–249. doi: 10.1016/0092-8674(80)90131-2. [DOI] [PubMed] [Google Scholar]
- Rybin V, Ullrich O, Rubino M, Alexandrov K, Silmon I, Seabra MC, Goody R, Zerial M. GTPase activity of Rab5 acts as a timer for endocytic membrane fusion. Nature. 1996;383:266–269. doi: 10.1038/383266a0. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fristch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RC. EFTu provides an internal kinetic standard for translation accuracy. Trends Biochem Sci. 1988;13:91–93. doi: 10.1016/0968-0004(88)90047-3. [DOI] [PubMed] [Google Scholar]
- Thompson RC, Dix DB, Karim AM. The reaction of ribosomes with elongation factor Tu ⋅ GTP complexes: Aminoacyl-tRNA-independent reactions in the elongation cycle determine the accuracy of protein synthesis. J Biol Chem. 1986;261:4868–4874. [PubMed] [Google Scholar]
- Toraño A, Sandoval A, Heredia CF. Soluble protein factors and ribosomal subunits from yeast. Interactions with aminoacyl-tRNA. Methods Enzymol. 1974;30:254–261. doi: 10.1016/0076-6879(74)30029-8. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose: Procedure and some applications. Proc Natl Acad Sci. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mollard GF, Stahl B, Li C, Südhof TC, Jahn R. Rab proteins in regulated exocytosis. Trends Biochem Sci. 1994;19:164–168. doi: 10.1016/0968-0004(94)90278-x. [DOI] [PubMed] [Google Scholar]
- Warner JR, Gorenstein C. The ribosomal proteins of Saccharomyces cerevisiae. Methods Cell Biol. 1978;20:45–60. doi: 10.1016/s0091-679x(08)62008-7. [DOI] [PubMed] [Google Scholar]
- Williams NP, Hinnebusch AG, Donahue TF. Mutations in the structural genes for eukaryotic translation initiation factors 2α and 2β of Saccharomyces cerevisiae disrupt translational control of GCN4 mRNA. Proc Natl Acad Sci. 1989;86:7515–7519. doi: 10.1073/pnas.86.19.7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Zeng K, Wilson IA, Balch WE. Structural insights into the function of the Rab GDI superfamily. Trends Biochem Sci. 1996;21:472–476. doi: 10.1016/s0968-0004(96)10062-1. [DOI] [PubMed] [Google Scholar]
- Yoon H, Donahue TF. The sui1 suppressor locus in Saccharomyces cerevisiae encodes a translation factor that functions during tRNAiMet recognition of the start codon. Mol Cell Biol. 1992;12:248–260. doi: 10.1128/mcb.12.1.248. [DOI] [PMC free article] [PubMed] [Google Scholar]