

Abstract
Candidate transcription factors involved in pancreatic endocrine development have been isolated using insulin gene regulation as a paradigm. The cell-type restricted basic helix–loop–helix (bHLH) gene, BETA2/NeuroD, expressed in pancreatic endocrine cells, the intestine, and the brain, activates insulin gene transcription and can induce neurons to differentiate. To understand the importance of BETA2 in pancreatic endocrine cell differentiation, mice lacking a functional BETA2 gene were generated by gene targeting experiments. Mice carrying a targeted disruption of the BETA2 gene developed severe diabetes and died perinatally. Homozygous BETA2 null mice had a striking reduction in the number of insulin-producing β cells and failed to develop mature islets. Islet morphogenesis appeared to be arrested between E14.5 and E17.5, a period characterized by major expansion of the β cell population. The presence of severe diabetes in these mice suggests that proper islet structure plays an important role in blood glucose homeostasis. In addition, secretin- and cholecystokinin-producing enteroendocrine cells failed to develop in the absence of BETA2. The absence of these two pancreatic secretagogs may explain the abnormal cellular polarity and inability to secrete zymogen granules in pancreatic acinar exocrine cells. The nervous system appeared to develop normally, despite abundant expression of BETA2 in differentiating neurons. Thus, BETA2 is critical for the normal development of several specialized cell types arising from the gut endoderm.
Keywords: Diabetes, pancreatic endocrine development, insulin gene regulation, BETA2, NeuroD, mice, enteroendocrine development
Mature pancreatic islets, which constitute 1%–2% of the total mass of the adult pancreas, are composed of four principal endocrine cell types: the α cells, β cells, δ cells, and PP cells, which produce the hormones glucagon, insulin, somatostatin, and pancreatic polypeptide, respectively. The β cells are the most abundant, comprising ∼65%–80% of the total number of endocrine cell and localize to the islet core. The remaining endocrine cell types tend to be distributed at the periphery of the islet. It has been suggested that the integrity of the islet structure is essential for the normal islet function of regulating glucose homeostasis (Halban et al. 1982; Orci 1982; Pipeleers et al. 1982; Lucas-Clerc et al. 1993).
Islet morphogenesis is a complex process that involves differentiation, proliferation, and migration of pancreatic endocrine cells, culminating in the formation of properly organized, three-dimensional, sphere-like structures. Early in pancreatic development, individual cells expressing glucagon, insulin, and peptide YY first appear in the dorsal buds of the foregut epithelium at embryonic day 9.5 (E9.5) (Gittes and Rutter 1992; Teitelman et al. 1993; Upchurch et al. 1994). Developing endocrine cells subsequently aggregate in interstitial clusters adjacent to the ductal epithelia beginning at about E14.5 (Pictet and Rutter 1972; Alpert et al. 1988; Herrera et al. 1991). At this time in pancreatic organogenesis, endocrine cell clusters do not exhibit the typical architecture of mature islets and do not contain all islet cell types. In the remaining 4 days of gestation, the endocrine cells detach from the exocrine matrix, increase in number, and reorganize to form mature islets. Morphologically distinct islets are first detected with the proper distribution of endocrine cell types at E17.5 (Pictet and Rutter 1972; Herrera et al. 1991). Although the molecular mechanisms that control islet formation are not known, it has been demonstrated recently that members of the cadherin family of cell adhesion molecules (CAMs) and neural CAM (N-CAM) are expressed in the pancreas and also appear to have a functional role in the aggregation and organization of the principal endocrine cell types (Rouiller et al. 1991; Moller et al. 1992; Hutton et al. 1993; Cirulli et al. 1994; Dahl et al. 1996).
Expression of the insulin gene is one of the hallmarks of β-cell differentiation in the developing pancreas. The insulin enhancer is complex, consisting of multiple cis-acting DNA elements interacting with distinct classes of transcription factors including basic helix–loop–helix (bHLH) proteins (Nelson et al. 1990; Walker et al. 1990; Cordle et al. 1991; German et al. 1991; Shieh and Tsai 1991; Peyton et al. 1994; Naya et al. 1995). A cell type-restricted bHLH protein, BETA2, was isolated as an important regulator of insulin gene expression and is expressed in pancreatic endocrine cells, the intestine, and the brain (Naya et al. 1995). In the intestine, BETA2 also activates transcription of the gene encoding the hormone secretin (Mutoh et al. 1997). The expression pattern of BETA2 suggests a role in endocrine pancreas development as well as the determination or differentiation of specialized cell types in the intestine and nervous system. Overexpression studies in frog embryos have implicated BETA2, termed NeuroD, as a differentiation factor in the developing nervous system, as it was shown to convert epidermal cells into neurons (Lee et al. 1995).
Gene targeting experiments in mice have demonstrated that bHLH transcription factors are instrumental in cell fate determination and differentiation of the muscle (Jan and Jan 1993; Weintraub 1993; Olson and Klein 1994), neuronal (Guillemot et al. 1993), lymphocytic (Bain et al. 1994; Zhuang et al. 1994), hematopoietic (Shivdasani et al. 1995), and mesenchymal (Chen and Behringer 1995) cell lineages. Given the central role bHLH factors have in the generation of a diverse array of cell types, bHLH proteins may function in a similar manner during pancreas development.
In this study we have inactivated the BETA2 gene using gene targeting in embryonic stem (ES) cells to understand the importance of BETA2 in pancreatic development. Mice homozygous for the BETA2 deletion developed severe diabetes and died 3–5 days after birth. Islet development was arrested in BETA2-deficient mice. In addition, examination of the mutant mice revealed failure to develop enteroendocrine cells expressing secretin and cholecystokinin and abnormal cellular polarity in pancreatic acinar cells. Although BETA2 is expressed abundantly in the developing nervous system, mutant mice did not exhibit any apparent neuronal defects.
Results
Targeted disruption of the BETA2 gene
A positive/negative-type targeting vector was constructed in which the bacterial lacZ gene containing a nuclear localization signal and a phosphoglycerate kinase–neomycin resistance (PGK–neoR) cassette replaced all but the first 66 amino acids of BETA2, resulting in a BETA2–LacZ fusion protein (Fig. 1A,B). This construction effectively deleted the bHLH and transactivation domains. Chimeras obtained from three correctly targeted ES cell clones were bred to C57BL/6 mice with resultant germ-line transmission. Genotype analysis of newborn litters from 129/C57 heterozygote intercrosses showed the following allelic frequencies: 22.7% wild-type (+/+), 55.7% heterozygote (+/−), and 21.6% homozygous mutant (−/−), indicating that deletion of the BETA2 gene did not result in embryonic lethality. Mice homozygous for the targeted disruption of BETA2 from three separate lines fed normally but were smaller and dehydrated by 2 days after birth compared with heterozygous littermates and died 3–5 days postpartum.
Figure 1.


Disruption of BETA2 gene by homologous recombination and genotype analysis of heterozygote intercrosses. (A) Maps of wild-type BETA2 locus, targeting vector, and disrupted BETA2 allele. I and II designate exons in the BETA2 gene. The 5′ external probe used for Southern blotting is shown (▪). The diagnostic restriction fragments with their expected sizes are indicated as lines. The bacterial lacZ gene was inserted in the same orientation as the BETA2 genomic locus. The neomycin resistance (neoR) gene was inserted in the opposite transcriptional orientation. The introduction of the lacZ–neoR cassette disrupted all [including the bHLH and transactivation (R. Stein, unpubl.) domains] but the first 66 amino acids of BETA2, resulting in a BETA2–LacZ fusion protein with a novel SacI site. (B) Southern analysis of SacI-digested DNA from the litter of a BETA2 +/− intercross. Sizes of wild-type and targeted alleles are indicated by 13 and 4 kb, respectively. Subsequent genotype analysis was performed by PCR on tail DNA using the primers B2.1, B2.2, and B2.lacZ. Arrows indicate the primers and hatched box designates the bHLH domain.
Diabetes in BETA2 mutant mice
To determine whether BETA2 is necessary for the normal islet function of maintaining glucose homeostasis, mice were examined for the presence of diabetes. At 2 days of age, mice lacking a functional BETA2 gene exhibited marked hyperglycemia (308.5 ± 85.1 mg/dl, n = 46) in contrast to age-matched +/+ (74.1 ± 13.7 mg/dl, n = 23) and +/− (95.9 ± 24.6 mg/dl, n = 30) animals. The presence of ketonuria in −/− animals further suggested the presence of severe diabetes and insulin deficiency resulting from abnormal β cell function. Attempts to rescue the diabetic phenotype by administration of insulin were unsuccessful, suggesting that the mutant animals were unable to respond to insulin, have become insulin resistant, or perhaps contained additional defects.
Defective pancreatic islet morphogenesis
To better understand the potential role of BETA2 in endocrine pancreatic differentiation, we characterized expression of BETA2 in the pancreas of +/− and −/− mice for expression of β-galactosidase by X-gal histochemistry at different stages of pancreatic development. At E9.5, a subset of cells in the pancreatic epithelium stained for β-galactosidase activity in both +/− and −/− pancreata (Fig. 2A, B). The majority of these cells expressed glucagon, indicating that BETA2 is present in the earliest islet precursors. By E14.5 β-gal-positive cells were located within or adjacent to the ductal epithelia in +/− (Fig. 2C) and −/− (Fig. 2D) dorsal and ventral lobes, consistent with the observation that endocrine cells originate from the pancreatic ducts (Pictet and Rutter 1972). Through a complex process, endocrine cells begin to cluster and subsequently organize into sphere-shaped pancreatic islets late in gestation at E17.5 (Pictet and Rutter 1972; Herrera et al. 1991). The organization of β-gal positive cells into easily identifiable pancreatic islets was first evident at E17.5 in +/− pancreas (Fig. 2E). β-Galactosidase-staining cells were localized exclusively to islets thereafter at all developmental stages through adulthood (data not shown). In striking contrast, morphologically distinct islets failed to develop at any stage of development in pancreata of −/− mice (Fig. 2F; data not shown), although β-gal-positive cells detached from the exocrine matrix and formed small clusters.
Figure 2.
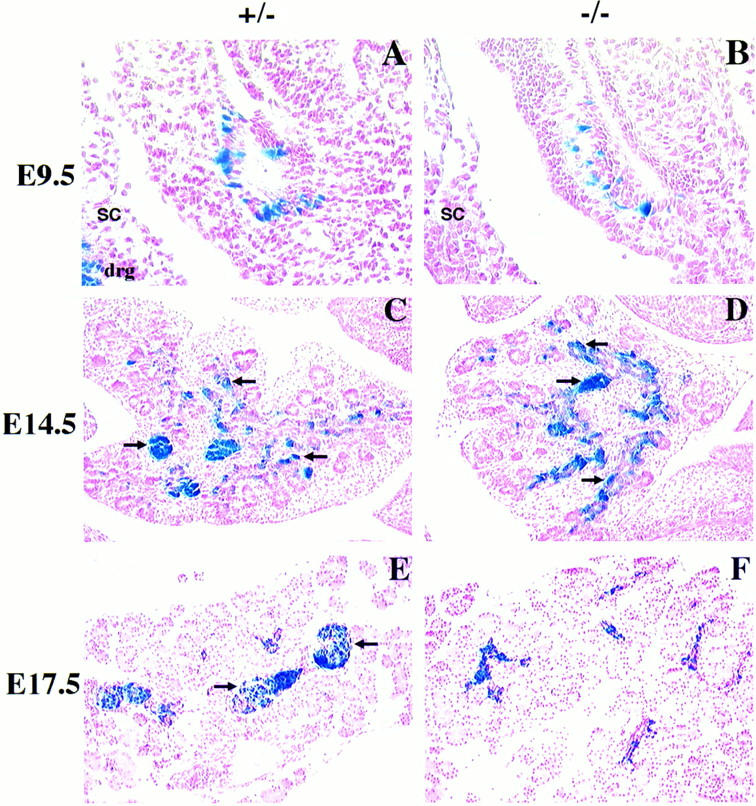
Abnormal pancreatic islet morphogenesis in BETA2 −/− mice. (A–D) Sagittal sections of BETA2 +/− (left panels) and BETA2 −/− (right panels) embryos at E9.5 (A,B) and pancreas at E14.5 (C,D) stained with X-gal. There were no obvious differences in morphology of the early developing pancreas or in the number of β-gal-positive cells. β-Gal-positive cells were similarly distributed in both +/− and −/− tissue. β-Gal expression was also detected in both the dorsal and ventral pancreas. (E,F) Pancreata of +/− and −/− mice at E17.5 stained with X-gal. At E17.5 (E) there was evidence of islet formation in the +/− pancreas, as indicated by β-gal-positive cells, which formed a sphere-shaped structure with a distinct border surrounded by acinar cells (arrows). In contrast, distinct islets were not present in −/− pancreas at E17.5 (F), although β-gal-positive cells were seen without distinctive shape or border. Sections were counterstained in nuclear fast red. (drg) Dorsal root ganglion; (sc) spinal cord. Original magnification, 500× (A,B); 250× (C–F).
Islets were examined further for the presence of β-galactosidase staining in insulin-, glucagon-, and somatostatin-expressing cells, the major islet cell types present in developing mice. Islets of +/− mice appeared identical to normal mice with the majority of cells expressing insulin in the islet core and smaller numbers of cells expressing glucagon and somatostatin distributed around the islet mantle (Figs. 3A,C,E). Examination of the same sections for β-galactosidase activity revealed that the disrupted BETA2 gene was expressed in each of these cell types but not in exocrine cells (Fig. 3, A,C, and E, and B,D, and F, respectively). This is the first in vivo demonstration that BETA2 expression is restricted to endocrine cells of the pancreas. In contrast, endocrine cells in the −/− pancreas failed to form mature islets at birth (Fig. 3J,L,N); rather, most of them formed clusters with a disproportionate distribution of endocrine cell types (Fig. 3I,K,M). Furthermore, cells expressing either peptide YY (PYY) or pancreatic polypeptide (not shown) were clearly seen in +/− (Fig. 3G,H) and −/− (Fig. 3O,P) animals using antisera that distinguish between the two. These results suggest that endocrine cells formed and aggregated as small clusters in the mutant pancreas but failed to organize properly into mature islets.
Figure 3.
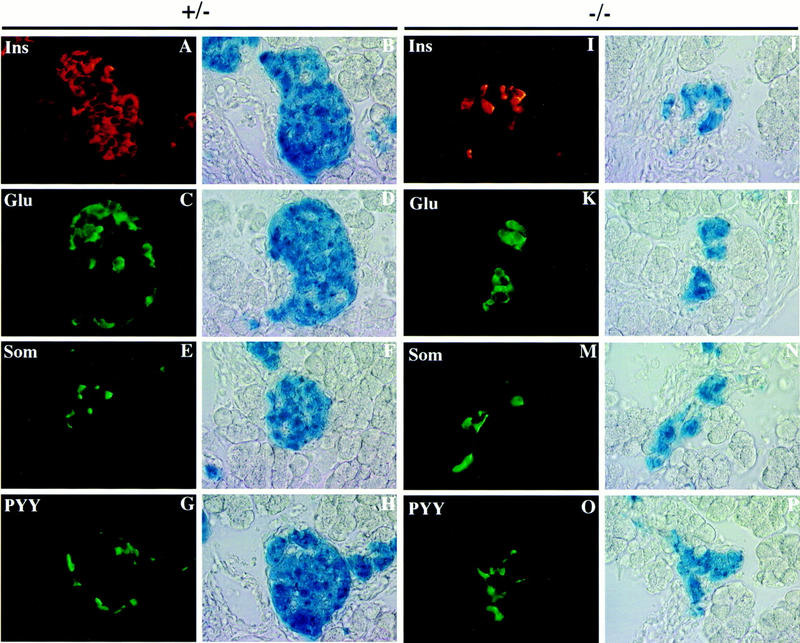
Multiple islet cell types coexpress BETA2. Localization of pancreatic hormones in BETA2-expressing cells of +/− (A–H) and −/− (I–P) by multiple labeling. Single sections of newborn mice were examined for β-galactosidase activity by X-gal-staining and for hormones by immunofluorescence. Well defined islets, heterogeneous in size were seen with X-gal-stained cells in +/− mice (B,D,F,H). X-Gal-stained cells within these islets showed immunofluorescent staining for insulin (A), glucagon (C), somatostatin (E), and peptide YY (G) visualized by Cy3-conjugated (A) and FITC-conjugated (C,E,G) secondary antibodies. Endocrine cell types in the +/− pancreas were distributed normally with the β cells (Ins) in the core of the islet and the other cell types in the periphery of the islet. (I–P) Localization of hormones and β-gal in the pancreatic endocrine cells of −/− mice by immunofluorescence (I,K,M,O) and X-gal staining (J,L,N,P) as in A–H. Endocrine cells in the −/− pancreas coexpressed β-gal but did not appear as organized islets. Each cell type appeared in clusters in contrast to their distribution in +/− mice. Original magnification, 1000×.
PDX-1, a homeodomain protein, has been shown to be essential for pancreas development. PDX-1-deficient mice are not embryonic lethal but fail to develop a pancreas and die perinatally (Jonsson et al. 1994; Offield et al. 1996). Recent evidence suggests that BETA2 increases PDX-1 gene expression (Sharma et al. 1997). Thus, decreased expression of PDX-1 in BETA2 −/− mice may explain the mutant phenotype. For this reason we carried out immunostaining of pancreas sections with PDX-1 antibodies. As shown in Figure 4, PDX-1-positive cells are present in the exocrine and endocrine cells of the E16.0 pancreas and there are no discernible differences between wild-type and mutant pancreas. However, decreased PDX-1 expression in the postnatal mutant pancreatic islet was observed (data not shown), possibly as a result of the loss of islet cells in the BETA2 −/− mice.
Figure 4.
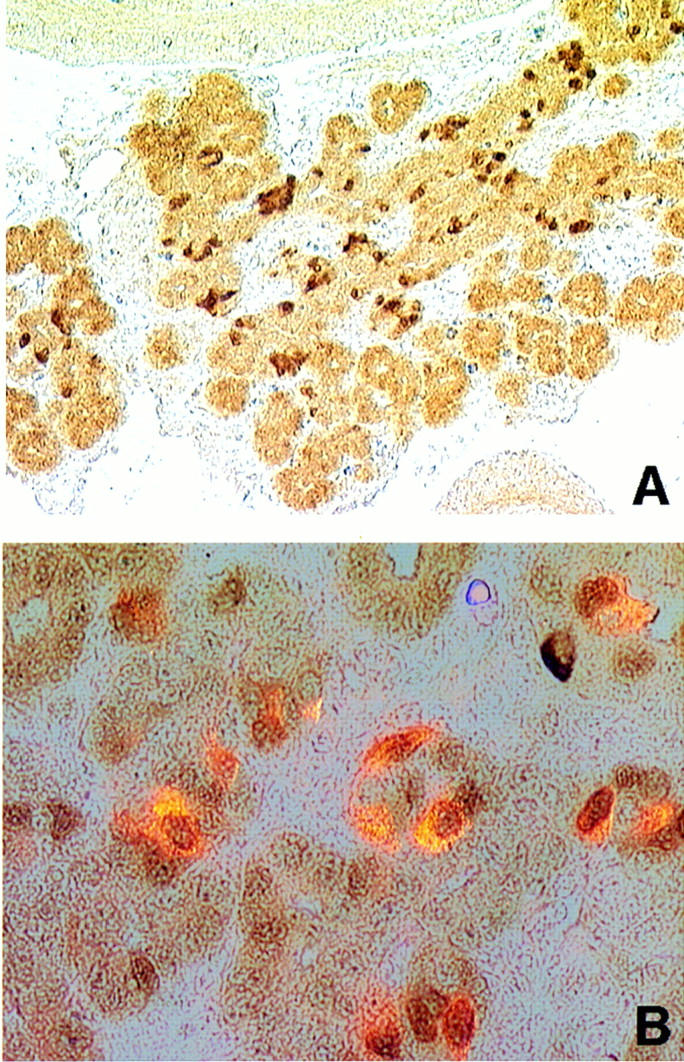
PDX-1 expression in BETA2 −/− pancreas. (A) At E16.0, PDX-1 was expressed throughout the pancreatic epithelium (original magnification, 250×); (B) coimmunolocalization of insulin (imunofluorescence, red, cytoplasmic) and PDX-1 (imunoperoxidase staining, brownish, nuclear) in −/− E16.0 pancreas (original magnification, 1000×). Most insulin-producing cells also coexpressed PDX-1.
Next, we examined whether differentiation of islet lineages from precursor endocrine cells was disrupted in the BETA2 −/− mice. The observed coexpression of multiple hormones in islet progenitor cells early in pancreatic endocrine differentiation suggests that the four lineages may arise from precursor cells that coexpress peptide YY, glucagon, and insulin, and that by E14.5, single cells that coexpress both insulin and glucagon are rarely seen (Teitelman et al. 1993; Upchurch et al. 1994; Guz et al. 1995). Endocrine cells coexpressing insulin and glucagon (data not shown) or insulin and somatostatin (not shown) were not seen in the −/− pancreas, suggesting that the developmental arrest occurred after the stage when α, β, and δ cells segregate.
In addition to defective morphogenesis, the number of β-gal-stained cells appeared reduced in −/− animals by E17.5 and at birth were ∼60% less abundant (Table 1A). However, the number of β-gal-stained cells appeared similar in the −/− mice compared to +/− animals at E9.5 and E14.5. Although the newborn −/− pancreas was similar in size to control tissue, examination of specific islet populations at this stage revealed that the number of β cells was reduced by nearly 75%, with an ∼40% and 20% reduction in α and δ cells, respectively (Table 1B). Furthermore, the −/− pancreas showed a substantial increase in the number of apoptotic cells (Fig. 5) with no obvious differences in cell proliferation as analyzed by staining with the proliferating cell nuclear antigen (PCNA; data not shown). Collectively, these results suggest that the defect in pancreatic islet morphogenesis in BETA2 −/− mice occurs after E14.5 but prior to E17.5 and that BETA2 is required for the survival of endocrine islet cells.
Table 1.
Reduction of the numbers of β-gal-expressing and endocrine cells in BETA2 −/− pancreas
| A. Endocrine cell distribution in BETA2 mutant mice | |||
|---|---|---|---|
| Stage
|
+/−
|
−/−
|
Percent reduction
|
| E9.5 | 56 | 54 | — |
| E14.5 | 567 | 540 | — |
| E17.5 | 2470 | 1050 | 57 |
| P0 | 2631 | 1092 | 59 |
| B. Endocrine cell distribution in BETA2 mutant mice | |||
| Cell type | +/+, +/− | −/− | Percent reduction |
| β | 1544 (60%) | 402 (37%) | 74 |
| α | 731 (29%) | 449 (41%) | 39 |
| δ | 288 (11%) | 236 (22%) | 18 |
| Total cells | 2563 | 1087 | 58 |
| β-gal | 2631 | 1092 | 59 |
(A) BETA2-expressing cells in pancreas development β-Gal-positive cells in +/− and −/− embryos at E9.5, E14.5, E17.5, and P0. Numbers are the sum of β-gal-positive cells in the pancreas sections, of approximate equal size, from three different embryos (E9.5 and E14.5) or from isolated pancreas sections from E17.5 and P0. (B) Endocrine cell number in BETA2 −/− pancreas. Endocrine cells were counted from immunostained sections of newborn (P0) pancreas estimated to be the same size. Data are shown as the sum of three separate pancreas sections from three different newborn mice for each group. Total number of cells is the sum of hormone positive cells from three pancreas sections.
Figure 5.

Increase in the number of apoptotic cells in the BETA2 −/− endocrine pancreas. X-Gal-stained E17.5 and P0 pancreatic sections were subjected to TUNEL assay. For each individual animal, the numbers of cells positive for apoptosis and/or X-gal were counted from five slide sets of four serial 7-μm sections, which included an average of five islet clusters per section. Results are the average of numbers from three different animals and are shown as percent of the number of endocrine cells; error bars represent standard deviation. At P0 there were ∼14-fold more (P < 0.00001, n = 3) apoptotic cells in the −/− endocrine pancreas (B,C) than in its +/− counterpart (A,C). However, at E17.5 (C), the −/− endocrine pancreas already showed a tendency of having fivefold more (P = 0.0204, n = 3) apoptotic cells than its +/− counterpart. Two-sided Student’s t-test was used to determine the significance of the differences.
Acinar cell defects in the exocrine pancreas
Although BETA2 is expressed abundantly in the nervous system, the brains of −/− mice revealed no obvious anatomic and histologic abnormalities. However, given the developmental arrest of islet endocrine cells, the exocrine pancreas was examined for possible defects. As illustrated in Figure 6, by 2 days of age, acinar cells in the −/− mice lacked the normal cellular polarity seen in +/− mice (Fig. 6C,F). Nuclei in −/− acinar cells were randomly distributed within the cell rather than in the basal region of the cell. Ultrastructural examination revealed a loss of the polarized distribution of organelles, overabundance of zymogen granules, and vacuolization characteristic of cellular degeneration in contrast to age-matched +/− animals (Fig. 6, cf. H and I with K and L). In addition, the acinar cell defect presumably occurred after birth, as there were no obvious morphological differences in the exocrine tissue at E17.5 (Fig. 6G, J). Consistent with the above findings, a two- to threefold increase in amylase expression was observed by Western analysis of protein extracts from postnatal day 2 (P2) −/− pancreas (Fig. 7). Similarly, a moderate increase in amylase levels was detected in mutant tissue immunostained with amylase antibodies (data not shown).
Figure 6.

Acinar cell abnormalities in BETA2 −/− mice. (A–F) Hematoxylin and eosin stain of pancreas of E17.5, P0, and P2 BETA2 +/− and −/− mice (original magnification, 400×). Acinar cell nuclei were randomly distributed within the cellular compartment of −/− mice (F) in contrast to +/− mice (C). Acinar cells in the −/− mice (F) lacked the distinct cellular polarity as well as the intense basophilic staining in the basal portion of acinar cells and acidophilic staining at the apical surface (arrows) evident at this developmental stage in +/− pancreas (C). (G–L) Ultrastructural analysis of exocrine pancreas. Polarization was evident in +/− pancreas at birth (P0) and at 2 at days of age (H,I) (original magnification, 2400×) but not in −/− pancreas at comparable stages (K,L) (original magnification, 3000×). Note the abundance of secretory granules and vacuoles in mutant pancreas at 2 days of age (L). (v) Secretory vesicles; (n) nuclei.
Figure 7.

Western blot analysis of α-amylase in the P2-pancreas. A representative result is shown. Equal amounts of total cellular extracts (40 μg) from each pancreas examined were subjected to SDS-PAGE; BETA2 −/− (lane 1), +/− (lane 2), and +/+ (lane 3). Western blot analysis was performed by using anti-α-amylase antibody.
Abnormal enteroendocrine cell differentiation
The abundance of zymogen granules in the neonatal, mutant acinar cells raised the possibility that secretion was compromised as a consequence of arrested islet development or a deficit in the production of the primary pancreatic secretatogogs secretin and cholecystokinin (CCK). The latter notion would be consistent with the demonstration that BETA2 is expressed in the intestine and regulates secretin gene expression (Naya et al. 1995; Mutoh et al. 1997). Hence, the intestinal tract of −/− mice were examined for the presence of secretin and CCK. Secretin-expressing enteroendocrine cells were absent in −/− mice in contrast to the +/− animals, where single secretin cells appeared scattered throughout the mucosa of the proximal small intestine (Fig. 8A,B). In addition, BETA2 −/− mice failed to develop CCK-expressing enteroendocrine cells (Fig. 8C,D), suggesting a previously unappreciated developmental relationship between secretin- and CCK-producing cells. Nuclear β-galactosidase activity was readily demonstrable in secretin cells of BETA2 +/− animals (Fig. 8G). Despite the absence of cells showing secretin and CCK immunoreactivity in BETA2 −/− mice, individual mucosal epithelial cells showed nuclear β-galactosidase staining, indicating the presence of enteroendocrine cells expressing the disrupted BETA2 gene (Fig. 8H). In contrast, the number of serotonin-expressing enteroendocrine cells appeared to be relatively unaffected in the −/− intestine (Fig. 8E,F). Similarly, enteroendocrine populations expressing glucagon, peptide YY, neurotensin, substance P, gastric inhibitory polypeptide, and somatostatin were relatively unaffected (not shown). These results suggest that BETA2 is an important regulator of secretin and CCK gene expression in the intestine.
Figure 8.
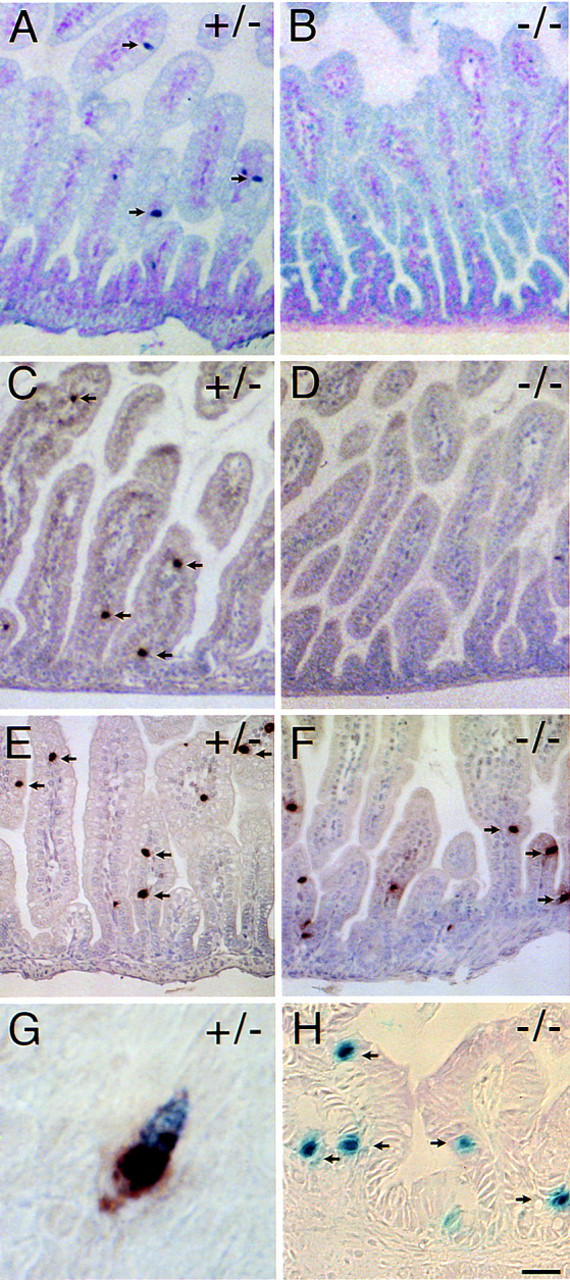
Absence of enteroendocrine cells expressing secretin and cholecystokinin in BETA2 −/− mice. Immunostaining for secretin (A,B), CCK (C,D), and serotonin (E,F) (arrows) in the small intestine of 2 day old BETA2 +/− (left panels) and BETA2 −/− (right panels) mice. Note the absence of secretin- and CCK-staining cells in the −/− mice (B,D) compared to +/− mice (A,C). Nuclear β-galactosidase staining in +/− (G) and −/− (H). G shows a single cell with brown cytoplasmic staining for secretin and blue nuclear X-gal staining from an adult BETA2 +/− mouse. Several isolated mucosal epithelial cells (arrows) in the small intestine of a −/− mouse showing nuclear β-galactosidase activity. Bar, 100 μm for A–D; 50 μm for E,F; 5 μm for G; and 20 μm for H. Sections were stained using the ABC method with True Blue peroxidase substrate counterstained with contrast red (A,B) or DAB substrate counterstained with hematoxylin (C–H).
Discussion
The phenotype of BETA2-deficient mice clearly demonstrates that BETA2 is required for normal pancreatic development and glucose homeostasis. Mice carrying a targeted disruption of the BETA2 gene developed severe, early-onset diabetes and died perinatally. Detailed examination of the pancreas revealed an arrest in the expansion of the pancreatic β-cell population, as well as other islet cell types, and remaining endocrine cells failed to develop into islets of Langerhans. Furthermore, enteroendocrine cells expressing secretin and CCK were also absent in the mutant mice, indicating a role for BETA2 in the differentiation of additional endocrine cell types that arise from the gut endoderm.
The role of BETA2 in pancreatic islet morphogenesis
The most striking phenotype of BETA2 mutant mice was a severe reduction in the number of insulin-expressing β cells and the failure of the remaining endocrine cells to form well-organized, mature pancreatic islets consistent with the islet-specific expression of BETA2. Islet cells aggregated and formed small clusters resembling the primordial endocrine clusters in the developing pancreas, indicating that BETA2 has a specific function in islet morphogenesis.
The reduction in β-cell number is not sufficient to explain the severe hyperglycemia and ketonuria, as animals can maintain glucose homeostasis with removal of 90% of the pancreas (Bonner-Weir et al. 1983). However, the reduced insulin content in combination with either impaired insulin secretion and/or defective glucose response may account for the severe diabetes. Expression of the β cell-specific glucose transporter GLUT-2 is largely unaffected (J. Lee, pers. comm.) indicating, in part, that this aspect of glucose-mediated insulin secretion is not defective. The inability to organize endocrine cells may also explain the hyperglycemic phenotype, as a perturbation in islet structure has been observed in humans with diabetes and in animal models of the disease (Gepts and Lecompte 1981; Gomez Dumm et al. 1990; Tokuyama et al. 1995). It is worthy to mention that the BETA2 gene maps to chromosome 2q31–32.1 (Tamimi et al. 1996; F.J. Naya and M.-J. Tsai, unpubl.), a region implicated in the susceptibility to type I diabetes (Copeman et al. 1995).
Although the molecular mechanisms involved in islet morphogenesis have not been studied in great detail, there is evidence that CAMs may have a role in this process (Rouiller et al. 1991; Cirulli et al. 1994; Dahl et al. 1996). Perhaps changes in the levels and timing of expression of CAMs are responsible for this defect. Preliminary results demonstrate that cadherins (data not shown) and N-CAM (J. Lee, pers. comm.) are present in the endocrine cells of the BETA2 mutant pancreas, suggesting that these cells are not defective in their ability to express these adhesion molecules. This raises the possibility that there may be additional cell-surface markers, perhaps novel CAMs, that play a greater role in islet cell sorting and organization. Alternatively, given the substantial increase in the number of apoptotic cells in −/− animals, our data would suggest that a critical mass of endocrine cells must be attained to effectively organize into mature islets.
It is intriguing that insulin was expressed in the −/− pancreas, as we demonstrated previously that BETA2 is an important regulator of the insulin gene (Naya et al. 1995). The simplest interpretation is that additional BETA2-related bHLH factors, expressed in pancreatic islets, partially compensate for the absence of BETA2, resulting in the differentiation of endocrine cells and activation of the insulin gene. It has been demonstrated that two bHLH genes closely related to BETA2, neurogenin 3 (ngn3) (Sommer et al. 1996) and IB1 (C. Bonny, P. Nicod, and G. Waeber, pers. comm.), are expressed in pancreatic islets; however, the role of these factors in insulin gene regulation has not been addressed. In a similar fashion the ubiquitous bHLH factors, which bind the insulin E box as homodimers, may account for the observed insulin gene expression. Our results would also suggest that BETA2 is required but not essential for insulin expression and that PDX1 might be sufficient to maintain expression in −/− animals. Nevertheless, the inability of ngn3, IB1, or the ubiquitous bHLH genes to complete the islet differentiation program indicates that BETA2 has a specific, nonoverlapping function in the development of the endocrine pancreas.
Various endocrine markers were used to determine the point at which islet development was arrested. We demonstrated that insulin and glucagon as well as insulin and somatostatin were not coexpressed, indicating that the developmental arrest occurred after the segregation of the α, β, and δ cell lineages. However, islet ontogeny remains a controversial issue and lack of endocrine hormone coexpression in the −/− pancreas cannot be used as conclusive evidence for the state of differentiation of these endocrine cells. Given the lack of available molecular markers for intermediate stages of islet endocrine cell development, if indeed they exist, it would be difficult to demonstrate the extent to which these islet cells have differentiated. Ultrastructural studies revealed the presence of secretory granules containing either insulin or glucagon in −/− pancreas (data not shown) indicating maturation of the secretory apparatus (Pictet and Rutter 1972). Collectively, our results demonstrate that BETA2 acts at a relatively late stage in islet cell differentiation consistent with its proposed role as a terminal differentiation factor in neurogenesis (Lee et al. 1995).
The apparent lack of a neuronal defect would suggest that additional neural-specific bHLH factors exist in the nervous system that compensate for the loss of BETA2. Recently, BETA2-related family members NEX-1/MATH-2 and NeuroD2 have been isolated and exhibit overlapping patterns of expression with BETA2 in the developing nervous system (Bartholoma and Nave 1994; Shimizu et al. 1995; Kume et al. 1996; McCormick et al. 1996; Yasunami et al. 1996). In contrast, NEX-1/MATH-2 and NeuroD2 are not expressed in the pancreas (data not shown). Therefore, contrary to the hypothesized neuronal function, BETA2 is dispensable for neural development and absolutely required for islet morphogenesis.
The role of BETA2 in enteroendocrine and pancreatic acinar cell differentiation
We have shown previously that the secretin gene is a second target for transcriptional activation by BETA2. In cell lines, nearly 75% of the E box-dependent transcription is mediated by BETA2. Futhermore, BETA2 immunoreactivity can be readily localized to secretin-expressing enteroendocrine cells of the mouse small intestine (Mutoh et al. 1997) as well as in CCK-expressing enteroendocrine cells (data not shown). The failure to develop secretin- and CCK-expressing enteroendocrine cells in the the BETA2 −/− mouse suggests that this bHLH protein is critical for expression of these peptides in normal, intestinal endocrine cells in addition to cell lines. The rat CCK gene contains several E-box sequences (Haun and Dixon 1990); however, they have not been tested thus far for their ability to interact with BETA2 to functionally activate CCK expression. The complete absence of cells expressing either secretin or CCK from the entire length of the small intestine contrasts findings in pdx −/− mice, in which the number of secretin, CCK, and serotonin cells was reduced by ∼65% in a short segment of the rostral duodenum but not in more caudal segments (Offield et al. 1996).
The presence of isolated mucosal intestinal epithelial cells expressing the lacZ gene suggests cells that normally express BETA2 are present in −/− mice. Thus, BETA2 may promote terminal differentiation of secretin- and CCK-expressing enteroendocrine cells by activating transcription of the genes encoding these two peptides. Expression of other neuroendocrine gene products in enteroendocrine cells does not appear to require BETA2. We have shown previously that ∼10% of intestinal secretin and CCK cells coexpress both peptides (Lopez et al. 1995). The absence of these two cell types in BETA2 −/− mice may indicate a previously unappreciated, shared developmental relationship.
The role of BETA2 in differentiation of pancreatic islets and enteroendocrine cells further supports the existence of a developmental relationship between the β cell and S-type enteroendocrine cell. These results are consistent with earlier observations that secretin is expressed in a subset of β cells in the developing pancreas (Wheeler et al. 1992) and that transgenic mice expressing the SV40 large T antigen driven by 1.6 kb of the secretin promoter developed insulin-producing tumors in the pancreas in addition to neuroendocrine tumors of the small intestine (Lopez et al. 1995).
The histological and ultrastructural abnormalities seen in acinar cells of BETA2 −/− mice does not appear to be a direct result of BETA2 deficiency, as this protein is not appreciably expressed in the exocrine pancreas. One possibility is that normal islet structure and/or function may contribute to the normal polarity and structural integrity of pancreatic acinar cells. Insulin is required for normal acinar cell function in the mature pancreas (Williams and Goldfine 1985). It is more likely that the acinar cell abnormalities result from the failure to express CCK and secretin. CCK is a potent secretagog for the secretion of protein-rich pancreatic juice from acinar cells. Secretin appears to further increase CCK-stimulated acinar cell secretion in addition to stimulating bicarbonate secretion by duct cells. Thus, in the absence of these two hormones, zymogen granules may accumulate in acinar cells and leak digestive enzymes into the cellular compartment, perhaps contributing to deterioration of the exocrine pancreas.
Transcriptional control of pancreatic development
BETA2 represents the first bHLH factor to have a specific function in pancreatic development. Recently, the roles of the homeodomain transcription factors Isl-1, PDX-1 and the paired box factor Pax4, in pancreatic development were determined by gene targeting experiments in mice. Targeted ablation of PDX-1 resulted in mice completely lacking a pancreas demonstrating an important function for PDX-1 in the initial stages of pancreas development (Jonsson et al. 1994; Offield et al. 1996). PDX-1 −/− mice develop small pancreatic duct structures containing insulin- and glucagon-expressing cells. Isl-1-deficient pancreatic epithelia grown in culture failed to generate α, β, and δ cells, suggesting an important function for Isl-1 in islet cell differentiation (Ahlgren et al. 1997). Inactivation of the Pax4 gene resulted in growth retardation and dehydration similar to BETA2 −/− mice (Sosa-Pineda et al. 1997). Mice fail to develop either β or δ cells in the absence of Pax4 and instead show increased numbers of α cells. The presence of both pancreatic acinar cells and the principal endocrine cell types in BETA2 −/− mice suggests that BETA2 likely acts in a regulatory cascade downstream of PDX-1, Isl-1, and Pax4. The observed phenotype does not appear to arise purely from reduced insulin gene expression. Recent evidence suggests that BETA2 increases PDX-1 gene expression (Sharma et al. 1997). However, cells staining for PDX-1 are present in BETA2 −/− mice, making it unlikely that the observed diabetes results from the failure to express PDX-1.
It will be of particular interest to determine whether mutations in the human BETA2 gene are associated with susceptibility to diabetes mellitus. Furthermore, the BETA2 knockout mouse is a useful model for the identification of specific genes involved in pancreatic islet cell morphogenesis.
Materials and methods
Targeting construct
A 14-kb NotI fragment containing the entire BETA2 gene, isolated from a 129/Sv mouse genomic library (a kind gift of P. Soriano, University of Washington, Seattle), was subcloned into pBSII-KS(+) (Stratagene) and mapped using a combination of PCR, Southern analysis, and restriction enzyme digests. The targeting vector was assembled as follows: The XmaI–NotI (blunt) fragment of the bacterial lacZ gene (pPD46.21) with a nuclear localization signal (obtained from E. Olson, University of Texas Southwestern Medical Center, Dallas) was subcloned into the XmaI–EcoRV sites of pBSII-KS(+). The 2.5-kb XbaI (blunt) fragment was subcloned into the SmaI site of pBSII-KS(+)–lacZ. The BamHI–XhoI fragment containing the 2.5-kb BETA2 5′ genomic sequence and lacZ was subcloned into the BamHI–XhoI site of the herpes simplex virus–thymidine kinase (HSV–TK) vector. The 4.5-kb EcoRI fragment of BETA2 3′ genomic sequences was subcloned into EcoRI of PGK–neo. An XhoI fragment consisting of BETA2 3′ sequences and neo was cloned into XhoI of HSV–TK containing 5′ BETA2 sequences and lacZ. The final targeting vector was CsCl purified and linearized with BamHI before electroporation into the ES cell line, AB 2.2 (provided by A. Bradley, Baylor College of Medicine, Houston, TX).
ES cell electroporations, cell culture, and blastocyst injections
ES cells were dispersed into single cells and resuspended at a density of 1 × 107 cells/ml in PBS. AB 2.2 cells were electroporated in a volume of 0.9 ml with 25 μg of targeting vector at 230 V and 500 μF. Cells were seeded onto 100 mm gelatinized plates in the presence of G418-resistant embryonic fibroblasts (STO cells) in ES medium. After 24 hr, medium was supplemented with FIAU and G418. Cells were cultured for a total of 10 days, after which colonies were picked and expanded. Duplicate clones were screened for the presence of disrupted BETA2 gene by Southern analysis. Mutant ES cells were injected into C57BL/6 embryos at the blastocyst stage using standard techniques. Chimeric male mice were obtained and mated to C57BL/6 females to obtain heterozygous mice.
Glucose levels
Glucose levels were measured with the One Touch Glucose Monitoring kit (Johnson & Johnson) using 10 μl of peripheral blood or urine from 2-day-old (P2) mice. Blood glucose values are represented as the average ± standard deviation. Urine glucose levels also indicated hyperglycemia (not shown). Ketone levels were measured with Ketostix reagent strips (Bayer) using 10 μl of urine from P2 mice. Semiquantitative ketone concentration was determined by color reaction after 15 sec.
Histochemistry and electron microscopy
Pancreas and brain were removed from newborn mice (P0) and were fixed and stained in X-gal solution as described in Joyner (1995). Fixed tissues were embedded in paraffin and sectioned at 6–7 μm. E9.5 embryos were sectioned at 4 μm; E14.5 and E17.5 embryos were sectioned at 6–7 μm. For immunohistochemistry, tissues were fixed in Bouin’s fixative and embedded in paraffin as described previously (Upchurch et al. 1994). Pancreatic tissue for electron microscopy was fixed in 3% glutaraldehyde in 0.1 m sodium cacodylate buffer (pH 7.4) for 1 hr at room temperature, washed in PBS, and stored in 70% ethanol. Fixation of pancreatic tissue for immunolabeling was prepared as described in Teitelman et al. (1993).
Immunohistochemistry
Primary antibodies were used at the following dilutions: rabbit anti-insulin 1:80 (Incstar); rabbit anti-glucagon 1:40 (Incstar); rabbit anti-somatostatin 1:20 (Incstar); guinea pig anti-C-peptide insulin 1:40 (Linco), rabbit anti-secretin 1:6000 (R25, W. Chey, University of Rochester, NY); rabbit anti-amylase 1:300 (Sigma); rabbit anti-PDX-1 1:500 (a gift from Dr. Palle Serup, Hagedorn Research Institute, Gentofte, Denmark), rabbit anti-CCK 1:2500 (RPZ7.1, W. Chey); rabbit anti-serotonin 1:40,000 (Incstar). Antisera against substance P, peptide YY, neurotensin, and gastric inhibitory polypeptide (GIP) were used as described previously (Upchurch et al. 194). Primary incubations were performed at room temperature for 1–2 hr or overnight at 4°C. Primary antibodies were detected either by immunofluorescent labeling with FITC-conjugated (1:160) or Cy3-conjugated (1:400) donkey anti-rabbit or anti-guinea pig IgG secondary antibodies immunoabsorbed for multiple labeling (Jackson ImmunoResearch Laboratories) or by immunoperoxidase using a Vectastain ABC kit (Vector Labs) using DAB or TrueBlue (Kierkegarrd & Perry) for detection. Dual-color immunohistochemistry was performed as described previously (Bukovsky et al. 1995). The first primary antibody (anti-PDX-1) was visualized by immunoperoxidase staining as described above, and the slides were then incubated with the second primary antibody (anti-insulin) and visualized with a Cy3-conjugated secondary antibody with a fluorescence microscope. Controls for specificity included nonimmune primary sera, mismatched primary and secondary antisera, known positive sections, and absorption with specific and heterologous antigens.
TUNEL assay
X-Gal-stained BETA2 +/− and −/− pancreatic tissues from E17.5 embryos and newborn mice (P0) were used in the TUNEL assay as described previously (Gavrieli et al. 1992).
Western blot analysis
Pancreatic tissues for protein isolation were frozen and stored at −70°C. Total pancreatic protein was prepared by homogenization in 50 mm Tris (pH 7.2), 5 mm MgCl2, 1 mm CaCl2, 10 mm dithiothreitol, 1% NP-40, 100 μg/ml of DNase I, 50 μg/ml of RNase A, and 75 μg/ml of phenylmethylsulphonyl fluoride. Protein concentration was determined using protein assay reagents (Bio-Rad). Forty micrograms of protein from pancreas was fractionated on a 10% SDS–polyacrylamide gel, transferred to nitrocellulose by electroblotting, incubated in a sequential order with a blocking solution, anti-α-amylase antibody 1:10000 (Sigma), and HRP-conjugated secondary antibody (Bio-Rad). The blots were then visualized using an ECL system (Amersham). Porcine α-amylase (Sigma) and HeLa cell extracts were used as positive and negative controls, respectively.
Acknowledgments
We thank T. Spencer, F. Pereira, A. Cooney, and S. Tsai for critical reading of the manuscript and M. Liu and P. Samora for helpful discussions. We also thank L. Hadsell for blastocyst injections, D. Turner for assistance with electron microscopy, and Dr. Palle Serup for the PDX-1 antibody. This work was supported by National Institutes of Health (NIH) grants HD17379 to M.J.T. and DK43673 and the GRASP Digestive Disease Center DK34928 to A.B.L.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL mtsai@condor.bcm.tmc.edu; FAX (713) 798-8227.
References
- Ahlgren U, Pfaff SL, Jessell TM, Edlund T, Edlund H. Independent requirement for ISL1 in formation of pancreatic mesenchyme and islet cells. Nature. 1997;385:257–260. doi: 10.1038/385257a0. [DOI] [PubMed] [Google Scholar]
- Alpert S, Hanahan D, Teitelman G. Hybrid insulin genes reveal a developmental lineage for pancreatic endocrine cell and imply a relationship with neurons. Cell. 1988;53:295–308. doi: 10.1016/0092-8674(88)90391-1. [DOI] [PubMed] [Google Scholar]
- Bain G, Maandag ECR, Izon DJ, Amsen D, Kruisbeek AM, Weintraub BC, Krop I, Schlissel MS, Feenyey AJ, Roon MV. E2A proteins are required for proper B cell development and initiation of immunoglobulin gene rearrangements. Cell. 1994;79:885–892. doi: 10.1016/0092-8674(94)90077-9. [DOI] [PubMed] [Google Scholar]
- Bartholoma A, Nave K A. NEX-1: A novel brain specific helix loop helix protein with autoregulation and sustained expression in mature cortical neurons. Mech Dev. 1994;48:217–228. doi: 10.1016/0925-4773(94)90061-2. [DOI] [PubMed] [Google Scholar]
- Bonner-Weir S, Trent D, Weir GC. Partial pancreatectomy in the rat and subsequent defect in glucose-induced insulin secretion. J Clin Invest. 1983;71:1544–1553. doi: 10.1172/JCI110910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukovsky A, Caudle MR, Keenan JA, Wimalasena J, Foster JS, Van Meter SE. Quantative evaluation of the cell cycle-related retinoblastoma protein and location of Thy-1 differentiation protein and macrophage during follicular development and atresia, and in human corpora lutea. Biol Reprod. 1995;52:776–792. doi: 10.1095/biolreprod52.4.776. [DOI] [PubMed] [Google Scholar]
- Chen Z-F, Behringer RR. twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes & Dev. 1995;9:686–699. doi: 10.1101/gad.9.6.686. [DOI] [PubMed] [Google Scholar]
- Cirulli V, Baetens D, Rutishauser U, Halban PA, Orci L, Rouiller DG. Expression of neural cell adhesion molecule (N-CAM) in rat islets and its role in islet cell type segregation. J Cell Sci. 1994;107:1429–1436. doi: 10.1242/jcs.107.6.1429. [DOI] [PubMed] [Google Scholar]
- Copeman JB, Cucca F, Hearne CM, Cornall RJ, Reed PW, Ronningen KS, Undlien DE, Nistico L, Briggeti R, Tosi R, et al. Linkage disequilibrium mapping of a type I diabetes susceptibility gene (IDDM7) to chromosome 2q31-q33. Nature Genet. 1995;9:80–85. doi: 10.1038/ng0195-80. [DOI] [PubMed] [Google Scholar]
- Cordle SR, Henderson E, Masucha H, Weil PA, Stein R. Pancreatic β-cell-type-specific transcription of the insulin gene is mediated by basic helix-loop-helix DNA-binding proteins. Mol Cell Biol. 1991;11:1734–1738. doi: 10.1128/mcb.11.3.1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl U, Sjodin A, Semb H. Cadherins regulate aggregation of pancreatic β cells in vivo. Development. 1996;122:2895–2902. doi: 10.1242/dev.122.9.2895. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman T, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragment. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gepts W, Lecompte PM. The pancreatic islets in diabetes. Am J Med. 1981;70:105–115. doi: 10.1016/0002-9343(81)90417-4. [DOI] [PubMed] [Google Scholar]
- German M S, Blanar M A, Nelson C, Moss L G, Rutter W J. Two related helix-loop-helix proteins participate in separate cell-specific complexes that bind the insulin enhancer. Mol Endocrinol. 1991;5:292–299. doi: 10.1210/mend-5-2-292. [DOI] [PubMed] [Google Scholar]
- Gittes GK, Rutter WJ. Onset of cell-specific gene expression in the developing mouse pancreas. Proc Natl Acad Sci. 1992;89:1128–1132. doi: 10.1073/pnas.89.3.1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez Dumm CLA, Semino MC, Gagliardino JJ. Sequential morphological changes in pancreatic islets of spontaneously diabetic rats. Pancreas. 1990;5:533–539. doi: 10.1097/00006676-199009000-00007. [DOI] [PubMed] [Google Scholar]
- Guillemot F, Lo LC, Johnson JE, Auerbach A, Anderson DJ, Joyner AL. Mammalian achaete-scute homolog 1 is required for the early development of olfactory and autonomic neurons. Cell. 1993;75:463–476. doi: 10.1016/0092-8674(93)90381-y. [DOI] [PubMed] [Google Scholar]
- Guz Y, Montminy MR, Stein R, Leonard J, Gamer LW, Wright CVE, Teitelman G. Expression of murine STF-1, a putative insulin gene transcription factor in b cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development. 1995;121:11–18. doi: 10.1242/dev.121.1.11. [DOI] [PubMed] [Google Scholar]
- Halban P, Wollheim C, Blondel B, Meda P, Niesor EN, Mintz DH. The importance of contact between pancreatic islets cells for the control of insulin release. Endocrinology. 1982;111:86–94. doi: 10.1210/endo-111-1-86. [DOI] [PubMed] [Google Scholar]
- Haun RS, Dixon JE. A transcriptional enhancer essential for the rat cholecystokinin gene contains a sequence identical to the -296 element of the human c-fos gene. J Biol Chem. 1990;265:15455–15463. [PubMed] [Google Scholar]
- Herrera PL, Huarte J, Sanvito F, Meda P, Orci L, Vassalli J-D. Embryogenesis of the murine endocrine pancreas; early expression of pancreatic polypeptide gene. Development. 1991;113:1257–1265. doi: 10.1242/dev.113.4.1257. [DOI] [PubMed] [Google Scholar]
- Hutton JC, Christofori G, Chin WY, Edman U, Guest PC, Hanahan D, Kelly RB. Molecular cloning of mouse pancreatic R-cadherin: Differential expression in endocrine and exocrine tissue. Mol Endocrinol. 1993;7:1151–1160. doi: 10.1210/mend.7.9.8247017. [DOI] [PubMed] [Google Scholar]
- Jan YN, Jan LY. HLH proteins, fly neurogenesis, and vertebrate myogenesis. Cell. 1993;75:827–830. doi: 10.1016/0092-8674(93)90525-u. [DOI] [PubMed] [Google Scholar]
- Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin promoter factor 1 is required for pancreas development in mice. Nature. 1994;371:606–609. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- Joyner AL. Gene targeting: A practical approach. Oxford, UK: Oxford University Press; 1995. [Google Scholar]
- Kume HK, Maruyama T, Tomita T, Iwatsubo T, Saido TC, Obata K. Molecular cloning of a novel basic helix loop helix protein from the rat brain. Biochem Biophys Res Commun. 1996;219:526–530. doi: 10.1006/bbrc.1996.0267. [DOI] [PubMed] [Google Scholar]
- Lee JL, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by neuroD, a basic helix loop helix transcription factor. Science. 1995;268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- Lopez MJ, Upchurch BH, Rindi G, Leiter AB. Studies in transgenic mice reveal potential relationships between secretin producing cells and other endocrine cell types. J Biol Chem. 1995;270:885–891. doi: 10.1074/jbc.270.2.885. [DOI] [PubMed] [Google Scholar]
- Lucas-Clerc C, Massart C, Campion JP, Launois B, Nicol M. Long-term culture of human pancreatic islets in an extracellular matrix; morphological and metabolic effects. Mol Cell Endocrinol. 1993;94:9–20. doi: 10.1016/0303-7207(93)90046-m. [DOI] [PubMed] [Google Scholar]
- McCormick MB, Tamimi RA, Snider L, Asakura A, Bergstrom D, Tapscott SJ. Neurod2 and neurod3: Distinct expression patterns and transcriptional activation potentials within the neurod gene family. Mol Cell Biol. 1996;16:5792–5800. doi: 10.1128/mcb.16.10.5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller CJ, Christgau S, Williamson MR, Madsen OD, Zhan-Po N, Bock E, Baekkeskov S. Differential expression of neural cell adhesion molecule and cadherins in pancreatic islets, glucagonomas, and insulinomas. Mol Endocrinol. 1992;6:1332–1342. doi: 10.1210/mend.6.8.1406710. [DOI] [PubMed] [Google Scholar]
- Mutoh H, Fung BP, Naya FJ, Tsai MJ, Nishitani J, Leiter AB. The basic helix loop helix transcription factor BETA2/NeuroD is expressed in enteroendocrine cells and activates secretin gene expression. Proc Natl Acad Sci. 1997;94:3560–3564. doi: 10.1073/pnas.94.8.3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naya FJ, Stellrecht CMM, Tsai M-J. Tissue specific regulation of the insulin gene by a novel basic helix loop helix transcription factor. Genes & Dev. 1995;9:1009–1019. doi: 10.1101/gad.9.8.1009. [DOI] [PubMed] [Google Scholar]
- Nelson C, Shen L-P, Meister A, Fodor E, Rutter W J. Pan: A transcriptional regulator that binds chymotrypsin, insulin, and AP-4 enhancer motifs. Genes & Dev. 1990;4:1035–1043. doi: 10.1101/gad.4.6.1035. [DOI] [PubMed] [Google Scholar]
- Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, Hogan BLM, Wright CVE. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122:983–995. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]
- Olson EN, Klein WH. bHLH factors in muscle development: Dead lines and commitments, what to leave in and what to leave out. Genes & Dev. 1994;8:1–8. doi: 10.1101/gad.8.1.1. [DOI] [PubMed] [Google Scholar]
- Orci L. Macro- and micro-domains in the nedocrine pancreas. Diabetes. 1982;31:538–565. doi: 10.2337/diab.31.6.538. [DOI] [PubMed] [Google Scholar]
- Peyton M, Moss LG, Tsai M-J. Two distinct class A helix-loop-helix transcription factors, E2A and BETA1, form separate DNA binding complexes on the insulin gene E box. J Biol Chem. 1994;269:25936–25941. [PubMed] [Google Scholar]
- Pictet RL, Rutter W J. Handbook of physiology. American Physiology Society; 1972. Development of the embryonic endocrine pancreas. D.F.SteinerN.Frenkel1, section 7, Williams and Wilkins, Washington, DC. [Google Scholar]
- Pipeleers DP, In’T Veld P, Maes E, Van De Winkel M. Glucose-induced insulin release depends on functional cooperation between islet cells. Proc Natl Acad Sci. 1982;79:7322–7325. doi: 10.1073/pnas.79.23.7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouiller DG, Cirulli V, Halban PA. Uvomorulin mediates calcium dependent aggregation of islet cells, whereas calcium independent cell adhesion molecules distinguish between islet cell types. Dev Biol. 1991;148:233–242. doi: 10.1016/0012-1606(91)90332-w. [DOI] [PubMed] [Google Scholar]
- Sharma S, Jhala US, Johnson T, Ferreri K, Leonard J, Montminy M. Hormonal regulation of an islet-specific enhancer in the pancreatic homeobox gene STF-1. Mol Cell Biol. 1997;17:2598–2604. doi: 10.1128/mcb.17.5.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh S-Y, Tsai M-J. Cell-specific and ubiquitous factors are responsible for the enhancer activity of the rat insulin II gene. J Biol Chem. 1991;266:16707–16714. [PubMed] [Google Scholar]
- Shimizu C, Akazawa C, Nakanishi S, Kageyama R. MATH-2 a mammalian helix loop helix factor structurally related to the product of Drosophila proneural gene atonal is specifically expressed in the nervous system. Eur J Biochem. 1995;229:239–248. doi: 10.1111/j.1432-1033.1995.tb20461.x. [DOI] [PubMed] [Google Scholar]
- Shivdasani RA, Mayer EL, Orkin SH. Absence of blood formation in mice lacking the T-cell leukemia oncoprotein tal-1/SCL-1. Nature. 1995;373:432–434. doi: 10.1038/373432a0. [DOI] [PubMed] [Google Scholar]
- Sommer L, Ma Q, Anderson DJ. Neurogenins, a novel family of atonal-related bHLH transcription factors, are putative mammalian neuronal determination genes that reveal progenitor cell heterogeneity in the developing CNS and PNS. Mol Cell Neurobiol. 1996;8:221–241. doi: 10.1006/mcne.1996.0060. [DOI] [PubMed] [Google Scholar]
- Sosa-Pineda B, Chowdury K, Torres M, Oliver G, Gruss P. The Pax4 gene is essential for differentiation of insulin-producing β cells in the mammalian pancreas. Nature. 1997;386:399–402. doi: 10.1038/386399a0. [DOI] [PubMed] [Google Scholar]
- Tamimi R, Steingrimsson E, Copeland N G, Dyer-Montgomery K, Lee JE, Hernandez R, Jenkins NA, Tapscott SJ. The NEUROD gene maps to human chromosome 2q32 and mouse chromosome 2. Genomics. 1996;34:418–421. doi: 10.1006/geno.1996.0306. [DOI] [PubMed] [Google Scholar]
- Teitelman G, Alpert S, Polak JM, Martinez A, Hanahan D. Precursor cells of mouse endocrine pancreas coexpress insulin, glucagon and the neuronal proteins tyrosine hydroxylase and neuropeptide Y, but not pancreatic polypeptide. Development. 1993;118:1031–1039. doi: 10.1242/dev.118.4.1031. [DOI] [PubMed] [Google Scholar]
- Tokuyama Y, Sturis J, DePaoli AM, Takeda J, Stoffel M, Tang J, Sun X, Polonsky KS, Bell GI. Evolution of β cell dysfunction in the male Zucker diabetic fatty rat. Diabetes. 1995;44:1447–1457. doi: 10.2337/diab.44.12.1447. [DOI] [PubMed] [Google Scholar]
- Upchurch BH, Aponte G W, Leiter AB. Expression of peptide YY in all four islet cell types in the developing mouse pancreas suggests a common peptide YY producing progenitor. Development. 1994;120:245–252. doi: 10.1242/dev.120.2.245. [DOI] [PubMed] [Google Scholar]
- Walker M D, Park C W, Rosen A, Aronheim A. A cDNA from a mouse pancreatic β-cell encoding a putative transcription factor of the insulin gene. Nucleic Acids Res. 1990;18:1159–1166. doi: 10.1093/nar/18.5.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H. The MyoD family and myogenesis: Redundancy, networks, and thresholds. Cell. 1993;75:1241–1244. doi: 10.1016/0092-8674(93)90610-3. [DOI] [PubMed] [Google Scholar]
- Wheeler MB, Nishitani J, Buchan A J, Kopin AS, Chey WY, Chang T-A, Leiter AB. Identification of a transcriptional enhancer important for enteroendocrine and pancreatic islet cell specific expression of the secretin gene. Mol Cell Biol. 1992;12:3531–3539. doi: 10.1128/mcb.12.8.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Goldfine ID. The insulin-pancreatic acinar axis. Diabetes. 1985;34:980–986. doi: 10.2337/diab.34.10.980. [DOI] [PubMed] [Google Scholar]
- Yasunami M, Suzuki K, Maruyama H, Kawakami H, Nagai Y, Hagiwara M, Okubo H. Molecular cloning and characterization of a cDNA encoding a novel basic helix loop helix protein structurally related to neuroD/BHF1. Biochem Biophys Res Commun. 1996;220:754–758. doi: 10.1006/bbrc.1996.0476. [DOI] [PubMed] [Google Scholar]
- Zhuang Y, Soriano P, Weintraub H. The helix loop helix gene E2A is required for B cell formation. Cell. 1994;79:875–884. doi: 10.1016/0092-8674(94)90076-0. [DOI] [PubMed] [Google Scholar]