

Abstract
Postsynthesis mismatch repair is an important contributor to mutation avoidance and genomic stability in bacteria, yeast, and humans. Regulation of its activity would allow organisms to regulate their ability to evolve. That mismatch repair might be down-regulated in stationary-phase Escherichia coli was suggested by the sequence spectrum of some stationary-phase (“adaptive”) mutations and by the observations that MutS and MutH levels decline during stationary phase. We report that overproduction of MutL inhibits mutation in stationary phase but not during growth. MutS overproduction has no such effect, and MutL overproduction does not prevent stationary-phase decline of either MutS or MutH. These results imply that MutS and MutH decline to levels appropriate for the decreased DNA synthesis in stationary phase, whereas functional MutL is limiting for mismatch repair specifically during stationary phase. Modulation of mutation rate and genetic stability in response to environmental or developmental cues, such as stationary phase and stress, could be important in evolution, development, microbial pathogenicity, and the origins of cancer.
Keywords: Mismatch repair, MutL, mutation, adaptive mutation, genetic instability, evolution, stationary phase
In Escherichia coli mismatch repair is the single largest contributor to avoidance of mutations due to DNA polymerase errors in replication (Radman 1988; Modrich 1991). Mismatch repair also promotes genetic stability by editing the fidelity of genetic recombination and transposon excision, and by the involvement of its component proteins in transcription-coupled DNA repair and very-short-patch repair (Radman 1988; Modrich 1991; Lieb and Shehnaz 1995; Mellon and Champe 1996). The mismatch repair proteins are highly conserved throughout evolution and appear to play roles in simple and complex eukaryotes similar to those that they play in bacteria (Reenan and Kolodner 1992; Modrich 1994; Baker et al. 1995, 1996; de Wind et al. 1995; Datta et al. 1996; Hunter et al. 1996; Kolodner 1996). These proteins act on incorrectly paired and unpaired bases in DNA that arise via DNA synthesis errors, recombination of diverged sequences, and DNA damage. In all of these circumstances, mismatch repair enforces genetic stability. The consequences of failing to maintain this enforcement are profound for speciation (Rayssiguier et al. 1989; Radman and Wagner 1993; Matic et al. 1995; Hunter et al. 1996; Zahrt and Maloy 1997) and for formation of cancers (Modrich 1994, 1995; Kolodner 1996).
Four proteins play critical roles in mismatch repair in E. coli (for review, see Modrich 1991). MutS binds to DNA base mismatches, to insertion/deletion single-strand loops of four or fewer nucleotides, which are intermediates in frameshift mutation, and probably also to sites of DNA damage (Mello et al. 1996). MutL interacts with MutS after mismatch binding and is thought to coordinate MutS with MutH. MutH endonuclease then nicks the unmethylated (new) DNA strand of a nearby hemimethylated GATC sequence. GATCs are hemimethylated during DNA replication because the new strand is transiently unmethylated. MutU helicase enters DNA at the single-strand nick and displaces the nicked strand, which may or may not be degraded during displacement (see R.S. Harris, K.J. Ross, M.-J. Lombardo, and S.M. Rosenberg, unpubl.). DNA polymerase III then resynthesizes the DNA, thus correcting the sequence. In recombination, the mismatches or small loops are caused by formation of heteroduplex DNA between nonidentical sequences rather than by replication errors as just described.
Although central to maintenance of genetic stability, little is known about whether the mismatch repair system might be regulated (but see Stahl 1988; Rayssiguier et al. 1989; Hastings and Rosenberg 1992; Rosenberg 1994, 1997; Longerich et al. 1995; Rosenberg et al. 1995, 1996 for hypotheses). If mismatch repair were regulated, then cells would regulate their potential to evolve. Two lines of work from E. coli have converged on the first evidence in any organism suggesting that mismatch repair proteins might be regulated and, more specifically, have suggested the down-regulation of mismatch repair during the differentiated states of stationary phase and nutritional stress.
First, stationary-phase reversions of a lac +1 frameshift mutation in E. coli (Cairns and Foster 1991) appear to be DNA polymerase errors that escape mismatch repair. The stationary-phase reversion mechanism in the lac frameshift assay system is distinct from growth-dependent Lac reversion (Rosenberg 1994, 1997; Rosenberg et al. 1995, 1996) in that the former includes homologous recombination (Harris et al. 1994, 1996; Foster et al. 1996) and produces mutations with a highly distinctive DNA sequence spectrum, mostly single-base deletions in small mononucleotide repeats (Foster and Trimarchi 1994; Rosenberg et al. 1994). This mutation spectrum is different from growth-dependent reversions of the same allele (Foster and Trimarchi 1994; Rosenberg et al. 1994) but is identical to growth-dependent reversions in mismatch repair–null mutant strains (Longerich et al. 1995). Thus, depressed mismatch repair could be responsible for the unique stationary-phase mutation spectrum. Most stationary-phase mutants are not heritably mismatch repair-defective (Longerich et al. 1995) [although some are (Torkelson et al. 1997)], indicating that any loss of mismatch repair during stationary-phase mutation must be transient. Such transient loss could occur either by down-regulation of the mismatch repair system or by a block at the DNA level, for example, either by under- or overmethylation of DNA sites required for operation of this methyl-directed repair system (Longerich et al. 1995).
Second, the recent discovery that MutS and MutH mismatch repair protein levels decrease in stationary phase and starving bacterial cells appears to support the hypothesis of down-regulation of mismatch repair at the protein level (Feng et al. 1996). However, although MutS and MutH protein levels decrease during stationary phase and starvation, so does DNA replication. Therefore, it is possible that the proper ratio of these proteins to replication errors is preserved, leaving mismatch repair functional in stationary-phase, starving cells.
If MutS, MutH, or any mismatch repair protein became limiting for mismatch repair function during stationary-phase mutation, then overproduction of the limiting mismatch repair protein might restore mismatch repair function and thereby decrease stationary-phase mutation. We report that overproduction of MutL has this effect, that overproduction of MutS does not, and that overproduction of MutL does not act indirectly by preventing the stationary-phase decline of MutS or MutH protein levels. Overproduction of MutL does not depress growth-dependent Lac reversion. The data imply that functional MutL protein becomes limiting specifically during stationary-phase mutation and that the decreased levels of MutS and MutH observed in stationary phase are appropriate and not limiting for the amount of DNA synthesis in stationary phase. These results imply a loss of mismatch repair function, at the level of MutL protein, specifically during the differentiated state of stationary phase. This provides the first evidence in any organism that mismatch repair function is not constitutive but, rather, can be modulated by cell physiology and differentiation. Environmental change could promote genetic change by this route.
Results
The term stationary-phase mutation is used here to refer to what has also been called “adaptive mutation” (for review, see Foster 1993). This process occurs in stressed, starving cells and so may reflect stress responses in general. In the assay system used here—reversion of a lac frameshift mutation in E. coli—the stationary-phase mutations are also mechanistically distinct from growth-dependent reversions of the same allele. Unlike growth-dependent Lac reversion, the stationary-phase mutation mechanism (1) requires homologous recombination (Harris et al. 1994, 1996; Foster et al. 1996); (2) includes DNA double-stand breaks that are implicated as a molecular intermediate in the mutagenesis (Harris et al. 1994); and (3) produces mutations that are nearly all single-base deletions in small mononucleotide repeats, whereas growth-dependent Lac reversions are heterogeneous (Foster and Trimarchi 1994; Rosenberg et al. 1994). Furthermore, (4) this unique sequence spectrum is identical to that observed in mismatch repair-defective cells, implying failure of mismatch repair in stationary-phase mutation (Longerich et al. 1995); and (5) although this question has not been addressed for the growth-dependent mutations, the stationary-phase mutations have been shown to occur genome-wide, in a hypermutable subpopulation of the stressed cells (Torkelson et al. 1997). This specific mutation mechanism is called recombination-dependent stationary-phase mutation. It is proposed to occur by DNA polymerase errors occurring during DNA synthesis primed by recombinational strand-exchange intermediates, which form at DNA double-strand breaks, in cells with attenuated mismatch repair (for review, see Rosenberg 1994, 1997; Rosenberg et al. 1995, 1996) or in DNA substrates that are refractory to mismatch repair (Kuzminov 1995; Longerich et al. 1995).
Key features of recombination-dependent mutation are seen in some (Taddei et al. 1995) but not all other stationary-phase mutation systems assayed (e.g., Foster and Trimarchi 1995; Galitski and Roth 1995, 1996; Hall 1995; Radicella et al. 1995). For those systems with no known features distinct from growth-dependent mutation (e.g., Foster and Trimarchi 1995; Galitski and Roth 1995, 1996; Radicella et al. 1995), the stationary-phase mutations may occur by the same mechanism as growth-dependent mutation and not be a distinct phenomenon. For example, the cells may be growing or replicating DNA cryptically.
We tested whether mismatch repair proteins MutS, MutL, or both become limiting during recombination-dependent stationary-phase reversion of a lac frameshift mutation. If the mismatch repair proteins become limiting transiently during stationary-phase mutation, then overproduction of those proteins from plasmids might be expected to restore mismatch repair function and inhibit formation of stationary-phase Lac+ revertants. To measure stationary-phase reversions of the lac frameshift mutation, the frameshift-bearing cells are spread on minimal lactose plates (see Materials and Methods; Cairns and Foster 1991; Harris et al. 1994, 1996). At about 2 days of incubation, growth-dependent revertant colonies appear. These are followed by stationary-phase revertant colonies that accumulate during the next several days (Cairns and Foster 1991; Foster 1993) and form via the different, recombination-dependent molecular mechanism (for review, see Rosenberg 1994, 1997; Rosenberg et al. 1995, 1996). Cells carrying a null mutation in the recombination gene recA form the initial growth-dependent mutations but then fail to accumulate any recombination-dependent Lac+ reversions over the next several days (Harris et al. 1994; see also Fig. 1A, below).
Figure 1.
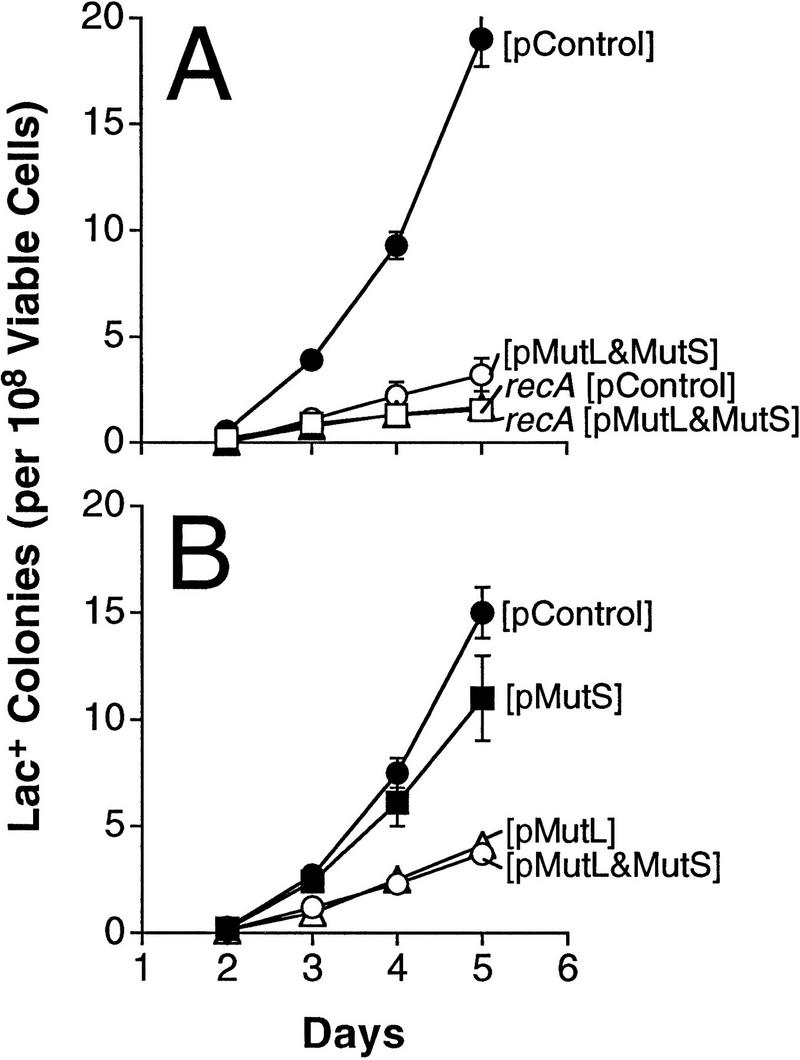
Overproduction of MutL alone, or with MutS, depresses stationary-phase Lac+ reversion. Plasmids [pControl], [pMutL&MutS], [pMutS], and [pMutL] are pSL4, pSL7, pSL6, and pSL5, respectively (Materials and Methods). Error bars represent one standard error of the mean (s.e.m.) and are smaller than the data point where not visible.
Overproduction of MutL inhibits stationary-phase Lac+ mutation
Data in Figure 1 show that overproduction of MutL mismatch repair protein from a multicopy plasmid depresses stationary-phase Lac reversion by about fourfold relative to that seen with a strain carrying a control plasmid. The results of multiple experiments of this type are compiled in Table 1. The depression is seen when MutL is overproduced either alone or in combination with MutS and is not observed when only MutS is overproduced (Fig. 1B; Table 1). This indicates that MutL is limiting, or stabilizes another protein that is limiting, during stationary-phase mutation. The plasmid producing both MutL and MutS was used in many of the experiments reported here. However, the results in Figure 1B and Table 1 demonstrate that overproduction of MutL alone is sufficient to depress mutation.
Table 1.
MutL overproduction diminishes stationary-phase mutation
| Mismatch repair protein overproduced from plasmid
|
Experiment no.
|
Cumulative no. of Lac+ colonies by day 5 per 108 viable cellsa (mean ± s.e.m.)b
|
Decrease in stationary-phase mutation relative to control plasmid-bearing strain
|
|
|---|---|---|---|---|
| within each experiment
|
average (mean ± s.e.m.)
|
|||
| None (control plasmid) | 1 | 3.3 ± 0.2 | 1 | 1 |
| 2 | 11 ± 2.1 | 1 | ||
| 3 | 19 ± 1.3 | 1 | ||
| 4 | 5.8 ± 1.9 | 1 | ||
| 5 | 89 ± 8.6 | 1 | ||
| 6 | 15 ± 1.2 | 1 | ||
| 7 | 23 ± 2.6 | 1 | ||
| 8 | 18 ± 2.2 | 1 | ||
| 9 | 37 ± 3.6 | 1 | ||
| MutL | 5 | 14 ± 2.0 | 6.4 | 4.0 ± 0.7 |
| 6 | 4.1 ± 0.4 | 3.7 | ||
| 7 | 7.5 ± 0.7 | 3.1 | ||
| 8 | 8.0 ± 0.7 | 2.3 | ||
| 9 | 7.9 ± 0.8 | 4.7 | ||
| MutS | 5 | 56 ± 5.2 | 1.6 | 1.3 ± 0.2 |
| 6 | 11 ± 2.0 | 1.4 | ||
| 7 | 20 ± 2.4 | 1.2 | ||
| 8 | 23 ± 2.3 | 0.8 | ||
| 9 | 22 ± 5.8 | 1.7 | ||
| MutL & MutS | 1 | 1.1 ± 0.2 | 3.0 | 3.9 ± 0.7 |
| 2 | 1.8 ± 0.4 | 6.1 | ||
| 3 | 3.2 ± 0.8 | 5.9 | ||
| 4 | 1.8 ± 0.2 | 3.2 | ||
| 5 | 15 ± 1.7 | 5.9 | ||
| 6 | 3.7 ± 0.5 | 4.1 | ||
| 7 | 19 ± 1.9 | 1.2 | ||
| 9 | 20 ± 2.2 | 1.9 | ||
In each experiment the mean number of Lac+ colonies was determined from 8 to 12 independent cultures of each strain. Jackpots of growth-dependent mutants were excluded from the calculations. Jackpots were defined as cultures containing a number of mutants greater than two standard deviations above the mean number of mutants calculated without that culture.
(s.e.m.) One standard error of the mean.
Overproduction of MutL does not inhibit growth-dependent Lac+ mutation
To assess whether the effect of MutL overproduction on mutation is specific to stationary-phase mutation, the effects of MutL (and MutS) overproduction on growth-dependent reversion of the same lac frameshift allele were assessed in two ways.
First, the mechanism of stationary-phase, but not growth-dependent, Lac+ reversion in these strains requires recombination genes (Harris et al. 1994, 1996; Foster et al. 1996), including functional recA. Thus, one may examine growth-dependent Lac+ reversion in the absence of any contribution of the recombination-dependent mutation mechanism by using a recA null mutant strain. Data in Figure 1A show that the recA cells overproducing MutL (and MutS) display no decrease in recA-independent Lac+ mutation relative to cells carrying the control plasmid.
Second, data in Table 2 (experiments 1–3) show that growth-dependent Lac reversion rates are unaffected by overproducing MutL (and MutS), relative to the mutation rates in strains bearing the control plasmid.
Table 2.
Mismatch repair proteins are not limiting during growth-dependent Lac+ mutation
| Relevant genotypea
|
Number of hours to form a colonyb
|
Experiment
|
Number of independent culturesc
|
Mutation rate (Lac+/cell per generation) ×10−10 c
|
Mean ± s.d.
|
|---|---|---|---|---|---|
| rec+ [pControl] | 52 | 1 | 40 | 3.0 | 8.0 ± 5.6 |
| 56 | 2 | 40 | 7.0 | ||
| 67 | 3 | 40 | 14 | ||
| rec+ [pMutSL] | 71 | 1 | 40 | 11 | 8.9 ± 1.9 |
| 58 | 2 | 39 | 7.2 | ||
| 67 | 3 | 40 | 8.6 | ||
| ΔrecA [pControl] | 64 | 1 | 40 | 3.0 | 2.5 ± 1.4d |
| 55 | 2 | 40 | 0.98 | ||
| 67 | 3 | 40 | 3.6 | ||
| ΔrecA [pMutSL] | 72 | 1 | 32 | 5.8 | 5.4 ± 0.6d |
| 58 | 2 | 40 | 5.8 | ||
| 67 | 3 | 40 | 4.7 | ||
| recD [pControl] | 54 | 4 | 40 | 16 | 19 ± 11e |
| 62 | 5 | 40 | 11 | ||
| 52 | 6 | 40 | 34 | ||
| 52 | 7 | 40 | 13 | ||
| recD [pMutSL] | N.D. | 4 | N.D. | N.D. | 13 ± 2.5e |
| 70 | 5 | 40 | 9.6 | ||
| 61 | 6 | 31 | 14 | ||
| 77 | 7 | 40 | 14 | ||
| recD ΔrecA [pControl] | 54 | 4 | 40 | 6.2 | 5.3 ± 0.7e |
| 58 | 5 | 40 | 4.5 | ||
| 53 | 6 | 40 | 5.1 | ||
| 69 | 7 | 40 | 5.3 | ||
| recD ΔrecA [pMutSL] | 60 | 4 | 40 | 5.7 | 9.6 ± 7.1e |
| 61 | 5 | 40 | 2.8 | ||
| 58 | 6 | 38 | 11 | ||
| 79 | 7 | 40 | 19 |
(N.D.) Not done.
See Materials and Methods for names and constructions of plasmids.
Determined with 10–12 different Lac+ revertants of each genotype (except in experient 4 in which six different revertants were used) as a t50, the time at which half of the colony-forming units have produced visible colonies under experimental conditions.
Growth-dependent Lac+ reversions were measured (Harris et al. 1996) and mutation rates calculated by the method of the median (Lea and Coulson 1949; von Borstel 1978).
The apparent decrease in growth-dependent mutation in recA compared with rec+ is not statistically significant (see standard deviations on data from isogenic recA and rec+ strain pairs). Even with the large variance on these numbers, however, it is clear that there is no mutation-depressing effect of MutL overproduction in these growth-dependent reversions (compare overproducing versus control plasmid in each otherwise isogenic pair) as there is in stationary-phase reverison (Table 1).
The recD strain displays stationary-phase Lac+ hypermutation (Harris et al. 1994) and also appears hypermutable in growth-dependent Lac+ reversion here. The apparent elevation of growth-dependent mutation in the stationary-phase hypermutable recD strain appears to be attributable to spillover of postplating, RecA-dependent stationary-phase revertants into growth-dependent revertant colony counts. This is indicated by the finding that the increase in recD is entirely recA+-dependent (experiments 4–7).
These data appear to contrast with results from a previous study in which co-overproduction of MutS and MutL appeared to inhibit growth-dependent Lac reversion relative to a control plasmid-bearing strain (Foster et al. 1996). However, in that study all growth-dependent mutants were scored after 2 days. We found that cells carrying the MutS and MutL-overproducing plasmid take longer than 2 days to form colonies (Table 2). Therefore we scored the growth-dependent mutants of each strain after an experimentally determined incubation time specific for that strain (Table 2). When controlled for speed of colony formation in this way, no difference in growth-dependent reversion rates is detected between control and overproducing strains (Table 2). These results allow us to infer that mismatch repair protein MutL is limiting specifically during stationary-phase and not growth-dependent Lac reversion.
Mutants that display stationary-phase hypermutation show greater MutL-promoted depression of stationary-phase mutation
The apparent deficiency of functional MutL during stationary-phase Lac reversion (Fig. 1B; Table 1) might be partial rather than absolute. If so, then strains that are hypermutable for stationary-phase Lac mutation by virtue of creating more DNA polymerase errors might reduce mismatch repair protein levels further by titrating away the limiting protein. Because stationary-phase Lac reversion uses recombination functions, whereas growth-dependent reversion does not (Harris et al. 1994, 1996; Foster et al. 1996), this idea can be tested using rec mutants that are hypermutable specifically in stationary-phase Lac reversion. A hyper-recombinagenic and stationary-phase hypermutable recD mutant strain (Harris et al. 1994) was used. The data in Figure 2 show that overexpression of MutL (with MutS) causes a dramatic 15-fold reduction of stationary-phase Lac reversion in this strain, whereas growth-dependent Lac reversion is unaffected (Fig. 2B, recA recD results; Table 2, experiments 4–7).
Figure 2.

Inhibition of stationary-phase Lac+ reversion in a hypermutable recD strain. Plasmids [pControl], and [pMutL&MutS] are pSL4, and pSL7, respectively (Materials and Methods). Error bars as in Fig. 1.
A stationary-phase hypermutable recG strain (Foster et al. 1996; Harris et al. 1996) was affected similarly by overproduction of MutS plus MutL (Foster et al. 1996; data not shown). Foster et al. (1996) postulated a direct protein–protein interaction between RecG and the MutS and MutL proteins. Our results showing a similarly large depression of stationary-phase Lac reversion by overproducing MutL and MutS in a recD strain argue against this interpretation because RecD and RecG act at different stages in recombination, on different DNA intermediates (Rosenberg and Hastings 1991; Kowalczykowski et al. 1994; Myers and Stahl 1994; West 1992, 1994). The data support the idea that the hypermutable recD and recG strains simply provide more recombination intermediates that are hypothesized to prime the DNA synthesis with polymerase errors, which leads to stationary-phase Lac reversion (Harris et al. 1994, 1996; Foster et al. 1996). The increased errors would reduce effective MutL levels further by titration.
This latter interpretation, that MutL mismatch repair protein is only partially limiting during stationary-phase mutation, is consistent with the observation that a mutL-defective strain shows recA-dependent stationary-phase hypermutation (Harris et al. 1997).
An alternative explanation for the observation that overproduction of MutL inhibits stationary-phase but not growth-dependent Lac+ mutation could be that MutL somehow inhibits homologous recombination, which is required for the stationary-phase mutation only (Harris et al. 1994, 1996; Foster et al. 1996). Measurements of the efficiencies of phage P1 transductional recombination showed no such inhibition by the MutL-overproducing plasmid (data not shown), thereby discouraging this alternative.
MutL overproduction does not inhibit mutation by preventing MutS or MutH decline during stationary phase and starvation
We wished to address the possibility that MutL overproduction might inhibit stationary-phase mutation by preventing the reported declines in either MutS or MutH proteins in starving, stationary-phase cells (Feng et al. 1996). Therefore, we used quantitative Western blots to determine the levels of MutL, MutS, and MutH proteins in cells overproducing MutL, MutS, and MutL plus MutS during stationary-phase starvation on lactose medium and during growth.
The Western blots were as performed previously (Feng et al. 1996) with modifications (Materials and Methods; see Fig. 3 for representative blots). Figure 4 summarizes quantification of the amounts of MutL, MutS, and MutH proteins in growing and starved stationary-phase cells carrying the control and overproducing plasmids. The data are from three to five independent experiments for each determination. The results can be summarized as follows.
Figure 3.

Representative quantitative Western immunoblots. Data from multiple experiments of this type are summarized and described in Fig. 4. (A) MutL levels. (Lanes 1–4) Quantification standards (Feng et al. 1996; see Materials and Methods; legend to Fig. 4): 0, 4.5, 9, and 18 ng of His6–MutL protein, respectively; (lanes 5–10) MutL in cultures grown exponentially (9.9 ng) and at days 0 (12.9 ng), 2 (12.5 ng), 4 (12.1 ng), 6 (9.6 ng), and 8 (9.9 ng), respectively. (B) MutS levels. (Lanes 1–5) Quantification standards: 0, 2.5, 5, 10, and 20 ng of His6–MutS protein, respectively; (lanes 6–11) MutS in cultures grown exponentially (15.4 ng) and at days 0 (10.8 ng), 2 (7.7 ng), 4 (6.2 ng), 6 (4.8 ng), and 8 (4.3 ng), respectively. (C) MutH levels. (Lanes 1–4) Quantification standards: 0, 0.25, 0.5, and 1.0 ng of His6–MutH protein, respectively; (lanes 5–10) MutH in cultures grown exponentially (0.78 ng) and at days 0 (0.54 ng), 2 (0.52 ng), 4 (0.44 ng), 6 (0.47 ng), and 8 (0.36 ng), respectively.
Figure 4.

Amounts of MutL, MutS, and MutH proteins in growing, stationary-phase, and starved cells carrying MutL- and MutS-overproducing plasmids. Data are summaries of quantifications from Western blots performed as in Feng et al. (1996; see Materials and Methods). Days 0–8 indicate days after plating the stationary-phase lac− cells on lactose medium (Materials and Methods). (E) Exponential cultures. At least three experiments were performed. Each histogram bar represents the mean (error bars, s.e.m.). (Solid bars) pControl; (open bars) pMutL; (hatched bars) pMutS; (shaded bars) pMutL and pMutS. (A) MutL levels. (B) MutS levels. For MutH, two different sets of experiments are shown. (C) The first was measured in nanograms of MutH protein per 150 μg of total cellular protein. (D) The second set was measured as numbers of MutH monomers per cell [see Materials and Methods for the recalibration of the number of MutH monomers per cell with respect to previous results (Feng et al. 1996). This is discussed further in the text.
First, we see that the plasmids constructed and used to overproduce MutL do so by ∼20- to 30-fold (Fig. 4A, open bars and shaded bars). Those overproducing MutS do so by 60- to 130-fold (Fig. 4B, hatched bars and shaded bars). Less than 10 percent breakdown products were observed in the overproducing strain (data not shown). This demonstration that MutS is overproduced allows us to rule out the possibility that the MutS plasmid did not inhibit stationary-phase mutation (Fig. 1B; Table 1) because of a failure to overproduce MutS protein. We conclude that MutS overproduction does not inhibit stationary-phase Lac reversion.
Second, in strains carrying the control plasmid, we observe declines in MutS and MutH proteins early in stationary phase and on prolonged exposure of the lac− cells to starvation on lactose minimal medium (Fig. 4B–D, solid bars). This is similar to results reported previously with plasmid-free cells (Feng et al. 1996).
Third, we note that overproduction of MutS plus MutL may have a small stabilizing effect on MutH, preventing MutH decline during prolonged starvation (Fig. 4C). This can be assessed only in the experiment measuring MutH as nanograms per micrograms of total cellular protein (Fig. 4C) and not by measuring MutH monomers per cell, because we observe that cells overproducing MutS plus MutL are at least twice as long as normal cells (data not shown). The mechanism of this enlargement is unknown. However, it may suggest an interaction between MutS and MutL and prokaryotic cell cycle control similar to that observed with eukaryotic mismatch repair and eukaryotic cell cycle regulation (Hawn et al. 1995; Anthoney et al. 1996). If the increase in MutH seen in Figure 4C is significant, then MutS plus MutL might make direct contact with MutH such that their overabundance could prevent MutH loss. If, for example, MutH were a target of a stationary phase-specific protease (Gottesman and Maurizi 1992; Miller 1996), contact with MutS plus MutL might protect against such proteolysis. Alternatively, overproduction of MutS and MutL might titrate such a protease directly. Other explanations are possible.
Finally, neither stabilization of MutS, nor significant stabilization of MutH is seen when MutL is overproduced alone (Figs. 4B–D, open bars). For MutH, two different sets of experiments are shown (Fig. 4C,D). The first, measured in nanograms of MutH protein per 150 μg of total cellular protein (Fig. 4C), shows a slight but statistically insignificant trend in prevention of MutH decline by overproducing MutL. The second set is measured as numbers of MutH monomers per cell. This is may be a more relevant measure, as the protein composition of cells changes dramatically in stationary phase, mostly because of loss of ribosomes (Davis et al. 1986; Bremer and Dennis 1987). These data show no significant prevention of MutH loss by MutL overproduction (Fig. 4D, open bars). We conclude that stabilization of MutH does not correlate with the depression of stationary-phase Lac reversion which is seen in cells producing MutL alone (Fig. 1B; Table 1). Thus, the depressing effect of MutL on stationary-phase mutation cannot be explained by stabilization of either MutS or MutH levels during starvation.
Discussion
The results reported here imply that during recombination-dependent, stationary-phase mutation, mismatch repair activity is diminished by a decrease in the level of functional MutL protein. The data also imply that the observed declines in MutS and MutH proteins (Fig. 4B–D; Feng et al. 1996) are proportional to decreased replication during starvation and stationary phase and do not cause a loss of mismatch repair function.
Significance
As far as we know, the results reported here represent the first natural circumstance in which mismatch repair activity has been shown to be modulated, not constitutive. This is significant because of the powerful effect of the mismatch repair system on maintenance of genetic and genomic stability in organisms from bacteria to humans. Cells that lack mismatch repair have 1000-fold higher spontaneous mutation rates (Modrich 1991), recombine sequences of only partial identity (Rayssiguier et al. 1989; Worth et al. 1994; Baker et al. 1995, 1996; de Wind et al. 1995; Matic et al. 1995, 1996; Chambers et al. 1996; Zahrt and Maloy 1997) causing genome rearrangements (Petit et al. 1991), and manifest microsatellite instability and cancer (for review, see Modrich 1994, 1995; Radman et al. 1995).
Mismatch repair deficiency is associated with successful bacterial pathogenesis and with nonpathogenic commensal bacteria of the human gut (LeClerc et al. 1996; Matic et al. 1997), implying that a mutator phenotype is selected in the war between pathogens and the host immune system and in nonpathogenic commensalism. But because the majority of the successful pathogens, natural isolates, and survivors of selection in general (Mao et al. 1997; Sniegowski et al. 1997) were not heritably mismatch repair-defective, these may have succeeded by a transient mismatch repair deficiency.
Mismatch repair also prevents interspecies recombination (Rayssiguier et al. 1989; Matic et al. 1995, 1996; Hunter et al. 1996; Zahrt and Maloy 1997), and its component proteins participate in transcription-coupled repair of damaged DNA (Mellon and Champe 1996) and in very-short-patch repair (Jones et al. 1987a,b; Lieb 1987; Raposa and Fox 1987; Zell and Fritz 1987). Diminished mismatch repair in response to environmental signals would mean that mutation, improper recombination, genome rearrangements, and genetic instability might vary in response to the environment of a cell. This would have profound consequences for evolution, development, microbial pathogenesis, cancer formation, tumor progression, and acquisition of drug resistance in tumors and pathogens.
Mechanism
The lack of functional MutL implied by the data reported here cannot be attributed simply to a decrease in the amount of MutL protein in stationary phase, as MutL levels do not appear to change during stationary phase and starvation (Fig. 4A; Feng et al. 1996). Several explanations for functional MutL deficiency are possible.
First, MutL protein might be modified or processed in stationary phase to a nonfunctional form.
Second, though present in abundant quantity, MutL might become limiting by means of titration by a DNA substrate formed during stationary phase. Creation of such a substrate could be a regulation mechanism. If it occurs, it is demonstrably a natural part of starvation and not artificially induced in this system.
Evidence for reduction of mismatch repair activity by titration was found first in the Hex mismatch repair system of Streptococcus, which can be titrated by DNA substrates during natural transformation (Guild and Shoemaker 1974; Humbert et al. 1995; Tiraby et al. 1975). Mismatch repair in E. coli can also be saturated artificially: by excess polymerase errors of a proofreading-defective mutant DNA polymerase (Schaaper 1988; Damagnez et al. 1989; Schaaper and Radman 1989); by mutagens thought to increase polymerase error (Cupples et al. 1990); or by overproduction of single-stranded DNA with regions of secondary structure containing mismatched bases (Maas et al. 1994, 1996). Thus, DNA substrates can titrate out mismatch repair proteins in E. coli in growing cells. Overproduction of Vsr protein, which interacts with MutL and MutS in very-short-patch DNA repair, can also titrate out mismatch repair activity (Doiron et al. 1996; Macintyre et al. 1997). The limiting proteins titrated out were MutL or MutH (Schaaper and Radman 1989; Macintyre et al. 1997) or MutS alone (Maas et al. 1996). Reduction of MutL by titration is compatible with the results of Schaaper and Radman (1989) and those of Macintyre et al. (1997) but is not obviously so with those of Maas et al. (1996). The titration hypothesis need not conflict with the apparent abundance of MutL and scarcity of MutS and MutH in stationary phase, for MutL may be used as an expendable rather than a catalytic component of the reaction (Schaaper and Radman 1989). If so, spent MutL protein might be visible on Western blots though useless to the cell for mismatch repair. Along these lines, the down-regulation of a protein that rejuvenates used MutL would also be compatible with our results. Finally, although MutS is the known DNA-binding component, its interaction with MutL could allow MutL to be titrated out by DNA substrates.
Third, functional MutL could be reduced by stationary-phase up-regulation of a protein such as Vsr, whose overproduction has been shown to diminish methyl-directed mismatch repair and might titrate MutL directly, rather than via a DNA intermediate (Macintyre et al. 1997).
Fourth, overproduction of MutL might stabilize a mismatch repair protein, other than MutS or MutH, which normally declines in stationary phase.
Fifth, recent results indicate that stationary-phase Lac reversion in the system used here occurs as part of genome-wide hypermutation in a subpopulation of the cells exposed to starvation (Torkelson et al. 1997). The size of the mutagenic subpopulation was estimated to be between 10−4 and 10−5 of all of the cells starved on lactose. Thus, MutL levels might decline only in cells of the subpopulation, which would be undetectable in Western analyses of the whole population. Any of the four previous mechanisms might also occur in the subpopulation or the whole population. Work to distinguish these possible mechanisms is in progress.
Why MutL? MutL is thought to coordinate the mismatch-bound MutS with the MutH endonuclease in a complex of all three proteins (for review, see Modrich 1991). Others have reported saturated mismatch repair that could be restored by MutL or MutH (discussed above; Schaaper and Radman 1989; Macintyre et al. 1997). As we were unable to overexpress MutH in our experiments (data not shown), this is possible in our assay system too. One possibility is that MutH might be a reusable endonuclease but that each reuse might require interaction with a fresh MutL, and MutL might be unable to recycle at all. If so, then overexpression of either protein would restore mismatch repair. Thus, we suggest that MutL is not only a coordinator of MutS with MutH but also a sensitive step at which regulation of the system could occur.
Regulation or random breakdown of mismatch repair in stationary phase?
Is the mechanism of MutL dysfunction a regulated response or, in the case of the fifth hypothesis, might it represent random loss of MutL protein in the subpopulation? One nonrandom aspect of the results is that only MutL (not other mismatch repair proteins) appears to become limiting (though it is possible that MutL or MutH might be limiting as discussed above). Ninio (1991) suggested a random model in which the normal error rates of replication, transcription, and translation should lead to transient and heritable mutator subpopulations, for example, by faulty synthesis of (MutL or) any protein involved in replication fidelity. Ninio’s model predicts frequencies of heritable mutator mutants to be found among cells carrying mutations that are far greater than those observed in this system (Torkelson et al. 1997), suggesting the possibility that the mutator state is not randomly achieved but, rather, is programmed. However, the error rates used in Ninio’s calculations may not apply during starvation.
Whether the loss of MutL function is accidental or programmed, it occurs in response to environmental conditions. Environmental influence over genetic stability could be important for reasons discussed above.
Implications for stationary-phase mutation
Bacteria differentiate and execute specific developmental programs to deal with stationary phase and starvation (Siegele and Kolter 1992; Kolter et al. 1993) during which they generate special mutants with the ability to prevail under limiting conditions (Zambrano and Kolter 1996). The stationary-phase mutation-specific loss of mismatch repair function reported here could be part of a developmental program for generating such mutants.
Stationary-phase reversions of the lac frameshift mutation in the system used here have been shown to result from a genome-wide hypermutable state in a subpopulation of the starved cells (Torkelson et al. 1997). MutL deficiency might or might not be the special feature that makes the subpopulation different. For example, the whole population might be MutL-deficient but only the subpopulation might, for example, perform the recombination necessary for Rec-dependent stationary-phase mutation. The subpopulation might be differentiated by possessing the DNA double-strand breaks that initiate recombination (Harris et al. 1994; Rosenberg 1994, 1997; Rosenberg et al. 1995, 1996).
Also (Torkelson et al. 1997), stationary-phase mutations are found not to be directed, in a Lamarckian manner, to the gene under selection (lac) but, rather, to occur in multiple unselected genes in all replicons in the cell (for evidence of unselected mutation, see also Foster 1997). This supports Darwinian models for stationary-phase mutation that include random mutation followed by selection for the adaptive mutation. However, the implication of the findings reported here—that mutation rates could be altered significantly by decreasing mismatch repair in response to environmental cues—suggests that the cells may generate the variation on which selection acts more vigorously when they “need” to evolve. Where such a mutagenic mode fits in the continuum between Lamarckian and Darwinian mutation models will probably be a subject of continuing discussion. Down-regulation of mismatch repair could provide a molecular mechanism for achieving rapid genetic change when selection is present. On the microscopic scale of changes in bacterial genotype, this could contribute to mechanisms producing the punctuations in punctuated equilibria (Eldredge and Gould 1972).
Materials and methods
Plasmids
Plasmids were modified from pBR322 (Bolivar et al. 1977) to carry kanamycin resistance (KanR) (pSL4) and the following E. coli genes regulated by their natural promoters: mutL (pSL5); mutS (pSL6); and mutS and mutL (pSL7). These genes are overexpressed because of the high copy number of pBR322. In pSL4 the HindIII–BamHI fragment of pBR322 is replaced by the KanR-conferring HindIII–BamHI fragment of pKC31 (R.N. Rao, described by, e.g., Rosenberg 1988). pSL5 contains a PstI–HindIII fragment of pAL51 carrying E. coli mutL (Lu et al. 1984) and the HindIII–BamHI fragment of pKC31 replacing the PstI–BamHI fragment of pBR322. In pSL6 the pBR322 ClaI–BamHI fragment is replaced by the mutS-containing ClaI–HindIII fragment of pMS312 (Su and Modrich 1986) and the HindIII–BamHI fragment of pKC31. In pSL7 the ClaI–PstI fragment of pBR322 is replaced by the ClaI–BglII fragment of pSL6, the BamHI–HindIII fragment of pKC31, and the HindIII–PstI fragment of pAL51. All plasmid genotypes were confirmed by restriction mapping and by complementation of the mutator phenotype (assayed as by Longerich et al. 1995; Torkelson et al. 1997) of mutS and/or mutL-defective E. coli strains.
Lac+ mutation assays
For Lac+ frameshift reversion studies, derivatives of a strain carrying the lacI33 allele (Cairns and Foster 1991) and carrying the plasmids described above were used. This strain is deleted for the chromosomal lac operon and bears an F′ episome carrying a lacI–lacZ fusion gene with a +1 frameshift mutation in lacI that is polar on lacZ (Cairns and Foster 1991). Procedures for measurement of stationary-phase Lac+ mutation and for measurements of growth-dependent Lac+ mutation rates were modified from Harris et al. (1996) as follows: Kanamycin (at 50 μg/ml) was included in the minimal glycerol broth, and 5 μg/ml of kanamycin in the minimal lactose plates and top agar. The scavenger cell strain is as before except that it carries the KanR-conferring control plasmid pSL4.
Western analyses
For measuring protein levels during starvation on lactose medium, it is important that Lac+ revertants do not accumulate in the population assayed. Thus, the strain background analyzed was FC29 (Cairns and Foster 1991), which is isogenic to the lac frameshift-bearing strain used except for the absence of the lac operon. The absence of this operon makes it nonrevertible. Derivatives of this strain carrying the plasmids described here were used. Western analyses were as described (Feng et al. 1996) with the following changes. (1) Bacteria were grown in media as described above for Lac reversion studies, washed, and spread on minimal lactose plates with 5 μg/ml of kanamycin plates as described above, but with no top agar. Plates were incubated for 8 days at 37°C. Every second day cells were washed off 10 plates with M9 salts and protein samples were prepared. Exponential and day 0 samples were prepared using liquid cultures at a density of about 30 Klett units, and the saturated culture, respectively. (2) MutS and MutH antisera were affinity-purified: One-milliliter columns containing ∼2 mg of His6–MutS or His6–MutH (Feng and Winkler 1995) coupled to CNBr-activated Sepharose 4B (Pharmacia Biotech, Uppsala, Sweden), prepared according to instructions (Pharmacia), were washed sequentially with 15 ml of 6 m guanidine HCl, 25 ml of buffer A (50 mm Tris-HCl at pH 7.4), 25 ml of Buffer B (buffer A + 4.5 m MgCl2 and 1.0 mg/ml of BSA), and equilibrated with 50 ml of buffer A. Ten milliliters of antisera were run through the columns, which were washed with 40 ml of 1 m guanidine-HCl and 20 ml of buffer A and eluted with 10 ml of buffer B. Elutions were dialyzed against 3 liters of PBS, and 3 liters PBS plus 35% glycerol. MutL antiserum was a gift from P. Modrich (Duke University, Durham, NC).
The absolute amount of MutH per cell has been recalibrated with respect to previous experiments (Feng et al. 1996) based on our finding that the standard MutH protein preparation to which calibration was performed previously had aggregated when the His6 affinity tag was cleaved off MutH. This aggregation was not detected until later experiments in which large amounts of standard were prepared. The new estimate for MutH in cells growing exponentially in enriched minimal salts glucose (EMMG) medium (Feng et al. 1996) is 34 ± 7 monomers per cell.
Acknowledgments
We are grateful to P. Modrich for gifts of plasmids and for his generous gift of MutL polyclonal antibody. We thank T. Palzkill for providing protocols for antisera affinity purification, C. Cupples, F. Taddei, and M. Radman for helpful discussion, and P.J. Hastings, M.-J. Lombardo, and J. Torkelson for comments on the manuscript. R.S.H. and S.L. were enrolled in the graduate degree program of the Department of Biological Sciences University of Alberta. This work was supported by grant NP-926 from the American Cancer Society to M.E.W., a National Institutes of Health grant to S.M.R., a grant from the National Cancer Institute of Canada with funds provided by the Canadian Cancer Society (to S.M.R.), an Alberta Heritage Foundation for Medical Research graduate studentship (R.S.H.), and an honorary Izaac Walton Killam fellowship (R.S.H.). S.M.R. was a Medical Research Council of Canada Scientist and an Alberta Heritage Senior Medical Scholar.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL smr@bcm.tmc.edu; FAX: (713) 798-5386.
References
- Anthoney DA, McIlwrath AJ, Gallagher WM, Edlin ARM, Brown R. Microsatellite instability, apoptosis, and loss of p53 function in drug-resistant tumor cells. Cancer Res. 1996;56:1374–1381. [PubMed] [Google Scholar]
- Baker SM, Bronner CE, Zhang L, Plug AW, Robatzek M, Warren G, Elliott EA, Yu J, Ashley T, Arnheim N, Flavell RA, Liskay RM. Male mice defective in DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell. 1995;82:309–319. doi: 10.1016/0092-8674(95)90318-6. [DOI] [PubMed] [Google Scholar]
- Baker SM, Plug AW, Prolla TA, Bronner CE, Harris AC, Yao X, Christie D-M, Monell C, Arnheim N, Bradley A, Ashley T, Liskay RM. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nature Genet. 1996;13:336–342. doi: 10.1038/ng0796-336. [DOI] [PubMed] [Google Scholar]
- Bolivar F, Rodriguez RL, Greene PJ, Betlach MC, Heyneker HL, Boyer HW, Crosa JH, Falkow S. Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. Gene. 1977;2:95–113. [PubMed] [Google Scholar]
- Bremer H, Dennis PP. Modulation of chemical composition and other parameters of the cell by growth rate. In: Neidhardt FC, Ingraham JL, Low KB, Magazanik B, Schaecter M, Umbarger E, editors. Escherichia coli and Salmonella typhimurium: Cellular and molecular biology. Washington, D.C.: American Society for Microbiology; 1987. pp. 1527–1542. [Google Scholar]
- Cairns J, Foster PL. Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics. 1991;128:695–701. doi: 10.1093/genetics/128.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SR, Hunter N, Louis EJ, Borts RH. The mismatch repair system reduces meiotic homeologous recombination and stimulates recombination-dependent chromosome loss. Mol Cell Biol. 1996;16:6110–6120. doi: 10.1128/mcb.16.11.6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupples CG, Cabrera M, Cruz C, Miller JH. A set of lacZ mutations in Escherichia coli that allow rapid detection of specific frameshift mutations. Genetics. 1990;125:275–280. doi: 10.1093/genetics/125.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damagnez V, Doutriaux M-P, Radman M. Saturation of mismatch repair in the mutD5 mutator strain of Escherichia coli. J Bacteriol. 1989;171:4494–4497. doi: 10.1128/jb.171.8.4494-4497.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta A, Adjiri A, New L, Crouse GF, Jinks-Robertson S. Mitotic crossovers between diverged sequences are regulated by mismatch repair proteins in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:1085–1093. doi: 10.1128/mcb.16.3.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BD, Luger SM, Tai PC. Role of ribosome degradation in the death of starved Escherichia coli cells. J Bacteriol. 1986;166:439–445. doi: 10.1128/jb.166.2.439-445.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wind N, Dekker M, Berns A, Radman M, te Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82:321–330. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- Doiron KM, Viau S, Koutroumanis M, Cupples CG. Overexpression of vsr in Escherichia coli is mutagenic. J Bacteriol. 1996;178:4294–4296. doi: 10.1128/jb.178.14.4294-4296.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldredge N, Gould SJ. Punctuated equilibria: An alternative to phyletic gradualism. In: Schopf TJM, editor. Models in paleobiology. San Francisco, CA: Freeman, Cooper and Co.; 1972. pp. 82–115. [Google Scholar]
- Feng G, Winkler ME. Single-step purifications of His6-MutH, His6-MutL and His6-MutS repair proteins of Escherichia coli K-12. BioTechniques. 1995;19:956–965. [PubMed] [Google Scholar]
- Feng G, Tsui H-CT, Winkler ME. Depletion of the cellular amounts of the MutS and MutH methyl-directed mismatch repair proteins in stationary-phase Escherichia coli K-12 cells. J Bacteriol. 1996;178:2388–2396. doi: 10.1128/jb.178.8.2388-2396.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. Adaptive mutation: The uses of adversity. Annu Rev Microbiol. 1993;47:467–504. doi: 10.1146/annurev.mi.47.100193.002343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Nonadaptive mutations occur in the F′ episome during adaptive mutation conditions in Escherichia coli. J Bacteriol. 1997;179:1550–1554. doi: 10.1128/jb.179.5.1550-1554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Trimarchi JM. Adaptive reversion of a frameshift mutation in Escherichia coli by simple base deletions in homopolymeric runs. Science. 1994;265:407–409. doi: 10.1126/science.8023164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Adaptive reversion of an episomal frameshift mutation in Escherichia coli requires conjugal functions but not actual conjugation. Proc Natl Acad Sci. 1995;92:5487–5490. doi: 10.1073/pnas.92.12.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Trimarchi JM, Maurer RA. Two enzymes, both of which process recombination intermediates, have opposite effects on adaptive mutation in Escherichia coli. Genetics. 1996;142:25–37. doi: 10.1093/genetics/142.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galitski T, Roth JR. Evidence that F′ transfer replication underlies apparent adaptive mutation. Science. 1995;268:421–423. doi: 10.1126/science.7716546. [DOI] [PubMed] [Google Scholar]
- ————— A search for a general phenomenon of adaptive mutability. Genetics. 1996;143:645–659. doi: 10.1093/genetics/143.2.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman S, Maurizi MR. Regulation of proteolysis: Energy-dependent proteases and their targets. Microbiol Rev. 1992;56:592–621. doi: 10.1128/mr.56.4.592-621.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guild WR, Shoemaker NB. Intracellular competition for a mismatch recognition system and marker-specific rescue of transforming DNA from inactivation by ultraviolet irradiation. Mol & Gen Genet. 1974;128:291–300. doi: 10.1007/BF00268517. [DOI] [PubMed] [Google Scholar]
- Hall BG. Genetics of selection-induced mutations: I. uvrA, uvrB, uvrC, and uvrD are selection-induced specific mutator loci. J Mol Evol. 1995;40:86–93. doi: 10.1007/BF00166599. [DOI] [PubMed] [Google Scholar]
- Harris RS, Longerich S, Rosenberg SM. Recombination in adaptive mutation. Science. 1994;264:258–260. doi: 10.1126/science.8146657. [DOI] [PubMed] [Google Scholar]
- Harris RS, Ross KJ, Rosenberg SM. Opposing roles of the Holliday junction processing systems of Escherichia coli in recombination-dependent adaptive mutation. Genetics. 1996;142:681–691. doi: 10.1093/genetics/142.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Bull HJ, Rosenberg SM. A direct role for DNA polymerase III in adaptive reversion of a frameshift mutation in Escherichia coli. Mutat Res. 1997;375:19–24. doi: 10.1016/s0027-5107(96)00244-8. [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Rosenberg SM. Gene conversion. In: Roitt IM, Delves PJ, editors. Encyclopedia of immunology. London, UK: Academic Press; 1992. pp. 602–605. [Google Scholar]
- Hawn MT, Umar A, Carretthers JM, Marra G, Kunkel TA, Bowland RC, Koi M. Evidence for a connection between the mismatch repair system and the G2 cell cycle checkpoint. Cancer Res. 1995;55:3721–3725. [PubMed] [Google Scholar]
- Humbert O, Prudhomme M, Hakenbeck R, Dowson CG, Claverys J-P. Homeologous recombination and mismatch repair during transformation in Streptococcus pneumoniae: Saturation of the Hex mismatch repair system. Proc Natl Acad Sci. 1995;92:9052–9056. doi: 10.1073/pnas.92.20.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter N, Chambers SR, Louis EJ, Borts RH. The mismatch repair system contributes to meiotic sterility in an interspecific yeast hybrid. EMBO J. 1996;15:1726–1733. [PMC free article] [PubMed] [Google Scholar]
- Jones M, Wagner R, Radman M. Mismatch repair and recombination in E. coli. Cell. 1987a;50:621–626. doi: 10.1016/0092-8674(87)90035-3. [DOI] [PubMed] [Google Scholar]
- ————— Mismatch repair of deaminated 5-methyl-cytosine. J Mol Biol. 1987b;193:155–159. doi: 10.1016/0022-2836(87)90724-8. [DOI] [PubMed] [Google Scholar]
- Kolodner R. Biochemistry and genetics of eukaryotic mismatch repair. Genes & Dev. 1996;10:1433–1442. doi: 10.1101/gad.10.12.1433. [DOI] [PubMed] [Google Scholar]
- Kolter R, Siegele DA, Tormo A. The stationary phase of the bacterial life cycle. Annu Rev Microbiol. 1993;47:855–874. doi: 10.1146/annurev.mi.47.100193.004231. [DOI] [PubMed] [Google Scholar]
- Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM. Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev. 1994;58:401–465. doi: 10.1128/mr.58.3.401-465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A. Collapse and repair of replication forks in Escherichia coli. Mol Microbiol. 1995;16:373–384. doi: 10.1111/j.1365-2958.1995.tb02403.x. [DOI] [PubMed] [Google Scholar]
- Lea DE, Coulson CA. The distribution of the numbers of mutants in bacterial populations. J Genet. 1949;49:264–285. doi: 10.1007/BF02986080. [DOI] [PubMed] [Google Scholar]
- LeClerc JE, Li B, Payne WL, Cebula TA. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science. 1996;274:1208–1211. doi: 10.1126/science.274.5290.1208. [DOI] [PubMed] [Google Scholar]
- Lieb M. Bacterial genes mutS, mutL, and dcm participate in repair of mismatches at 5-methylcytosine sites. J Bacteriol. 1987;169:5241–5246. doi: 10.1128/jb.169.11.5241-5246.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb M, Shehnaz R. Very short patch repair of T:G mismatches in vivo: Importance of context and accessory proteins. J Bacteriol. 1995;177:660–666. doi: 10.1128/jb.177.3.660-666.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longerich S, Galloway AM, Harris RS, Wong C, Rosenberg SM. Adaptive mutation sequences reproduced by mismatch repair deficiency. Proc Natl Acad Sci. 1995;92:12017–12020. doi: 10.1073/pnas.92.26.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A-L, Welsh K, Clark S, Su S-S, Modrich P. Repair of DNA base-pair mismatches in extracts of Escherichia coli. Cold Spring Harbor Symp Quant Biol. 1984;49:589–596. doi: 10.1101/sqb.1984.049.01.066. [DOI] [PubMed] [Google Scholar]
- Maas WK, Wang C, Lima T, Zubay G, Lim D. Multicopy single-stranded DNAs with mismatched base pairs are mutagenic in Escherichia coli protein. Mol Microbiol. 1994;14:437–441. doi: 10.1111/j.1365-2958.1994.tb02178.x. [DOI] [PubMed] [Google Scholar]
- Maas WK, Wang C, Lima T, Hach A, Lim D. Multicopy single-stranded DNA of Escherichia coli enhances mutation and recombination frequencies by titrating MutS protein. Mol Microbiol. 1996;19:505–509. doi: 10.1046/j.1365-2958.1996.392921.x. [DOI] [PubMed] [Google Scholar]
- Macintyre, G., K.M.J. Doiron, and C.G. Cupples. 1997. The Vsr endonuclease of Escherichia coli: An efficient DNA repair enzyme and a potent mutagen. J. Bacteriol. (in press). [DOI] [PMC free article] [PubMed]
- Mao EF, Lane L, Lee J, Miller JH. Proliferation of mutators in a cell population. J Bacteriol. 1997;179:417–422. doi: 10.1128/jb.179.2.417-422.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matic I, Rayssiguier C, Radman M. Interspecies gene exchange in bacteria: The role of SOS and mismatch repair systems in evolution of species. Cell. 1995;80:507–515. doi: 10.1016/0092-8674(95)90501-4. [DOI] [PubMed] [Google Scholar]
- Matic I, Taddei F, Radman M. Genetic barriers among bacteria. Trends Microbiol. 1996;4:69–73. doi: 10.1016/0966-842X(96)81514-9. [DOI] [PubMed] [Google Scholar]
- Matic, I., M. Radman, F. Taddei, B. Picard, C. Doit, E. Bingen, E. Denamur, and J. Elion. 1997. High polymorphism of mutation rates in commensal and pathogenic Escherichia coli natural isolates. Science (in press). [DOI] [PubMed]
- Mello JA, Acharya S, Fishel R, Essigmann JM. The mismatch repair protein hMSH2 binds selectively to DNA adducts of the anticancer drug cisplatin. Chem Biol. 1996;3:579–589. doi: 10.1016/s1074-5521(96)90149-0. [DOI] [PubMed] [Google Scholar]
- Mellon I, Champe GN. Products of DNA mismatch repair genes mutS and mutL are required for transcription-coupled nucleotide-excision repair of the lactose operon in Escherichia coli. Proc Natl Acad Sci. 1996;93:1292–1297. doi: 10.1073/pnas.93.3.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CG. Protein degradation and proteolytic modification. In: Neidhardt FC, editor. Escherichia coli and Salmonella cellular and molecular biology. Washington, D.C.: ASM Press; 1996. pp. 938–954. [Google Scholar]
- Modrich P. Mechanisms and biological effects of mismatch repair. Annu Rev Genet. 1991;25:229–253. doi: 10.1146/annurev.ge.25.120191.001305. [DOI] [PubMed] [Google Scholar]
- ————— Mismatch repair, genetic stability, and cancer. Science. 1994;266:1959–1960. doi: 10.1126/science.7801122. [DOI] [PubMed] [Google Scholar]
- ————— Mismatch repair, genetic stability and tumour avoidance. Phil Trans R Soc London B. 1995;347:89–95. doi: 10.1098/rstb.1995.0014. [DOI] [PubMed] [Google Scholar]
- Myers RS, Stahl FW. χ and RecBCD enzyme of Escherichia coli. Annu Rev Genet. 1994;28:49–70. doi: 10.1146/annurev.ge.28.120194.000405. [DOI] [PubMed] [Google Scholar]
- Ninio J. Transient mutators: A semiquantitative analysis of the influence of translation and transcription errors on mutation rates. Genetics. 1991;129:957–962. doi: 10.1093/genetics/129.3.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit M-A, Dimpfl J, Radman M, Echols H. Control of large chromosomal deletions in Escherichia coli by the mismatch repair system. Genetics. 1991;129:327–332. doi: 10.1093/genetics/129.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radicella JP, Park PU, Fox MS. Adaptive mutation in Escherichia coli: A role for conjugation. Science. 1995;268:418–420. doi: 10.1126/science.7716545. [DOI] [PubMed] [Google Scholar]
- Radman M. Mismatch repair and genetic recombination. In: Kucherlapati R, Smith GR, editors. Genetic recombination. Washington, D.C.: American Society for Microbiology; 1988. pp. 169–192. [Google Scholar]
- Radman M, Wagner R. Mismatch recognition in chromosomal interactions and speciation. Chromosoma. 1993;102:369–373. doi: 10.1007/BF00360400. [DOI] [PubMed] [Google Scholar]
- Radman M, Matic I, Halliday JA, Taddei F. Editing DNA replication and recombination by mismatch repair: From bacterial genetics to mechanisms of predisposition to cancer in humans. Phil Trans R Soc London. 1995;347:97–103. doi: 10.1098/rstb.1995.0015. [DOI] [PubMed] [Google Scholar]
- Raposa S, Fox MS. Some features of base pair mismatch and heterology repair in Escherichia coli. Genetics. 1987;117:381–390. doi: 10.1093/genetics/117.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayssiguier C, Thaler DS, Radman M. The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch repair mutants. Nature. 1989;342:396–401. doi: 10.1038/342396a0. [DOI] [PubMed] [Google Scholar]
- Reenan RAG, Kolodner R. Isolation and characterization of two Saccharomyces cerevisiae genes encoding homologs of the bacterial HexA and MutS mismatch repair proteins. Genetics. 1992;132:963–973. doi: 10.1093/genetics/132.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SM. Chain-bias of Escherichia coli Rec-mediated λ patch recombinants is independent of the orientation of λ cos. Genetics. 1988;120:7–21. doi: 10.1093/genetics/120.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— In pursuit of a molecular mechanism for adaptive mutation. Genome. 1994;37:893–899. doi: 10.1139/g94-127. [DOI] [PubMed] [Google Scholar]
- ———. 1997. Mutation for survival. Curr. Opinion Genet. Dev. (in press). [DOI] [PubMed]
- Rosenberg SM, Hastings PJ. The split-end model for homologous recombination at double-strand breaks and at Chi. Biochimie. 1991;73:385–397. doi: 10.1016/0300-9084(91)90105-a. [DOI] [PubMed] [Google Scholar]
- Rosenberg SM, Longerich S, Gee P, Harris RS. Adaptive mutation by deletions in small mononucleotide repeats. Science. 1994;265:405–407. doi: 10.1126/science.8023163. [DOI] [PubMed] [Google Scholar]
- Rosenberg SM, Harris RS, Torkelson J. Molecular handles on adaptive mutation. Mol Microbiol. 1995;18:185–189. doi: 10.1111/j.1365-2958.1995.mmi_18020185.x. [DOI] [PubMed] [Google Scholar]
- Rosenberg SM, Harris RS, Longerich S, Galloway AM. Recombination-dependent mutation in non-dividing cells. Mutat Res. 1996;350:69–76. doi: 10.1016/0027-5107(95)00092-5. [DOI] [PubMed] [Google Scholar]
- Schaaper RM. Mechanism of mutagenesis in the Escherichia coli mutator mutD5: Role of DNA mismatch repair. Proc Natl Acad Sci. 1988;85:8126–8130. doi: 10.1073/pnas.85.21.8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaper RM, Radman M. The extreme mutator effect of Escherichia coli mutD5 results from saturation of mismatch repair by excessive DNA replication errors. EMBO J. 1989;8:3511–3516. doi: 10.1002/j.1460-2075.1989.tb08516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegele DA, Kolter R. Life after log. J Bacteriol. 1992;174:345–348. doi: 10.1128/jb.174.2.345-348.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski PD, Gerrish PJ, Lenski RE. Evolution of high mutation rates in experimental populations of E. coli. Nature. 1997;387:703–705. doi: 10.1038/42701. [DOI] [PubMed] [Google Scholar]
- Stahl FW. A unicorn in the garden. Nature. 1988;335:112–113. doi: 10.1038/335112a0. [DOI] [PubMed] [Google Scholar]
- Su S-S, Modrich P. Escherichia coli mutS-encoded protein binds to mismatched DNA base pairs. Proc Natl Acad Sci. 1986;83:5057–5061. doi: 10.1073/pnas.83.14.5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei F, Matic I, Radman M. cAMP-dependent SOS induction and mutagenesis in resting bacterial populations. Proc Natl Acad Sci. 1995;92:11736–11740. doi: 10.1073/pnas.92.25.11736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiraby G, Fox MS, Bernheimer H. Marker discrimination in deoxyribonucleic acid transformation of Pneumococcus strains. J Bacteriol. 1975;121:608–618. doi: 10.1128/jb.121.2.608-618.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torkelson J, Harris RS, Lombardo M-J, Nagendran J, Thulin C, Rosenberg SM. Genome-wide hypermutation in a subpopulation of stationary-phase cells underlies recombination-dependent adaptive mutation. EMBO J. 1997;16:3303–3311. doi: 10.1093/emboj/16.11.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Borstel RC. Measuring spontaneous mutation rates in yeast. Methods Cell Biol. 1978;20:1–24. doi: 10.1016/s0091-679x(08)62005-1. [DOI] [PubMed] [Google Scholar]
- West SC. Enzymes and molecular mechanisms of genetic recombination. Annu Rev Biochem. 1992;61:603–640. doi: 10.1146/annurev.bi.61.070192.003131. [DOI] [PubMed] [Google Scholar]
- ————— The processing of recombination intermediates: Mechanistic insights from studies of bacterial proteins. Cell. 1994;76:9–15. doi: 10.1016/0092-8674(94)90168-6. [DOI] [PubMed] [Google Scholar]
- Worth L, Jr, Clark S, Radman M, Modrich P. Mismatch repair proteins MutS and MutL inhibit RecA-catalyzed strand transfer between diverged DNAs. Proc Natl Acad Sci. 1994;91:3238–3241. doi: 10.1073/pnas.91.8.3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahrt, T.C. and S. Maloy. 1997. Barriers to recombination between closely related bacteria: MutS and RecBCD inhibit recombination between Salmonella typhimurium and Salmonella typhi. Proc. Natl. Acad. Sci.. (in press). [DOI] [PMC free article] [PubMed]
- Zambrano MM, Kolter R. GASPing for life in stationary phase. Cell. 1996;86:181–184. doi: 10.1016/s0092-8674(00)80089-6. [DOI] [PubMed] [Google Scholar]
- Zell R, Fritz H. DNA mismatch-repair in Escherichia coli counteracting the hydrolytic deamination of 5-methyl-cytosine residues. EMBO J. 1987;6:1809–1815. doi: 10.1002/j.1460-2075.1987.tb02435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]