

Abstract
Mice lacking the gene encoding poly(ADP-ribosyl) transferase (PARP or ADPRT) display no phenotypic abnormalities, although aged mice are susceptible to epidermal hyperplasia and obesity in a mixed genetic background. Whereas embryonic fibroblasts lacking PARP exhibit normal DNA excision repair, they grow more slowly in vitro. Here we investigated the putative roles of PARP in cell proliferation, cell death, radiosensitivity, and DNA recombination, as well as chromosomal stability. We show that the proliferation deficiency in vitro and in vivo is most likely caused by a hypersensitive response to environmental stress. Although PARP is specifically cleaved during apoptosis, cells lacking this molecule apoptosed normally in response to treatment with anti-Fas, tumor neurosis factor α, γ-irradiation, and dexamethasone, indicating that PARP is dispensable in apoptosis and that PARP−/− thymocytes are not hypersensitive to ionizing radiation. Furthermore, the capacity of mutant cells to carry out immunoglobulin class switching and V(D)J recombination is normal. Finally, primary PARP mutant fibroblasts and splenocytes exhibited an elevated frequency of spontaneous sister chromatid exchanges and elevated micronuclei formation after treatment with genotoxic agents, establishing an important role for PARP in the maintenance of genomic integrity.
Keywords: PARP inactivation, aggregation of embryos, stress response, apoptosis, recombination, sister chromatid exchange
NAD+:protein (ADP-ribosyl)transferase (polymerizing) (PARP or ADPRT, EC2.4.2.30) is an abundant, chromatin-associated protein, which is highly conserved and present in most eukaryotes except yeast. In response to DNA damage caused by environmental genotoxic agents and endogenous cellular reactions, PARP binds rapidly to DNA strand breaks and catalyzes the transfer of ADP-ribose from its substrate NAD+ to a number of nuclear acceptors, mainly to itself (for review, see Althaus and Richter 1987; de Murcia and Menessier-de Murcia 1994). Poly(ADP-ribosyl)ation is thought to play a multifunctional role in numerous cellular processes including proliferation and replication, stress response, cell toxicity and apoptosis, DNA repair and recombination, as well as the maintenance of chromosomal stability (for review, see Lindahl et al. 1995; Shall 1995).
Given the characteristic feature of the binding of PARP to DNA ends in response to DNA damage, much effort has been made to understand the role of PARP in cell death and apoptosis. Cells treated with inhibitors of PARP have been reported to become resistant to various genotoxic agents, such as UVB- and γ-irradiation, tumor necrosis factor (TNF), alkylating agents, and free radicals (Malorni et al. 1995; McGowan et al. 1996). In contrast, other studies showed that inhibition or a dominant-negative mutation of PARP sensitizes cells to cell death induced by oxidative stress, alkylating agents, γ-irradiation, or heat shock (Nosseri et al. 1994; Küpper et al. 1995; Schreiber et al. 1995; Shah et al. 1996). Although results obtained from these studies are contradictory and the mechanisms involved in such processes are complex, the function of PARP is well documented in free radical-induced cell toxicity in neurons and pancreatic islet cells (Zhang et al. 1994; Heller et al. 1995).
The association of PARP function with cell death is inferred from recent studies demonstrating that PARP is rapidly and specifically cleaved during apoptosis (Lazebnik et al. 1994; Nicholson et al. 1995; Tewari et al. 1995). Apoptosis is a highly conserved mechanism that is controlled by a hierarchical set of genes including mammalian homologs of the Caenorhabditis elegans genes, such as ced-3, ced-4, and ced-9. ced-3 is related to a family of mammalian cysteine proteases, called interleukin-1β-converting enzyme (ICE)-related proteases or caspases, that have been identified recently as the central players of apoptosis. PARP has been proposed to act as a critical death substrate, because it is rapidly cleaved by caspase-3/CPP32/Yama/apopain, a mammalian ICE-related protease involved in apoptosis (Nicholson et al. 1995; Tewari et al. 1995). Following induction of apoptosis by TNF or agonistic anti-Fas antibodies, CPP32 is rapidly activated, resulting in PARP cleavage into 85- and 29-kD polypeptides (Enari et al. 1995; Los et al. 1995; Nicholson et al. 1995; Tewari et al. 1995). CPP32 activation and subsequent PARP cleavage have also been observed during cell death induced by various other apoptotic stimuli, such as chemotherapeutic drugs, γ-irradiation, and viral infection (for review, see Patel et al. 1996; Nagata 1997). However, the physiological relevance and functional consequences of PARP and its cleavage in apoptosis have not yet been addressed.
Numerous studies suggest a role for PARP in DNA repair and recombination as well as the maintenance of genomic stability. Poly(ADP-ribosyl)ation modifies various nuclear proteins, including histones, lamins, topoisomerases, and DNA polymerases (see Althaus and Richter 1987). Upon genotoxic damage, PARP is activated and binds to interrupted DNA strands, and following subsequent automodification, PARP is released from DNA. This event permits access for other components of the DNA repair machinery and thereby may prevent unwanted recombination (Satoh and Lindahl 1992; for review, see also Lindahl et al. 1995; Shall 1995). A role for PARP in DNA end binding and rejoining has been demonstrated by studies in which cells treated with PARP inhibitors display an impaired ability to rejoin DNA strand breaks (Ding et al. 1992) and to integrate foreign DNA into their genome (Farzaneh et al. 1988; Gäken et al. 1996). The same treatment, however, facilitates immunoglobulin class switching in B lymphocytes (Shockett and Stavnezer 1993). In addition, inhibition of poly(ADP-ribose) synthesis, by either enzyme inhibitors or a dominant-negative mutation, results in an increase in recombination frequency and genomic instability (Waldman and Waldman 1991; Schreiber et al. 1995). Further support for a role of PARP in maintaining genomic stability stems from studies using 3-AB, which increases micronuclei formation in response to various DNA damaging agents (Catena et al. 1994; Steirum et al. 1995).
To define the biological function of PARP, we generated mice lacking the molecule using homologous recombination (Wang et al. 1995). Our previous study showed that although young PARP−/− mice display no phenotypic abnormalities, older mice originating from a mixed genetic background (129/Sv × C57BL/6) are susceptible to epidermal hyperplasia and obesity (Wang et al. 1995). Although embryonic fibroblasts isolated from mutant mice exhibit normal excision repair of damaged DNA following treatment with UV irradiation and alkylating agents, they proliferate more slowly in vitro than wild-type controls. In the present study we further investigated the function of PARP in stress response, apoptosis, and DNA recombination, as well as genomic stability.
Results
PARP depletion results in impaired cell proliferation in vitro and in vivo
We reported previously that primary wild-type and heterozygous (PARP+/−) cells exhibit no difference in their proliferation and that homozygous mutant (−/−) fibroblasts grow more slowly than their wild-type controls, although all three genotypes are morphologically indistinguishable (Wang et al. 1995). Cell cycle analysis revealed a normal distribution of cells in different cell cycle phases and showed no differences in cell cycle entry and progression (data not shown; Agarwal et al. 1997). Because PARP has been shown to play a role in stress responses, we reasoned that this proliferation deficiency might be attributable to an enhanced stress response to the in vitro culture. Cells were challenged in culture by increasing temperature from 37°C to 39°C. When primary embryonic fibroblasts from the three genotypes were cultured at 37°C, PARP mutant cells grew more slowly compared with wild-type (not shown) and heterozygous (+/−) controls (Fig. 1A). Raising the culture temperature to 39°C resulted in slower cell proliferation in all genotypes, but the rate of growth reduction was more pronounced in PARP−/− fibroblasts than in normal controls (Fig. 1B). To investigate whether the apparent growth deficiency of PARP−/− fibroblasts was attributable to a lack of soluble factors and thus could be rescued by factors secreted from wild-type cells, we mixed wild-type and mutant primary fibroblasts at passage 2 at 1:1 ratio and cultured them at 39°C. The proliferation capacity of each genotype was monitored by genotyping genomic DNA extracted from the co-culture mixtures at different passages. The ratio of PARP−/− to wild-type cells decreased over the course of the culture as depicted by the intensity of the bands on a Southern blot (Fig. 1C). These results indicate that mutant fibroblasts are hypersensitive to heat stress and that this defect cannot be corrected by factors secreted from or by contact with wild-type cells.
Figure 1.



Proliferation of PARP mutant cells in response to experimental stress. (A) PARP heterozygous (+/−) and homozygous (−/−) primary fibroblasts were cultured at 37°C and 39°C for 14 days. At each time point the number of cells was counted from triplicate culture. (B) The ratio of number of cells at 37°C vs. 39°C indicates the magnitude of proliferation reduction of each genotype. (C) Southern blot analysis of genomic DNA extracted from the co-cultures of PARP−/− (mutant) and wild-type cells at different passages from passage 2 (P2) through 7 (P7). DNA was digested by PvuII and hybridized with a specific probe (see Materials and Methods). Control DNAs (Co.) are from wild-type (+/+), heterozygous (+/−) and homozygous (−/−) mouse tail biopsies respectively. The presence of mutant cells in cocultured mixtures is depicted by the intensity of the bands on the Southern blot.
Although PARP−/− fibroblasts proliferate more slowly in culture, mutant embryos develop normally and show no difference in body size compared with wild-type and heterozygous littermates (Wang et al. 1995; Z.-Q. Wang and E.F. Wagner, unpubl.). To further investigate whether the impaired proliferation of mutant cells could be observed in vivo, we devised an in vivo proliferation assay (Fig. 2A). To this end, wild-type (+/+) morulae were aggregated with mutant (−/−) morulae and cultivated to the blastocyst stage before implantation into foster mothers. To avoid the possible influence of variations in genetic background, all donor embryos used were derived from the same mouse family. The proliferation status of mutant cells in the context of the whole embryo was reflected by the contribution of mutant (−/−) cells to various organs as visualized by quantitative Southern blot analysis of genomic DNA (Fig. 2A; see also Materials and Methods). After aggregation and transfer, 21 E17.5 fetuses were obtained and 8 tissues were dissected from each embryo for DNA analysis. Apart from two fetuses that were completely derived from PARP−/− morulae, 19 embryos exhibited variable but underrepresented contribution from −/− cells (0%–50%) in their organs (Fig. 2B) compared with the theoretical contribution from both origins, which should be equal if these cells have no defect in proliferation. Because these organs are derived from different developmental germ layers, the reduced contribution is not specific for a particular cell type but rather reflects a general defect caused by PARP deficiency. Thirteen chimeric fetuses generated from aggregation of wild-type (+/+) and heterozygous (+/−) morulae exhibited equal contribution of both cell origins in eight organs analyzed (data not shown). These results demonstrate that PARP−/− cells participate less efficiently in embryonic development when they encounter wild-type cells. Taken together, data from in vitro and in vivo experiments indicate that cells lacking PARP are more sensitive to environmental stress and are at a disadvantage when competing with wild-type cells in a given cellular compartment.
Figure 2.
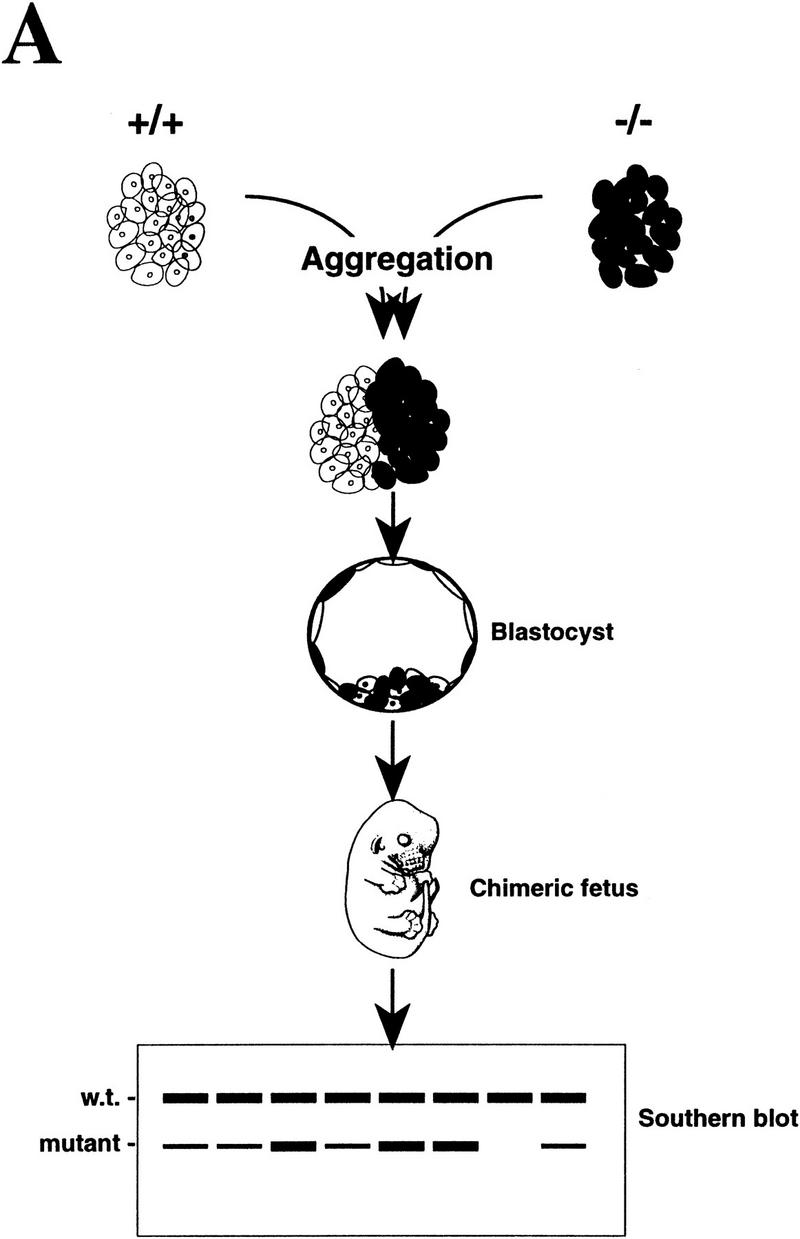
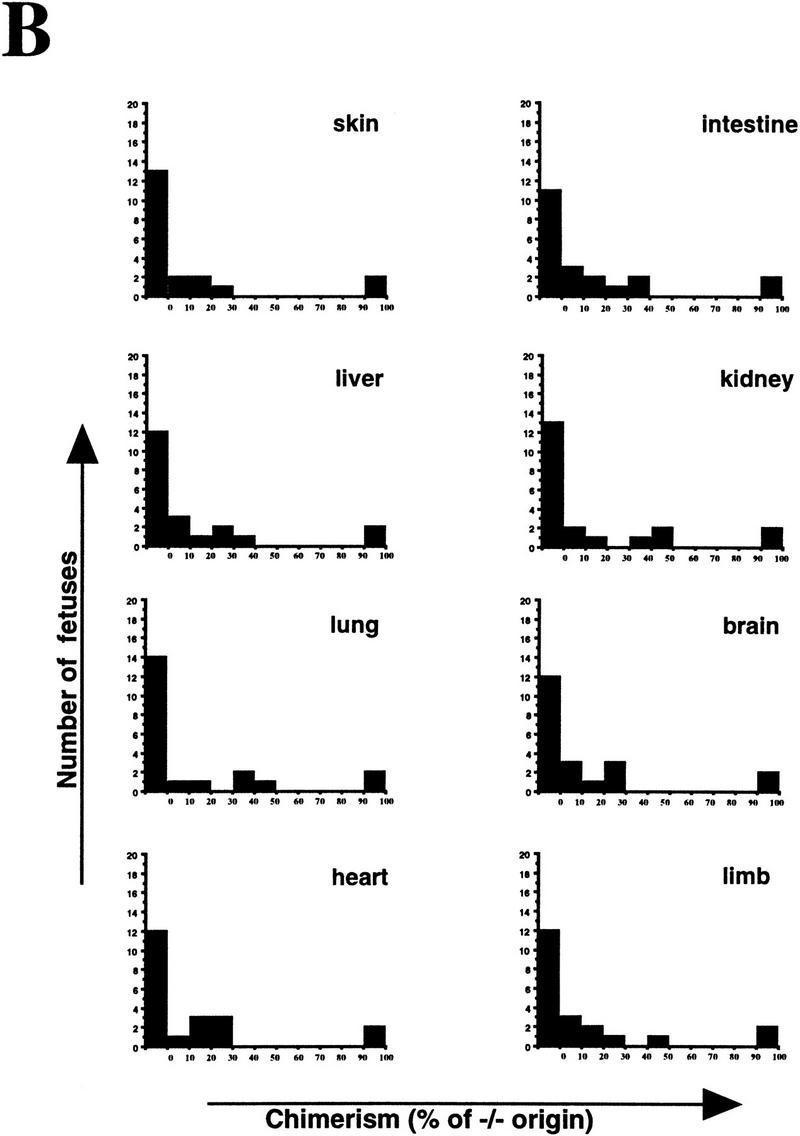
Proliferation of PARP mutant cells in vivo. (A) The diagram shows the experimental approach to examine the proliferation capacity and competence of PARP mutant cells. Wild-type (w.t., +/+) and PARP mutant (−/−) embryos were aggregated at the morula stage. The proliferation status of mutant cells in chimeric embryos was monitored by the contribution of each origin, which was determined by quantitative Southern blot analysis using DNA isolated from different organs (see also Materials and Methods). (B) A total of 21 E17.5 fetuses was obtained and eight tissues from each were analyzed by Southern blotting. The contribution from PARP−/− origin is represented by the percentage along with the number of fetuses that contained the same range of contribution in a given organ.
TNFα and anti-Fas antibody induce apoptosis in PARP−/− cells
The proliferation defect in PARP−/− cells may be caused by altered apoptosis response, because PARP has been shown to be an apoptosis-relevant substrate for CPP32 and other ICE-related proteases during apoptosis. To study the functional importance of PARP during apoptosis, we investigated the sensitivity of PARP−/− fibroblasts toward apoptosis induced by TNF and Fas receptor ligation. Primary fibroblasts were first sensitized by actinomycin D and then treated with different concentrations of TNF or agonistic anti-Fas antibodies. The proportion of dead cells from all three genotypes was similar and increased with increasing concentrations of the apoptotic agents (Fig. 3A). More than 90% of cells from all three genotypes died at a concentration of 800 IU/ml of TNF, and more than 80% of cells died at a concentration of 1000 ng/ml of anti-Fas. Western blot analysis of extracts from these cells showed that PARP was cleaved in wild-type (+/+) and heterozygous (+/−) cells after treatment with anti-Fas antibody or TNF (Fig. 3B). Interestingly, there was no reduction of PARP at the protein level in heterozygous samples compared with wild-type samples (Fig. 3B). Furthermore, the absence of PARP protein in mutant cells was noted, which confirms a null mutation at the PARP locus (Fig. 3B). The cell death induced by TNF and anti-Fas was through a programmed cell death pathway as revealed by the appearance of characteristic DNA ladders in all DNA samples (data not shown). TNF and anti-Fas induced apoptosis was also confirmed by an ex vivo study where lymphocytes freshly isolated from spleens and lymph nodes of different genotypes underwent apoptosis at the same efficiency (data not shown). These results clearly demonstrate that the absence of PARP does not interfere with the apoptotic process and, moreover, that the cleavage of PARP is not a signal triggering downstream apoptotic events.
Figure 3.

Apoptosis of PARP mutant cells. (A) Primary embryonic fibroblasts were treated for 18 hr with different concentrations of TNF or anti-Fas antibody and the viability was monitored by the MTT assay. (B) Detection of PARP cleavage: Primary embryonic fibroblasts of wild-type (+/+), heterozygous (+/−), and homozygous (−/−) mice were incubated for 8 hr with anti-Fas (α-Fas) or TNF in the presence of actinomycin D (Act D). Total cell extracts were prepared, subjected to immunoprecipitation with an PARP-specific antiserum, and analyzed by SDS-PAGE and immunoblotting. Cleavage of PARP (116 kD) into the characteristic 85-kD fragment was visualized with anti-PARP antibodies. Note the absence of PARP in homozygous (−/−) cells.
PARP−/− thymocytes undergo normal apoptosis induced by γ-irradiation and dexamethasone
Because PARP is activated by DNA breaks and is thought to be involved in repairing double-strand breaks (DSBs) we investigated the sensitivity of cells of mice lacking PARP to ionizing radiation. Thymocytes were prepared from 6-week-old mice and exposed to different doses of γ-irradiation. After 24 hr, cells were stained with propidium iodide and their viability determined by flow cytometry. The percentage of nonviable cells in each population was normalized to that of untreated cells of the same genotype. Thymocytes from all three genotypes exhibited a similar profile of cell death in a dose-dependent manner (Fig. 4A; +/− not shown) and >80% of cells died at doses of 5–7 Gy. Following exposure to 5-Gy irradiation, we checked cell viability at different time points and found that the kinetics of cell death were similar between wild-type and mutant cells (Fig. 4B), indicating that PARP−/− thymocytes have normal sensitivity to ionizing radiation. Furthermore, there was no difference between PARP−/− and wild-type littermates in their short- and long-term lifespans following 4.5-Gy γ-irradiation (data not shown; see also Wang et al. 1995). These results demonstrate that the lack of PARP has no direct effect on thymocyte repair of DSBs induced by γ-irradiation nor is PARP an essential molecule in the γ-irradiation-induced apoptotic pathway. An alternate apoptotic pathway was also examined in thymocytes by treating cells with the glucocorticoid dexamethasone, which is believed to activate endonucleases leading to the DNA fragmentation (Walker et al. 1991). Again, we found the same kinetics and magnitude of cell death in all three genotypes, indicating that PARP is not involved in apoptosis induced by this agent (Fig. 4C).
Figure 4.

Apoptosis of thymocytes from wild-type (+/+) and PARP mutant (−/−) mice following the treatment with γ-irradiation and dexamethasone. (A) Cells were irradiated with different doses of γ-ray and their viability was measured after 24 hr. (B) After treatment with 5 Gy of γ-irradiation, cell viability was monitored at different time points. (C) Induction of cell death following treatment of thymocytes with dexamethasone.
PARP−/− splenocytes carry out normal immunoglobulin class switch and V(D)J recombination
Both immunoglobulin heavy chain isotype switch recombination and V(D)J recombination in lymphocyte development involve DNA breakage and rejoining, processes believed to activate PARP. Therefore, we examined these processes in PARP−/− lymphocytes. Freshly isolated splenocytes were treated with bacterial lipopolysaccharide (LPS) and transformation growth factor β (TGFβ), and immunoglobulin heavy chain isotype switching to IgA was determined by flow cytometry using antibodies against B220, a pan B cell marker, and IgA. No difference was observed in the number of IgA-positive cells derived from wild-type and PARP−/− mice after stimulation (Fig. 5). Similar results were obtained when cells were costained with anti-IgM and anti-IgA antibodies (data not shown). These results indicate that the absence of PARP does not affect class switch recombination.
Figure 5.

IgA switching in wild-type (+/+) and PARP mutant (−/−) mice. Splenocytes were treated with TGFβ to induce expression of surface IgA. After 4 days, IgA-positive B lymphocytes were determined by costaining using anti-IgA and anti-B220 antibodies. The results from 11 mice per genotype are depicted.
We used a PCR-based method (Ehlich et al. 1994) to detect V(D)J recombination events in genomic DNA prepared from both wild-type and PARP mutant splenocytes. Analyses of the PCR products by agarose gel and Southern blot showed a normal pattern of V(D)J rearrangements in PARP−/− cells (data not shown). Sequencing analysis of these PCR products showed that 10 of the 15 clones were in-frame and had productive rearrangements (Table 1). Furthermore, sequence analyses of RT–PCR products using specific T-cell receptor (TCR) γ and β primers confirmed that TCR rearrangement had occurred normally in mutant thymocytes (data not shown). These results indicate that lymphocytes lacking PARP can carry out normal immunoglobulin class switching and V(D)J recombination, and, thus, this enzyme is not essential in these processes.
Table 1.
V(D)J junctions in PARP mutant splenocytes
| VH sequence
|
P,N
|
DH sequence
|
P,N
|
JH sequence
|
DH
|
JH
|
Prod.
|
|---|---|---|---|---|---|---|---|
Sequence analysis of PCR products derived from PARP−/− splenocytes. The conserved cysteine 92 (C92) was used for orientation. Productive (p) nonproductive (n.p.) joints were defined from C92 relative to the J region.
PARP−/− splenocytes exhibit a high frequency of sister chromatid exchange
To further investigate the role of PARP in DNA recombination and stabilization of chromatin structures, we examined the frequency of sister chromatid exchange (SCE) in PARP−/− splenocytes because SCE is thought to be consequence of recombination or recombination repair (Kuzminov 1996). For scoring SCE, 25–50 metaphase chromosome spreads (∼40 chromosomes per spread) were counted per genotype. One of three independent experiments is presented in Figure 6A. Although the majority of wild-type (+/+) cells contained ∼8 SCEs per genome, PARP−/− cells contained ∼20 SCEs, a two- to threefold increase (Fig. 6A). When cells were treated with mitomycin C (MMC), a DNA cross-linking agent, the number of SCEs was increased in both wild-type and mutant splenocytes (Fig. 6A). The higher spontaneous SCE rate observed in PARP−/− cells indicates that the absence of PARP gives rise to a higher recombination activity in cells, and further suggests that mice lacking PARP have an unstable genome.
Figure 6.
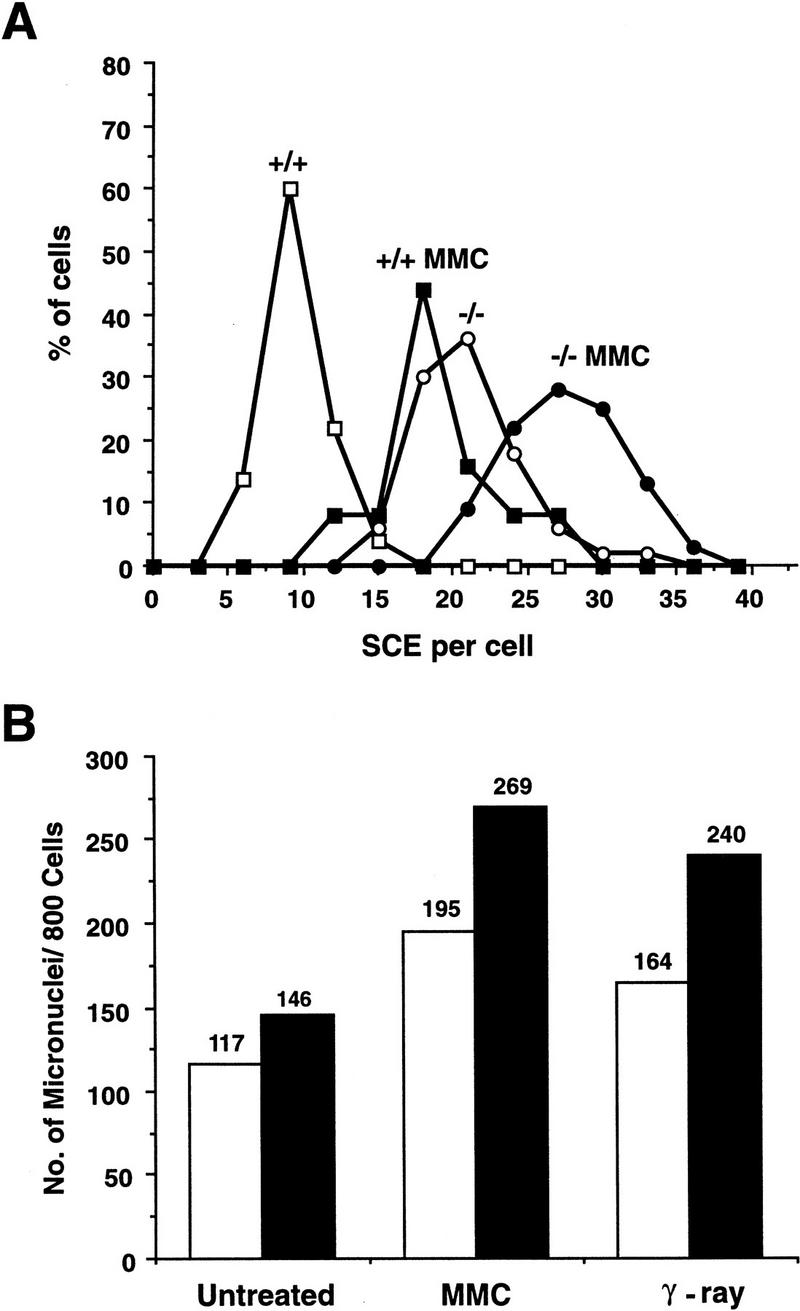
(A) SCE rate in splenocytes freshly isolated from wild-type (+/+) and PARP mutant (−/−) mice. Cells treated and nontreated with MMC were counted for SCE. Whereas 50 chromosomal spreads were counted for the untreated groups, 25 spreads were counted after MMC treatment. (B) Micronuclei formation in primary embryonic fibroblasts isolated from wild-type (open bars) and mutant (solid bars) mice. A total of 800 cells was counted for the presence of micronuclei with or without treatment with γ-irradiation or MMC. Although untreated cells showed no significant difference in the number of micronuclei between +/+ and −/− cells, after treatment the number of micronuclei in −/− cells was significantly higher than in +/+ cells (P 0.01).
PARP mutant fibroblasts contain more micronuclei following genotoxic treatment
To substantiate the role of PARP in genomic stability, micronuclei were enumerated in primary embryonic fibroblasts, as the presence of micronuclei is a biomarker for genomic stability. Cells from three different genotypes were either untreated or treated with γ-irradiation or MMC. Figure 6B shows a representative result from five independent experiments. In the untreated group, PARP−/− cells contained a slightly higher number of micronuclei compared with wild-type controls; however, the difference was not significant by χ2 analysis (Fig. 6B). After treatment with γ-irradiation or MMC, the number of micronuclei in both wild-type and mutant cells was increased and under these conditions PARP−/− cells contained a significantly higher number of micronuclei compared with wild-type controls (P 0.01) (Fig. 6B). It was noted that the number of micronuclei in heterozygous cells was similar to that of wild-type cells (data not shown). These results further indicate that the lack of PARP or poly(ADP-ribosyl)ation affects the genomic stability of cells following experimental stress.
Discussion
In view of numerous publications that implicate PARP in many cellular processes, such as proliferation, apoptosis, DNA repair and recombination, as well as chromosomal stability, it was surprising that mice carrying a null mutation for this molecule are devoid of severe phenotypic abnormalities and that cells derived from these animals function normally in DNA excision repair. The present study demonstrates that PARP-deficient cells are more sensitive to heat stress and exhibit compromised proliferation ability both in vitro and in vivo. Unexpectedly, embryonic fibroblasts and lymphoid cells display normal apoptotic sensitivity after treatment with various apoptotic agents, indicating that PARP is not required for the execution of the apoptotic program. Primary fibroblasts and splenocytes showed an increased number of spontaneous SCE and elevated micronuclei formation following γ-irradiation and MMC treatment, indicating a higher rate of recombination or recombination repair in mutant cells and a role for PARP in the maintenance of genomic stability. These results demonstrate that this enzyme functions in many cellular processes, some of which may be dispensable and others in which PARP appears to be essential.
Cells lacking PARP are more sensitive to experimental stress in vitro and in vivo
Our previous study showed that cells lacking PARP exhibited a deficiency in proliferation in tissue culture (Wang et al. 1995). Here we demonstrate that this deficiency is more pronounced when temperature insult is applied, suggesting that mutant cells have an altered stress response. This defect cannot be rescued by co-culture with wild-type cells and, more important, is also evident in vivo when mutant cells are mixed with wild-type cells by embryo aggregation. This is in agreement with previous findings showing that the inhibition of PARP decreases the recovery of cells after oxidative stress and heat shock (Nosseri et al. 1994; Shah et al. 1996). These results suggest that cells lacking PARP are hypersensitive to environmental insults and slow down their proliferation. However, we did not observe impaired cell cycle progression and/or re-entry to the cell cycle in mutant cells following serum depletion (data not shown), γ-irradiation, or addition of adriamycin or other genotoxic agents (Agarwal et al. 1997). This finding contradicts a recent observation that 3-AB treatment represses G1 arrest and augments G2 arrest induced by γ-irradiation (Masutani et al. 1995). However, we cannot rule out the possibility that the difference in the cell proliferation curve escaped detection in our cell cycle assays. It is also possible that poly(ADP-ribosyl)ation may be required for DNA metabolism enzymes to become fully active (Simbulan et al. 1993). Another explanation for the proliferation defect may be that the increased genomic instability of mutant cells leading to DNA amplification and/or loss of chromatin (Fig. 6B; see also Catena et al. 1994; Ding and Smulson 1994; Küpper et al. 1996) may further contribute to the delay in proliferation. It is surprising that although proliferation deficiency and hypersensitivity to stress appeared in PARP mutant cells in vitro and in vivo, PARP−/− fetuses develop normally in the same litter where wild-type and heterozygous embryos are present. A likely explanation is that there is no direct competition between mutant and wild-type cells within individual embryos in contrast to the competitive situation in aggregation chimeras. Finally, the proliferation defect could not be attributable to the fact that PARP−/− cells are prone to apoptosis because we have not observed higher apoptotic rates in spontaneous culture (data not shown) and these cells exhibit normal apoptotic responses (see below).
PARP is dispensable for apoptosis
Recent studies demonstrated that PARP is cleaved by ICE-related proteases, in particular caspase-3/CPP32, which is activated in cells following treatment with various apoptotic agents. In the present study we show that fibroblasts and lymphoid cells from PARP−/− mice exhibit a normal apoptotic response after treatment with anti-Fas antibody, TNF, γ-irradiation, and dexamethasone. These results demonstrate clearly that PARP is not an essential molecule in the apoptotic cascade and suggest that PARP cleavage is most likely a consequence rather than an integral component of the apoptotic process. However, it remains unclear why cells cleave PARP during apoptosis. One explanation could be that the cleavage of PARP is a step by which cells committing self-destruction switch off all possible protective mechanisms, including DNA repair. Poly(ADP-ribosyl)ation may be one of these mechanisms because PARP senses DNA strand interruptions and catalyzes poly(ADP-ribosyl)ation upon binding to the breaks, an energy-consuming process, which depletes the precursors of ATP generation (see Zhang et al. 1994). Early cleavage of PARP and perhaps other DNA end-binding proteins might be required to prevent protection of DNA ends by these proteins in a cell destined for deletion. In agreement with this hypothesis is the observation that DNA-dependent protein kinase (DNA-PK), another DNA break binding protein involved in DNA double-stranded break repair (Jackson and Jeggo 1995), is also specifically cleaved during apoptosis (Casciola-Rosen et al. 1996; Song et al. 1996). However, the present study could not address whether the cleavage of PARP or the cleaved fragments play a role in apoptosis because mutant mice carry a null mutation at the PARP locus. To answer this question, we are generating mice carrying a mutant PARP in which the specific cleavage site (DEVD) is altered so that the molecule cannot be cleaved by CPP32 but retains the ability to bind to DNA breaks and catalyze poly(ADP-ribosyl)ation.
PARP is not essential for DNA double-stranded break repair induced by γ-irradiation in thymocytes
Our previous study showed that the null mutation in the PARP gene has no influence on excision repair of DNA damaged by UV-radiation and treatment with alkylating agents (Wang et al. 1995). The present study further demonstrates that thymocytes lacking PARP are not hypersensitive to ionizing radiation, suggesting that PARP is dispensable for DNA double-stranded break repair induced by γ-irradiation. These findings are surprising in view of the fact that repression of PARP activity by either inhibitors or transdominant mutations sensitizes cells to γ-ray treatment (Küpper et al. 1995). These results may be a result of nonspecific effects of the chemical inhibitors or cell types used in these studies. It is also possible that the dominant-negative form of PARP binds DNA ends but cannot be released, thereby impeding the access of the DNA repair machinery. Because PARP is absent in our system, the repair machinery can act at DNA ends without interference to fix the lesions.
The lack of PARP does not affect immunoglobulin class switch or V(D)J recombination
Although numerous studies imply a role for PARP in repairing DNA breaks, cells lacking PARP carry out a normal immunoglobulin class switch recombination and V(D)J recombination, processes involving DNA breakage and rejoining. This indicates that either PARP is not involved in these processes or that alternative pathways compensate for the lack of this enzyme. Moreover, other DNA rejoining processes, such as integration of viral DNA into the genome, are also not affected, because no difference in colony formation was observed after retroviral infection into PARP mutant and wild-type fibroblasts (Z.-Q. Wang, L. Stingl, and E.F. Wagner, unpubl.). These results contradict previous findings that pharmacological inhibition of PARP facilitates immunoglobulin class switching (Shockett and Stavnezer 1993) and blocks efficient integration of foreign DNA into the mammalian genome (Farzaneh et al. 1988; Gäken et al. 1996). The discrepancy between these results and ours may be attributable to alterations in other cellular processes caused by side effects of the PARP inhibitors or the systems used. Although PARP seems to be dispensable in immunoglobulin class switch and V(D)J recombination, PARP may still be involved in recombination processes by either interacting with other DNA end-binding proteins or acting as an antirecombinogenic molecule. Indeed, a recent study from our laboratory demonstrates that PARP depletion can partially rescue V(D)J recombination blocked in scid mice that is caused by a mutation in the carboxy terminus of DNA break-activated protein kinase catalytic subunit (DNA-PKcs), suggesting a genetic interaction of PARP and DNA-PK (C. Morrison, G.C.M. Smith, L. Stingl, S.P. Jackson, E.F. Wagner, and Z.-Q. Wang, in prep.).
The role of PARP for other recombination events and in chromosomal stability
Although PARP is dispensable in DNA excision repair, DSB repair induced by γ-irradiation, immunoglobulin class switch, and V(D)J recombination, splenocytes from mutant mice exhibit elevated spontaneous SCE. Whereas the mechanism involved in SCE is not entirely clear, many studies point to the involvement of recombination or recombination repair in this process (for review, see Shall 1995; Kuzminov 1996). Because SCE occurs during and soon after DNA replication, it is conceivable that SCE happens when the replication fork encounters unrepaired lesions and affected repair components (Cleaver et al. 1996). It has been shown that PARP is present in newly replicated chromatin and in DNA replication forks and that chemical inhibition and dominant-negative mutation of PARP stimulates SCE formation following DNA damage (Schreiber et al. 1995), presumably as a result of inefficient removal of PARP (wild-type or mutant) from DNA lesions. Although our findings seem to be consistent with these results, different mechanisms may be involved because these mice are devoid of PARP. One explanation for our results is that PARP functions as an antirecombinogenic factor at DNA ends, which is in agreement with the model proposed by others, for example, Shall (1995) and Lindahl et al. (1995). Therefore, our results indicate that PARP plays an important role in genomic recombination and stability. However, the role of this molecule in homologous recombination is not clear. Although chemical inhibition of PARP activity increases chromosomal homologous recombination frequency in mammalian cells (Waldman et al. 1996), a transient extrachromosomal recombination study showed that PARP−/− cells can efficiently repair a mutated luciferase reporter gene via homologous recombination (Morrison and Wagner 1996).
High levels of spontaneous SCE and micronuclei in PARP−/− cells after treatment with MMC or γ-irradiation might also be attributable to a structural function of PARP, because numerous studies demonstrate the nuclear abundance of PARP and its involvement in chromatin conformation. Moreover, many structural nuclear proteins have been shown to be acceptors for poly(ADP-ribosyl)ation. Further evidence to support the structural functions of PARP in chromatin may be the finding that cells lacking PARP are not more sensitive to genotoxic agents, such as MMC, with respect to the SCE frequencies. However, we cannot exclude the possibility that the increased SCE and micronuclei are attributable not only to the absence of PARP as a structural protein but also to the lack of PARP activity in response to DNA breaks, or the combinatorial effects of both. Nevertheless, elevation of micronuclei numbers following treatment with γ-irradiation and MMC is consistent with previous findings showing that inhibition of PARP results in increased micronuclei formation (Catena et al. 1994; Steirum et al. 1995). Taken together, these results demonstrate that PARP plays a role in the maintenance of chromosomal integrity.
Although PARP mutant mice display elevated SCE and micronuclei in response to genotoxic treatment, they never developed malignancies in over 2 years of observation and their lifespans are comparable to those of wild-type and heterozygous controls. This is surprising because micronuclei and SCE are well-known biomarkers for genomic stability that is thought to play a critical role in tumor development. One possibility is that environmental insults do not induce genetic changes that can be fixed by tissues and cells in mice. In fact, we observed normal chromosomal karyotypes and in vivo micronuclei frequency in PARP−/− mice (I.-D. Adler, pers. comm.). Another explanation is that an unstable genome per se may not be sufficient for tumorigenesis. Genetic changes, such as mutations in oncogenes or in tumor suppressors, are required as the “first hit” in the process of multistep tumorigenesis, and an unstable genome may then allow the acquisition of subsequent genetic and epigenetic changes necessary for neoplastic transformation. To test this hypothesis, we are introducing genetic predispositions for tumor formation in PARP null mice to analyze whether the loss of genomic integrity can contribute to tumor development.
Materials and methods
Isolation and culture of primary embryonic fibroblasts
Embryonic fibroblasts were isolated and cultured essentially as described previously (Wang et al. 1995). All experiments were carried out with cells derived from embryos of the same litter, and two populations of primary embryonic fibroblasts of each genotype at early passages (passages 2–3) were used. All cell-culture experiments were performed in triplicate and repeated at least two times. Cells (1 × 105) were plated into six-well plates in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal calf serum (FCS) and cultured at either 37°C or 39°C in a 5% CO2 incubator. The medium was changed every 2 days, and cells were counted using a hemocytometer. For the co-culture experiments, passage 2 PARP+/+ and −/− cells were mixed at 1:1 ratio. To monitor the content of cell origins in the culture mixture, DNA was isolated at each passage and subjected to Southern blot analysis. The density of wild-type (4.7-kb) and mutant (1.7-kb) bands after PvuII digestion and hybridization with a specific probe (see Wang et al. 1995) was visualized.
Morula aggregation
For aggregation, embryos at morula stage [2.5 days postcoitum (d.p.c.)] were derived from either PARP−/− or wild-type intercrosses. All parents used were from the same family of mouse colony and of a mixed genetic background (129/Sv × C57BL/6). Zona pellucida was removed by acidic tyrode solution, and one PARP−/− morula was aggregated with one wild-type (+/+) morula in M16 medium in a microdrop culture dish. After 24–36 hr, the aggregates developed to blastocysts that were then transferred into pseudopregnant females. At the embryonic stage E17.5, eight organs were dissected from each fetus and DNA was extracted for Southern blot analysis. To determine the contribution of PARP−/− cells in these organs, the density of wild-type and mutant bands on the Southern blot was visualized and quantified using a PhosphorImager (Molecular Dynamics, Inc.).
Apoptosis induction and assays
Anti-Fas and TNFα were used to induce apoptosis in fibroblasts and peripheral lymphocytes. Fibroblasts were treated with different concentrations of anti-Fas antibody (Jo2; Pharmingen, San Diego, CA) or with mouse TNFα in the presence of 0.25 μg/ml of actinomycin D. The MTT detection method was used to measure dead cells after 18 hr in culture with DMEM medium (Schulze-Osthoff et al. 1994). To determine the cleavage of PARP, fibroblasts were treated with either anti-Fas antibody (1 μg/ml) or TNF (40 ng/ml) in the presence of actinomycin D. After 8 hr, cells were lysed by repeated freeze/thaw cycles in 50 mm Tris at pH 8.0, 2 mm MgCl2, 2 mm dithiothreitol (DTT), 1 mm PMSF, and 100 μm TLCK. The extracts were subjected to immunoprecipitation with an anti-human PARP antibody (kind gift of Dr. G. de Murcia, Ecole Supérièure de Biotechnologie, Strasbourg, France). After electrophoresis on a 7.5% SDS-PAGE and transfer onto a nitrocellulose membrane, PARP was visualized using anti-PARP antibody followed by enhanced chemoluminescent staining.
γ-Irradiation and dexamethasone treatment of thymocytes
Thymocytes were isolated from wild-type, heterozygous, and homozygous mutant mice at 6 weeks of age and were exposed to different doses of γ-ray from a dual 137Cs source (Gammacell 40; Nordion, Kanata, Canada) at a rate of 1.2 Gy/min. Thymocytes were cultivated for 24 hr in Iscove’s modified Dulbecco’s medium (IMEM) medium containing 10% FCS before staining with propidium iodide (PI) and analysis by flow cytometry (FACScan, Becton Dickinson, San Jose, CA) as described by Wang et al. (1995). The surviving fraction of cells was calculated relative to the untreated group of the same genotype. In a separate experiment, thymocytes were irradiated with 5 Gy and then cultured for various periods followed by staining with PI to determine the fraction of dead cells. For dexamethasone treatment, freshly isolated thymocytes were incubated with a final concentration of 1 μm dexamethasone and cultured for different periods before PI staining and FACS analysis.
Induction of immunoglobulin class switching
Total splenic cell suspensions were prepared and erythrocytes were lysed. Lymphocytes were resuspended at 1 × 106/ml in DMEM medium (day 0). For each experiment, 4 × 106 cells were stimulated with Salmonella typhimurium lipopolysaccharide (LPS) (Sigma, St. Louis, MO) at 20 μg/ml. To induce class switching to IgA, human TGFβ1 (Genentech, San Francisco, CA) was added to the medium to a concentration of 2 ng/ml and added daily until day 4 when cells were analyzed. Cells were washed and resuspended in PBS containing 3% FCS. Cells were costained at 4°C using goat anti-mouse IgA conjugated with R-phycoerythrin (PE) (Harlan Sera-lab, Crawley Down, UK) and rat anti-mouse CD45R/B220 conjugated with fluorescein isothiocyanate (FITC) (RA3-6B2; Pharmingen), or goat anti-mouse IgM–FITC (Harlan, Sera-lab). After staining, cells were analyzed by flow cytometry.
V(D)J recombination assay
The immunoglobulin V(D)J recombination assay used in this study was based on the method described by Ehlich et al. (1994). Briefly, DNA was prepared from spleen and brain (as a control) and amplified by PCR reaction (see Wang et al. 1992). The primers for PCR analysis are listed below: (VHA) -GCGAAGCTTA(AG)GCCTGGG(AG)CTTCAGTGAAG-; (MBJH1) -AGTGTGGCAGATGGCCT; (MBJH2) TAGTCCTTCATGACCTG-; (MBJH3) -TTGATTCCCGTTTGCAG-; (JH4E) -AGGCTCTGAGATCCCTAGACAG-; (JH4A) -GGGTCTAGACTCTCAGCCGGCTCCCTCAGGG-. Reaction products from amplification with VHA and each of the JH primers were purified by Wizard PCR Prep (Promega, Madison, WI) and subcloned into pT7Blue T-vector (Novagen, Madison, WI). Plasmids containing insert were sequenced using an automatic sequencer (model 377, Applied Biosystems, Foster City, CA). Sequences were analyzed by comparison with those published (Kabat 1991). Productive and nonproductive joints were defined from the conserved cysteine 92 (TGT) relative to the J region.
Sister chromatid exchange and micronucleus assays
For SCE examination, splenocytes were isolated from PARP+/+, +/−, and −/− sibling mice at 4–6 months of age and stimulated to proliferate with ConA. Cells were cultivated and metaphase spreads were prepared as described basically by Gustashaw (1991) and Sharma and Sharma (1994). Briefly, cells were cultivated at 2 × 106 cells/ml in minimum essential medium containing 15% FCS and 1.5 μg/ml of ConA (Sigma, St. Louis, MO). After 24 hr in culture, the cells were treated either with 3 × 10−8 m MMC for 2 hr in the presence of 10 μm bromodeoxyuridine (BrdU) (Boehringer Mannheim, Germany) or with BrdU alone. After another 69 hr in culture, colcemid was added to a final concentration of 20 ng/ml for 2 hr before harvest and preparation of chromosome spreads. The differentially stained chromatids were then counted for chromatid exchanges under a microscope.
For enumeration of micronuclei, early passage (p2 or p3) primary embryonic fibroblasts of three genotypes from the same litter were used. Cells (1.3 × 105) were seeded onto gelatin-coated glass chamber slides (Nunc, Denmark) in complete DMEM containing 10% FCS on the day before treatment. Treatment consisted of exposure to 3 Gy γ-ray or exposure for 2 hr at 37°C to 3 × 10−7 m MMC. After treatment, cultures were continued for 24 hr before washing the slides in PBS and fixing cells for 5 min in 2% paraformaldehyde in PBS, permeabilizing in 0.5% Triton X-100 and 70% ethanol for 10 min. After a final PBS rinse, slides were overlaid with Mowiol (Hoechst, Germany) containing 1 μg/ml of DAPI (Sigma), coverslipped, and viewed with a fluorescence microscope. Duplicate slides were prepared for each cell type/treatment group and 800 cells were counted for each sample. The treatment of cells and screening of micronuclei were carried out by a double-blind approach.
Acknowledgments
We thank Hans-Christian Theussl for the whole-body irradiation experiments and Robert Kurzbauer for technical assistance. We are also grateful to Drs. I.-D. Adler, B. Auer, M. Cotten, P. Hainaut, and Z. Herceg for helpful comments and discussions and to Dr. G. de Murcia for providing anti-PARP antibody. This research was supported in part by the Austrian Industrial Research Promotion Fund.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
We recently performed whole-body irradiation of PARP−/− mice and found that these mice are highly sensitive to 8 Gy γ-irradiation, as 16 out of 16 PARP−/− mice died 10 days postirradiation, whereas all wild-type controls survived. This result was consistent in two independent experiments using mice of either 129/Sv or 129/BL6 (129/Sv × C57BL/6) mixed genetic background, and is in agreement with a recent report by de Murcia et al. (Proc. Natl. Acad. Sci. 1997 94: 7303–7307).
Footnotes
E-MAIL zqwang@iarc.fr; FAX +33-4-7273 8329.
References
- Agarwal, M., A. Agarwal, W.R. Taylor, Z.-Q. Wang, E.F. Wagner, and G. Stark. 1997. Defective induction but normal activation and function of p53 in mouse cells lacking poly-ADP-ribose polymerase. Oncogene (in press). [DOI] [PubMed]
- Althaus FR, Richter C. ADP-ribosylation of proteins—Enzymology and biological significance. Mol Biol Biochem Biophys. 1987;37:1–125. [PubMed] [Google Scholar]
- Casciola-Rosen L, Nicholson DW, Chong T, Rowan KR, Thornberry NA, Miller DK, Rosen A. Apopain/CPP32 cleaves proteins that are essential for cellular repair: A fundamental principle of apoptotic death. J Exp Med. 1996;183:1957–1964. doi: 10.1084/jem.183.5.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catena C, Villani P, Conti D, Righi E. Micronuclei and 3AB index in X-irradiated human lymphocytes in G0 and G1 phases. Mutat Res. 1994;311:231–237. doi: 10.1016/0027-5107(94)90181-3. [DOI] [PubMed] [Google Scholar]
- Cleaver JE, Mitchell DL, Feeney L, Afzal V. Chromatid exchanges may be induced by damage in sites of transcriptional activity. Mutagenesis. 1996;11:183–187. doi: 10.1093/mutage/11.2.183. [DOI] [PubMed] [Google Scholar]
- de Murcia G, Menessier-de Murcia J. Poly(ADP-ribose) polymerase: A molecular nick-sensor. Trends Biochem Sci. 1994;19:172–176. doi: 10.1016/0968-0004(94)90280-1. [DOI] [PubMed] [Google Scholar]
- Ding R, Smulson M. Depletion of nuclear poly(ADP-ribose) polymerase by antisense RNA expression: Influences on genomic stability, chromatin organization, and carcinogen cytotoxicity. Cancer Res. 1994;54:4627–4634. [PubMed] [Google Scholar]
- Ding R, Pommier Y, Kang VH, Smulson M. Depletion of poly(ADP-ribose) polymerase by antisense RNA expression results in a delay in DNA strand break rejoining. J Biol Chem. 1992;267:12804–12812. [PubMed] [Google Scholar]
- Ehlich A, Martin V, Muller W, Rajewsky K. Analysis of the B-cell progenitor compartment at the level of single cells. Curr Biol. 1994;4:573–583. doi: 10.1016/s0960-9822(00)00129-9. [DOI] [PubMed] [Google Scholar]
- Enari M, Hug H, Nagata S. Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature. 1995;375:78–81. doi: 10.1038/375078a0. [DOI] [PubMed] [Google Scholar]
- Farzaneh F, Panayotou GN, Bowler LD, Hardas BD, Broom T, Walther C, Shall S. ADP-ribosylation is involved in the integration of foreign DNA into the mammalian cell genome. Nucleic Acids Res. 1988;16:11319–11326. doi: 10.1093/nar/16.23.11319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gäken JA, Tavassoli M, Gan SU, Vallian S, Giddings I, Darling DC, Galea-Lauri J, Thomas MG, Abedi H, Schreiber V, Menissier-de Murcia J, Collins MK, Shall S, Farzaneh F. Efficient retroviral infection of mammalian cells is blocked by inhibition of poly(ADP-ribose) polymerase activity. J Virol. 1996;70:3992–4000. doi: 10.1128/jvi.70.6.3992-4000.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustashaw KM. Chromosome stains. In: Barch M, editor. The ACT cytogenetics laboratory manual. 2nd ed. New York, NY: Raven Press; 1991. pp. 244–246. [Google Scholar]
- Heller B, Wang Z-Q, Wagner EF, Radons J, Bürkle A, Fehsel K, Burkart V, Kolb H. Inactivation of the poly(ADP-ribose) polymerase gene affects oxygen radical and nitric oxide toxicity in islet cells. J Biol Chem. 1995;270:11176–11180. doi: 10.1074/jbc.270.19.11176. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Jeggo PA. DNA double-strand break repair and V(D)J recombination: involvement of DNA-PK. Trends Biochem Sci. 1995;20:412–415. doi: 10.1016/s0968-0004(00)89090-8. [DOI] [PubMed] [Google Scholar]
- Kabat E, Wu T, Perry HM, Gottesman KS, Foeller C. Sequences of proteins of immunological interest. 5th ed. Washington, DC.: U.S. Dept. of Health and Human Services; 1991. [Google Scholar]
- Kuzminov A. Recombinational repair in eukaryotes. In: Kuzminov A, editor. Recombinational repair of DNA damage. New York, NY: Springer; 1996. pp. 185–203. [Google Scholar]
- Küpper JH, Müller M, Jacobson MK, Tatsumi-Miyajima J, Coyle DL, Jacobson EL, Bürkle A. Trans-dominant inhibition of poly(ADP-ribosyl)ation sensitizes cells against γ-irradiation and N-methyl-N′-nitro-N-nitrosoguanidine but does not limit DNA replication of a polyomavirus replicon. Mol Cell Biol. 1995;15:3154–3163. doi: 10.1128/mcb.15.6.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küpper JH, Müller M, Bürkle A. Trans-dominant inhibition of poly(ADP-ribosyl)ation potentiates carcinogen induced gene amplification in SV40-transformed Chinese hamster cells. Cancer Res. 1996;56:2715–2717. [PubMed] [Google Scholar]
- Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- Lindahl T, Satoh MS, Poirier GG, Klungland A. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem Sci. 1995;20:405–411. doi: 10.1016/s0968-0004(00)89089-1. [DOI] [PubMed] [Google Scholar]
- Los M, Van de Craen M, Penning LC, Schenk H, Westendorp M, Baeuerle PA, Dröge W, Krammer PH, Fiers W, Schulze-Osthoff K. Requirement of an ICE/CED-3 protease for Fas/APO-1-mediated apoptosis. Nature. 1995;375:81–83. doi: 10.1038/375081a0. [DOI] [PubMed] [Google Scholar]
- Malorni W, Rivabene R, Straface E, Rainaldi G, Monti D, Salvioli S, Cossarizza A, Franceschi C. 3-Aminobenzamide protects cells from UV-B-induced apoptosis by acting on cytoskeleton and substrate adhesion. Biochem Biophys Res Commun. 1995;207:715–724. doi: 10.1006/bbrc.1995.1246. [DOI] [PubMed] [Google Scholar]
- Masutani M, Nozaki T, Wakabayashi K, Sugimura T. Role of poly(ADP-ribose) polymerase in cell-cycle checkpoint mechanisms following γ-irradiation. Biochimie. 1995;77:462–465. doi: 10.1016/0300-9084(96)88161-2. [DOI] [PubMed] [Google Scholar]
- McGowan AJ, Ruiz-Ruiz MC, Gorman AM, Lopez-Rivas A, Cotter TG. Reactive oxygen intermediate(s) (ROI): Common mediator(s) of poly(ADP-ribose)polymerase (PARP) cleavage and apoptosis. FEBS Lett. 1996;392:299–303. doi: 10.1016/0014-5793(96)00838-1. [DOI] [PubMed] [Google Scholar]
- Morrison C, Wagner E. Extrachromosomal recombination occurs efficiently in cells defective in various DNA repair systems. Nucleic Acids Res. 1996;24:2053–2058. doi: 10.1093/nar/24.11.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, Munday NA, Raju SM, Smulson ME, Yamin T-T, Yu VL, Miller DK. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Nosseri C, Coppola S, Ghibelli L. Possible involvement of poly(ADP-ribosyl) polymerase in triggering stress-induced apoptosis. Exp Cell Res. 1994;212:367–373. doi: 10.1006/excr.1994.1156. [DOI] [PubMed] [Google Scholar]
- Patel T, Gores GJ, Kaufmann SH. The role of proteases during apoptosis. FASEB J. 1996;10:587–597. doi: 10.1096/fasebj.10.5.8621058. [DOI] [PubMed] [Google Scholar]
- Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356:356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- Schreiber V, Hunting D, Trucco C, Gowans B, Grunwald D, de Murcia G, de Murcia JM. A dominant-negative mutant of human poly(ADP-ribose) polymerase affects cell recovery, apoptosis, and sister chromatid exchange following DNA damage. Proc Natl Acad Sci. 1995;92:4753–4757. doi: 10.1073/pnas.92.11.4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Osthoff K, Krammer PH, Dröge W. Divergent signalling via APO-1/Fas and the TNF receptor, two homologous molecules involved in physiological cell death. EMBO J. 1994;13:4587–4596. doi: 10.1002/j.1460-2075.1994.tb06780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah GM, Poirier D, Desnoyers S, Saint-Martin S, Hoflack JC, Rong P, ApSimon M, Kirkland JB, Poirier GG. Complete inhibition of poly(ADP-ribose) polymerase activity prevents the recovery of C3H10T1/2 cells from oxidative stress. Biochim Biophys Acta. 1996;1312:1–7. doi: 10.1016/0167-4889(96)00004-3. [DOI] [PubMed] [Google Scholar]
- Shall S. ADP-ribosylation reactions. Biochimie. 1995;77:313–318. doi: 10.1016/0300-9084(96)88140-5. [DOI] [PubMed] [Google Scholar]
- Sharma AK, Sharma A. Sister chromatid exchange. In: Sharma AK, Sharma A, editors. Chromosome techniques, a manual. Harwood Academic Publishers; 1994. pp. 74–81. [Google Scholar]
- Shockett P, Stavnezer J. Inhibitors of poly(ADP-ribose) polymerase increase antibody class switching. J Immunol. 1993;151:6962–6976. [PubMed] [Google Scholar]
- Simbulan CM, Suzuki M, Izuta S, Sakurai T, Savoysky E, Kojima K, Miyahara K, Shizuta Y, Yoshida S. Poly(ADP-ribose) polymerase stimulates DNA polymerase α by physical association. J Biol Chem. 1993;268:93–99. [PubMed] [Google Scholar]
- Song Q, Lees-Miller SP, Kumar S, Zhang Z, Chan DW, Smith GC, Jackson SP, Alnemri ES, Litwack G, Khanna KK, Lavin MF. DNA-dependent protein kinase catalytic subunit: A target for an ICE-like protease in apoptosis. EMBO J. 1996;15:3238–3246. [PMC free article] [PubMed] [Google Scholar]
- Steirum RH, van Herwijnen MH, Pasman PC, Hageman GJ, Kleinjans JC, van Agen B. Inhibition of poly(ADP-ribose) polymerase increases (+/−)-anti-benzo [a]pyrene diolepoxide-induced micronuclei formation and p53 accumulation in isolated human peripheral blood lymphocytes. Carcinogenesis. 1995;16:2765–2771. doi: 10.1093/carcin/16.11.2765. [DOI] [PubMed] [Google Scholar]
- Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32 b, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- Wang Z-Q, Ovitt C, Grigoriadis AE, Möhle-Steinlein U, Rüther U, Wagner EF. Bone and haematopoietic defects in mice lacking c-fos. Nature. 1992;360:741–745. doi: 10.1038/360741a0. [DOI] [PubMed] [Google Scholar]
- Wang Z-Q, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M, Wagner EF. Mice lacking PARP and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes & Dev. 1995;9:509–520. doi: 10.1101/gad.9.5.509. [DOI] [PubMed] [Google Scholar]
- Waldman AS, Waldman BC. Stimulation of intrachromosomal homologous recombination in mammalian cells by an inhibitor of poly(ADP-ribosylation) Nucleic Acids Res. 1991;19:5943–5947. doi: 10.1093/nar/19.21.5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldman BC, O’Quinn JR, Waldman AS. Enrichment for gene targeting in mammalian cells by inhibition of poly(ADP-ribosylation) Biochim Biophys Acta. 1996;1308:241–250. doi: 10.1016/0167-4781(96)00111-x. [DOI] [PubMed] [Google Scholar]
- Walker PR, Smith C, Youdale T, Leblanc J, Whitfield JF, Sikorska M. Topoisomerase II-reactive chemotherapeutic drugs induce apoptosis in thymocytes. Cancer Res. 1991;51:1078–1085. [PubMed] [Google Scholar]
- Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]