

Abstract
During development of the central nervous system, oligodendrocyte progenitor cells (O-2A) undergo an orderly pattern of cell proliferation and differentiation, culminating in the ability of oligodendrocytes to myelinate axons. Here we report that p27Kip1, a cyclin-dependent kinase inhibitor, is an important component of the decision of O-2A cells to withdraw from the cell cycle. In vitro, accumulation of p27 correlates with differentiation of oligodendrocytes. Furthermore, only a fraction of O-2A cells derived from p27-knockout mice differentiate successfully compared to controls. Inability to differentiate correlates with continued proliferation, suggesting that p27 is an important component of the machinery required for the G1/G0 transition in O-2A cells. In vivo, expansion of O-2A precursors before withdrawal, in part, leads to a greater number of oligodendrocytes. Together these data indicate a role for p27 during the decision to withdraw from the cell cycle in the oligodendrocyte lineage.
Keywords: Oligodendrocyte progenitor cells, cell proliferation, differentiation, CKI p27Kip1
Differentiation of cells requires withdrawal from the cell cycle and induction of a novel program of gene expression leading to elaboration of a specialized phenotype (Hofbauer and Denhardt 1991; Duronio and O’Farrell 1994; Lassar et al. 1994; Boulikas 1995). This process requires inductive signals from the environment coincident with or after cell cycle withdrawal. Failure to coordinate growth arrest and differentiation may result in a failure to form normal tissues because cells either continue to proliferate (Kipreos et al. 1996) or undergo apoptosis (Boudreau et al. 1996). Furthermore, growth arrest within the context of differentiation may be separated into two events: one in which the proliferating cell withdraws from the cell cycle to allow changes in gene expression and the other in which the differentiated cell is prevented from reentering the cell cycle. It is unclear what changes in the expression of cell cycle regulatory molecules reflects the establishment of the growth arrest state, or its maintenance, or both.
Progression of a cell through G1 and into S phase is dependent on the coordinated activation of two cyclin-dependent kinases (CDKs), CDK4/6 and CDK2 (Resnitzky and Reed 1995; Sherr and Roberts 1995). Interference of either kinase is sufficient to lead to growth arrest because regulatory networks coordinate CDK activity and entry into S phase (Koff 1995). Activation of each kinase is a highly regulated process beginning with the association of a CDK subunit with a positive regulatory subunit, cyclin E with CDK2 (Koff et al. 1992) and cyclin D with CDK4/6 (Matsushime et al. 1992; Xiong et al. 1992; Meyerson and Harlow 1994), and followed by phosphorylation of the CDK subunit by the CDK-activating kinase (Fisher and Morgan 1994; Matsuoka et al. 1994). The activation process may be regulated negatively by other phosphorylation events on the CDK subunit (Morgan 1995) and by the interaction of the cyclin/CDK complex with other proteins (Sherr and Roberts 1995), specifically the CDK inhibitors (CKI), Inks (p15, p16, p18, and p19), and Kips (p21, p27, and p57). Ink proteins are specific inhibitors of the cyclin D/CDK complex and interfere with the activity either in ternary complexes (Reynisdóttir et al. 1997) or by competing with cyclin D for the CDK subunit (Hirai et al. 1995). Kip proteins are strong inhibitors of both CDKs in vitro (Harper et al. 1993; Polyak et al. 1994; Toyoshima and Hunter 1994; Lee et al. 1995; Matsuoka et al. 1995). However, p27 may be more selective for CDK2 compared to CDK4 in vivo (Soos et al. 1996; Hauser et al. 1997). The solution of ternary cyclin A/CDK2/p27 crystals suggests that p27 prevents ATP access or the ability to coordinate ATP (Russo et al. 1996).
Differentiation is associated with a reduction in the overall amount of G1 CDK activity. Ectopic expression of D-type cyclins can prevent differentiation in some cell lines derived from muscle (Skapek et al. 1995) and hematopoietic (Kato and Sherr 1993) lineages. Reciprocally, the introduction of p21 or p27 into neuronal (Kranenburg et al. 1995), hematopoietic (Liu et al. 1996), and muscle (Guo et al. 1995) precursor cells enhances their differentiation. Consistent with these observations, the accumulation of CKIs has been noted in many terminally differentiated cell types in mice (Matsuoka et al. 1995; Parker et al. 1995). Expression of p21 mRNA is greatest in the liver, muscle, and epithelium of the intestine and stomach, and p57 mRNA levels are highest in the kidney, muscularis mucosae in the stomach, and muscle. p27 mRNA expression studies are of limited utility as post-transcriptional mechanisms regulate the expression of this protein (Pagano et al. 1995; Agrawal et al. 1996; Hengst and Reed 1996; Millard et al. 1997). We and others detected p27 protein in a subset of developing thymocytes (Hoffman et al. 1996), in neurons (Lee et al. 1996), in luteal cells of the ovary (Kiyokawa et al. 1996), in mature T-cells (Firpo et al. 1994), and in protein extracts from all organ systems—although not necessarily in all cell types in each tissue (Kiyokawa et al. 1996; Nakayama et al. 1996). Consistent with a role in the development of all organs, mice deficient for the CDK inhibitory function of p27 display enhanced growth (Fero et al. 1996; Kiyokawa et al. 1996; Nakayama et al. 1996). Growth is not attributable to alterations in the level of growth hormones, insulin-like growth factor I (IGF-I) and IGF-II, or adrenocorticotropic hormone (ACTH), endocrine factors known to affect large numbers of target tissues. Rather, growth correlates with an increase in the percentage of cycling cells in tissues undergoing maturation, suggesting that loss of p27 might lead to additional cell divisions before withdrawal from the cell cycle (Kiyokawa et al. 1996). However, it was unclear whether the increase in proliferation was a consequence of failure to withdraw from the cell cycle and differentiate in a timely fashion. No obvious cell cycle defects were observed in fibroblasts obtained from embryonic p27−/− mice (Fero et al. 1996; Nakayama et al. 1996). Recently, others have described hyperproliferative phenotypes in Caenorhabditis elegans and Drosophila melanogaster and have mapped these to the cul-1 (Kipreos et al. 1996) and dacapo (Lane et al. 1996) loci, respectively. Although there is no significant homology between cul-1 and p27, dacapo has strong homology in the cyclin/CDK-binding domain. Consistent with this, the loss of function phenotypes are more similar for dacapo and p27 than for cul-1 and p27.
O-2A bipotential precursor cells have the capacity to differentiate into either oligodendrocytes or astrocytes (Temple and Raff 1985; Raff 1989). These cells can be isolated from the cortex of neonatal mice and maintained in a proliferative state in medium conditioned by the B104 neuroblastoma cell line (Temple and Raff 1985; Barres et al. 1994; Casaccia-Bonnefil et al. 1996). Addition of basal fibroblast growth factor (bFGF) and platelet-derived growth factor (PDGF) can replace the B104-conditioned medium. O-2A cells are identified readily by bipolar morphology, reactivity to A2B5 antibodies (Raff et al. 1983), and reactivity to NG2 antibodies (Nishiyama et al. 1996). In vitro, in the presence of thyroid hormone and mitogens, these cells divide and the amount of p27 increases with each division; eventually, the cells stop proliferating and differentiate into oligodendrocytes (Durand et al. 1997). In addition, culture of O-2A cells in serum-free conditions with thyroid hormone induces growth arrest and differentiation into oligodendrocytes (Barres et al. 1994). Oligodendrocytes are detected as cells with highly branched processes that express myelin basic protein (MBP) and galactocerebroside (GalC) (Raff et al. 1983), but do not express NG2 (Nishiyama et al. 1996).
CG-4 cells, an immortalized cell line derived from a bipotential rat O-2A cell, differentiate along the type II astrocyte lineage when deprived of conditioned medium but maintained in serum (Louis et al. 1992). Differentiation correlated with accumulation of p27 and a concomitant loss of cyclin E/CDK2 kinase activity suggesting that p27 may play an important role in the commitment decision of glial progenitors (Tikoo et al. 1997). However, these cells differentiated poorly into oligodendrocytes and we were unable to address whether p27 had an obligatory role in oligodendrocyte differentiation.
To investigate the consequence of loss of p27 function in the glial lineage, we followed the differentiation of O-2A cells obtained from p27−/− mice into oligodendrocytes. We report that under conditions that promote differentiation of wild-type cells, cells obtained from p27−/− mice have impaired growth arrest after mitogen removal. This defect in growth arrest was not sufficient to eliminate the oligodendrocyte lineage as a fraction of cells continue to form oligodendrocytes morphologically indistinguishable from those of wild-type cells. Impaired growth arrest correlated with continued progression of O-2A cells into S-phase, directly demonstrating for the first time that p27 is part of the circuitry deciding whether cells should commit to the cell cycle or withdraw—the restriction point. Consistent with the hypothesis that continued cycling of the O-2A cells will expand the number of precursors before differentiation, we detected a substantial increase in the number of oligodendrocytes and type I astrocytes in animals early in postnatal development. This correlated with increased MBP and proteolipid (PLP) production. These data suggest that p27 is an important component of the machinery that regulates withdrawal of O-2A cells during G1 traverse.
Results
p27 expression correlates with withdrawal of O-2A cells from the cell cycle
Environmental signals control the decision of cells to commit to a round of cell division or withdraw from the cell cycle and undergo differentiation. The commitment decision is regulated positively by the activity of G1 CDKs and negatively by the concentration of stoichiometric CDK inhibitory proteins Inks and Kips (Sherr and Roberts 1995). To determine the changes in the cell cycle machinery that occur during mitogen withdrawal-induced differentiation of O-2A cells into oligodendrocytes, we examined the expression of various cyclins, CDKs, and CKI during differentiation of primary O-2A precursors.
We obtained O-2A precursor cells by selective shaking of mixed glial cultures and maintained them in a proliferative state by culturing in B104-conditioned medium (McCarthy and DeVellis 1980). More than 90% of the shaken cells were identified positively as oligodendrocyte precursors on the basis of their characteristic bipolar morphology and immunoreactivity with the O-2A markers A2B5 and NG2. The contaminating cells were MBP- and glial fibrillary acidic protein (GFAP)-positive astrocytes. After 5 days of serum-free culture in the presence of thyroid hormone, 90–95% of the cells had a highly branched morphology, were not reactive with either A2B5 or NG2 antibodies, and were reactive with GalC (O1)- and MBP-specific antibodies. These cells were classified as oligodendrocytes (Casaccia-Bonnefil et al. 1996).
After differentiation to oligodendrocytes, we examined the expression of G1 CDKs (CDK2, CDK4, CDK5), CKIs (p16, p21, p27), and cyclins (D-type cyclins and cyclin E) by immunoblotting. Overall the amount of CDK2 and CDK4 and all G1 cyclins decreased, whereas the amounts of CDK5, p21, and p27 increased (Fig. 1A). The level of p16 did not change (Fig. 1A). These data suggest that in agreement with the CG-4 cell model, the differentiation of primary glial precursors correlates with increased expression of the CDK inhibitor p27 and a loss of G1 cyclins and CDKs. Together these events might conspire to establish the nonproliferative state, maintain the nonproliferative state, or both.
Figure 1.
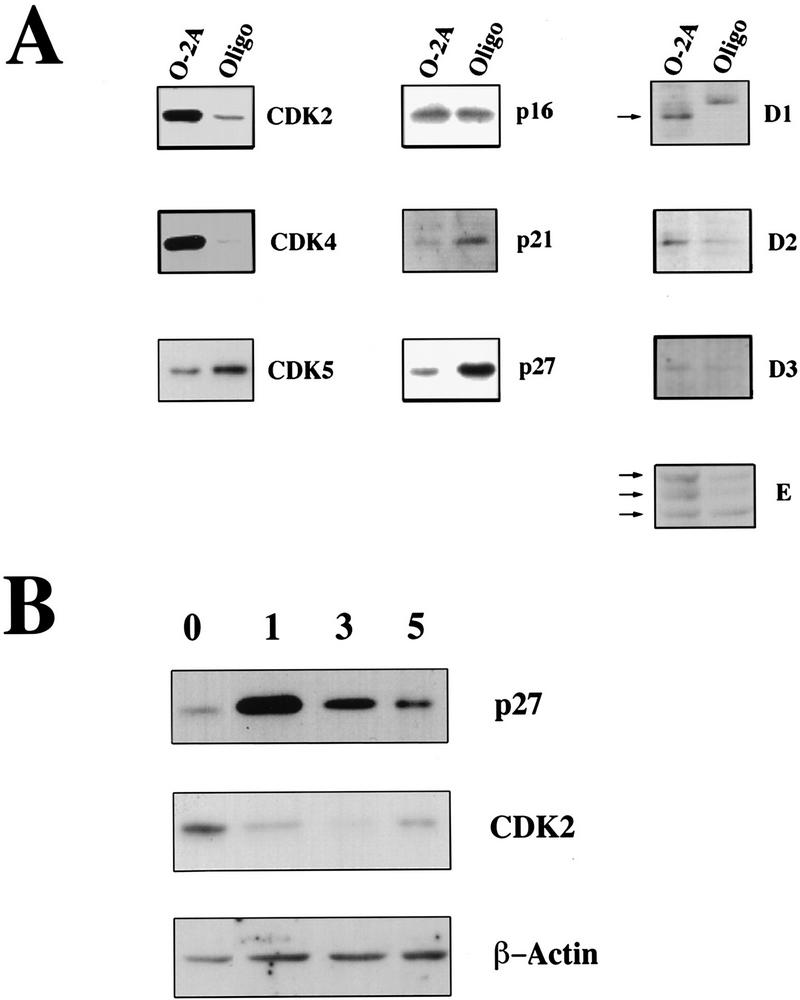
Expression of cell cycle proteins in oligodendroglial progenitors and differentiated oligodendrocytes. (A) Changes in the amount of G1 regulatory proteins during oligodendrocyte differentiation. Protein extracts (20–80 μg, depending on the antibody used) obtained from proliferating cortical O-2A precursors (O-2A) or oligodendrocytes maintained in differentiation medium for 5 days (Oligo) were analyzed by Western blot analysis using antibodies indicated on the right of each panel. (B) The protein levels of p27 change during oligodendrocyte differentiation. Protein extracts isolated from O-2A precursors (0) and oligodendrocytes kept in differentiation medium for 1, 3, or 5 days were assayed by Western blot analysis using anti-p27 or anti-CDK-2 antibodies. β-Actin was used as control for protein loading.
To determine whether p27 was involved in either the establishment of growth arrest, or its maintenance (or both), we examined the expression of p27 and CDK2 protein during the differentiation process and correlated this with the expression of an oligodendroycte differentiation marker GalC. Expression of p27 was lowest in the proliferating O-2A cells, maximal the first day of serum-free culture, and decreased thereafter (Fig. 1B). On the other hand, we observed a rapid loss of CDK2 and its expression remained low throughout the time course. As a control for protein loading we measured the amount of actin. Consistent with the rapid increase in p27 and loss of CDK2, we found that after 24 hr in serum-free medium the percentage of cycling cells, as judged by incorporation of bromodeoxyuridine (BrdU), dropped from 80% ± 11% (n = 6) to 15.2% ± 1.6% (n = 3) within a day and was at its lowest by 5 days (n = 3). We detected GalC in 2.6% ± 2% of the cycling precursor cell population (n = 6) and this did not increase concomitantly with growth arrest. At 24 hr (n = 3) only 3.5 ± 1.8% of the cells were GalC positive. However, after 5 days of serum deprivation 87 ± 1.2% of the cells were positive (n = 3). Together these data suggest that the increase in p27 represented an early event establishing withdrawal of cells from the cell cycle.
Commitment decisions in O-2A precursors deficient of p27
These data suggested that p27 might participate in establishing growth arrest of O-2A cells. Previous work from our laboratory reported that mice lacking p27 displayed an increase in the number of many cell types, attributable in part to an increase in the proliferation of cells undergoing postnatal maturation (Fero et al. 1996; Kiyokawa et al. 1996; Nakayama et al. 1996). However, we were unable to demonstrate that the increase in S-phase cells, in either developing thymocytes or the cells of the intermediate lobe of the pituitary, occurred by altering the efficiency of the G1 → G0 transition. Consequently, we investigated the process of O-2A cell differentiation further to determine whether loss of p27 affects the commitment function. Does the loss of p27 lead to inappropriate proliferation at the expense of differentiation?
We obtained mixed glial cultures from cortical dissections of neonatal mice and after 7–10 days of culture we separated O-2A precursors from the type I astrocytes by differential shaking. Equal numbers of O-2A cells from wild-type and knockout animals were plated and induced to differentiate by withdrawing mitogens. First, we determined the percentage of cycling cells by BrdU labeling the day of isolation. At the beginning of the experiment, a greater percentage of p27−/− cells passed through S-phase over a 6-hr period (Table 1; Fig. 2a,b; p27−/−, n = 3; p27+/+, n = 5), suggesting that the loss of p27 increased the fraction of S-phase cells. We did not detect a change in cell size (cf. the phase contrast in Fig. 2c,d). To confirm that the cycling cells were O-2A precursors and not a novel cell type specifically enriched in p27−/− cultures, we double-stained cells for BrdU and NG2, a marker for O-2A precursors. Of the BrdU positive cells, 85–90% were also NG2 positive indicating that they were O-2A precursors (Fig. 2e,f).
Table 1.
Percentage of BrdU-incorporating cells
| Genotype
|
Day 0
|
Day 5
|
|---|---|---|
| +/+ | 31 ± 6 | 10 ± 3 |
| −/− | 48 ± 7 | 51 ± 15 |
Increased proliferation of p27−/− cells. Cultures received a 6-hr pulse with 10 μm BrdU either the day of plating (day 0) or after 5 days in serum-free differentiation medium (day 5). Labeled cells were identified by BrdU immunoreactivity. The percentage of cycling cells was calculated as number of BrdU-positive cells vs. total cell number (n = 3, p27−/−; n = 5, p27+/+).
Figure 2.
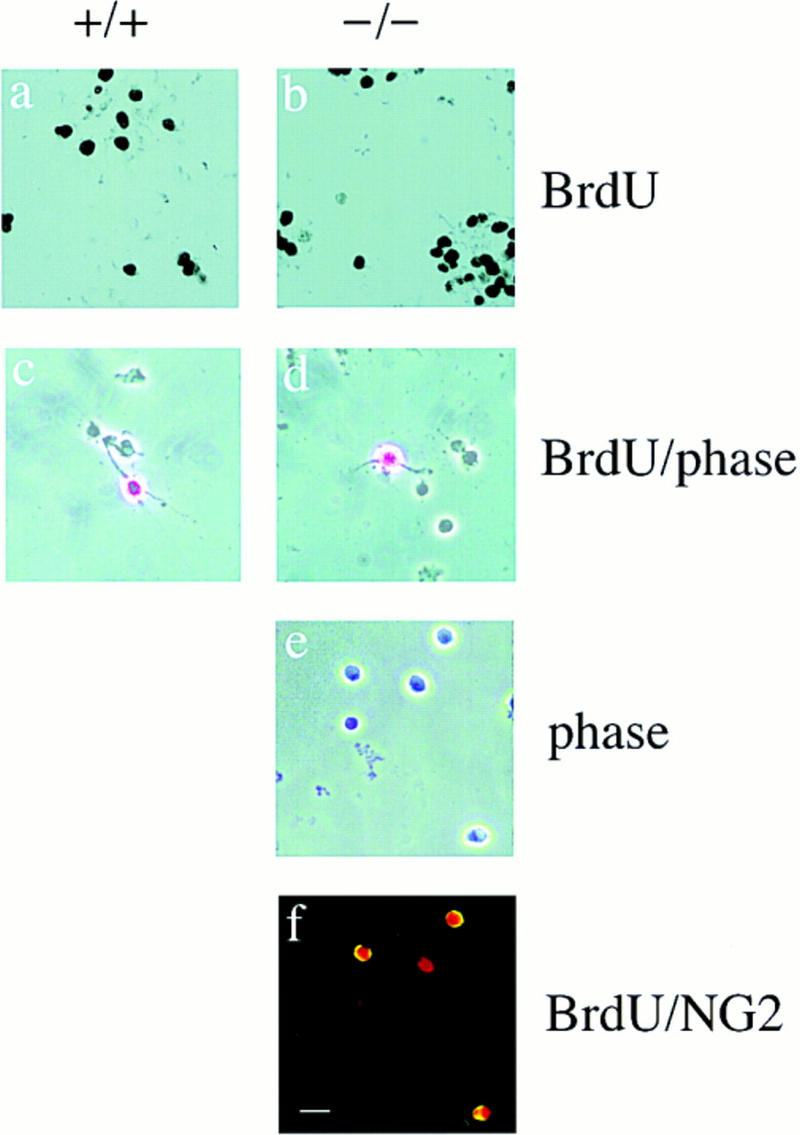
Enhanced proliferation of O-2A progenitors in p27 null cultures on day 0. Bright field (a,b) and phase contrast (c,d) photomicrographs of p27+/+ and p27−/− cultures pulsed with 10 μm BrdU for 6 hr on day 0 of plating. Labeled cells were visualized by immunocytochemistry using anti-BrdU antibody. The phase contrast appearance of p27−/− and p27+/+ BrdU-positive cells reveals no difference in cell size. On day 0 of plating, the majority of the cells were round with simple or no processes (c–e). The identity of the cycling cells as proliferating O-2A progenitors was determined by double labeling with BrdU (Texas red) and NG2 (FITC) (f). Bar, 40 μm.
Next, we measured the proliferation of cells 5 days after withdrawing mitogens. By this time, there was a dramatic decrease in the proportion of cycling cells in the wild-type cultures, which did not occur in the cultures derived from p27-deficient mice (Table 1; Fig. 3a,b). The majority of cells in the wild-type culture were differentiated oligodendrocytes as determined by the following three criteria: (1) they did not incorporate BrdU (Fig. 3c); (2) they displayed a highly branched morphology (Fig. 4a,c); and (3) they were MBP positive (Fig. 5k).
Figure 3.
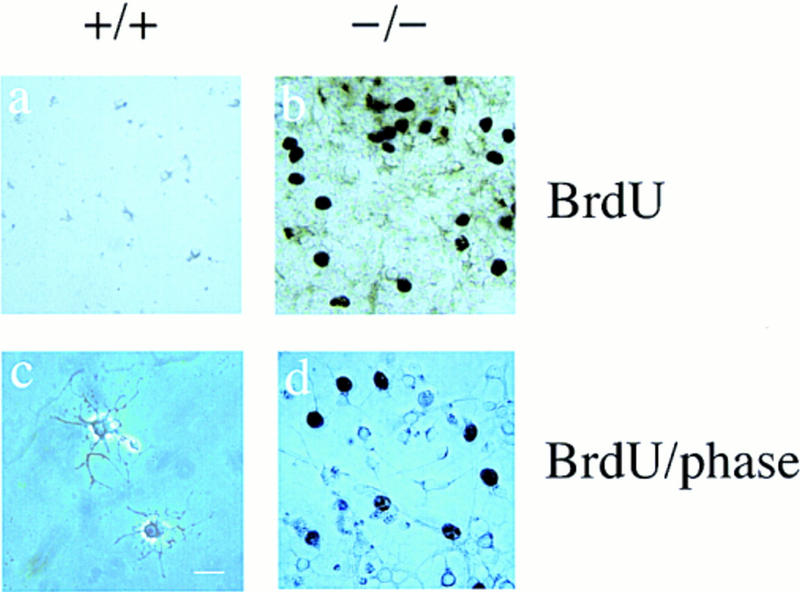
Enhanced proliferation of O-2A progenitors in p27 null cultures after 5 days of mitogen withdrawal. Bright field (a,b) and phase contrast (c,d) photomicrographs of p27+/+ (+/+) and p27−/− (−/−) cells cultured for 5 days in the absence of serum and mitogens. After a 6-hr pulse with 10 μm of BrdU, cells were fixed, stained with antibodies against BrdU, and visualized using diaminobenzidine as substrate. Cycling cells were still observed in the p27−/− cultures (b). Phase contrast reveals that these p27−/− cycling cells have a simple, bipolar morphology (d), whereas the highly branched cells in either p27+/+ (c) or p27−/− (d) cultures do not incorporate BrdU. Bar, 40 μm.
Figure 4.
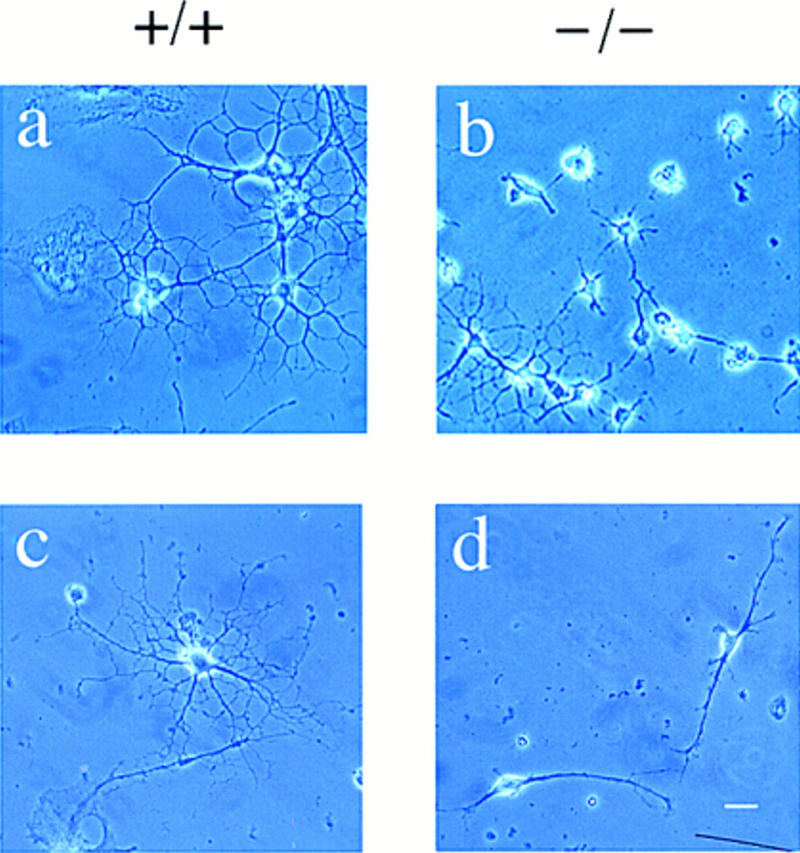
Impaired differentiation of p27−/− oligodendrocyte progenitors. Phase contrast photomicrographs of cultures obtained from p27+/+ (a,c) and p27−/− mice (b,d) maintained in serum-free differentiation medium for 5 days. The majority of p27+/+ precursors differentiate into mature oligodendrocytes, identified by the characteristic highly branched morphology (a,c). In contrast, the majority of p27−/− cultures are characterized by cells with few simple processes (b,d). (a,b) Bar, 30 μm; (c,d) bar, 40 μm.
Figure 5.
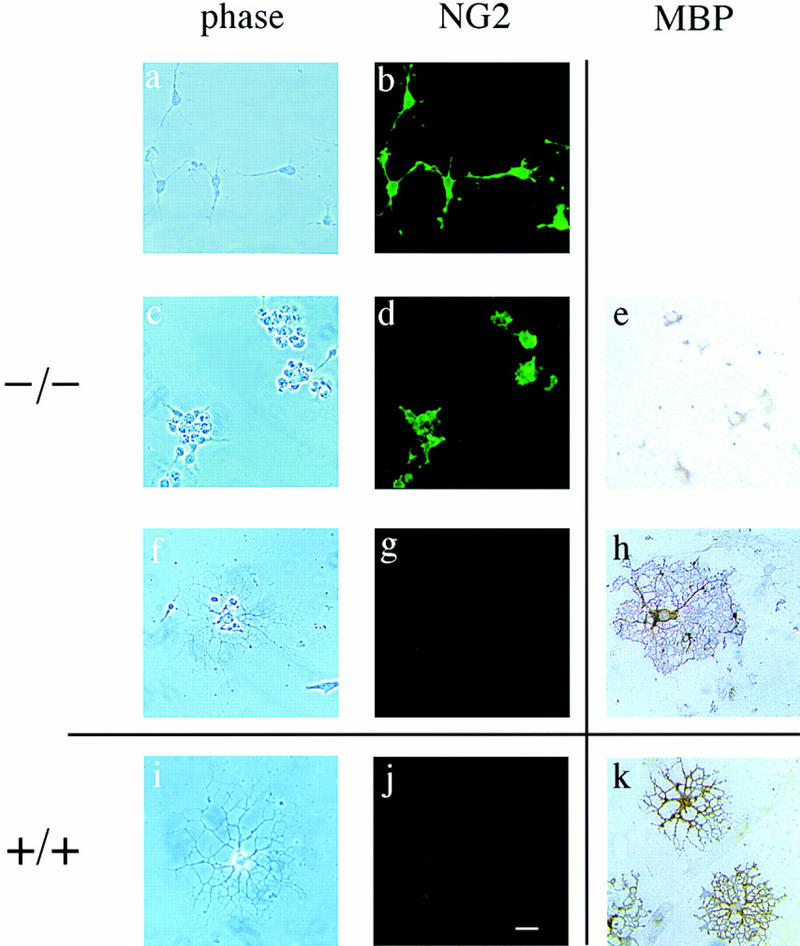
Immunocytochemistry of p27−/− cultures reveals primarily oligodendrocyte progenitors after 5 days in differentiation conditions. Cells from p27−/− (−/−) and p27+/+ (+/+) mice were cultured for 5 days in serum-free differentiation medium and then stained with antibodies against the precursor marker NG2 (b,d,g,j) or the differentiation marker MBP (e,h,k). Phase-contrast photomicrographs are shown only for the cultures stained with anti-NG2 antibodies (a–d,f–g,i–j). The majority of p27−/− cells growing either in isolates (a) or in clusters (c) were oligodendrocyte precursors, as indicated by the simple morphology, NG2 immunoreactivity (b,d), and lack of MBP expression (e). Differentiated oligodendrocytes in the p27−/− (f–h) and in the p27+/+ (i–k) cultures were identified by loss of NG2 immunoreactivity (g,j) and positive MBP immunoreactivity (h,k). Bar, 40 μm.
In contrast, there were three distinct cell types in p27−/− cultures: bipolar cells, small round cells growing either isolated or in cultures (Figs. 4, b and d, and 5, a and c), and highly branched cells with elaborate processes (Fig. 4b; Fig. 5f). Of these cell types, only the bipolar and small round cells incorporated BrdU (Fig. 3d). To determine the identity of these cells, we examined the expression of NG2 and MBP, markers of O-2A precursors and oligodendrocytes, respectively. The highly branched cells that did not incorporate BrdU were unreactive to NG2 antiserum and expressed MBP consistent with identification as oligodendrocytes (Fig. 5i–k). The small round cells bearing few or no processes labeled with the anti-NG2 antibody and were unreactive to the MBP antibody (Fig. 5a–h), consistent with identification as oligodendrocyte precursors. Some of these cells incorporated BrdU and some did not. In summary, although it is clear, by both morphological and immunocytochemical criteria, that the BrdU-positive cells are O-2A precursors that failed to differentiate, the BrdU-negative and NG2-positive cells might represent an early stage of oligodendrocyte differentiation or a population of cells in which differentiation was delayed.
Next, we assessed the percentage of differentiated cells in wild-type and p27−/− cultures kept for 5 days after mitogen withdrawal by counting the number of O-2A precursors (NG2-positive cells bearing no or few processes) and mature oligodendrocytes (highly branched MBP-positive cells). In wild-type cultures, only a small fraction were precursors (Table 2). In contrast, p27−/− cultures consisted predominantly of O-2A precursors (Table 2). We conclude that in p27−/− cultures the majority of cells were still at a precursor stage and the ability of p27−/− cells to withdraw from the cell cycle was perturbed. Regardless, it is clear that the commitment event was not abrogated completely by p27 disruption as some cells did differentiate. We speculate that p27 is only part of the commitment machinery and that other events such as the down-regulation of CDK2 might be involved.
Table 2.
Progenitors in cultures differentiated for 5 days
| Genotype
|
Number of precursors counted per field
|
Percentage of cells that are precursors
|
|---|---|---|
| +/+ | 1.0 ± 0.3* | 15 ± 5** |
| −/− | 10.5 ± 3.9* | 62 ± 8** |
The majority of p27 −/− cells in cultures differentiated for 5 days are oligodendrocyte precursors. Cultures were maintained in serum-free differentiation medium for 5 days, and the number of process-bearing cells was quantitated on each coverslip (n = 17 cultures from 8 p27 +/+ mice; n = 15 cultures from 6 p27 −/− mice). Progenitors were defined by the presence of simple short processes and NG2 immunoreactivity (Figs. 4 and 5). The raw numbers of progenitors counted per field are shown in the first column. Percentages were calculated by normalizing the number of progenitors to the total number of process-bearing cells and are shown in the third column. The results are expressed as mean ± s.e.m. The P value indicates statistical significance using Student’s t-test. (*) P = 0.02; (**) P = 0.0001.
Increased numbers of glial cells in p27−/− mice
Next, we examined the numbers of neuronal and glial cells in the brain of p27−/− mice. To accomplish this we examined sagittal brain sections of adult mice (p27−/−, n = 3; p27+/+, n = 3) and counted the numbers of cells expressing either mitogen-associated protein 2 (MAP2) or GFAP, markers of neuronal and astrocytic cells, respectively. Consistent with the reports of others, the numbers of MAP2-staining neurons in the CA1 region of the hippocampus increased 30% ± 8%. The numbers of GFAP-staining cells, type I astrocytes, was 250% ± 10% higher in the hippocampus and in the cerebellum of the same animals.
It is difficult to quantitate the numbers of oligodendrocytes in these regions of adult brain because available antibodies stain the cell processes and not the nucleus. To circumvent this problem we counted the number of toluidine blue staining cells in 1 μm transverse sections of the optic nerve cut 1 mm in front of the optic chiasm in both p27−/− and control mice (Fig. 6). Glial cell bodies are the primary cell type in this region of the optic nerve. We quantitated the cell number in random fields of several sections. Surprisingly, in 6-day-old mice (n = 4) the number of glial cells was 262% ± 31% higher than in controls; however, at 9 weeks (n = 8) glial cells were only 50% ± 10% greater in p27−/− compared to controls. Consequently, the loss of p27 led to an increase in the numbers of glial cells in vivo.
Figure 6.
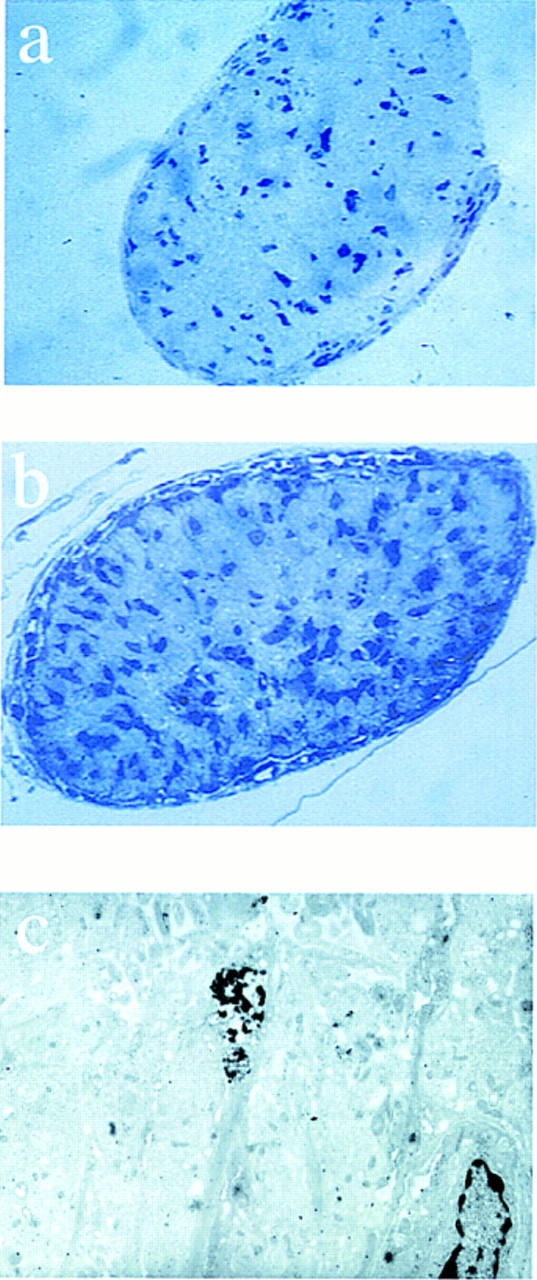
Glial cell number is increased in the optic nerve. Semithin sections (a,b) and electron microscopy of optic nerve from 6-day-old neonatal mice (c). Sections were cut 1 mm anterior of the optic chiasm and stained with toluidine blue and the number of nuclei were counted. Representative sections are shown for p27+/+ (a) and p27−/− mice (b). Electron microscopy of ultrathin sections from p27−/− mice revealed the presence of several apoptotic nuclei. A representative field (c) showing two nuclei (lower right and center of the field), one of which is undergoing apoptosis (center of the field).
The finding that oligodendrocyte differentiation efficiency was decreased in vitro, but oligodendrocyte number increased in vivo might be explained by either accumulation of oligodendrocytes that do not express MBP in vivo (what we define as nonfunctional), or increased proliferation of the O-2A precursor pool before differentiation. To address these alternatives we examined the expression of MBP in adult mouse brain and the numbers of O-2A cells.
Myelination in p27-deficient mice
To address the functional state of oligodendrocytes we examined MBP expression by Western blot and immunohistochemistry. We prepared crude brain homogenates and measured the steady state amount of myelin-specific proteins MBP and PLP by immunoblotting after normalization of proteins by weight. In p27−/− mice both MBP and PLP increased in the membrane fractions (Fig. 7). This increase might reflect either increased numbers of functional oligodendrocytes or increased biosynthetic activity of the oligodendrocytes.
Figure 7.

Expression of MBP and PLP are increased in p27−/− mice. Western blot analysis of cytosolic (s, supernatant) and membrane fractions (p, pellet) obtained by differential centrifugation of whole brain homogenates from p27 null (−/−) mice and wild-type (+/+) controls. Equal amount of proteins (100 μg) were loaded and separated by SDS-PAGE. Purified rat myelin (10 μg) was used as a positive control.
Consistent with Western blotting, we observed increased MBP immunoreactivity in brain sections from 6-day-old wild-type and p27−/− mice (n = 3 for each group). We observed an increase in MBP expression in the brainstem and cerebellum (data not shown). Similarly, we observed increased MBP immunoreactivity in 9-week-old animals (n = 4 for each group) (Fig. 8). Electron micrographs of the optic nerve revealed normal patterns of myelination (data not shown). Together these data suggest that oligodendrocytes formed in p27−/− mice were normal with regard to myelination.
Figure 8.
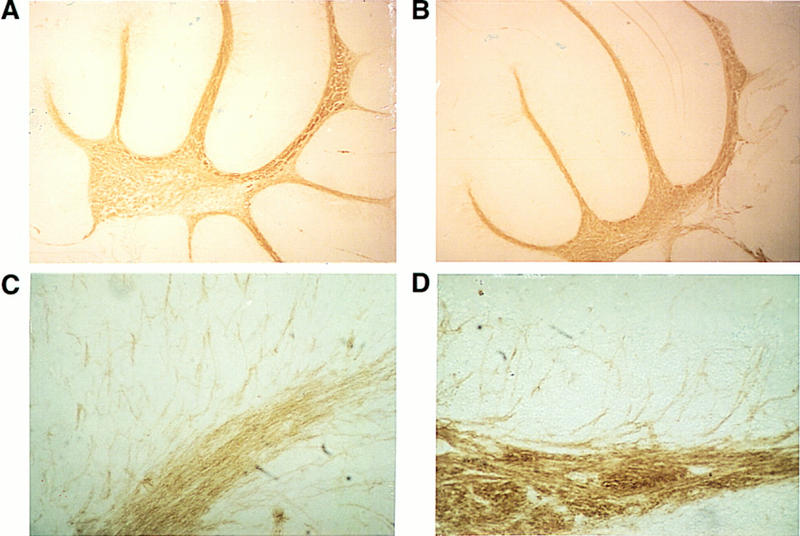
Enhanced MBP immunoreactivity in p27−/− mice. Immunohistochemical analysis of sagittal brain sections from 9-week-old p27+/+ (A,B) and p27−/− (C,D) mice. Increased MBP expression in the cerebellum at low (A,C) and high (B,D) magnification. (A,C) Bar, 300 μm; (B,D) bar, 42.5 μm.
Another possibility was that the absence of p27 may affect neuronal/glial communication making the oligodendrocytes less dependent on neuronal survival signals. However, this is unlikely because we observed apoptotic cells in electron micrographs from the 6-day-old optic nerve of p27−/− mice (Fig. 6c) and numbers of glial cells normalized to neuronal number by 9 weeks. This observation is consistent with the proposal that the number of oligodendrocytes is determined by axonal contact in the optic nerve (Raff et al. 1993; Burne et al. 1996) and in the absence of these factors oligodendrocytes are likely to be eliminated by programmed cell death. Together these data suggest that oligodendrocytes formed in p27−/− mice are normal with regard to their requirement for neuronal survival factors.
The increase in oligodendrocytes is attributed to an increase in the number of precursor cells
Another mechanism that may explain the early postnatal increase in oligodendrocyte number is an expansion of the O-2A precursor population. In this case, the reduction in differentiation efficiency with continued cycling, observed in vitro, might lead to an increase in the size of the precursor pool in vivo. To address whether the number of O-2A precursor cells was affected in p27−/− mice, we counted the O-2A and type I astrocytes in mixed glial cultures obtained from the brains of animals. We identified O-2A precursors in the mixed glial cultures by three criteria: (1) positive staining for A2B5; (2) distinctive bipolar morphology; and (3) the absence of reactivity to antibodies specific for GFAP and GalC. Cultures obtained from p27−/− mice had a sevenfold increase in O-2A precursors compared to wild-type mice (p27−/−, n = 6; p27+/+, n = 10) (Fig. 9). In addition, the numbers of GFAP-staining cells, representing type I astrocytes, increased 2.8-fold. The magnitude of this increase was similar to that observed for the astrocytes counted in sagittal brain sections. Consequently, the loss of p27 affects the proliferation of O-2A cells, both in vivo and in vitro.
Figure 9.

The number of glial cells is increased in p27−/− mice. The bar graph indicates the number of O-2A cells (counted after differential shaking) and type I astrocytes (isolated from the same mixed glial culture), normalized to the number of cells counted immediately after dissection. Solid bars indicate p27+/+; open bars, p27−/− cells.
Discussion
Growth arrest and differentiation of muscle, neuronal, and hematopoietic cells require down-regulation of CDK activities (Kato and Sherr 1993; Guo et al. 1995; Kranenburg et al. 1995; Skapek et al. 1995; Liu et al. 1996). O-2A precursor cells differentiate to oligodendrocytes either by following an intrinsic counting mechanism that is coupled to cell division (“intrinsic clock”) (Raff et al. 1985, 1988), or by removing the mitogens required for continued proliferation (Barres et al. 1994). Recently, in cells undergoing the intrinsic program, Durand and colleagues (1997) correlated oligodendrocyte differentiation with an increase in p27 protein. At present, we demonstrate that the CDK inhibitor p27 facilitates the differentiation of oligodendrocytes; in the absence of p27, O-2A cells differentiate poorly when deprived of mitogens. The failure to differentiate correlated with continued cycling of O-2A precursor cells leading to an expansion of these cells. In vivo we detected myelinated axons and an increase in the amount of myelin components, attributable to an increase in the number of oligodendrocytes. The discrepancy, vis-á-vis terminal differentiation, between the in vivo and in vitro observations can be reconciled by postulating that an increase in O-2A precursors (as a result of increased cycling before terminal differentiation) occurs in vivo, and that the in vitro conditions are not able to substitute fully for inductive or survival signals normally present in vivo. In addition to delayed withdrawal of the O-2A precursor, increased seeding of the O-2A compartment by a progenitor cell also impacts on the number of O-2A precursors (P. Casaccia-Bonnefil, M.V. Chao, and A. Koff, unpubl.). Hence, we can conclude that p27 plays an important role establishing growth arrest to allow differentiation in mitogen-deprived O-2A cells, and that other pathways impinge on this process. These results indicate for the first time that loss of p27 affected proliferation of cells at the expense of differentiation. Hence, p27 is involved in the machinery that controls withdrawal of cells from the cell cycle, at least in O-2A cells.
p27 is likely to be an effector of the differentiation program of oligodendrocytes
In O-2A cells, precursors to oligodendrocytes, differentiation may be induced either by withdrawing mitogens, or by an intrinsic program (a “clock”) that is activated after a discrete number of divisions. The clock postulates the existence of a molecular component that can count the number of divisions. The “counter” is linked to an effector, which initiates cell cycle arrest and differentiation. The relationship between these pathways is obscure and mitogens might suppress the effector, rather than being on a completely distinct pathway. The similarities of the clock and replicative senescence, and of mitogen withdrawal to G1 arrest make O-2A cells an attractive model for studying the roles of CKI in differentiation. There is a substantial amount of data supporting the involvement of the p16 and p21 proteins in senescence (Noda et al. 1994; Stein and Dulic 1995; Tahara et al. 1995; Hara et al. 1996). Thus, we predict that the clock might incorporate pathways shared by p16 and p21. Furthermore, we predict that if the clock and mitogen pathways are related, the effector component of the clock would be shared between the mechanisms.
Of three CDK inhibitors we measured during oligodendrocyte differentiation, only p27 expression was changed dramatically. We were surprised that we did not observe a change in p16 levels as mutation of p16 is associated with malignant glioma and the overexpression of p16 can prevent U87 glioblastoma proliferation (Medema et al. 1995; Poulos et al. 1996). The lack of an increase in p16 suggests that it may be involved in mediating the intrinsic program. The intrinsic program relies on the experimental observation that O-2A cells divide a limited number of times before differentiating into oligodendrocytes (Barres et al. 1994; Bögler and Noble 1994). This is reminiscent of cells undergoing a programmed senescence and suggests that those proteins involved in senescence, such as p21 and p16 (Noda et al. 1994; Stein and Dulic 1995; Tahara et al. 1995; Hara et al. 1996), may play a greater role in the establishment of growth arrest and terminal differentiation by the intrinsic mechanism.
Several approaches have concluded that p27 levels increase during oligodendrocyte differentiation (this study; Durand et al. 1997). Durand et al. (1997) have shown that p27 levels are elevated as O-2A cells age in culture, correlating with the eventual cessation of growth and ultimately, differentiation of cells. Furthermore, we have shown that mitogen withdrawal correlates with induction of p27, cessation of growth and differentiation, and the absence of p27 affects the process of growth arrest. To accomodate these observations we suggest that p27 might be a nodal point where the differentiation of oligodendrocytes, whether by mitogen-induced or intrinsically programmed pathways, come together (Fig. 10). We speculate that p27 is part of the effector mechanism, and that mitogens suppress the induction of the p27 and block the clock program, suggesting that other changes in the cell cycle—or the machinery that controls p27 expression—might be the actual counting mechanism.
Figure 10.

p27 has a central role in differentiation of oligodendrocytes. This model postulates that p27 plays a role in both the intrinsic clock and the mitogen withdrawal differentiation pathway.
The role of p27 in oligodendrocyte differentiation
Consistent with an important role for p27 in the differentiation of oligodendrocytes we observe that this process is perturbed in the O-2A cells obtained from p27−/− mice. At a time when growth arrest and differentiation occur in wild-type cultures, a large proportion of cells obtained from p27−/− mice are still cycling, as evidenced by incorporation of BrdU. Other cells have clearly withdrawn from the cell cycle but have not elaborated markers specific to the oligodendrocyte lineage, such as MBP, and a small percentage have stopped dividing and undergo differentiation. Although we are unable to establish the direct linkage between these three cell types, it seems likely that the “noncycling, MBP-negative, NG2-positive” cells are delayed in differentiation (Gard and Pfeiffer 1989, 1990), and the culture conditions used may not provide sufficient factor to sustain these cells longer. Alternatively, these cells might represent a differentiation dead-end, one not normally observed because p27 prevents cells from embarking on this pathway.
If the loss of p27 delays the eventual withdrawal of cells from the cell cycle, then one predicts that there would be an expansion of precursor cells before differentiation. This expansion might allow an increase in the number of cells elaborating the terminally differentiated phenotype in the animal. Consistent with this, we observed an increased number of oligodendrocytes in the optic nerve in vivo and increased myelination in the optic nerve (data not shown) and in other regions of the central nervous system. Perhaps in vivo other signals acting on other p27-independent pathways preserve the differentiation functions not preserved in the in vitro system. Ultimately, homeostatic mechanisms can normalize cell number in organ systems. For example, neuronal cells may provide inductive stimuli. It has been proposed that the number of oligodendrocytes is determined by axonal contact providing signals that promote their growth and survival (Raff et al. 1993).
The studies in O-2A cells documented here suggest that p27 is an important component of the commitment machinery. p27 may operate within the cell cycle to establish the rate of G1 passage and alterations in G1 duration can affect the ability of cells to respond to anti-mitogenic signals that withdraw cells from the cell cycle. Overexpression of Cln3 in yeast (Cross 1988), overexpression of cyclin E in mammalian cells (Ohtsubo and Roberts 1993), cul-1 deficiencies in C. elegans (Kipreos et al. 1996), deficiency of p27 in mice (Fero et al. 1996; Kiyokawa et al. 1996; Nakayama et al. 1996), deficiency of p27 in 3T3 fibroblasts (Coats et al. 1996), and deficiency of dacapo, a p27-related CDK inhibitor in Drosophilia (Lane et al. 1996), diminish the response of cells to environmental signals. Thus, the gain of rate-limiting regulators of S-phase entry, the cyclins, and the loss of p27 yield similar proliferation and differentiation phenotypes. However, overexpression of cyclins or deletion of cul-1 also affects cell size, whereas the loss of p27 or dacapo do not, suggesting that the cyclins and p27 might have discrete as well as overlapping functions. We speculate that p27 might be rate limiting for withdrawal from the cell cycle, and communication between rate-limiting cyclins and rate-limiting p27 occurs as the cells traverse the restriction point. Consistent with this, p27 is both a substrate and an inhibitor of the cyclin E-associated kinase (Sheaff et al. 1997). It is tempting to speculate that growth arrest eventually occurs in p27−/− mice because down-regulation of cyclin/CDK2 occurs, allowing cells to default into the differentiation program. Elucidation of epistasis relationships between the various cyclins and inhibitors will clarify this process further.
Materials and methods
Cell culture
Primary cortical cultures (postnatal day zero) of O-2A precursors were obtained by differential shaking of mixed glial cultures that were kept in M15 media (MEM containing 15% fetal calf serum) for 7 days. Immediately after shaking, precursor cells were plated on poly-lysine coated dishes in M15 medium. After 6–18 hr, precursor cells were either kept proliferating in MM medium [DMEM containing 30% B104-conditioned medium + N1 supplements (50 μg/ml of transferrin, 16 μg/ml of putrescine, 60 ng/ml of progesterone, 50 ng/ml of selenium, and 5 μg/ml of insulin)] or differentiated to oligodendrocytes in differentiation medium, consisting of BME:F12 (1:1) supplemented with 100 μg/ml of transferrin, 20 μg/ml of putrescine, 12.8 ng/ml of progesterone, 10.4 ng/ml of selenium, 25 μg/ml of insulin, 0.8 μg/ml of thyroxine, 0.6% glucose, and 6.6 mm glutamine. After the shake-off, astrocytes (GFAP+) were maintained in M15 medium.
Bromodeoxyuridine incorporation
Cells were given a 6-hr pulse with 10 μm 5-bromo-2′-deoxyuridine and then fixed in ice cold acetone/methanol (1:1). After 30 min in 1 n HCl, cells were incubated in anti-BrdU antibody (1:100) in PBS + 0.3% Tween 20 + 3% horse serum at 4°C overnight. After 1 hr of incubation at room temperature in anti-mouse secondary antibody solution (1:500 in PBS), cells were stained using Vectastain Elite ABC, according to manufacturer’s instructions, and visualized using diaminobenzidine as substrate.
Cell extracts
Cells were detached from culture plates by scraping, and then collected in Hank’s salt solution (GIBCO). Cells were washed in PBS, and then resuspended in HKM buffer [30 mm HEPES-KOH (pH 7.4), 7.5 mm MgCl2], 0.5 mm dithiothreitol (DTT), 2 mm PMSF, 5 μg/ml of leupeptin, and 3.5 μg/ml of aprotinin. After lysis by sonication, cells were subjected to centrifugation to separate the cell pellet. Protein extracts were adjusted to 0.1 n NaCl and then stored at −70°C.
Tissue extracts
Immediately after dissection, a 10% brain homogenate was obtained in PBS containing 320 mm sucrose and protease inhibitors. The homogenate was subjected to low speed centrifugation (10 min at 800g at 4°C) and the supernatant spun for 20 min at 9000g at 4°C. The pellet was then resuspended in PBS containing freshly prepared protease inhibitors and further spun at 165,000g for 120 min at 4°C to obtain membrane (pellet) and cytosolic (supernatant) fractions.
Western blotting
Protein extracts (20–100 μg/lane) were analyzed by SDS–polyacrylamide gel electrophoresis. Proteins were transferred to a polyvinylidene fluoride (PVDF) membrane. The membrane was then blocked with 5% nonfat milk (Carnation) and incubated with primary antibodies. Most antibodies were obtained from Santa Cruz, except for p27 (Soos et al. 1996), anti-MBP, and anti-PLP (gift from Drs. L. Pedraza and D. Colman, Rockefeller University, New York, NY). After incubating with anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibody (Boehringer/Mannheim), proteins were visualized using an enhanced chemiluminescence system (Amersham).
Immunocytochemistry/immunohistochemistry
Cells were fixed for 10 min at room temperature in 4% paraformaldehyde in PBS (pH 7.4). Brains were fixed in 4% paraformaldehyde in PBS (pH 7.4) for 12–16 hr at 4°C, dehydrated, and then paraffin embedded. Ten-micrometer sections were deparaffinized and then processed for immunohistochemistry. Primary antibodies were used at 1:500 for NG2 (a gift from Joel Levine, Mt. Sinai School of Medicine, New York, NY), 1:2000 for GFAP (Dako Corporation), 1:250 for MAP2 (Boehringer/Mannheim), and 1:2000 for MBP (a gift from Liliana Pedraza). For A2B5 and GalC staining, the antibodies were used undiluted on live cells for 30 min at room temperature and then fixed in 4% paraformaldehyde. After 1 hr of incubation at room temperature in secondary antibody solution, cells were stained using Vectastain Elite ABC, according to manufacturer’s instructions, and visualized using diaminobenzidine as substrate.
Electron microscopy
Optic nerves were immersion fixed in 2.5% glutaraldehyde in 0.1 m cacodylate buffer (pH 7.4) at 4°C for 12–16 hr and then postfixed in 1% OsO4, 1.5% K4Fe(CN)6 in 0.1 m cacodylate buffer for 1 hr at 4°C. After serial dehydration steps in alcohol, samples were embedded in Epon. Semithin sections (1 μm) were cut exactly 1 mm anterior to the optic chiasm and stained with toluidine blue. Glial cell nuclei were counted. Ultrathin sections (60 nm) were cut in a Sorvall MT5000 ultramicrotome, using a Diatome diamond knife (Diatome, Fort Washington, PA). Sections were stained with lead citrate and examined in a JEOL 100 CX II electron miroscope (JEOL, USA, Peabody, MA) operating at 80 Kv and photographed on Kodak 4489 electron microscope film.
Acknowledgments
This work was supported in part by the National Institutes of Health (NIH) grants HD232315 and NS21072 to MVC and CA68425 and GM52597 to A.K. A.K. is also supported by a Pew Scholarship in Biomedical Sciences and the Frederick R. Adler Chair for Junior Faculty. This work was also supported by fellowships from the NIH (P.C.B.), the Charles Revson Foundation (R.T.), the DeWitt Wallace Research Fund of Memorial Sloan-Kettering Cancer Center (H.K.), and the Dorothy Rodbell Foundation (M.V.C.). We thank Joel Levine, Liliana Pedraza, and David Colman for generously providing antibodies; Leigh Cohen-Gould and Dilruba Khanam for technical assistance; and Martin Raff for communicating unpublished results. In addition, we thank the Sloan-Kettering Institute Cell Cycle Group for their advice during the course of this work.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL a-koff@ski.mskcc.org; FAX (212) 639-2861.
References
- Agrawal D, Hauser P, McPherson F, Dong F, Garcia A, Pledger WJ. Repression of p27Kip1 synthesis by platelet-derived growth factor in Balb/c 3T3 cells. Mol Biol Cell. 1996;8:4327–4336. doi: 10.1128/mcb.16.8.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres B, Lazar M, Raff M. A novel role for thyroid hormone, glucocorticoids, and retinoic acid in timing of oligodendrocyte development. Development. 1994;120:1097–1108. doi: 10.1242/dev.120.5.1097. [DOI] [PubMed] [Google Scholar]
- Bögler O, Noble M. Measurement of time in oligodendrocyte-type-2 astrocyte (O-2A) progenitors is a cellular process distinct from differentiation of division. Dev Biol. 1994;162:525–538. doi: 10.1006/dbio.1994.1106. [DOI] [PubMed] [Google Scholar]
- Boudreau N, Werb Z, Bissell MJ. Suppression of apoptosis by basement membrane requires three-dimensional tissue organization and withdrawal from the cell cycle. Proc Natl Acad Sci. 1996;93:3509–3513. doi: 10.1073/pnas.93.8.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulikas T. Phosphorylation of transcription factors and control of the cell cycle. Crit Rev Eukaryot Gene Expr. 1995;5:1–77. [PubMed] [Google Scholar]
- Burne JF, Staple JK, Raff MC. Glial cells are increased proportionally in transgenic optic nerves with increased numbers of axons. J Neurosci. 1996;16:2064–2073. doi: 10.1523/JNEUROSCI.16-06-02064.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Aibel L, Chao MV. Central glial and neuronal populations display differential sensitivity to ceramide-dependent cell death. J Neurosci Res. 1996;43:382–389. doi: 10.1002/(SICI)1097-4547(19960201)43:3<382::AID-JNR13>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Coats S, Flanagan WM, Nourse J, Roberts JM. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science. 1996;272:877–880. doi: 10.1126/science.272.5263.877. [DOI] [PubMed] [Google Scholar]
- Cross FR. DAF1, a mutant gene affecting size control, pheromone arrest, and cell cycle kinetics of Saccharomyces cerevisiae. Mol Biol Cell. 1988;8:4675–4684. doi: 10.1128/mcb.8.11.4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand B, Gao FB, Raff M. Accumulation of the cyclin-dependent kinase inhibitor p27/Kip1 and the timing of oligodendrocyte differentiation. EMBO J. 1997;16:306–317. doi: 10.1093/emboj/16.2.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duronio RJ, O’Farrell PH. Developmental control of a G1–S transcriptional program in Drosophilia. Development. 1994;120:1503–1515. doi: 10.1242/dev.120.6.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, Kaushansky K, Roberts JM. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996;85:733–744. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- Firpo EJ, Koff A, Solomon MJ, Roberts JM. Inactivation of a Cdk2 inhibitor during interleukin 2-induced proliferation of human T lymphocytes. Mol Cell Biol. 1994;14:4889–4901. doi: 10.1128/mcb.14.7.4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RP, Morgan DO. A novel cyclin associates with MO15/CDK7 to form the CDK activating kinase. Cell. 1994;78:713–724. doi: 10.1016/0092-8674(94)90535-5. [DOI] [PubMed] [Google Scholar]
- Gard AL, Pfeiffer SE. Oligodendrocyte progenitors isolated directly from developing telencephalon at a specific phenotypic stage: Myelinogenic potential in a defined environment. Development. 1989;106:119–132. doi: 10.1242/dev.106.1.119. [DOI] [PubMed] [Google Scholar]
- ————— Two proliferative stages of the oligodendrocyte lineage (A2B5+O4− and O4+GalC−) under different mitogenic control. Neuron. 1990;5:615–625. doi: 10.1016/0896-6273(90)90216-3. [DOI] [PubMed] [Google Scholar]
- Guo K, Wang J, Andres V, Smith RC, Walsh K. MyoD-induced expression of p21 inhibits cyclin-dependent kinase activity upon myocyte terminal differentiation. Mol Cell Biol. 1995;15:3823–3829. doi: 10.1128/mcb.15.7.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol. 1996;16:859–867. doi: 10.1128/mcb.16.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- Hauser PJ, Agrawal D, Flanagan M, Pledger WJ. The role of p27Kip1 in the in vitro differentiation of murine keratinocytes. Cell Growth Differ. 1997;8:203–211. [PubMed] [Google Scholar]
- Hengst L, Reed SI. Translational control of p27Kip1 during the cell cycle. Science. 1996;271:1861–1864. doi: 10.1126/science.271.5257.1861. [DOI] [PubMed] [Google Scholar]
- Hirai H, Roussel MF, Kato JY, Ashmun RA, Sherr CJ. Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol Cell Biol. 1995;15:2672–2681. doi: 10.1128/mcb.15.5.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer R, Denhardt DT. Cell cycle-regulated and proliferation stimulus-responsive genes. Crit Rev Eukaryot Gene Expr. 1991;1:247–300. [PubMed] [Google Scholar]
- Hoffman ES, Passoni L, Crompton T, Leu TMJ, Schatz DG, Koff A, Owen MJ, Hayday AC. Productive T-cell receptor β-chain gene rearrangement: Coincident regulation of cell cycle and clonality during development in vivo. Genes & Dev. 1996;9:948–962. doi: 10.1101/gad.10.8.948. [DOI] [PubMed] [Google Scholar]
- Kato JY, Sherr CJ. Inhibition of granulocyte differentiation by G1 cyclins D2 and D3 but not D1. Proc Natl Acad Sci. 1993;90:11513–11517. doi: 10.1073/pnas.90.24.11513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipreos ET, Lander LE, Wing JP, He WW, Hedgecock EM. cul-1 is required for cell cycle exit in C. elegans and identifies a novel gene family. Cell. 1996;86:829–839. doi: 10.1016/s0092-8674(00)81267-2. [DOI] [PubMed] [Google Scholar]
- Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27Kip1. Cell. 1996;85:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- Koff A. The regulation of G1 progression in mammalian cells. In: DeBellis A, Shipani E, editors. Future trends in endocrinology. Rome, Italy: Ares-Serona Symposia Publications; 1995. pp. 207–216. [Google Scholar]
- Koff A, Giordano A, Desai D, Yamashita K, Harper JW, Elledge S, Nishimoto T, Morgan DO, Franza BR, Roberts JM. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science. 1992;257:1689–1694. doi: 10.1126/science.1388288. [DOI] [PubMed] [Google Scholar]
- Kranenburg O, Scharnhost V, van der Eb AJ, Zantema A. Inhibition of cyclin-dependent kinase activity triggers neuronal differentiation of mouse neuroblastoma cells. J Cell Biol. 1995;131:227–234. doi: 10.1083/jcb.131.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane ME, Sauer K, Wallace K, Jan YN, Lehner CF, Vaessin H. Dacapo, a cyclin-dependent kinase inhibitor, stops cell proliferation during Drosophilia development. Cell. 1996;87:1225–1235. doi: 10.1016/s0092-8674(00)81818-8. [DOI] [PubMed] [Google Scholar]
- Lassar AB, Skapek SX, Novitch B. Regulatory mechanisms that coordinate skeletal muscle differentiation and cell cycle withdrawal. Curr Opin Cell Biol. 1994;6:788–794. doi: 10.1016/0955-0674(94)90046-9. [DOI] [PubMed] [Google Scholar]
- Lee MH, Reynisdóttir I, Massagué J. Cloning of p57Kip2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes & Dev. 1995;9:639–649. doi: 10.1101/gad.9.6.639. [DOI] [PubMed] [Google Scholar]
- Lee MH, Nikolic M, Baptista CA, Lai E, Tsai LH, Massaguá J. The brain-specific activator p35 allows Cdk5 to escape inhibition by p27Kip1 in neurons. Proc Natl Acad Sci. 1996;93:3259–3263. doi: 10.1073/pnas.93.8.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Lee MH, Cohen M, Bommakanti M, Freedman LP. Transcriptional activation of the Cdk inhibitor p21 by vitamin D3 leads to the induced differentiation of the myelomonocytic cell line U937. Genes & Dev. 1996;10:142–153. doi: 10.1101/gad.10.2.142. [DOI] [PubMed] [Google Scholar]
- Louis J, Magal E, Muir D, Manthorpe M, Varon S. CG-4, a new bipotential glial cell line from rat brain is capable of differentiating in vitro into either nature oligodendrocytes of type-2 astrocytes. J Neurosci Res. 1992;31:193–204. doi: 10.1002/jnr.490310125. [DOI] [PubMed] [Google Scholar]
- Matsuoka M, Kato JY, Fisher RP, Morgan DO, Sherr CJ. Activation of cyclin-dependent kinase 4 (cdk4) by mouse MO15-associated kinase. Mol Cell Biol. 1994;14:7265–7275. doi: 10.1128/mcb.14.11.7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Edwards MC, Bai C, Parker S, Zhang P, Baldini A, Harper JW, Elledge SJ. p57Kip2, a structurally distinct member of the p21Cip1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes & Dev. 1995;9:650–662. doi: 10.1101/gad.9.6.650. [DOI] [PubMed] [Google Scholar]
- Matsushime H, Ewen ME, Strom DK, Kato JY, Hanks SK, Roussel MF, Sherr CJ. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992;71:323–334. doi: 10.1016/0092-8674(92)90360-o. [DOI] [PubMed] [Google Scholar]
- McCarthy, K.D. and DeVellis, J. 1980. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J. Cell. Biol. 85: 890–902. [DOI] [PMC free article] [PubMed]
- Medema RH, Herrera RE, Lam F, Weinberg RA. Growth suppression by p16INK4 requires functional retinoblastoma protein. Proc Natl Acad Sci. 1995;92:6289–6293. doi: 10.1073/pnas.92.14.6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson M, Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol. 1994;14:2077–2086. doi: 10.1128/mcb.14.3.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard SS, Yan JS, Nguyen H, Pagano M, Kiyokawa H, Koff A. Enhanced ribosomal association of p27Kip1 mRNA is a mechanism contributing to accumulation during growth arrest. J Biol Chem. 1997;272:7093–7098. doi: 10.1074/jbc.272.11.7093. [DOI] [PubMed] [Google Scholar]
- Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–134. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY, Nakayama K-i. Mice lacking p27Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Lin X-H, Giese N, Heldin C-H, Stallcup WB. Co-localization of NG2 proteoglycan and PDGF alpha receptor on O-2A progenitor cells in the developing rat brain. J Neurosci Res. 1996;43:299–314. doi: 10.1002/(SICI)1097-4547(19960201)43:3<299::AID-JNR5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res. 1994;211:90–98. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- Ohtusbo M, Roberts JM. Cyclin-dependent regulation of G1 in mammalian fibroblasts. Science. 1993;259:1908–1912. doi: 10.1126/science.8384376. [DOI] [PubMed] [Google Scholar]
- Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M. Role of the ubiquitin proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- Parker SB, Eichele G, Zhang P, Rawls A, Sands AT, Bradley A, Olson EN, Harper JW, Elledge SJ. p53-independent expression of p21Cip1 in muscle and other terminally differentiating cells. Science. 1995;267:1024–1027. doi: 10.1126/science.7863329. [DOI] [PubMed] [Google Scholar]
- Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massagué J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- Poulos NL, Farmer AA, Chan KW, Stanbridge EJ. Design of a novel bicistronic expression vector with demonstration of a p16INK4-induced G1-S block. Cancer Res. 1996;56:1719–1723. [PubMed] [Google Scholar]
- Raff M. Glial cell diversification in the rat optic nerve. Science. 1989;243:1450–1455. doi: 10.1126/science.2648568. [DOI] [PubMed] [Google Scholar]
- Raff MC, Miller RH, Noble M. A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on the culture medium. Nature. 1983;303:390–396. doi: 10.1038/303390a0. [DOI] [PubMed] [Google Scholar]
- Raff MC, Abney ER, Fok-Seang J. Reconstruction of a developmental clock in vitro: A critical role for astrocytes in the timing of oligodendrocyte differentiation. Cell. 1985;42:61–69. doi: 10.1016/s0092-8674(85)80101-x. [DOI] [PubMed] [Google Scholar]
- Raff MC, Lillien LE, Richardson WD, Burne JF, Noble MD. Platelet-derived growth factor from astrocytes drives the clock that times oligodendrocyte development in culture. Nature. 1988;333:562–565. doi: 10.1038/333562a0. [DOI] [PubMed] [Google Scholar]
- Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival: Lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- Resnitzky D, Reed SI. Different roles for cyclin D1 and E in regulation of the G1-to-S transition. Mol Cell Biol. 1995;15:3463–3469. doi: 10.1128/mcb.15.7.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynisdóttir I, Massagué J. The subcellular locations of p15Ink4b and p27Kip1 coordinate their inhibitory interactions with cdk4 and cdk2. Genes & Dev. 1997;11:492–503. doi: 10.1101/gad.11.4.492. [DOI] [PubMed] [Google Scholar]
- Russo AA, Jeffrey PD, Patten AK, Massagué J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E-Cdk2 is a regulator of p27Kip1. Genes & Dev. 1997;11:1464–1478. doi: 10.1101/gad.11.11.1464. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes & Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- Skapek SX, Rhee J, Spicer DB, Lassar AB. Inhibition of myogenic differentiation in proliferating myoblasts by cyclin D1-dependent kinase. Science. 1995;267:1022–1024. doi: 10.1126/science.7863328. [DOI] [PubMed] [Google Scholar]
- Soos TJ, Kiyokawa H, Yan JS, Rubin MS, Giordano A, DeBlasio A, Bottega S, Wong B, Mendelsohn J, Koff A. Formation of p27-CDK complexes during the human mitotic cell cycle. Cell Growth Differ. 1996;7:135–146. [PubMed] [Google Scholar]
- Stein GH, Dulic V. Origins of G1 arrest in senescent human fibroblasts. Bioessays. 1995;17:537–543. doi: 10.1002/bies.950170610. [DOI] [PubMed] [Google Scholar]
- Tahara H, Sato E, Noda A, Ide T. Increase in expression level of p21sdi1/cip1/waf1 with increasing division age in both normal and SV40-transformed human fibroblasts. Oncogene. 1995;10:835–840. [PubMed] [Google Scholar]
- Temple S, Raff MC. Differentiation of a bipotential glial progenitor cell in single cell microculture. Nature. 1985;313:223–225. doi: 10.1038/313223a0. [DOI] [PubMed] [Google Scholar]
- Tikoo R, Casaccia-Bonnefil P, Chao MV, Koff A. Changes in cyclin-dependent kinase 2 and p27Kip accompany glial cell differentiation of central glial-4 cells. J Biol Chem. 1997;272:442–447. doi: 10.1074/jbc.272.1.442. [DOI] [PubMed] [Google Scholar]
- Toyoshima T, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994;78:67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Zhang H, Beach D. D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell. 1992;71:504–514. doi: 10.1016/0092-8674(92)90518-h. [DOI] [PubMed] [Google Scholar]