

Abstract
Maternal-fetal genotype (MFG) incompatibility is an interaction between the genes of a mother and offspring at a particular locus that adversely affects the developing fetus, thereby increasing susceptibility to disease. Statistical methods for examining MFG incompatibility as a disease risk factor have been developed for nuclear families. Because families collected as part of a study can be large and complex, containing multiple generations and marriage loops, we create the Extended-MFG (EMFG) Test, a model-based likelihood approach, to allow for arbitrary family structures. We modify the MFG test by replacing the nuclear-family based “mating type” approach with Ott’s representation of a pedigree likelihood and calculating MFG incompatibility along with the Mendelian transmission probability. In order to allow for extension to arbitrary family structures, we make a slightly more stringent assumption of random mating with respect to the locus of interest. Simulations show that the EMFG test has appropriate type-I error rate, power, and precise parameter estimation when random mating holds. Our simulations and real data example illustrate that the chief advantages of the EMFG test over the earlier nuclear family version of the MFG test are improved accuracy of parameter estimation and power gains in the presence of missing genotypes.
Keywords: maternal-fetal interaction, gene by environment, family based association, pedigree likelihood
INTRODUCTION
Studies show that prenatal environment may contribute to disease risk in offspring later in their lives [Cannon, 1997; Cantor-Graae et al., 2000; Louey and Thornburg, 2005; McKinney et al., 1999]. One example is Maternal-Fetal Genotype (MFG) incompatibility, which arises through an interaction between maternal and fetal gene products [Laing et al., 1995; Marcelis et al., 1998; Ober, 1998]. The underlying genetic basis for this incompatibility is what allows us to study it even decades after the adverse environment has passed.
Sinsheimer et al. [2003] developed the MFG incompatibility test, a likelihood-based, affected-only statistical method for examining MFG incompatibility as a disease risk factor. The original method uses parent-offspring trios in an adaptation of Weinberg’s log-linear method [Weinberg et al., 1998] for estimating genotypic relative risks for maternal and offspring main effects. The MFG test allows the user to jointly model offspring allelic effects, maternal allelic effects, and maternal-offspring genotype interactions, such as maternal-fetal genotype incompatibility. Kraft et al. [2004, 2005] extended the MFG test to allow multiple siblings per nuclear family and to model locus-specific effects of maternal immunological processes, such as prior exposure. Other researchers developed the MFG test variations for use with nuclear families or with case-mother control-mother data [Chen et al., 2005, 2009; Cordell et al., 2004; Li et al., 2009]. Through the MFG and related tests, MFG incompatibility has been tested as a potential risk factor for diseases as diverse as autism, schizophrenia, and rheumatoid arthritis (RA) [Chen et al., 2005, 2009; Hsieh et al., 2006a; Insel et al., 2005; Palmer et al., 2002, 2006, 2008; Zandi et al., 2006].
Although the MFG test is powerful and adept at jointly modeling effects without confounding them, the restriction to nuclear families forces researchers who have extended family data to make a difficult choice. They must either risk losing power by selecting a single nuclear family per extended pedigree or risk introducing biases by partitioning their extended pedigrees into a number of nuclear families treated as independent. For this reason, we have created the Extended-MFG (EMFG) test to handle arbitrary pedigree structures in testing hypotheses about maternal allelic effects, offspring allelic effects, and maternal-offspring genotype incompatibilities. Before we develop the EMFG test, we review the current, nuclear family MFG test. We then use simulations to illustrate the properties and potential advantages and disadvantages of the EMFG test when compared to the nuclear family MFG test. Finally, we provide a simple illustration of the EMFG test on a real data set consisting of a single large pedigree.
MFG INCOMPATIBILITY CONCEPTS
Table I illustrates how the MFG test works in general for MFG incompatibility at a bi-allelic locus. There are seven possible combinations of mother and offspring’s genotypes (Table I, Columns 1 and 2). The most general model (Table I, Column 3) allows the offspring’s disease risk to differ for each MFG combination [Sinsheimer et al., 2003]. In Table I, these risks are designated by δij, where i and j represent the number of variant alleles (allele 2) present in the mother and offspring, respectively. Genotype interaction (MFG incompatibility) is reflected by the lack of constraints on δij that force the multiplicative model δij = ρi ηj and make maternal and offspring effects independent.
TABLE I.
Seven MFG combinations and the offspring risk with a bi-allelic system
| Mother | Offspring | Generalized risk | RHD | NIMA |
|---|---|---|---|---|
| 2/2 | 2/2 | δ22 | β | βρ2 |
| 2/2 | 1/2 | δ21 | β | βρ1 |
| 1/2 | 2/2 | δ12 | β | βρ2 |
| 1/2 | 1/2 | δ11 | β | βρ1 |
| 1/2 | 1/1 | δ10 | β | βμ |
| 1/1 | 1/2 | δ01 | βμ | βρ1 |
| 1/1 | 1/1 | δ00 | β | β |
When prior evidence exists for a particular maternal-fetal genotype incompatibility mechanism, the number of parameters can be reduced. Two examples are provided in Table I and Figure 1A, B. Consider RHD incompatibility (Table I, Column 4 and Fig. 1A). In this case, allele 2 corresponds to the antigen-coding allele (often coded as D) and allele 1 corresponds to the null allele (often coded as d). RHD incompatibility occurs when the immune system of a mother with genotype 1/1 recognizes the fetus’ protein product from the 1/2 genotype as foreign and mounts an immune response that can be detrimental to her offspring. The potentially detrimental genotype combination has an increased risk of μ over the baseline risk of β. All other maternal-offspring genotype combinations have the baseline risk.
Fig. 1.

(A) Pedigree depiction of RHD incompatibility (B) Pedigree depiction of non-inherited maternal antigen (NIMA) incompatibility
The second example (Table I, Column 5 and Fig. 1B) is derived from RA and HLA-DRB1, where offspring allelic effects are highly significant and non-inherited maternal antigens (NIMA) have been implicated to increase risk of disease in offspring [Harney et al., 2003; Newton et al., 2004; van der Horst-Bruinsma et al., 1998]. Offspring with 1/2 or 2/2 genotypes are at increased risk (ρ1 and ρ2, respectively) over the baseline risk. NIMA effects can occur to 1/1 offspring whose mother has genotype 1/2. This MFG combination carries increased risk to the offspring.
THE NUCLEAR FAMILY MFG TEST
We start our model development with the affected-only, nuclear family MFG [Kraft et al., 2004, 2005], which allows for multiple siblings in a family. The MFG test conditional likelihood controls for ascertainment and has the following form for a completely genotyped family:
| (1) |
Here, G = (G1,…,Gk) represents the observed genotypes of the k-affected offspring in the family, and Gr and Gs represent the genotypes of the mother and father. The denominator sums over all possible ordered (phased) genotypes for offspring and parents (g, gr, gs). The vector D = (D1,…,Dk) denotes the k offspring in the family, with Dc equal to 1 for the k-affected offspring. There are six mating types (MT) to consider for a bi-allelic locus (Table II). Using mating types controls for non-random mating at the trait locus.
TABLE II.
Mating types for a bi-allelic locus
| MT | Parental genotype pair | Frequency (ψMT) under random mating |
|---|---|---|
| 1 | {2/2, 2/2} | P(2/2)2 |
| 2 | {2/2, 1/2} | 2P(2/2)P(1/2) |
| 3 | {2/2, 1/1} | 2P(2/2)P(1/1) |
| 4 | {1/2, 1/2} | P(1/2)2 |
| 5 | {1/2, 1/1} | 2P(1/2)P(1/1) |
| 6 | {1/1, 1/1} | P(1/1)2 |
The MFG test is an affected-only analysis, however, the genotypes of unaffected or phenotype unknown offspring contribute if there are missing parental data. The conditional likelihood (1) must be summed over all possible parental genotypes (gr, gs) consistent with the observed genotypes in the family, which now include the genotypes of unaffected or phenotype unknown offspring [Hsieh et al., 2006b; Kraft et al., 2004; Minassian et al., 2006; Palmer et al., 2002]. The denominator remains the same. The likelihood for the entire sample is the product of the likelihoods for separate and independent families.
THE EMFG TEST
Reliance on mating types limits the traditional EMFG test to nuclear family analysis and burdens the model with nuisance parameters. In extended pedigrees, it is unclear how to weight matings, where one or both parents are non-founders. Assuming random mating at the locus of interest avoids mating types. Random mating takes place when selection of a mate is independent of marker genotype. Thus, the probability of a mating type equals the product of the genotype frequencies (Table II, Column 3). The model now depends on founder genotypes. Based on this assumption, the conditional likelihood of any pedigree can be evaluated by taking the ratio of two unconditional pedigree likelihoods [Ott, 1974].
The MFG likelihood for a pedigree with n members is defined as the conditional likelihood L(G|D) of the observed genotypes G given the trait phenotypes D in the pedigree. The affected-only analysis is achieved by treating all unaffected individuals as phenotype unknown. Hence, the vector D omits the disease phenotypes of pedigree founders. In contrast, G includes the marker genotypes of founders when these genotypes are known. If users have affected founders, they can be included in the analysis by introducing their parents without phenotype or genotype data. If genotypes are missing for some individuals or genotype phases are unknown, the likelihood is summed over the possible ordered genotypes g that are consistent with the observed genotypes G in the family.
The conditional likelihood for the maternal-fetal genotype incompatibility effects is:
| (2) |
The probability of disease in offspring c is a function of both her and her mother’s genotype. Prior(gj) represents founder j’s genotype frequency. These founder population genotype frequencies are estimated by maximizing the likelihood along with the risk parameters. The second term in the numerator, Pr(Gi|gi), is 1 if the proposed genotype for i, gi, is consistent with the observed genotype Gi, and 0 if it is inconsistent. When Gi is missing, Pr(Gi|gi) = 1. This term is calculated for each family member, regardless of affection status. Offspring and maternal allelic effects, and maternal-fetal genotype interactions are parameterized through Pr(Dc|gc,gr), which is calculated with Trans(gc|gr,gs), the transmission probability for offspring, mother, and father triple (c,r,s). The denominator sums over all possible ordered (phased) genotypes for the n family members, and is similar to standard ascertainment correction [Lange, 2002]. When parental genotypes are unavailable for founders, we treat them as phenotype unknown.
To illustrate the adaptability of Pr(Dc|gc,gr), we consider a few pertinent examples. First, consider RHD incompatibility and hemolytic disease of the newborn where disease risk differs for boys and girls.
| (3) |
MFG incompatibility occurs when the mother’s genotype is 1/1 and the offspring’s is 1/2.
In our second example we modify Pr(Dc|gc,gr) to allow for both NIMA and offspring allelic effects as follows:
| (4) |
Parameters ρ1 and ρ2 represent the relative risk of disease for an offspring who carries one or two copies of a risk allele at the locus of interest compared to an offspring who carries no copies of a risk allele. MFG incompatibility occurs when the mother’s genotype is 1/2 and the offspring’s is 1/1. Since Di is considered missing if i is unaffected or phenotype unknown and the baseline population prevalence β cancels from the conditional likelihood, we fix β = 1, which is equivalent to δ00 = 1 (Table I, Column 3).
IMPLEMENTING THE EMFG TEST
The EMFG test is implemented in the Mendel software [Lange et al., 2001] for pedigrees of variable size and structure. The current program handles the generalized risk model for a bi-allelic locus (Table I, Column 3). Parameters under specific hypotheses are estimated by placing restrictions on this model. RHD incompatibility without gender effects imposes δ00 = δ10 = δ11 = δ12 = δ21 = δ22 = 1 and estimates only δ01. NIMA sets δ00 = 1 and imposes δ01 = δ11 = δ21 and δ12 = δ22, accounting for the offspring allelic effects. Thus, there are three free parameters, two for offspring allelic effects and one (δ10) accounting for the NIMA effect. Nested models are compared using a likelihood ratio (LR) test. For example, to test for a significant NIMA effect in the presence of an offspring allelic effect, one can compare a full model with three free parameters (NIMA and offspring allelic effects) to a restricted model with two free parameters (offspring allelic effects) using a one-degree of freedom LR test.
SIMULATION STUDY: OBJECTIVES AND CONDITIONS
The simulations are designed for three purposes. In Simulation Study 1, we demonstrate the statistical properties of the EMFG test under realistic effect sizes for RHD and NIMA incompatibility. In Simulation Study 2, we compare three possible study designs using (a) extended pedigrees in their entirety, (b) one nuclear family per extended pedigree [Palmer et al., 2008], and (c) all nuclear families available from extended pedigrees and treated as independent. We anticipate that design (a) will be superior to (b) in power and (c) in accuracy. In Simulation Study 3, we determine the effects of violating the random mating assumption on parameter estimation, power, and type-I error rate of the EMFG test.
In our simulation studies, we characterize the properties of the EMFG test in terms of its rejection rates and estimation accuracy. Unless otherwise mentioned, founder genotypes are simulated according to Hardy Weinberg Equilibrium (HWE) with allele frequencies P(1) = 0.33 and P(2) = 0.67. Non-founder genotypes are simulated using Mendel’s gene dropping option [Lange et al., 2001]. Samples are simulated with no MFG effect and with detrimental MFG effects ranging from 1.5 to 2.5 in 0.1 increments (results only shown for certain values). We chose this range because of previous estimates of MFG effects [Insel et al., 2005; Kraft et al., 2004; Palmer et al., 2008]. We refer to these simulation conditions as scenarios. For each scenario, 1,000 data sets are simulated. Parameter estimates and their standard errors are averaged over the data sets. The number of pedigrees within a data set varies. For each model, rejection rate and coverage are shown. Coverage is the proportion of 95% confidence intervals that contain the MFG relative risk’s true value. The rejection rate is the proportion of samples where the LR test rejects the null hypothesis of no MFG effect at a significance level of 0.05. In all scenarios, two-sided tests are used. Since each performance statistic p is a proportion, its standard error is .
RESULTS
SIMULATION STUDY 1: PROPERTIES OF THE EMFG TEST
For Simulation Study 1, each of 1,000 simulated data sets contain 200 three-generational pedigrees. Only individuals 7 and 8 are affected (with dark shading in Fig. 2). First, we use the EMFG Test to estimate the MFG effect in simulated samples with no gender-specific MFG effect (Table III, Scenarios A and B). When μ = 1, the type-I error rate of 0.040 is close to the desired level of 0.05. The EMFG test has 0.816 power to detect an MFG effect of 1.7 when there are no gender effects (Scenario B, model μm = μf). When these same data sets are analyzed under a model that allows for gender differences (μm≠μf), power is reduced to 0.724. In all scenarios, the parameter estimates are close to the true MFG effect. Founder genotype frequencies and their standard errors are the same across models, and are equal to their expected values under HWE (P̂(1/1) = 0.109, SE = 0.012; P̂(1/2) = 0.443, SE = 0.018; P̂(2/2) = 0.448, SE = 0.018). Accurate results are also obtained for data simulated with 1.5≤μ≤2.5 (data not shown).
Fig. 2.
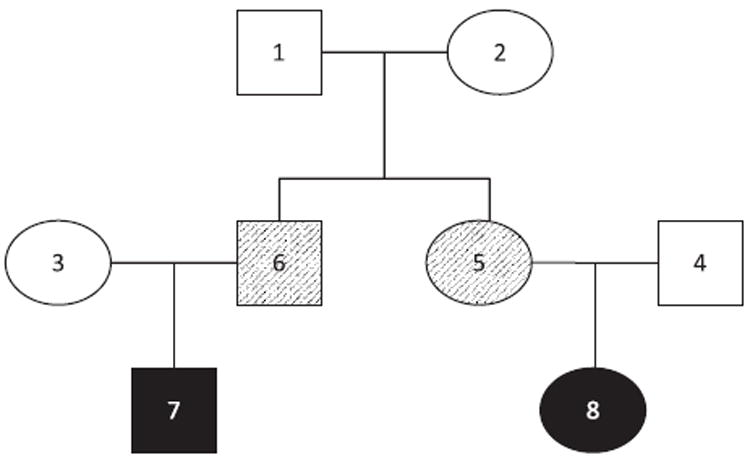
Pedigree structure used for Simulation Studies 1 and 2. Dark shading represents individuals who are affected in both simulations (Simulation Studies 1 and 2). Hatching represents individuals who are only affected in Simulation Study 2.
TABLE III.
Properties of the EMFG test simulation study 1—RHD incompatibility
| Scenarioa | Modelb constraints | μ̂m(SE) | Coverage of μm(SE) | μ̂f (SE) | Coverage of μf (SE) | Rejection rate (SE) | P̂(1/1)(SE) | P̂(1/2) (SE) | P̂(2/2)(SE) |
|---|---|---|---|---|---|---|---|---|---|
| A:μm = 1.0 | μm = μf | 0.989 | 0.956 | = μ̂m | = μ̂m | 0.040 | 0.109 | 0.443 | 0.448 |
| μf = 1.0 | (0.211) | (0.006) | (0.006) | (0.012) | (0.018) | (0.018) | |||
| B:μm = 1.7 | μm = μf | 1.698 | 0.953 | = μ̂m | = μ̂m | 0.816 | 0.109 | 0.442 | 0.449 |
| μf = 1.7 | (0.305) | (0.007) | (0.012) | (0.012) | (0.018) | (0.018) | |||
| None | 1.676 | 0.953 | 1.685 | 0.952 | 0.724 | 0.109 | 0.442 | 0.449 | |
| (0.417) | (0.007) | (0.393) | (0.007) | (0.014) | (0.012) | (0.018) | (0.018) | ||
| C:μm = 1.7 | μf = 1.0 | 1.677 | 0.950 | = 1 | N/A | 0.536 | 0.109 | 0.443 | 0.448 |
| μf = 1.0 | (0.413) | (0.007) | (0.016) | (0.012) | (0.018) | (0.018) | |||
| None | 1.677 | 0.949 | 0.963 | 0.953 | 0.450 | 0.109 | 0.443 | 0.448 | |
| (0.415) | (0.007) | (0.279) | (0.007) | (0.016) | (0.012) | (0.018) | (0.018) |
Scenario refers to the conditions used to simulate a particular dataset.
Model constraints column lists the constraints (if any) used to fit a particular statistical model to the data.
Several studies found that MFG incompatibility effects are confined to a single gender [Insel et al., 2005; Palmer et al., 2006, 2008]. To mimic this scenario, samples are simulated with males at increased risk of disease but females at baseline risk (μf = 1.0). Under a correct model constraining the female effect to 1, power to detect the MFG effect is 0.536 (Table III, Scenario C). Power decreases because each family has exactly one affected male and one affected female, so there are now only half the individuals contributing to μm’s estimation. Under a model that allows gender differences, parameter estimates are close to the true values and power is reduced to 0.45. Genotype frequency estimates and standard errors are the same as those observed in the previous scenarios. Similar results are obtained for data simulated with any value of μm between 1.5 and 2.5 (data not shown).
To determine type I error, power, coverage, and parameter estimation of the EMFG when offspring allelic effects are present, we simulated data with NIMA MFG incompatibility and offspring allelic effects (Table I, Column 5). A total of 15% of RA patients do not carry the shared epitope encoded by certain HLA-DRB1 alleles [Harney et al., 2003], so bi-allelic gene frequencies are P(1) = 0.39 and P(2) = 0.61 where allele 2 represents the shared epitope. Data are simulated without NIMA (μ = 1) or offspring effects (ρ1 = ρ2 = 1) (Table IV, Scenario A), and with μ = 2.5, ρ1 = 2 and ρ2 = 2.5 (Scenario B). In both scenarios, we fit a full model that estimates NIMA and offspring allelic effects. We also fit three reduced models (results not shown), that represent the null models of (a) no offspring allelic or NIMA effects (μ = ρ1 = ρ2 = 1.0), (b) no offspring allelic effects (ρ1 = ρ2 = 1.0), and (c) no NIMA effect (μ = 1.0). Using LR tests, these three models allow us to test for (a) either offspring or NIMA effects, (b) offspring effects in the presence of NIMA effects, and (c) NIMA effects in the presence of offspring effects.
TABLE IV.
Properties of the EMFG tests simulation study 1—NIMA incompatibility
| Coverage | Rejection Rate | |||
|---|---|---|---|---|
| μ̂(SE) | μ(SE) | μ = 1.0(SE) | P̂(1/1)(SE) | |
| ρ̂1(SE) | ρ1(SE) | ρ1 = ρ2 = 1.0(SE) | P̂(1/2)(SE) | |
| Scenario | ρ̂2(SE) | ρ2(SE) | μ = ρ1 = ρ2 1.0(SE) | P̂(2/2)(SE) |
| A: μ = 1.0, | 1.001 (0.279) | 0.941 (0.007) | 0.048 (0.007) | 0.156 (0.016) |
| ρ1 = 1.0, | 1.014 (0.246) | 0.956 (0.006) | 0.047 (0.007) | 0.475 (0.018) |
| ρ2 = 1.0 | 1.011 (0.276) | 0.943 (0.007) | 0.060 (0.008) | 0.369 (0.021) |
| B: μ = 2.5, | 2.570 (0.940) | 0.950 (0.007) | 0.818 (0.012) | 0.152 (0.017) |
| ρ1 = 2, | 2.058 (0.708) | 0.944 (0.007) | 0.771 (0.013) | 0.476 (0.018) |
| ρ2 = 2.5 | 2.575 (0.939) | 0.949 (0.007) | 0.797 (0.013) | 0.372 (0.021) |
When simulated under the null hypothesis (μ = ρ1 = ρ2 = 1.0), the parameter estimates (1.001–1.014), coverages (0.941–0.956), and type-I error rates (0.048–0.060) are appropriate. When μ = 2.5, ρ1 = 2.0, and ρ2 = 2.5, parameter estimates are close to their true values (μ̂ = 2.57, ρ̂1 = 2.058, ρ̂2 = 2.575) and coverage is appropriate (0.94–0.95). There is ~80% power to detect all three effects (rejection rate = 0.818 of μ = 1.0, rejection rate = 0.771 of ρ1 = ρ2 = 1.0, and rejection rate = 0.797 of μ = ρ1 = ρ2 = 1.0). Founder genotype frequencies are equal to their expected values under HWE.
SIMULATION STUDY 2: COMPARISON OF THE EXTENDED PEDIGREE AND NUCLEAR FAMILY STUDY DESIGNS
To avoid potential biases in analyzing related and sometimes overlapping nuclear families, the current recommended practice when using the MFG test is to select nuclear families from an extended pedigree so that the offspring in different nuclear families are no more related than second cousins [Palmer et al., 2008]. To examine the impact on power, we consider three simulation scenarios involving 80 three-generational pedigrees with affected siblings and first cousins (Fig. 2, individuals 5–8). Table V reports parameter estimates, standard errors, coverage, and rejection rates using the EMFG test. The 80 three-generational pedigrees are analyzed as full pedigrees (Scenario A), as 80 selected nuclear families by removing individuals 3, 4, 7, and 8 (Scenario B), and as 240 independent nuclear families (Scenario C). Regardless of which we use, all the three approaches give reasonable parameter estimates (μ̂ range: 0.979–0.993), type-I error rates (~0.060), and coverage (range: 0.942–0.965) when μ = 1.0.
TABLE V.
Extended pedigrees in their entirety vs. partitioning pedigrees into nuclear families
| Scenario | Number of families | μ | μ̂(SE) | Coverage (SE) | Rejection rate (SE) |
|---|---|---|---|---|---|
| Completely genotyped families EMFG test (parameterized using genotypes) | |||||
| A | 80 extended pedigreesa | 1.0 | 0.991 (0.219) | 0.942 (0.007) | 0.059 (0.007) |
| 1.7 | 1.691 (0.300) | 0.953 (0.007) | 0.825 (0.012) | ||
| 2.5 | 2.532 (0.412) | 0.954 (0.007) | >0.999 | ||
| B | 80 nuclear familiesb | 1.0 | 0.979 (0.277) | 0.965 (0.006) | 0.058 (0.007) |
| 1.7 | 1.598 (0.338) | 0.948 (0.007) | 0.607 (0.015) | ||
| 2.5 | 2.231 (0.421) | 0.900 (0.009) | 0.991 (0.003) | ||
| C | 240 nuclear familiesc | 1.0 | 0.993 (0.211) | 0.947 (0.007) | 0.058 (0.007) |
| 1.7 | 1.623 (0.268) | 0.932 (0.008) | 0.800 (0.013) | ||
| 2.5 | 2.356 (0.347) | 0.913 (0.009) | >0.999 | ||
| MFG test (parameterized using mating types) | |||||
| D | 240 nuclear families | 1.7 | 1.636 (0.301) | 0.940 (0.008) | 0.731 (0.014) |
| 2.5 | 2.376 (0.396) | 0.926 (0.008) | >0.999 | ||
| Second generation allowed to be missing (see Figure II and Simulation Study II text) | |||||
| E | 80 extended pedigreesa | 1.0 | 0.993 (0.252) | 0.952 (0.007) | 0.055 (0.007) |
| 1.7 | 1.697 (0.339) | 0.959 (0.006) | 0.736 (0.014) | ||
| 2.5 | 2.536 (0.464) | 0.957 (0.006) | >0.999 | ||
| F | 80 nuclear familiesb | 1.0 | 1.001 (1.923) | 0.953 (0.007) | 0.047 (0.007) |
| 1.7 | 1.612 (1.585) | 0.974 (0.005) | 0.222 (0.013) | ||
| 2.5 | 2.310 (0.974) | 0.972 (0.005) | 0.549 (0.016) | ||
| G | 180 nuclear familiescd | 1.0 | 0.972 (0.680) | 0.961 (0.006) | 0.057 (0.007) |
| 1.7 | 1.624 (0.504) | 0.960 (0.006) | 0.359 (0.015) | ||
| 2.5 | 2.306 (0.627) | 0.939 (0.008) | 0.853 (0.011) | ||
Extended pedigrees with all non-founders affected are simulated and analyzed.
A single nuclear family is chosen per extended family. See the text for the selection strategy
All nuclear families with at least one genotyped parent and one affected child from an extended family.
The reduction in the number of families from scenario C is due to the missing parents.
When μ = 1.7, analyzing pedigrees in their entirety produces the most accurate result (μ̂ = 1:691, coverage = 0.953). Analyzing either 80 nuclear families or all 240 nuclear families slightly underestimates the MFG effect (Scenario B: μ̂ = 1.598, coverage = 0.948; Scenario C: μ̂ = 1.623, coverage = 0.932). As expected, analyzing pedigrees in their entirety is more powerful than analyzing one nuclear family per pedigree, but analyzing all 240 nuclear families recovers the power (Scenario A: rejection rate = 0.825; Scenario B: rejection rate = 0.607; and Scenario C: rejection rate = 0.80). More striking trends are seen when μ = 2.5 where there is plenty of power to detect the MFG effect with all three study designs (power > 0.999). Analyzing pedigrees in their entirety gives the most accurate estimates (μ̂ = 2.532, coverage = 0.954). Surprisingly, we see decided underestimation and reduced coverage when analyzing either one nuclear family per pedigree or analyzing all nuclear families (Scenario B: μ̂ = 2.231, coverage = 0.900; Scenario C: μ̂ = 2.356, coverage = 0.913).
To investigate if the parameter underestimation observed in Scenarios B and C results from the random mating assumption, we analyzed the same samples simulated in Scenario C (μ = 1.7 and 2.5) using the original mating type based nuclear family MFG test (Scenario D). Under both values of μ, the parameters are again underestimated (μ̂ = 1.636, coverage = 0.940; μ̂ = 2.376, coverage = 0.926). Since we continue to see underestimation of the MFG parameter even when we do not assume random mating, we can rule it out as a contributing factor. When μ = 1.7, the rejection rate is slightly lower than in Scenario C, and this difference proves to be statistically significant according to McNemar’s test (P-value < 0.001). This suggests that assuming random mating when it holds increases the power of the MFG test.
Instead, the underestimation is the result of the loss of information regarding MFG incompatibility that occurs when analyzing extended pedigrees as nuclear families. For our simulations, in order for individual 8 to be MFG incompatible, her mother (individual 5) must have genotype 1/1, meaning that the individual 5 cannot be MFG incompatible with her mother (individual 2). As a result, we expect individual 5 to carry the 1/1 genotype more often and carry the 1/2 genotype less often than individual 6, whose child (individual 7) can be incompatible with his mother (individual 3) even when the individual 6 is incompatible with his mother (individual 2). We can make our intuition more concrete by calculating the average frequencies for individuals 5 and 6 over all the simulations under Scenario A where μ = 2.5. The frequency of the 1/1 genotype for individual 5 (f1/1 = 0.166) is greater than the frequency for individual 6 (f1/1 = 0.109), and the frequency of the 1/2 genotype for individual 5 (f1/2 = 0.523) is less than the frequency for individual 6 (f1/2 = 0.664). When analyzed as a nuclear family, individual 6’s decreased tendency to be incompatible with individual 2, leads to an underestimation in μ. On the other hand, the extended pedigree’s likelihood correctly accounts for the relationships among the three generations.
There are additional situations where exploiting extended pedigrees has advantages. For example, if a parent of an affected individual is unavailable, we expect an increase in power if we genotype the parents of the missing parent. Using the 80 three-generational pedigrees from Table V, Scenario A, we deleted genotypes in the second generation so that 40 families are missing individuals 3–6, 20 families are missing individuals 5 and 6, 10 families are missing individual 5, and 10 families are missing individual 6. After analyzing the 80 extended pedigrees with missing genotypes (Table V, Scenario E), we conducted two nuclear family analyses. In the first analysis (Scenario F), we select a nuclear family with at least one genotyped affected offspring from each pedigree, resulting in 80 nuclear families, choosing trios over dyads, and dyads over singletons (individuals with two ungenotyped parents). If two nuclear families fit this criterion, one is chosen at random. Our final sample composition is 20 trios, 20 dyads, and 40 singletons. In the second nuclear family analysis (Scenario G), we analyze all nuclear families with at least one genotyped affected offspring, for a total of 180 nuclear families (40 trios, 60 dyads, and 80 singletons).
When μ = 1.0, parameter estimates μ̂ (0.972–1.001), type-I error rates (0.047–0.057), and coverage (0.952–0.961) are appropriate if extended pedigrees are analyzed in their entirety, if a single nuclear family per pedigree is analyzed, or if all allowable nuclear families are analyzed. The most dramatic difference from the complete genotype data analyses are the increases in the SEs for μ when the pedigrees with missing data are analyzed as nuclear families (Scenario F: SE = 1.923 vs. Scenario B: SE = 0.277; Scenario G: SE = 0.680 vs. Scenario C: SE = 0.211), whereas the SE for μ is only slightly increased when using an extended pedigree study design (Scenario E: SE = 0.252 vs. Scenario A: SE = 0.219).
When μ = 1.7, the EMFG test still yields reasonable parameter estimates and power in the presence of missing genotypes when extended pedigrees are analyzed in their entirety (μ̂ = 1.697, coverage = 0.959, rejection rate = 0.736). There is a slight power loss compared to the situation where all individuals are genotyped (Scenario A, rejection rate = 0.825). When a single nuclear family per pedigree is analyzed, there is a large reduction in power relative to Scenario B, and much larger standard errors (1.585), but μ is only slightly underestimated (μ̂ = 1.612, coverage = 0.974, rejection rate = 0.222). When all allowable nuclear families are analyzed (Scenario G), μ is slightly underestimated, and power is larger in comparison to using only one nuclear family per analysis, but is still much smaller than the power seen when analyzing extended pedigrees (μ̂ = 1.624, coverage = 0.960, rejection rate = 0.359). The SE of μ is still large (0.504) but not nearly as large as in Scenario F. Thus, analyzing extended pedigrees provides a dramatic increase in power over nuclear families when there are missing data.
When μ = 2.5 and pedigrees are analyzed in their entirety, parameter estimates are accurate, and power is very high (μ̂ = 2.536, coverage = 0.957, rejection rate ≥ 0.999). When nuclear families are analyzed, the MFG parameter is underestimated, standard errors are large, and power is reduced (Scenario F: μ̂ = 2.31, SE = 0.974, coverage = 0.972, rejection rate = 0.549; Scenario G: μ̂ = 2.306, SE = 0.627, coverage = 0.939, rejection rate = 0.853). Again the large standard errors for μ offset the bias so that the coverage is not significantly reduced from the nominal value of 0.95.
SIMULATION STUDY 3: POPULATION STRATIFICATION
Because the EMFG test assumes random mating it is important to investigate violations of this assumption. Violations can occur in different ways. For example, population stratification (PS) may be present. Sinsheimer et al. [2003] found that the original MFG test, which does not assume random mating, is unaffected by PS, but it is possible that the EMFG test may be sensitive to it.
To understand the impact of PS, we turn to additional simulations. Each scenario involves one of three levels (No PS, Moderate PS, and Large PS), 1,000 samples, and the null hypothesis of μ = 1.0 or the alternative hypothesis of μ = 2.5. The true MFG effect is the same in the population but the baseline disease risk differs between populations. To allow direct comparison to the nuclear family MFG test, we simulate 240 parent-offspring trios. In the No PS example, trios are sampled from one population using an average of RHD frequencies for Finland and Germany (P(1) = 0.37, P(2) = 0.63). In the second example (Moderate PS), half the trios have Finnish RHD frequencies [Palmer et al., 2002; P(1) = 0.33, P(2) = 0.67], and half have German RHD frequencies [Wagner et al., 1995; P(1) = 0.41, P(2) = 0.59]. In the third example (Large PS), half the trios have Finnish RHD frequencies, and half have Asian RHD frequencies [Reid and Lomas-Francis, 2004; P(1) = 0.10, P(2) = 0.90]. Results are shown in Table VI.
TABLE VI.
Parameter estimates, coverage, and rejection rates for Simulation Study 3
| Scenario | μ̂(SE) | Coverage (SE) | Rejection rate (SE) |
|---|---|---|---|
| A: No PS, μ = 1.0 | 0.983 (0.207) | 0.954 (0.007) | 0.050 (0.007) |
| B: Moderate PS, μ = 1.0 | 0.970 (0.256) | 0.953 (0.007) | 0.052 (0.007) |
| C: Large PS, μ = 1.0 | 0.804 (0.325) | 0.929 (0.008) | 0.085 (0.009) |
| D: No PS, μ = 2.5 | 2.506 (0.529) | 0.960 (0.006) | 0.988 (0.003) |
| E: Moderate PS, μ = 2.5 | 2.480 (0.523) | 0.949 (0.007) | 0.985 (0.004) |
| F: Large PS, μ = 2.5 assuming RM | 2.054 (0.663) | 0.900 (0.009) | 0.636 (0.015) |
| G: Large PS, μ = 2.5 not assuming RMa | 2.492 (0.893) | 0.970 (0.005) | 0.768 (0.013) |
Analyzed using the nuclear family MFG test [Kraft et al., [14]2004, 2005].
For the samples with μ = 1.0 and No PS or Moderate PS, coverage (0.954 and 0.953), type-I error rate (0.050 and 0.052), and parameter estimation (0.983 and 0.970) are appropriate. Under large PS, type-I error rate is inflated (0.085), coverage is low (0.929), and μ is underestimated (0.804). For samples with μ = 2.5 and No PS (μ̂ = 2.506, coverage = 0.960, power = 0.988) or Moderate PS (μ̂ = 2.48, coverage 50.949, power = 0.985), the results are very similar. In the Large PS scenario, MFG is underestimated (2.054), coverage is low (0.900), and power is greatly reduced (rejection rate = 0.636). If we analyze these same samples using the nuclear family MFG test [Kraft et al., 2004, 2005], we obtain an appropriate parameter estimate and coverage (μ̂ = 2.492, coverage = 0.970). However, power is reduced (rejection rate = 0.768), reflecting the small proportion of Asians who are affected because of MFG incompatibility.
Because the EMFG test is sensitive to random mating violation, we suggest testing for departures from this assumption in a randomly selected sample of control parent pairs from the same study population. One option is to test for departure from HWE. A better option is to test for random mating violation by comparing observed parental mating type frequencies to expected mating type frequencies assuming random mating (Table II). Let P(MTj)Ha and P(MTj)Ho represent the probability of mating type j under the null (random mating holds) and alternative (random mating violated) hypotheses. If we denote the corresponding multinomial mating type counts in the control data by Nj, then the appropriate test statistic is
| (5) |
This statistic is compared to a χ2 distribution with three degrees of freedom. When we apply the HWE and LR tests to 1,000 samples each comprised of 120 Finnish control parents and 120 Asian control parents (Table VI, Scenario C), non-random mating that leads to 20% underestimation of μ (Table VI, Scenario D) can be detected with approximately 70% power under the LR test (5) vs. 40% power under a HWE test. The random mating test is more powerful despite having more degrees of freedom and allows us to determine situations where the nuclear family MFG test may be preferable to the EMFG test.
APPLICATION TO RHD INCOMPATIBILITY
To illustrate the application of Mendel’s EMFG test and compare using an extended pedigree with nuclear families on actual data, we analyze a single, large pedigree from an internal isolate in Finland. This pedigree is part of a previously described genetic study of Finnish individuals affected with schizophrenia, schizoaffective psychosis disorder, and schizophrenia spectrum disorder [Hovatta et al., 1999; Ekelund et al., 2000, 2001; Paunio et al., 2001]. The pedigree spans 5 generations with 13 founders, 19 married-ins and 62 offspring; 14 individuals are known to be affected. A total of 51 of the 93 pedigree members have RHD genotypes. In previous studies, nuclear families were extracted from this pedigree and included in the data set for analysis to examine the question of whether RHD incompatibility is a risk factor for schizophrenia [Kraft et al., 2004; Palmer et al., 2002, 2008].
We estimate the RHD incompatibility effect in this large extended pedigree by using three study designs similar to those used in Simulation Study II: using the extended pedigree, using nuclear families without regard to individuals interrelatedness (11 Nuclear Families), and using only nuclear families that are distantly related to one another (7 Selected Nuclear Families). To treat the data as nuclear families without regard to their interrelatedness, we first use the TRIM_PEDIGREE option of Mendel software [Lange and Sinsheimer, 2004] to extract the 11 nuclear families with at least one genotyped affected sibling. To select the nuclear families that are not closely related, we first calculate the theoretical kinship matrix for the pedigree using the KINSHIP_MATRICES option of Mendel and then select the seven nuclear families where the between nuclear family relatedness of the offspring is no closer than second cousins.
The estimate of the MFG incompatibility parameter μ (~1.8) is similar for all three analyses (Table VII) and is lowest for the complete pedigree analysis and highest when using the selected nuclear families. The standard error and the P-value testing μ’s deviation from the null value of 1 are smallest in the complete pedigree analysis and largest in the selected nuclear families. The founder genotype frequency estimates are consistent with the published Finnish RHD genotype frequencies [Laakso and Toiviainen, 2009]. These results further illustrate the advantage of keeping extended pedigrees intact.
TABLE VII.
RHD incompatibility illustration
| Number genotyped | P̂(1/1)(SE) | P̂(1/2)(SE) | P̂(2/2)(SE) | μ̂(SE) | P-value | |
|---|---|---|---|---|---|---|
| Complete pedigree | 51 | 0.158 (0.114) | 0.508 (0.202) | 0.334 (0.158) | 1.827 (1.016) | 0.159 |
| 11 nuclear families | 48 | 0.171 (0.101) | 0.467 (0.126) | 0.361 (0.113) | 1.887 (1.260) | 0.355 |
| Seven selected nuclear families | 30 | 0.122 (0.117) | 0.466 (0.158) | 0.412 (0.158) | 1.944 (1.659) | 0.415 |
DISCUSSION
The nuclear family MFG test [Kraft et al., 2004, 2005; Sinsheimer et al. 2003] can be extended to accommodate arbitrary pedigree structures. The EMFG test, like its predecessor, is capable of estimating gender-specific MFG incompatibility effects, maternal allelic effects, and offspring allelic effects. Our simulation studies address EMFG test properties, such as accuracy of parameter estimation, coverage and rejection rates, and the sensitivity to model misspecification. Simulation Study 1 shows that the EMFG test has reasonable type-I error rate, coverage, and parameter estimates and adequate power to detect moderate effect sizes with as few as 200 affected cousin pairs in three-generation pedigrees.
Simulation Study 2 demonstrates the superior power advantage of the EMFG test over the nuclear family MFG test. It is hardly surprising that selecting one nuclear family per extended pedigree reduces the power. Additional power can also be gained by using genotypes rather than mating types. The most dramatic power gains occur when there are missing genotype data. In this setting, preserving pedigree structure allows genotype information to propagate from genotyped members to ungenotyped members. This information is lost when a pedigree is split into nuclear families. The EMFG test also gives accurate parameter estimates and reasonable type-I error rates in the presence of missing parental genotypes.
The EMFG test can decrease bias. The extent of bias caused by the loss of information that occurs from treating related nuclear families as independent can be large, as much as a 10.8% underestimation in our simulations. Analyzing pedigrees in their entirety avoids this problem without sacrificing power. Of course the degree of the bias depends on who is affected in the pedigree. In our simulations, we see notable underestimation of the RHD incompatibility parameter because both mother and daughter are affected. We would not expect to see the same degree of underestimation if only the grandchildren were affected. Furthermore, the degree of bias and its direction depend in part on the nature of the MFG incompatibility. For example, if MFG incompatibility requires that both mother and daughter share the 2/2 genotype, then we would expect to see overestimation when analyzing nuclear families taken from the same pedigree structure as Figure 2 (siblings and grandchildren affected).
In our analysis of a real data set, the EMFG test was capable of estimating the effects of RHD incompatibility on schizophrenia for a large family spanning five generations, where nearly half of the family members were missing genotypes. When analyzing only some nuclear families from this extended pedigree, precision was reduced. In practice, we do not recommend using the EMFG test on such a small sample size, but did so here to provide a particularly straightforward comparison between pedigree and nuclear family analyses in a more realistic way than was possible in our simulations.
Unlike the nuclear family MFG test, the EMFG test is sensitive to violations of random mating. When PS is present and large, the EMFG test loses power and underestimates the true MFG effect. Therefore, one should conduct a test of random mating prior to applying the EMFG test if violation is suspected. A test of HWE is not a substitute for a test of random mating comparing observed mating type frequencies to expected mating type frequencies. When a severe violation of random mating is observed, we recommend using the nuclear family version of the MFG test.
In spite of its sensitivity to violations of random mating, the EMFG test is an improvement over our previous test, particularly in ease of use. Although our simulation studies focus on specific examples of MFG incompatibility, the EMFG test software allows users to specify the risks under the general bi-allelic locus model (Table I). Our new EMFG test has been developed as part of the statistical genetics software program Mendel [Lange et al., 2001]. It is less data specific and more user friendly than our earlier software versions. The EMFG test will be released as an option in an upcoming version of Mendel.
Acknowledgments
We thank Drs. L. Peltonen, J. Lonnqvist, and J. Turunen for the use of the Finnish Pedigree. These data were collected with funding from the Center of Excellence in Disease Genetics, Academy of Finland, Biocentrum Helsinki Finland, and USPHS grant MH66001.
References
- Cannon TD. On the nature and mechanisms of obstetric influences in schizophrenia: a review and synthesis of epidemiological studies. Int Rev Psychiatry. 1997;9:387–397. [Google Scholar]
- Cantor-Graae E, Ismail B, McNeil TF. Are neurological abnormalities in schizophrenic patients and their siblings the result of perinatal trauma? Acta Psychiatr Scand. 2000;101:142–147. doi: 10.1034/j.1600-0447.2000.90054.x. [DOI] [PubMed] [Google Scholar]
- Chen DF, Hu Y, Yang F, Li Z, Wu B, Fang Z, Li J, Wang L. Cytochrome P450 gene polymorphisms and risk of low birth weight. Genet Epidemiol. 2005;28:368–375. doi: 10.1002/gepi.20067. [DOI] [PubMed] [Google Scholar]
- Chen J, Zheng H, Wilson ML. Likelihood ratio tests for maternal and fetal genetic effects on obstetric complications. Genet Epidemiol. 2009;33:526–538. doi: 10.1002/gepi.20405. [DOI] [PubMed] [Google Scholar]
- Cordell HJ, Barratt BJ, Clayton DG. Case/pseudocontrol analysis in genetic association studies: a unified framework for detection of genotype and haplotype associations, gene-gene and gene-environment interactions, and parent-of-origin effects. Genet Epidemiol. 2004;26:167–185. doi: 10.1002/gepi.10307. [DOI] [PubMed] [Google Scholar]
- Ekelund J, Lichtermann D, Hovatta I, Ellonen P, Suvisaari J, Terwilliger JD, Juvonen H, Varilo T, Ara-järvi R, Kokko-Sahin ML, Lönnqvist J, Peltonen L. Genome-wide scan for schizophrenia in the Finnish population: evidence for a locus on chromosome 7q22. Hum Mol Genet. 2000;9:1049–1057. doi: 10.1093/hmg/9.7.1049. [DOI] [PubMed] [Google Scholar]
- Ekelund J, Hovatta I, Parker A, Paunio T, Varilo T, Martin R, Suhonen J, Ellonen P, Chan G, Sinsheimer JS, Sobel E, Juvonen H, Arajärvi R, Partonen T, Suvisaari J, Lönnqvist J, Meyer J, Peltonen L. Chromosome 1 loci in Finnish schizophrenia families. Hum Mol Genet. 2001;15:1611–1617. doi: 10.1093/hmg/10.15.1611. [DOI] [PubMed] [Google Scholar]
- Harney S, Newton J, Milicic A, Brown MA, Wordsworth BP. Non-inherited maternal HLA alleles are associated with rheumatoid arthritis. Rheumatology. 2003;42:171–174. doi: 10.1093/rheumatology/keg059. [DOI] [PubMed] [Google Scholar]
- Hovatta I, Varilo T, Suvisaari J, Terwilliger JD, Ollikainen V, Arajärvi R, Juvonen H, Kokko-Sahin ML, Väisänen L, Mannila H, Lönnqvist J, Peltonen L. A genomewide screen for schizophrenia genes in an isolated Finnish subpopulation, suggesting multiple susceptibility loci. Am J Hum Genet. 1999;65:1114–1124. doi: 10.1086/302567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh HJ, Palmer CGS, Harney S, Newton JL, Wordsworth P, Brown MA, Sinsheimer JS. The v-MFG Test: investigating maternal, offspring, and maternal-fetal genetic incompatibility effects on disease and viability. Genet Epidemiol. 2006a;30:333–347. doi: 10.1002/gepi.20148. [DOI] [PubMed] [Google Scholar]
- Hsieh HJ, Palmer CGS, Sinsheimer JS. Allowing for missing data at highly polymorphic genes when testing for maternal, offspring, and maternal-fetal genotype incompatibility effects. Hum Hered. 2006b;62:165–174. doi: 10.1159/000096444. [DOI] [PubMed] [Google Scholar]
- Insel BJ, Brown AS, Bresnahan MA, Schaefer CA, Susser ES. Maternal-fetal blood incompatibility and the risk of schizophrenia in offspring. Schizophr Res. 2005;80:331–342. doi: 10.1016/j.schres.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Kraft P, Palmer CGS, Woodward JA, Turunen JA, Minassian S, Paunio T, Lonnqvist J, Peltonen L, Sinsheimer JS. RHD Maternal-fetal genotype incompatibility and schizophrenia: extending the MFG test to include multiple siblings and birth order. Eur J Hum Genet. 2004;12:192–198. doi: 10.1038/sj.ejhg.5201129. [DOI] [PubMed] [Google Scholar]
- Kraft P, Hsieh HJ, Cordell HJ, Sinsheimer JS. A conditional-on-exchangeable-parental-genotypes likelihood that remains unbiased at the causal locus under multiple-affected-sibling ascertainment. Genet Epidemiol. 2005;29:87–90. doi: 10.1002/gepi.20069. [DOI] [PubMed] [Google Scholar]
- Laakso S, Toiviainen W. Finnish Red Cross Blood Service in 2008. Helsinki: Libris; 2009. [Google Scholar]
- Laing P, Knight J, Wright P, Irving W. Disruption of fetal brain development by maternal antibodies as an etiological factor in schizophrenia. In: Mednick SA, Hollister JM, editors. Neural Development and Schizophrenia: Theory And Research. New York: Plenum Press; 1995. pp. 215–245. [Google Scholar]
- Lange K. Mathematical and statistical methods for genetic analysis. 2. New York: Springer-Verlag; 2002. [Google Scholar]
- Lange K, Sinsheimer JS. The pedigree trimming problem. Hum Hered. 2004;58:108–111. doi: 10.1159/000083031. [DOI] [PubMed] [Google Scholar]
- Lange K, Cantor R, Horvath S, Perola M, Sabatti C, Sinsheimer J, Sobel E. Mendel Version 4.0: a complete package for the exact genetic analysis of discrete traits in pedigree and population data sets. Am J Hum Genet. 2001;69S:A1886. [Google Scholar]
- Li S, Lu Q, Fu W, Romero R, Cui Y. A regularized regression approach for dissecting genetic conflicts that increase disease risk in pregnancy. Stat Appl Genet Mol Biol. 2009;8 doi: 10.2202/1544-6115.1474. Article 45. [DOI] [PubMed] [Google Scholar]
- Louey S, Thornburg KL. The prenatal environment and later cardiovascular disease. Early Hum Dev. 2005;81:745–751. doi: 10.1016/j.earlhumdev.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Marcelis M, van Os J, Sham P, Jones P, Gilvarry C, Cannon M, McKenzie K, Murray R. Obstetric complications and familial morbid risk of psychiatric disorders. Am J Med Genet. 1998;81:29–36. doi: 10.1002/(sici)1096-8628(19980207)81:1<29::aid-ajmg6>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- McKinney PA, Juszczak E, Findlay E, Smith K, Thomson CS. Pre- and perinatal risk factors for childhood leukaemia and other malignancies: a Scottish case control study. Br J Can. 1999;80:1844–1851. doi: 10.1038/sj.bjc.6690609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minassian SL, Palmer CGS, Turunen JA, Paunio T, Lonnqvist J, Peltonen L, Woodward JA, Sinsheimer JS. Incorporating serotypes into family based association studies using the MFG test. Ann Hum Genet. 2006;70:541–553. doi: 10.1111/j.1469-1809.2005.00243.x. [DOI] [PubMed] [Google Scholar]
- Newton JL, Harney SM, Wordsworth BP, Brown MA. A review of the MHC genetics of rheumatoid arthritis. Genes Immun. 2004;5:151–157. doi: 10.1038/sj.gene.6364045. [DOI] [PubMed] [Google Scholar]
- Ober C. HLA and pregnancy: the paradox of the fetal allograft. Am J Hum Genet. 1998;62:1–5. doi: 10.1086/301692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott J. Estimation of the recombination fraction in human pedigrees: efficient computation of the likelihood for human linkage studies. Am J Hum Genet. 1974;26:588–597. [PMC free article] [PubMed] [Google Scholar]
- Palmer CGS, Turunen JA, Sinsheimer JS, Minassian S, Paunio T, Lonnqvist J, Peltonen L, Woodward JA. RHD maternal-fetal-genotype incompatibility increases schizophrenia susceptibility. Am J Hum Genet. 2002;71:1312–1319. doi: 10.1086/344659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CGS, Hsieh HJ, Reed EF, Lonnqvist J, Peltonen L, Woodward JA, Sinsheimer JS. HLA-B maternal-fetal genotype matching increases risk of schizophrenia. Am J Hum Genet. 2006;79:710–715. doi: 10.1086/507829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CGS, Mallery E, Turunen JA, Hsieh H, Peltonen L, Lonnqvist J, Woodward JA, Sinsheimer JS. Effect of Rhesus D incompatibility on schizophrenia depends on offspring sex. Schizophr Res. 2008;104:135–145. doi: 10.1016/j.schres.2008.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paunio T, Ekelund J, Varilo T, Parker A, Hovatta I, Turunen JA, Rinard K, Foti A, Terwilliger JD, Juv-onen H, Suvisaari J, Arajärvi R, Suokas J, Partonen T, Lönnqvist J, Meyer J, Peltonen L. Genome-wide scan in a nationwide study sample of schizophrenia families in Finland reveals susceptibility loci on chromosomes 2q and 5q. Hum Mol Genet. 2001;10:3037–3048. doi: 10.1093/hmg/10.26.3037. [DOI] [PubMed] [Google Scholar]
- Reid ME, Lomas-Francis C. The Blood Group Antigen Facts Book. 2. New York: Elsevier Academic Press; 2004. [Google Scholar]
- Sinsheimer JS, Palmer CGS, Woodward JA. Detecting genotype combinations that increase risk for disease: the maternal-fetal genotype incompatibility test. Genet Epidemiol. 2003;24:1–13. doi: 10.1002/gepi.10211. [DOI] [PubMed] [Google Scholar]
- van der Horst-Bruinsma IE, Haxes JM, Schreuder GM, Radstake TR, Barrera P, van de Putte LB, Mustamu D, van Schaardenburg D, Breedveld FC, de Vries RR. Influence of non-inherited maternal HLA-DR antigens on susceptibility to rheumatoid arthritis. Ann Rheum Dis. 1998;57:672–675. doi: 10.1136/ard.57.11.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner FF, Kasulke D, Kerowgan M, Flegel WA. Frequencies of the blood groups ABO, Rhesus, D Category VI, Kell, and of clinically relevant high-frequency antigens in south-western Germany. Infus Trans. 1995;22:285–290. doi: 10.1159/000223144. [DOI] [PubMed] [Google Scholar]
- Weinberg CR, Wilcox AJ, Lie RT. A log-linear approach to case-parent-triad data: assessing effects of disease genes that act either directly or through maternal effects and that may be subject to parental imprinting. Am J Hum Genet. 1998;62:969–978. doi: 10.1086/301802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi PP, Kalaydjian A, Avramopoulos D, Shao H, Fallin MD, Newschaffer CJ. Rh and ABO maternal-fetal incompatibility and risk of autism. Am J Med Genet Part B. 2006;141B:643–647. doi: 10.1002/ajmg.b.30391. [DOI] [PubMed] [Google Scholar]