

Abstract
Recombination catalyzed by the Flp site-specific recombinase involves breakage and joining of four DNA strands between two target substrates. The reaction is carried out in two steps of pairwise strand exchanges by a DNA–protein assembly in which four Flp monomers act cooperatively to execute strand cleavage and joining. Two models for recombination have been proposed. In the trimer model, the two active sites required for each step are assembled from three Flp monomers. In the tetramer (or dimer of asymmetric dimers) model, the two active sites are assembled from four Flp monomers, two monomers each contributing one active site. Experiments in which the two models challenge each other reveal that, within the Flp tetramer arranged on a Holliday junction, the two active sites required for its resolution are derived from all four, rather than three, Flp monomers. Thus, the relative protein subunit configuration of the tetramer silences the trimers within it by excluding them from assembling a functional active site pair.
Keywords: Site-specific recombination, shared active site, active site silencing
Flp site-specific recombination provides a useful paradigm for mechanisms of strand breakage and exchange between two DNA molecules mediated by a protein assembly consisting of four recombinase monomers (Jayaram 1994; Sadowski 1995). An important issue is how the assembly of the recombination complex from two DNA substrates and four monomeric units of the recombinase is coupled to the spatial and temporal coordination of the ensuing phosphoryl transfer steps. Assuming that the cleavage/joining events at the four phosphodiester bonds are executed by identical active site pockets, there must be some mechanism built into the architecture of the DNA–protein complex to prevent undesirable strand exchanges. A broken strand may, in principle, be joined onto itself (nonrecombinant resealing), or transferred to one of the other three strands (recombinant joining). Thus, only one-third of the possible recombinant outcomes would be biologically meaningful. The relationship between the architecture of the Flp–DNA complex and its strand transfer potential has been brought into special focus by two alternative models proposed recently (Lee and Jayaram 1995; Qian and Cox 1995; Lee et al. 1996). The point of contention between the two models is whether the recombinationally relevant unit of Flp is a trimer or a tetramer.
The Flp protein catalyzes site-specific recombination by use of a mechanism that is common to recombinases of the Integrase family (Argos et al. 1986; Abremski and Hoess 1992; Stark et al. 1992; Sadowski 1995). The first step in the reaction is the cutting and swapping of one pair of strands at one end of the strand exchange region (or spacer) to form a Holliday junction that covalently unites the DNA partners in a four-stranded structure. The recombination complex then changes its configuration in an isomerization step so as to mediate cutting and swapping of the second pair of strands at the other end of the spacer. Thus, the Holliday junction is resolved, and a pair of reciprocally recombinant products are formed.
A monomer of the Flp protein has only a partial active site (Chen et al. 1992, 1993). The assembly of the complete active site requires the sharing of catalytic residues by two Flp monomers. Within the shared active site, three key catalytic residues (Arg-191, His-305, and Arg-308; the RHR triad) are supplied by one monomer, which orients the scissile phosphate adjacent to it for nucleophilic attack (activation in cis). The active site tyrosine (Tyr-343), which provides the nucleophile for strand cleavage, is supplied by a second Flp monomer bound across the spacer (DNA cleavage in trans). These four residues represent the invariant tetrad of the Int family recombinases (Argos et al. 1986; Abremski and Hoess 1992).
Within the context of the Flp tetramer, how are the two active sites for generating the Holliday intermediate and the two active sites for resolving it derived? Do all four Flp monomers contribute to each of the active site pairs, or will less than four Flp monomers suffice? The question is significant in light of the finding by Qian and Cox (1995) that a Holliday junction can be resolved by three Flp monomers bound to three of its arms. They incorporate this result into a model in which a Flp trimer is the chemically relevant unit of recombination. As illustrated in Figure 1A, the trimer is functionally asymmetric, with distinct catalytic contributions from the individual monomers. In contrast, based on their results on the catalytic contributions of individual Flp monomers towards recombination, Lee and Jayaram (1995) have proposed a model in which a Flp tetramer composed of two asymmetric dimers is the unit of recombination. In the asymmetric dimer, one monomer is responsible for the orientation of the labile phosphate (by use of the RHR domain), and the other monomer is responsible for effecting strand cleavage with Tyr-343 (Fig. 1B).
Figure 1.
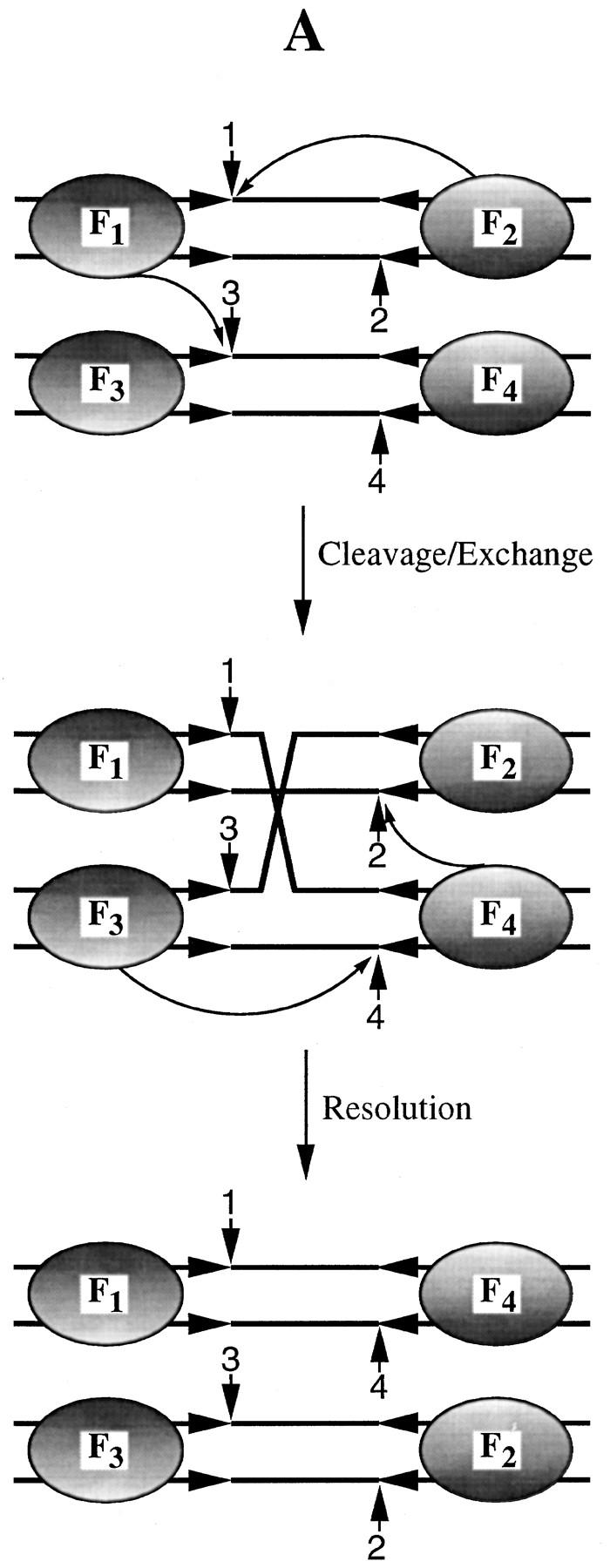
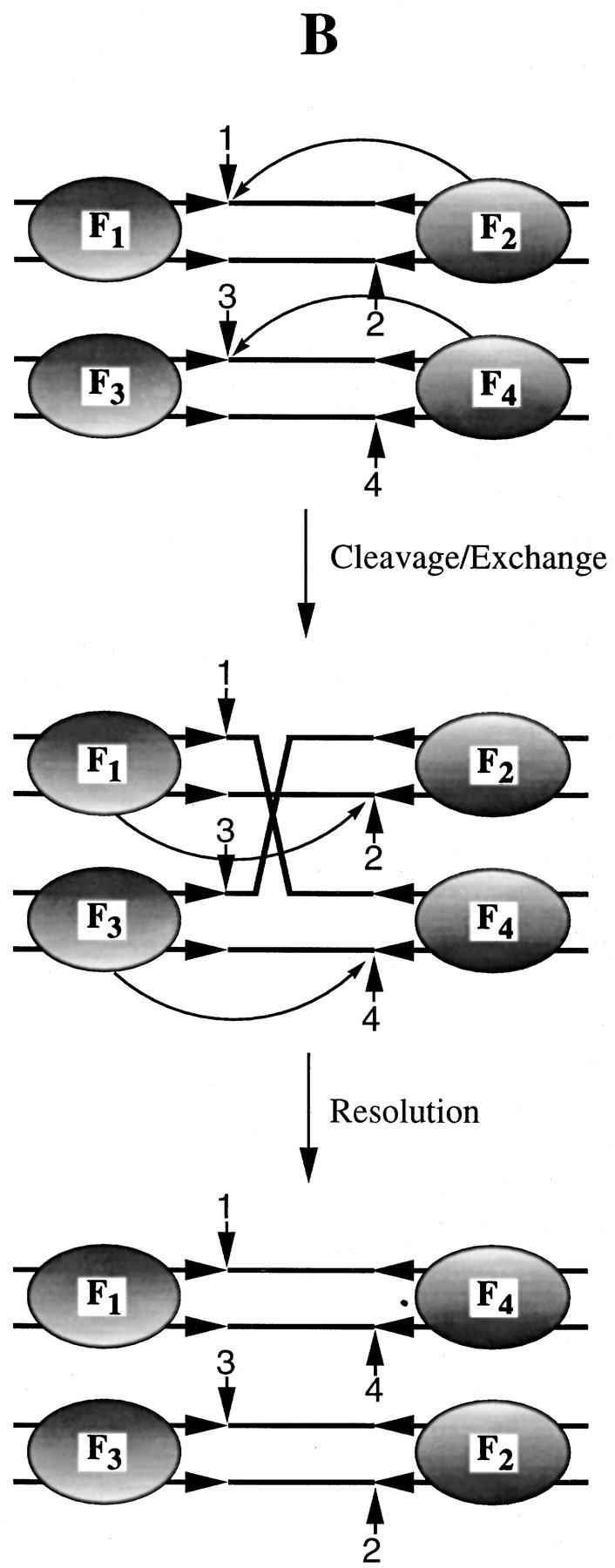
Two models for Flp site-specific recombination. The two models for Flp recombination proposed recently (Qian and Cox 1995; Lee and Jayaram 1996; Lee et al. 1996) are shown schematically to depict their modes of operation during the generation of a Holliday junction from two linear DNA duplexes, and the subsequent resolution of this junction. The phosphodiesters at the cleavage/exchange positions are numbered 1–4. The Flp monomers bound adjacent to each of these positions are indicated by the appropriate subscript (F1–F4). The horizontal parallel arrows represent Flp-binding elements. (A) In the asymmetric trimer model, catalytic contributions from three Flp monomers F1, F2, and F3 are sufficient to mediate strand cleavage and exchange at the left end of the strand exchange region (spacer). The directionality of Tyr-343 donation (or the cleavage mode; indicated by the curved arrow) conforms to trans-horizontal (F2–F1) plus trans-vertical (F1–F3), as defined by Chen et al. (1992). Note that F4–F1 (trans-diagonal) and F1–F3 (trans-vertical) cleavages would also yield exchange at the left. Resolution of the Holliday intermediate at the right may be mediated by the F1–F2–F4 set or the F2–F3–F4 set. (B) In the dimer of asymmetric dimers model, it is the whole tetramer of Flp (F1–F2–F3–F4) that is the catalytic unit in recombination. For strand exchange at the left, F1 and F3 provide the RHR triad domain, and F2 and F4 provide Tyr-343. Here, the cleavage is depicted to occur in the trans-horizontal fashion. The preponderance of experimental evidence supports this cleavage mode by Flp (Lee et al. 1994, 1996). For resolution at the right, the F1–F3 pair and the F2–F4 pair reciprocally reverse their catalytic contributions. Note that trans-diagonal mode of cleavage would also fit the model.
From a theoretical perspective, there is no good argument to choose one model over the other. Consistent with the shared active site of Flp, one can obtain the two catalytic pockets needed to execute each of the two pairwise strand exchanges with equal ease from the dimeric dimer or a pair of asymmetric trimers (see Fig. 1). In this paper, we describe experiments designed to distinguish between the two models in the context of a normal recombination complex consisting of two DNA partners and four Flp monomers. The results are readily explained by the dimer of asymmetric dimers model, but cannot be satisfied by the asymmetric trimer model.
Results
The two models for the Flp recombination reaction are shown in Figure 1. Their distinguishing features are highlighted with respect to the formation and resolution of the Holliday junction intermediate of recombination.
In the asymmetric trimer model (Fig. 1A; Qian and Cox 1995), three Flp monomers, two bound on the same side of the strand exchange region (or spacer) and one bound on the opposite side, mediate two strand breakage and joining events at one or the other end of the spacer. In the context of one round of recombination, the first and second pair of strand exchanges would be mediated by two sets of Flp trimers. The curved arrows indicate the Flp monomers that donate the active site tyrosine, or the directionality of strand cleavage. Note that in this model the two cleavage events are nonequivalent. The phosphate at position 1 is cleaved in the trans-horizontal mode (across the spacer) by F2, and that at position 3 is cleaved in the trans-vertical mode (top to bottom on the same side of the spacer) by F1. By convention, the DNA arms are arranged in a parallel fashion; the two left arms (or the two right arms) are arranged on the same side of the spacer (Chen et al. 1992). A variation of this model that utilizes trans-vertical (bottom to top; cleavage at 1) plus trans-diagonal (top to bottom across the spacer; cleavage at 3) can also be drawn.
In the dimer of asymmetric dimers model (Fig. 1B; Lee and Jayaram 1995), two dimers assembled from all four Flp monomers bound to the two DNA partners or to the Holliday junction mediate resolution. The asymmetry in the dimer results from their nonequivalent contributions to the active site: One monomer donates the RHR triad domain, and the other provides Tyr-343. During normal recombination, there must be a reversal of dimer asymmetry as the recombination complex switches from the Holliday-forming mode to the Holliday-resolving mode. Note that this model requires only a unitary mode of cleavage. In Figure 1B, the cleavage is shown to be trans-horizontal. The preponderance of evidence derived from substrates containing various arm configurations is consistent with this cleavage directionality (Chen et al. 1992; Lee et al. 1994, 1996). The model will accept the trans-diagonal mode equally well.
A Holliday junction substrate can be resolved by a complementing pair of Flp active site variants
In Figure 2, the results from a resolution assay by use of a synthetic Holliday junction substrate are displayed. Each pair of panels (A,B and C,D) represents a set of resolution reactions in which the cross-over point was mobile throughout the spacer (A,B), or was stationary at a given position (C,D). Each junction was assayed for resolution at the left or the right end of the spacer by end-labeling an appropriate strand. The products of the reactions were analyzed under denaturing conditions. Wild-type Flp acted efficiently on the junctions, yielding the strand cleavage (CL or CR) as well as the resolution product (RL or RR) (Fig. 2, lanes 2). Neither the triad double mutant Flp(R191S, H305L) nor the active site tyrosine mutant Flp(Y343F) was capable of strand cutting or resolution (absence of CL/CR and RL/RR in Fig. 2, lanes 3 and 4). In reactions containing a roughly equimolar mixture of the two step–arrest variants, resolution was evident (Fig. 2, lanes 5).
Figure 2.

Resolution of Holliday junctions by a pair of Flp step–arrest mutants. The synthetic Holliday junctions used in the assay are schematically represented at the top. The Flp-binding elements have the following sequence: 5′-GAAGTTCC TATAC-3′/3′-CTTCAAGGATATG-5′. In one (A,B), the junction was freely mobile through the spacer (5′-TTTCAGAA-3′/3′-AAAGTCTT-5′) on both DNA partners. In the other (C,D), the junction point was immobilized by flanking it with two positions of heterology on either side. This substrate mimicked a crossover between two spacers with the following sequences: 5′-TTTCAGAA-3′/3′-AAAGTCTT-5′ and 5′-TTCACAAA-3′/3′-AAGTGTTT-5′. The position of crossover was between nucleotide positions 4 and 5 of the spacer. The position of the 3′ end label in each experimental set (A–D) is indicated by the asterisk (*). The reactions were analyzed by electrophoresis in 10% denaturing polyacrylamide gels. The bands labeled RL and RR refer to resolution products from cleavage/exchange at the left and right ends, respectively. (CL and CR) Strand cleavage products from the left and right ends, respectively. (S) The labeled substrate band. Lanes 1–5 represent, respectively, reactions containing no added protein, wild-type Flp, Flp(Y343F), Flp(R191S, H305L), and an approximately equimolar mixture of the two Flp variants. Densitometric scans estimated the ratio of the intensities of the RR (or RL) bands in lanes 5 and 2 of each panel (A–D) to be ∼10%–15%.
Assuming that the four arms of the Holliday junction are equivalent in Flp binding and that the binding affinities of the two Flp variants are the same, there are 16 possible protein configurations in the quarternary complexes (protein occupancy at all four arms) formed by the variant proteins (24; see Fig. 3). According to the Qian and Cox model (Fig. 1A), none of these configurations can assemble two active sites at any one time. A trimer derived from the variant pair can have two triad domains and one Tyr-343, or one triad domain and two Tyr-343, but cannot have two triad domains and two Tyr-343. Hence, no resolution should have been observed. According to the Lee–Jayaram model (Fig. 1B), a tetramer consisting of two monomeric units of the Flp variants can yield two active sites. Only the configuration that arranges the active sites on the same side of the spacer, however, can mediate resolution. As a result, depending on whether the single-labeled end is on the right or the left of the spacer, only 1 of the 16 protein arrangements would produce scorable resolution events (a labeled recombinant strand) in the assay shown in Figure 2. Densitometric scans of the autoradiograph represented by Figure 2 revealed that the extent of resolution obtained with the complementing pair relative to that obtained with Flp (∼10%–15%; see legend to Fig. 2) was within reasonable limits of the expected value (1:8 or 12.5%; see legend to Fig. 3).
Figure 3.

Protein arrangements on the Holliday arms derived from an equimolar binary combination of Flp variants. The end label on one of the strands of the Holliday substrate is shown by the asterisk. This end labeling allows experimental monitoring of resolution at the right spacer end. The triad and Tyr-343 mutants of Flp are denoted by the shaded and unshaded ovals, respectively. Of 16 possible arrangements, only 1 tetramer can yield the two active sites required for resolution at the right (shaded box). None of the trimeric configurations can provide two active sites. For wild-type Flp, all 16 arrangements are catalytically functional. Assuming that the assembly of an active site pair at the left or at the right is equally likely, 8 out of the 16 would give resolution at the right.
A Y junction substrate cannot be resolved by a pair of complementing step–arrest Flp variants
The results from Holliday junction resolution demonstrate that, within the tetrameric form of Flp, a pair of active sites can be derived from a dimeric dimer. However, it does not address the question: Is a tetramer of Flp, then, the minimal oligomeric state of the protein that can provide two active sites? Or, will fewer than four monomers suffice? The experiments of Qian and Cox (1995), by use of a Holliday junction with only three good Flp binding arms, and those of Lee et al. (1996), by use of a Y junction substrate (that has only three Flp binding arms), suggest that a trimer of Flp may also yield two active sites. In these instances, however, the possibility that a fourth Flp monomer from solution could have contributed to catalysis without stably associating with the trimeric Flp bound to DNA cannot be excluded.
Therefore, we tested whether a Y junction can be resolved by a mixture of Flp(Y343F) and Flp(R191S, H305L). Because this substrate contains three Flp-binding arms, its resolution into a linear recombinant plus a hairpin recombinant requires the action of two active sites (Fig. 4). If the three DNA bound Flp monomers alone take part in its resolution, the pair of Flp variants will not be active. Among the heterotrimers derived from the two proteins, no more than one active site can be assembled, because there will be a shortage of one Tyr-343 residue or one triad unit. If an unbound fourth Flp monomer is also involved in the reaction, this deficit can be made up. As a result, the variant protein combination should be able to exhibit catalytic complementation in the resolution of Y. As shown in Figure 4, Flp(R191S, H305L), on pairing with Flp(Y343F), failed to yield the linear or the hairpin recombinant (R1,3 or H1,2, respectively; Fig. 4, lane 5). In contrast, both products were obtained in a reaction with wild-type Flp (Fig. 4, lane 2). The presence of the cleavage band C1 in the reaction with the mutant pair (Fig. 4, lane 5), as well as its absence in reactions with the individual mutants (Fig. 4, lanes 3,4), is significant. This result reveals that, given a three-armed substrate, the mutant combination can organize one, but not two, catalytic pockets.
Figure 4.
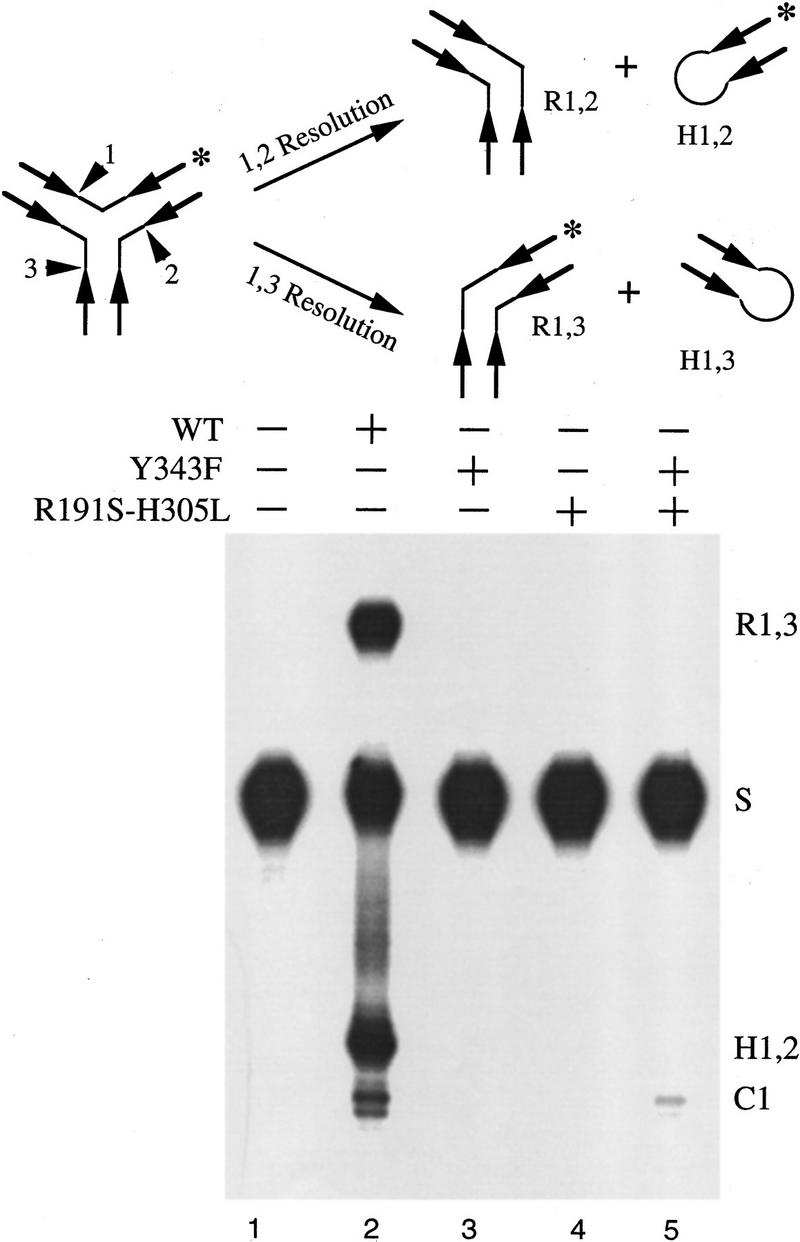
Resolution of a Y junction substrate by Flp or by paired step–arrest mutants of Flp. The Y junction substrate is the same as the Y1 substrate described previously (Lee et al. 1996) and was assembled from three synthetic DNA strands (left, right, and top). The spacer sequence on all three arms was 5′-TTTTAAAA-3′, facilitating the formation of a fully base-paired junction. Each of the three Flp binding elements had the sequence: 5′-GAAGTTCCTATTC-3′/3′-CTTCAAGGATAAG-5′. The top strand was labeled at the 3′ end (asterisk). The labile phosphodiester positions are numbered 1–3. Resolution of the Y by cleavage/exchange at 1/3 will give the labeled linear recombinant R1,3. Resolution at 1/2 will give the labeled hairpin recombinant H1,2. Strand cleavage at 1 will yield the cleavage product C1. The samples were fractionated by electrophoresis in 10% denaturing polyacrylamide. Assays in lanes 1–5 contained, respectively, no added protein, Flp, Flp(Y343F), Flp(R191S, H305L), and a roughly equimolar mixture of the two Flp variants. (S) The labeled strand from the substrate.
The failure of the catalytically complementing Flp mutant pair to resolve the Y junction, together with the ability of the same mutant pair to resolve the Holliday junction and to cleave the Y junction, shows that the oligomeric state of Flp on a DNA substrate is determined solely by the number of Flp-binding arms, with no contribution from Flp monomers in solution. On the Y substrate, the mutant pair can assemble only a trimer, and hence, may yield one, but no more than one, active site. On the Holliday substrate, they can assemble a tetramer and may yield two active sites. A trimer of wild-type Flp can simultaneously build two active sites, and thus resolve the Y junction.
Does Flp resolve a normal Holliday junction by use of a trimer or a dimeric dimer?
The finding that two active sites can be derived from either the trimeric state of Flp (resolution of Y substrate by wild-type Flp shown in Fig. 4), or from the tetrameric state (resolution of Holliday junction by the pair of Flp mutants shown in Fig. 2) highlights the critical questions: What is the reactive Flp entity that resolves a normal Holliday junction whose four arms are occupied by four Flp monomers? Is it a trimer within the tetramer or the whole tetramer itself? Although they agree with the dimeric dimer model, the experiments in Figure 2 fail to falsify the asymmetric trimer model, because the pair of Flp variants [Flp(Y343F) and Flp(R191S, H305L)] could not have assembled two active sites in their two heterotrimeric states.
Challenging the two models simultaneously would require an experimental design that ensures that all four arms of the Holliday junction are filled by Flp (i.e., a tetramer is in place), and that within this tetramer, a trimer has all the catalytic components to assemble two active sites. Furthermore, this design should be able to distinguish between the outcome of trimer-mediated resolutions from that of tetramer mediated resolutions. A strategy that satisfies these conditions is shown in Figure 5. Here, the substrate is a synthetic Holliday junction with one normal Flp-binding arm and three weakened binding arms (Lee et al. 1994, 1996). A monomer of Flp (or a catalytic Flp mutant) bound to the normal arm forms a stable protein–DNA complex with a long half-life compared with the time scale of the recombination reaction. Under appropriate salt conditions, it is possible, therefore, to establish a long-lived (with respect to the time of the resolution reaction) complex of a Flp(Y343F) monomer bound to the normal arm, and a short-lived complex of the protein bound to each of the other three arms. When an excess of wild-type Flp is added to the system, the predominant tetrameric DNA-bound protein state is expected to consist of three Flp monomers associated with the weak arms, and a Flp(Y343F) monomer associated with the normal arm. In the configuration represented by Figure 5A (two left arms and one of the two right arms bound by Flp), the trimer model predicts that resolution of the junction by cleavage and exchange at the left or at the right end of the spacer would be equally likely. Note that a trimer constituted by three wild-type Flp monomers, or one constituted by two Flp monomers and one Flp(Y343F) monomer, would be competent in resolution. Although Flp(Y343F) cannot provide the cleavage nucleophile, the protein can accept it from a partner Flp to promote cleavage/joining at the adjacent phosphodiester position. In contrast, the dimeric dimer model predicts that resolution would occur only at the right end. This is because the Flp, as well as the Flp(Y343F) monomer bound at the right, can orient the phosphodiesters adjacent to them, which can then be cleaved by Tyr-343 donated by the two Flp monomers bound at the left. Only one phosphodiester cleavage, but not the two cleavages required for resolution, can occur at the left end, because Flp(Y343F) bound at the right cannot contribute Tyr-343 to the reaction.
Figure 5.

Verification of the anticipated resolution events from the trimer and dimeric dimer models for a given arrangement of the three to one Flp to Flp(Y343F) tetramer. (A) The normal Flp-binding element in the Holliday junction had the sequence 5′-GAAGTTCCTATTC-3′/3′-CTTCAAGGATAAG-5′. In the weakened arms (large X), the CG pair at position 7 was replaced by an AT pair (Lee et al. 1996). The junction contained the native spacer sequence 5′-TTTCTAGA-3′/3′-AAAGATCT-5′. By means of a directed protein replacement strategy described earlier (Lee et al. 1994), one Flp(Y343F) monomer and three wild-type Flp monomers can be placed on the one normal Flp-binding arm and the three weakened arms, respectively. If a trimer within the tetramer is the active species, resolution of the junction at the left end of the spacer can occur by the action of the F1–F3–F4 trimer set. Similarly, resolution at the right end can occur by the action of the F2–F3–F4 trimer set. In this case, the reaction derives the two triad domains from F2 [Flp(Y343F)] and F4, and the two Tyr-343 residues from F3 and F4, respectively. The Tyr-343 is not essential for the strand joining reaction. Thus, the trimer model predicts equivalent resolutions at the left and right ends. If the tetramer (dimeric dimer) is the active species, resolution can only occur at the right. Left resolution is ruled out because Flp(Y343F) (F2) bound to the normal arm cannot provide the cleavage nucleophile. (B) Holliday junction was labeled at the 3′ end (asterisk) on two strands so as to monitor resolution at the left or the right end of the spacer. The short vertical arrows numbered 1–4 indicate the points of strand cleavage by Flp. In targeted protein placement experiments, the initial binding was done with Flp(Y343F) at ∼8–10 monomers per binding arm. The high molar ratio of protein to DNA ensured that virtually all of the normal arms in the substrate population were stably occupied by this protein. After 10-min preincubation at 30°C, an 8–10 molar excess [over Flp(Y343F)] of Flp (lanes 3–5) or Flp(R191S, H305L) (lanes 6–8) was added to the reaction mixture. Samples were analyzed at 10 min following the addition of the second protein. Lane 1 is a control reaction to which neither Flp nor a Flp variant was added. Lane 2 represents a 10 min reaction with wild-type Flp (∼8–10 protein monomers per binding arm) in 200 mm NaCl. The final NaCl concentrations were 100 mm in lanes 3 and 6, 150 mm in lanes 4 and 7, and 200 mm in lanes 5 and 8. The NaCl concentration prior to the addition of the second protein was 20 mm less in each case. The product bands corresponding to resolution at the left end (1,3 resolution) and the right end (2,4 resolution) are named RL and RR, respectively. CR is the radioactive cleavage band from the right (cleavage at 2); CL is from the left (cleavage at 1).
The results of a resolution experiment with a Holliday junction that conforms to Figure 5A are displayed in Figure 5B. In a reaction with wild-type Flp alone, the junction was resolved into recombinants to yield the radioactive strands RR (right resolution) and RL (left resolution) (Fig. 5B, lane 2). When Flp(Y343F) was targeted to the normal arm (top right) and Flp was targeted to the other three arms (X), the resolution reaction was biased markedly to the right (RR>>RL; Fig. 5B, lanes 3–5). The magnitude of the bias may be gauged as follows. In the Flp reaction in lane 2, the ratio of RL to RR is ∼2.0, representing the inherent leftward bias in the system. In the targeted reaction shown in lane 5, the ratio of RR to RL is ∼8.0. Thus, the odds in favor of right resolution increased 2 × 8.0 = 16-fold. This result agrees with the dimeric dimer model, and disagrees with the trimer model. If the reaction followed the trimer model, the ratio of RR to RL should have been the same as that seen with the wild-type Flp reaction. Given the protein arrangement in Figure 5A on the Holliday arms, recall that, the trimer model does not favor left over right resolution or vice versa. The small yield of RL (∼11%) and the corresponding weak cleavage seen at the left (at position 1; CL in lane 5) may be accounted for by complexes in which Flp replaced Flp(Y343F) on the normal Holliday arm (adjacent to cleavage position 2). Note that as only two strands of the Holliday substrate were labeled, only one cleavage at the left (position 1) and one at the right (position 2) could be monitored.
The above interpretation is valid if, and only if, the assumption that nearly all of the substrates contributing to the resolution reaction had undergone simultaneous protein replacement from Flp(Y343F) to Flp on all three weak arms could be upheld, because this is the only type of tetrameric complex in which either the trimeric or the tetrameric mode of resolution can, in principle, be operational. Protein replacement on two arms (and perhaps on one arm) in a fraction of the substrate population, however, was unavoidable under our assay conditions (see below). At least a subset of the double replacements could also yield Holliday resolution, because they would provide the required complement of two Tyr-343 residues. Hence, their contribution to the resolution must be estimated and subtracted from the reaction observed in lanes 3–5 of Figure 5B. Single replacements would be inactive in resolution and need not be considered further.
Among the three possible double replacements (Fig. 6A; Table 1), we need be concerned solely with the arrangement I. This is the only arrangement that can produce resolution at the right exclusively, in agreement with the experimentally observed strong bias in the directionality of resolution (Fig. 5B, lanes 3–5). Note that this reaction can occur only by a tetramer mechanism, and thus, fails to test the trimer model. Is the contribution of configuration I (Fig. 6A; Table 1) to the reaction observed in lanes 3–5 of Figure 5B significant enough to invalidate the basic premise of this experiment? To estimate the extent of this contribution, we repeated the protein-targeting experiment under identical reaction conditions, but with Flp(R191S, H305L) instead of Flp as the replacement protein (Fig. 5B, lanes 6–8). For this mutant, the double replacement represented by configuration I (Fig. 6B) is functionally equivalent to the wild-type configuration I (Fig. 6A). This arrangement will yield resolution only at the right via the dimeric dimer mode (see Table 1). Resolution was severely depressed in this experiment (Fig. 5B, lanes 6–8), to ∼15% of that obtained upon replacement with wild-type Flp (Fig. 5B, cf. RR in lanes 5 and 8). The disparity in resolution towards the right, however, was still evident (RR>>RL). Therefore, we conclude that the directional resolution of the Holliday junction by wild-type Flp (Fig. 5, lane 3–5) was predominantly caused by simultaneous triple replacements (Fig. 5A), with only a small contribution (no more than 15%) from the type I replacement (Fig. 6A, I).
Figure 6.
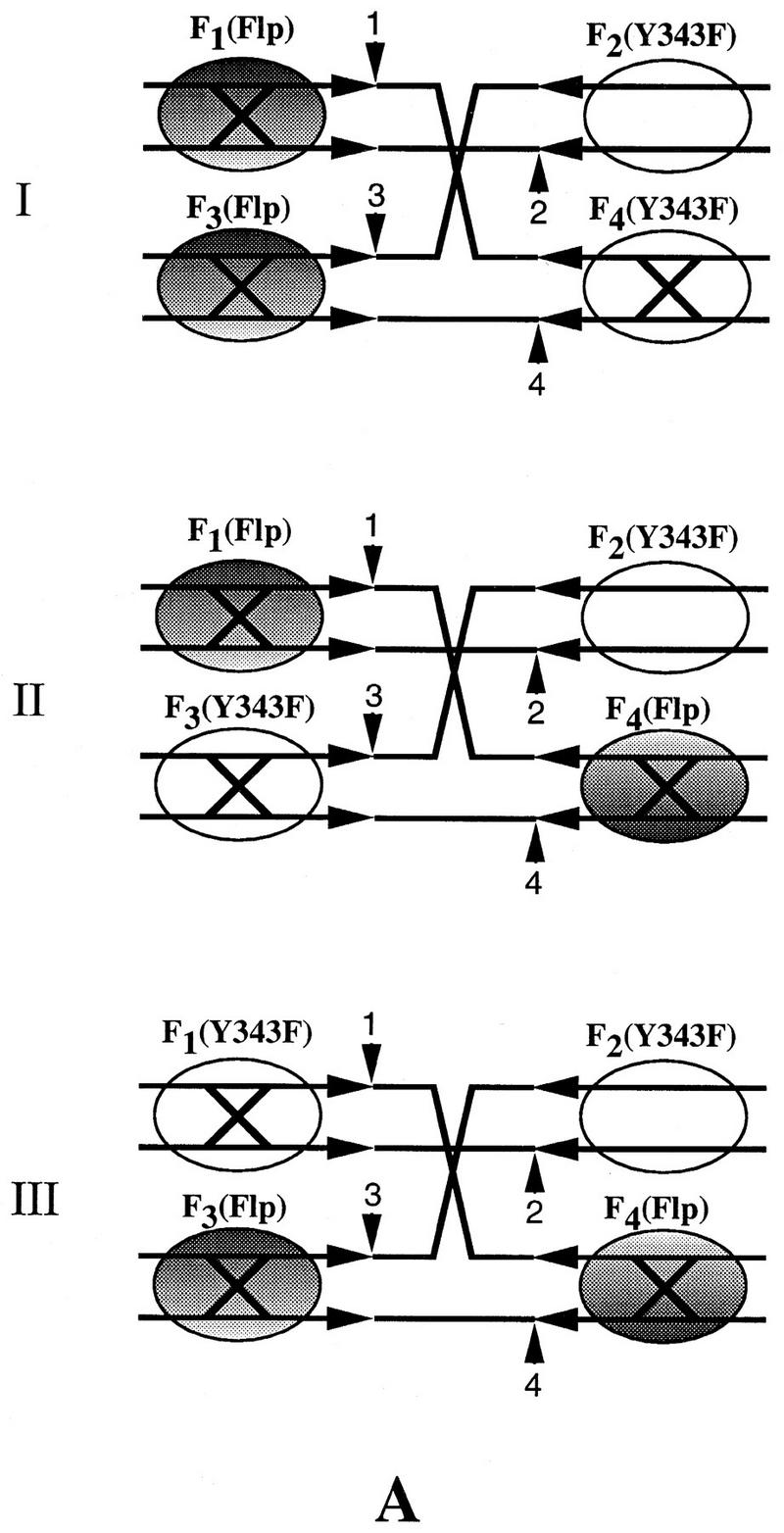
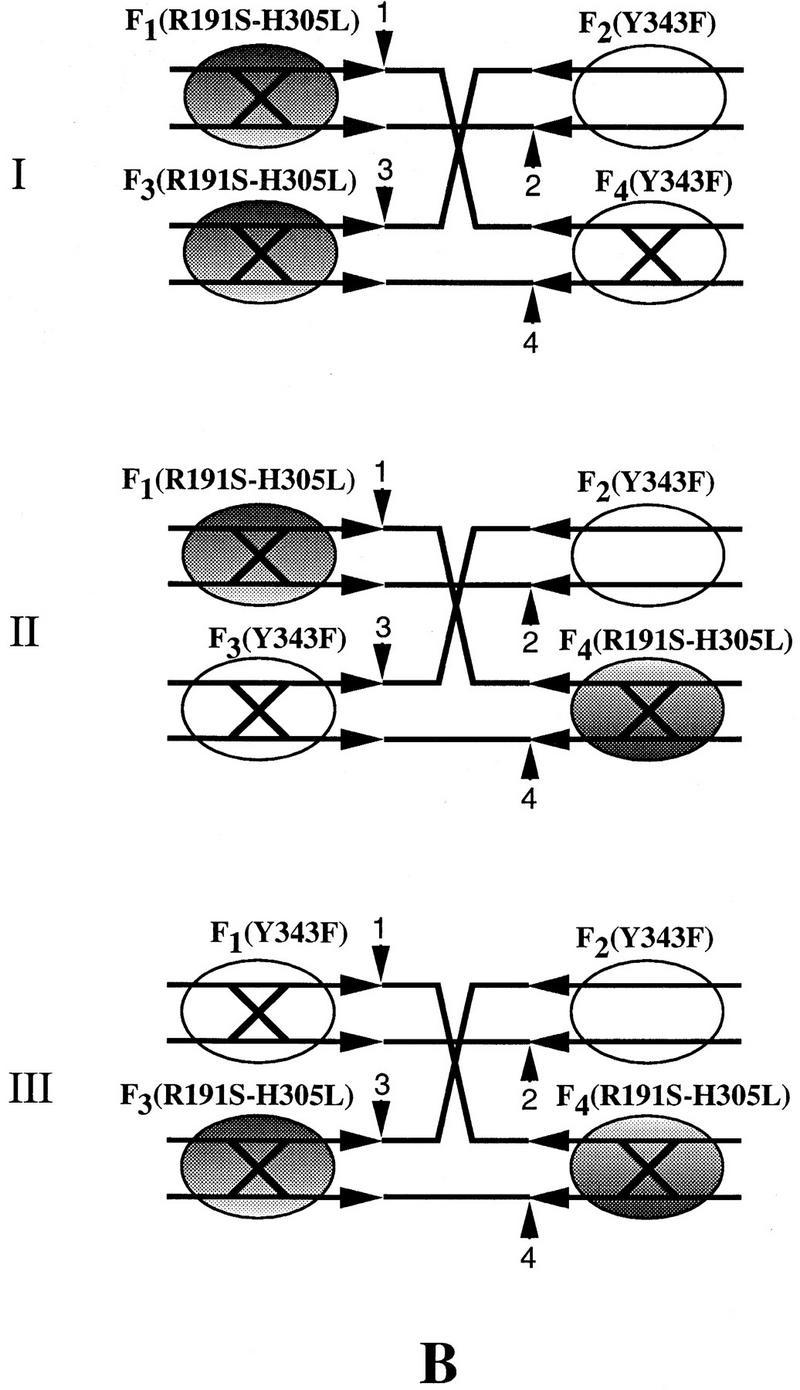
Resolution-competent arrangements of 2:2 tetramers between Flp(Y343F) and Flp or Flp(Y343F) and Flp(R191S, H305L) on a Holliday junction. (A) Replacement of two weak arms of the Holliday junction by Flp can give three possible arrangements I–III. Whereas I can give only right resolution by the tetramer mechanism, II and III should yield unbiased resolution by the trimer mechanism. (B) Only one of the three double replacements that can be obtained with Flp(R191S, H305L), I, can assemble two resolution-competent active sites. Resolution in this case will be at the right end via the tetramer.
Table 1.
Predicted directionality of Holliday junction resolution by pairwise combinations between Flp(Y343F) and Flp or Flp(R191S, H305L) following targeted protein placements
| Protein combinations
|
Strand exchange predictions
|
|
|---|---|---|
| dimeric dimer model
|
trimer model
|
|
| 3Flp:1Flp(Y343F)
|
RR > > RL
|
RR = RL
|
| 2Flp:2Flp(Y343F) | ||
| I | RR > > RL | inactive |
| II | inactive | RR = RLa |
| III | inactive | RR = RL |
| 2Flp(Y343F):2Flp (R191S, H305L) | ||
| I | RR > > RL | inactive |
| II | inactive | inactive |
| III | inactive | inactive |
The protein configuration for 3Flp:1Flp(Y343F) is shown in Fig. 5A. The three possible configurations, I-III, of 2Flp:2Flp(Y343F) and 2Flp(R191S, H305L):2Flp(Y343F) are shown in Fig. 6, A and B, respectively. Only the 3:1 protein arrangement between Flp and Flp(Y343F) can test the dimeric dimer and trimer models simultaneously. Each of the 2:2 arrangements in this protein combination precludes resolution by one of the two models. Arrangement III can resolve the junction by the cleavage modes depicted in Fig. 1 (one trans-horizontal cleavage plus one trans-vertical cleavage; Chen et al. 1992).
For resolution by II, one trans-diagonal cleavage and one trans-vertical cleavage (Chen et al. 1992) would be required. Note that distinction between trans-horizontal and trans-diagonal cleavages in a Holliday junction is not straightforward (Lee et al. 1996). The cleavage directionality will depend on the isomeric state assumed by a given junction (top strand crossed or bottom strands crossed). For the Flp(Y343F)–Flp(R191S, H305L) combination, only one of the three 2:2 configurations (I) is functional. (RR and RL refer to resolution at the right and left ends of the spacer, respectively).
The double replacements II and III (Fig. 6; Table 1) cannot vitiate our interpretation for the following reasons. For wild-type Flp (Fig. 6A; Table 1), they can only support the trimer mechanism and should produce unbiased resolution (similar to the Flp reaction in lane 2 of Fig. 5B). The observed result is the opposite of this prediction. Note that for configuration II to be active in resolution, one of the two requisite cleavages will have to be trans-diagonal (Chen et al 1992; see Table 1). For the triad mutant, neither the double replacements II and III (Fig. 6B; Table 1), nor the triple replacement will be active in resolution. Provided the relative abundance of three double replacements are equivalent (I = II = III), the failure to observe significant left resolution in the replacement reaction with Flp becomes even more revealing. Given that II (and probably III as well) will yield equivalent left or right resolution (Table 1), the near absence of left resolution further argues against the operation of the trimer mechanism within the Flp tetramer.
The highly biased resolution of a Holliday junction whose four arms are occupied by three Flp and one Flp(Y343F) monomers unequivocally proves that three monomers within the Flp tetramer cannot assemble two resolution competent active sites; it takes all four to do this.
Discussion
The shared active site of Flp poses a fundamental mechanistic dilemma: How many Flp monomers participate in the sharing of catalytic residues during the breakage and joining of one pair of DNA strands? Recall that the reaction takes place within a complex containing four Flp monomers, each bound to one of the Flp binding arms of the two DNA partners. Each of the two steps of recombination, the initiation step that generates a Holliday intermediate and the termination step that resolves it, requires the action of two active sites. Because the assembly of a Flp active site is achieved by the delivery of Tyr-343 from one Flp monomer to the RHR triad domain of a second Flp monomer (Chen et al. 1992, 1993), two active sites can be simultaneously derived, in principle, from two, three, or four Flp monomers. There is strong experimental evidence that excludes the assembly of two active sites in tandem by a Flp dimer (Qian et al. 1990; Qian and Cox 1995; Lee et al. 1996). The dilemma then reduces to the following: Is the active form Flp during a round of recombination a trimer within a tetramer or the whole tetramer itself (see Fig. 1)?
The catalytic unit within the recombination complex is not a trimer but the whole tetramer
The finding that three DNA-bound monomers of Flp can mediate the resolution of a three-armed or of a four-armed junction with one vacant arm (reactions requiring the coordinated functioning of two active sites) makes the trimer model a legitimate alternative to the dimeric dimer model (Qian and Cox 1995; Lee et al. 1996). In the context of the recombination mechanism, the trimer model has certain attractive features. Strand cleavage across partner substrates, as required by this model (Fig. 1), would be a useful attribute for a reaction whose physiological end is to join broken strands in the recombinant mode. The dimeric dimer model does not offer a simple obvious mechanism to accomplish this end (see Fig. 1). The trimer model also relates the Flp reaction to the λ integrase reaction. Nunes-Duby et al. (1989) showed that the first strand exchange in the Int mediated recombination requires not only the binding of two Int monomers adjacent to the scissile phosphodiester bonds, but also the binding of a third Int monomer on the far side of the strand exchange region. This protein arrangement would be equivalent to that postulated by the trimer model (Qian and Cox 1995). The requirement for interactions within a set of three Int monomers has been observed in the resolution of Holliday junction substrates as well (Franz and Landy 1990; Kho and Landy 1994). The apparent evolutionary conservation of the functional oligomeric states of two members of the Integrase family recombinases (Argos et al. 1986) would be pleasing.
Are the dimeric dimer and the asymmetric trimer model mutually exclusive? And which of the two represents the normal Flp reaction? The results of a test in which either a trimer or tetramer is given equal opportunity to form the active entity conform to the predictions of the dimeric dimer model, and contradict those of the trimer model. These findings also concur with the earlier observations that the strand exchange reaction between a half-site and a full-site requires that the half-site be able to dimerize in its Flp-associated form (Chen et al. 1993; Qian and Cox 1995). Thus, in the tetrameric state of Flp complexed with two recombination substrates, each of the two chemical steps that initiates and terminates the reaction (Holliday junction formation and resolution) involves the participation of all four Flp monomers. The Flp tetramer effectively silences the trimers within it.
What is the mechanism of silencing? It is clear that the presence of the fourth monomer in the Holliday–Flp tetramer complex disrupts catalytically productive trimer interactions that are feasible in its absence. We do not know whether this is simply the result of a steric effect resulting from protein occupancy of DNA, or requires more specific DNA–protein and/or protein–protein interactions.
Because the critical experiment in our study utilizes a resolution system that contains three Flp monomers and one Flp(Y343F) monomer (and not all four Flp monomers, as would be the case in the native reaction), skeptics might argue that the absence of the trimer reaction is a peculiarity of this artificial system. Given the highly conservative nature of the point mutation, however, and the history of its behavior in previous catalytic complementation experiments, this is a rather weak argument. It seems unlikely, for example, that the presence of one Flp(Y343F) monomer within the tetramer might force the Holliday structure to adopt a configuration that excludes one pair of strand cleavages, thus negating our test of the trimer model. Our conclusions must be deemed valid, therefore, until an experiment with all four Flp monomers (which at present appears to be impossible to design) disproves them.
Protein architecture of the catalytic unit and active site orientation in other recombination systems
A number of site-specific DNA recombination and DNA transposition systems utilize a tetramer of the recombinase or transposase as the active enzyme unit (for review, see Sadowski 1993, 1995). In several of these cases, the recombinase (or the transposase) exists as a monomer in solution, and oligomerizes into a functional tetramer only after binding to the DNA substrate. An example is the transposase of phage Mu (Mu A protein). Recent analyses have revealed parallels between Mu A and Flp in the logistics of the assembly and orientation of their active sites (Yang et al. 1995, 1996; Aldaz et al. 1996; Savilahti and Mizuuchi 1996). The main catalytic domain of Mu A bound to one end of the Mu genome, in fact, orients the cleavage nucleophile (in this case, a hydroxide anion) to execute strand breakage at the other end of the genome (Aldaz et al. 1996; Savilahti et al. 1996). The directionality of Mu cleavage may be considered as equivalent to the trans-horizontal cleavage executed by Tyr-343 of Flp (Lee et al. 1994).
Although the coupling of the oligomeric assembly of the recombinase (or transposase) to its chemical potential may be a general theme in DNA recombination reactions, the mechanism of this coupling may be specific to individual systems. In the case of Mu A, available evidence indicates that structural/catalytic contributions from all four monomers are essential for chemical competence of the tetramer. On the other hand, the simplest chemically competent state of Flp consistent with the dimeric dimer model would be a single DNA substrate bound by two Flp monomers, or one-half of the recombination complex (see Fig. 1). All published results, including the directionality of DNA cleavage by Flp, support this notion (Lee et al. 1994; Voziyanov et al. 1996). Because the Flp dimer assembles only one active site at a time, the cleavages within a DNA molecule are always single-stranded. In the absence of a receptive recombination partner, such cleavages can be quickly reversed by a strand-sealing reaction. On the other hand, cleavages occurring within the synapsed substrate pair associated with the Flp tetramer can be channeled into recombination. Thus, the chemically competent state appears to be a tetramer for Mu A, while it is a dimer for Flp. In contrast, the recombinationally competent state, capable of one pair of strand cleavages and strand transfers, is a tetramer for both proteins.
The shared active site: a mechanism for catalytic versatility
As has been pointed out previously (Lee and Jayaram 1993; Aldaz et al. 1996; Yang et al. 1996), the shared active site is an attractive mechanism for postponing the initiation of a multistep reaction until all the requisite components have been properly assembled. Furthermore, as shown for the Flp system in this study and in previous work, this feature can be utilized to impart flexibility in target selection, to confer directionality on the reaction, or to mediate suppression of certain reaction modes. Whereas the shared active site is especially suited for expanding the mechanistic possibilities of the recombination reaction, it is by no means unique to DNA recombinases. Several allosteric enzymes assemble functional active sites from partial active sites at the interface of protein dimers, one of the classic examples being aspartate transcarbamoylase (Wente and Schachman 1987). The crystal structure of β-galactosidase also reveals catalytic contributions from two monomers towards a single active site pocket (Jacobson et al. 1994). The partial active site of Flp, with the consequent trans mode of DNA cleavage, likely represents the evolutionary adaptation of a general control mechanism in enzyme regulation to meet the specific challenge of ordered breakage and joining of phosphodiester bonds during recombination.
Materials and methods
Purification of Flp and Flp variants
Wild-type Flp and Flp variants used in these experiments were ∼90% pure and were obtained by published procedures that included a final affinity purification step (Lee et al. 1996). Estimation of protein concentrations in these preparations has been described previously (Lee and Jayaram 1995).
Synthetic Holliday junction and Y junction substrates
Conditions for hybridization of appropriate oligonucleotides for substrate assembly were the same as detailed in Lee et al. (1996). The relevant features of the various substrates used are described in the Results, displayed in the figures, and explained in their legends. Nonspecific nucleotide segments of specific length that have no effect on Flp mediated reactions were present on either side of the recombination target site. The lengths of the strands were so chosen as to distinguish the products resulting from alternative strand cleavage and exchange events. In Holliday junctions, where directed protein replacement was required, the appropriate Flp-binding elements were weakened by altering a critical Flp contact position within each of them from a C–G to an A–T pair (Lee et al. 1994). The complete sequences of the substrates are available upon request.
The 3′ end of a deoxyoligonucleotide was labeled with 3′-α-32P-labeled cordycepin 5′-triphosphate by the terminal deoxynucleotide transferase reaction. The unreacted cordycepin phosphate was removed by spin-dialysis on a Sephadex G-25 column.
Resolution of Holliday and Y junction substrates
The conditions for resolution were essentially the same as those for recombination reactions described previously (Chen et al. 1992; Whang et al. 1994). Normally, the reactions contained ∼0.025 to 0.05 pmole of the substrate and an excess of Flp (8–10 pmoles of Flp per pmole of Flp-binding element) in a reaction volume of 30 μl. Following incubation for 5 min at 30°C, reactions were stopped by immersing samples in a boiling water bath for 5 min. After addition of SDS (0.1% final concentration) and proteinase K treatment (100 μg per sample for 1 hr at 37°C), samples were phenol-chloroform extracted and DNA was precipitated with ethanol. The DNA pellet was recovered by centrifugation, washed twice with 80% ethanol, and dried in vacuo. Strands were denatured in 95% formamide at 95°C, and samples were fractionated by electrophoresis in 10% denaturing polyacrylamide gels (acrylamide/bis-acrylamide 19:1).
Targeting of Flp to specific binding elements within the Holliday substrates
To ∼0.025 pmole of the labeled substrate, a Flp variant was added (at a molar ratio of protein monomer to binding element of ∼8 to 10) in recombination medium (Chen et al. 1992) containing three different concentrations of NaCl (80, 130, and 180 mm). The DNA–protein mixtures were preincubated at 30°C for 10 min. At time zero, ∼8- to 10-fold molar excess of Flp (over the Flp variant) or a second Flp variant was added to the assay mixtures, and incubation was continued at 30°C for 10 min. Addition of the second protein aliquot increased the NaCl concentration in each sample by 20 mm. Samples were processed as described under resolution assays.
General methods
Restriction enzyme digestions, isolation of plasmid DNA, and other miscellaneous procedures were done as described by Sambrook et al. (1989).
Acknowledgments
This work was supported by grants from the National Institutes of Health and the National Science Foundation. We acknowledge with gratitude all comments, criticisms and suggestions that helped mold the final version of this manuscript. In particular, David Sherratt suggested the inclusion of a table that simplifies the outcome of a critical but rather complex experiment.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jayaram@almach.cc.utexas.edu; FAX (512) 471-5546.
References
- Abremski KE, Hoess RH. Evidence for a second conserved arginine residue in the intergrase family of recombination proteins. Protein Eng. 1992;5:87–91. doi: 10.1093/protein/5.1.87. [DOI] [PubMed] [Google Scholar]
- Aldaz H, Schuster E, Baker TA. The interwoven architecture of Mu transposase couples DNA synapsis to catalysis. Cell. 1996;85:257–269. doi: 10.1016/s0092-8674(00)81102-2. [DOI] [PubMed] [Google Scholar]
- Argos P, Landy A, Abremski K, Egan JB, Haggard-Ljungquist E, Hoess RH, Kahn ML, Kalionis B, Narayana SVL, Pierson LS, III, Sternberg N, Leong JM. The Integrase family of site-specific recombinases: Regional similarity and global diversity. EMBO J. 1986;5:433–440. doi: 10.1002/j.1460-2075.1986.tb04229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JW, Lee J, Jayaram M. DNA cleavage in trans by the active site tyrosine during Flp recombination: Switching protein partners before exchanging strands. Cell. 1992;69:647–658. doi: 10.1016/0092-8674(92)90228-5. [DOI] [PubMed] [Google Scholar]
- Chen JW, Yang SH, Jayaram M. Tests for the fractional active-site model in Flp site-specific recombination: Assembly of a functional recombination complex in half-site and full-site strand transfer. J Biol Chem. 1993;268:14417–14425. [PubMed] [Google Scholar]
- Franz B, Landy A. Interactions between lambda Int molecules bound to sites in the region of strand exchange are required for efficient Holliday junction resolution. J Mol Biol. 1990;215:523–535. doi: 10.1016/s0022-2836(05)80165-2. [DOI] [PubMed] [Google Scholar]
- Jacobson RH, Zhang XJ, DuBose RF, Matthews BW. Three-dimensional structure of beta-galactosidase from E. coli. Nature. 1994;369:761–766. doi: 10.1038/369761a0. [DOI] [PubMed] [Google Scholar]
- Jayaram M. Mechanism of site-specific recombination: The Flp paradigm. Nucleic Acid Mol Biol. 1994;8:268–286. [Google Scholar]
- Kho SH, Landy A. Dissecting the resolution reaction of lambda integrase using suicide Holliday junction substrates. EMBO J. 1994;13:2711–2724. doi: 10.1002/j.1460-2075.1994.tb06562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Jayaram M. Mechanism of site-specific recombination: Logic of assembling recombinase catalytic site from fractional active sites. J Biol Chem. 1993;268:17564–17570. [PubMed] [Google Scholar]
- ————— Functional roles of individual recombinase monomers in strand breakage and strand union during site-specific DNA recombination. J Biol Chem. 1995;270:23203–23211. doi: 10.1074/jbc.270.39.23203. [DOI] [PubMed] [Google Scholar]
- Lee J, Whang I, Lee J, Jayaram M. Directed protein replacement in recombination full sites reveals trans-horizontal DNA cleavage by Flp recombinase. EMBO J. 1994;13:5346–5354. doi: 10.1002/j.1460-2075.1994.tb06869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Whang I, Jayaram M. The shared active site of the Flp site-specific recombinase: Active site assembly and cleavage directionality on two, three, and four armed DNA substrates. J Mol Biol. 1996;257:532–549. doi: 10.1006/jmbi.1996.0183. [DOI] [PubMed] [Google Scholar]
- Nunes-Duby SE, Matsumoto L, Landy A. Half-att site substrates reveal the homology independence and minimal protein requirements for productive synapsis in λ excisive recombination. Cell. 1989;59:197–206. doi: 10.1016/0092-8674(89)90881-7. [DOI] [PubMed] [Google Scholar]
- Qian XH, Cox MM. Asymmetry in active complexes of FLP recombinase. Genes & Dev. 1995;9:2053–2064. doi: 10.1101/gad.9.16.2053. [DOI] [PubMed] [Google Scholar]
- Qian XH, Inman RB, Cox MM. Protein-based asymmetry and protein-protein interactions in FLP recombinase-mediated site-specific recombination. J Biol Chem. 1990;265:21779–21788. [PubMed] [Google Scholar]
- Sadowski PD. Site-specific genetic recombination: Hops, flips, and flops. FASEB J. 1993;7:760–767. doi: 10.1096/fasebj.7.9.8392474. [DOI] [PubMed] [Google Scholar]
- ————— The Flp recombinase of the 2-micron plasmid of Saccharomyces cerevisiae. Prog Nucleic Acids Res Mol Biol. 1995;51:53–91. [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Savilahti H, Mizuuchi K. Mu transpositional recombination: Donor cleavage and strand transfer in trans by the Mu transposase. Cell. 1996;85:271–280. doi: 10.1016/s0092-8674(00)81103-4. [DOI] [PubMed] [Google Scholar]
- Stark WM, Boocock MR, Sherratt DJ. Catalysis by site-specific recombinases. Trends Genet. 1992;8:432–439. [PubMed] [Google Scholar]
- Voziyanov Y, Lee J, Whang I, Lee J, Jayaram M. Analyses of the first chemical step in Flp site-specific recombination: Synapsis may not be a pre-requisite for strand cleavage. J Mol Biol. 1996;256:720–735. doi: 10.1006/jmbi.1996.0120. [DOI] [PubMed] [Google Scholar]
- Wente SR, Schachman HK. Shared active sites in oligomeric enzymes: Model studies of aspartate transcarbamoylase produced by site-directed mutagenesis. Proc Natl Acad Sci. 1987;84:31–35. doi: 10.1073/pnas.84.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whang I, Lee J, Jayaram M. Active-site assembly and mode of DNA cleavage by Flp recombinase during full-site recombination. Mol Cell Biol. 1994;14:7492–7498. doi: 10.1128/mcb.14.11.7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JY, Kim K, Jayaram M, Harshey RM. A domain sharing model for active site assembly within the MuA tetramer during transposition: The enhancer may specify domain contributions. EMBO J. 1995;14:2374–2385. doi: 10.1002/j.1460-2075.1995.tb07232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JY, Jayaram M, Harshey RM. Positional information within the Mu transposase tetramer: Catalytic contributions of individual monomers. Cell. 1996;85:447–455. doi: 10.1016/s0092-8674(00)81122-8. [DOI] [PubMed] [Google Scholar]