

Abstract
Growth factors and hormones activate protein translation by phosphorylation and inactivation of the translational repressors, the eIF4E-binding proteins (4E-BPs), through a wortmannin- and rapamycin-sensitive signaling pathway. The mechanism by which signals emanating from extracellular signals lead to phosphorylation of 4E-BPs is not well understood. Here we demonstrate that the activity of the serine/threonine kinase Akt/PKB is required in a signaling cascade that leads to phosphorylation and inactivation of 4E-BP1. PI 3-kinase elicits the phosphorylation of 4E-BP1 in a wortmannin- and rapamycin-sensitive manner, whereas activated Akt-mediated phosphorylation of 4E-BP1 is wortmannin resistant but rapamycin sensitive. A dominant negative mutant of Akt blocks insulin-mediated phosphorylation of 4E-BP1, indicating that Akt is required for the in vivo phosphorylation of 4E-BP1. Importantly, an activated Akt induces phosphorylation of 4E-BP1 on the same sites that are phosphorylated upon serum stimulation. Similar to what has been observed with serum and growth factors, phosphorylation of 4E-BP1 by Akt inhibits the interaction between 4E-BP1 and eIF-4E. Furthermore, phosphorylation of 4E-BP1 by Akt requires the activity of FRAP/mTOR. FRAP/mTOR may lie downstream of Akt in this signaling cascade. These results demonstrate that the PI 3-kinase-Akt signaling pathway, in concert with FRAP/mTOR, induces the phosphorylation of 4E-BP1.
Keywords: Protein synthesis, phosphorylation, PI 3-kinase, protein kinase B, eIF4E, FRAP/mTOR
Numerous cellular processes are controlled by extracellular stimuli that activate signaling cascades. Many stimuli activate common pathways, such as the well-described Ras and phosphoinositide 3-kinase pathways. Phosphoinositide 3-kinase (PI 3-kinase) is activated by growth factor receptors after growth factor stimulation and induces cell proliferation and cell survival (for review, see Franke et al. 1997a; Vanhaesebroeck et al. 1997). Several downstream targets of PI 3-kinase have been identified, including p70 ribosomal protein S6 kinase (p70S6k) (Chou and Blenis 1995; Proud 1996), the GTPases Rac (Hawkins et al. 1995), certain protein kinase C isoforms (Nakanishi et al. 1993; Akimoto et al. 1996), and the serine/threonine kinase Akt (also known as protein kinase B-PKB) (Burgering and Coffer 1995; Franke et al. 1995). Upon activation by growth factors, PI 3-kinase phosphorylates the D3 position of phosphatidylinositols. These phospholipids act as second messengers that mediate the diverse cellular functions of PI 3-kinase, including activation of Akt (for review, see Franke et al. 1997a; Hemmings 1997). Wortmannin, a PI 3-kinase inhibitor, blocks activation of Akt after stimulation with growth factors, indicating that the activity of PI 3-kinase is an obligatory step in Akt activation by growth factors (Burgering and Coffer 1995; Franke et al. 1997b; Kennedy et al. 1997). Translocation of Akt to the plasma membrane through its pleckstrin homology (PH) domain is likely required for its activity (Hemmings 1997). Indeed, constitutive targeting of Akt to the plasma membrane is sufficient to promote its activation (Burgering and Coffer 1995; Franke et al. 1997b; Kennedy et al. 1997). Full activation of Akt by growth factors requires the phosphorylation of threonine and serine residues by upstream kinases (Alessi et al. 1997; Stokoe et al. 1997). Both the upstream activating kinases and the recruitment of Akt to the plasma membrane are thought to be dependent on the products of PI 3-kinase (Cohen et al. 1997). Akt mediates some of the PI 3-kinase cellular responses, including protection from apoptosis induced by serum and growth factor deprivation (Kennedy et al. 1997).
The immunosuppressive drug rapamycin, an inhibitor of G1 cell cycle progression, blocks the activation of p70S6k by growth factors and Akt (Chung et al. 1992; Price et al. 1992; Burgering and Coffer 1995). Rapamycin forms a complex with the immunophilin FKBP12 to generate a potent inhibitor of FRAP/mTOR (also termed RAFT1 or RAPT1) (Brown and Schreiber 1996). FRAP/mTOR activates p70S6k in vivo (Brown et al. 1995); it is unclear, however, whether FRAP/mTOR is a downstream effector in the PI 3-kinase and Akt signaling pathway. FRAP, which is the mammalian homolog of the Saccharomyces cerevisiae targets of rapamycin TOR1 and TOR2 (Hall 1996), is a member, together with ATM and DNA–PK, of a recently characterized family of phosphatidylinositol kinases-related (PIK-related) kinases. PIK-related kinases activation and mechanisms of action remain unclear (Hoekstra 1997). FKBP rapamycinassociated protein/mammalian target of rapamycin (FRAP/mTOR) could provide a link between cell cycle progression and the control of mRNA translation, as rapamycin, which blocks the cell cycle in G1, also causes a decrease in mRNA translation (Beretta et al. 1996; Brown and Schreiber 1996). Consistent with this finding, the yeast TOR has been demonstrated to regulate G1 progression through a translational mechanism (Barbet et al. 1996).
Regulation of protein translation is an important aspect of the control of cell growth. A rate-limiting step in this process is the binding of the mRNA to the small ribosomal subunit, a step mediated by the eIF4 group of initiation factors (for review, see Sonenberg 1996). eIF4F, through its smaller subunit eIF4E, recognizes the cap structure (m7GpppX, where X is any nucleotide) that is present at the 5′ end of all cellular, except organellar, mRNAs. eIF4F, in conjunction with eIF4B, is thought to unwind the secondary structure in the mRNA 5′-UTR to facilitate ribosome binding (Sonenberg 1996). Overexpression of eIF4E in rodent cells leads to cellular transformation and eIF4E has been implicated in cell cycle control (Lazaris-Karatzas et al. 1990; Sonenberg 1996). In addition, a role for eIF4E in cell survival has been proposed, as NIH 3T3 cells that express eIF4E ectopically are refractory to apoptosis induced by serum deprivation (Polunovsky et al. 1996). eIF4E is the target of a family of translational repressors termed the 4E-BPs (for eIF4E-Binding Proteins; also known as PHAS). These repressors bind to eIF4E and prevent its association with eIF4G and incorporation into the eIF4F complex, which leads to inhibition of cap-dependent, but not cap-independent, translation (Sonenberg 1996). Overexpression of 4E-BP1 or 4E-BP2 in cells transformed by eIF4E, Ha-v-ras or v-src partially reverts their transformed phenotypes (Rousseau et al. 1996).
Inhibition of translation by 4E-BPs is reversible. After treatment of cells with serum, growth factors, or hormones, 4E-BP1 (the prototype member of the family), is hyperphosphorylated in a wortmannin- and rapamycin-sensitive manner, and dissociates from eIF4E (Beretta et al. 1996; von Manteuffel et al. 1996, 1997). The rapid increase in 4E-BP1 phosphorylation after serum or growth factor stimulation provides a very attractive mechanism for explaining the increase in translation rates of several mRNAs after stimulation.
Because phosphorylation of 4E-BP1 is wortmannin sensitive, and mutants in the PDGF receptor that fail to activate PI 3-kinase also fail to phosphorylate 4E-BP1 after PDGF treatment (Beretta et al. 1996; von Manteuffel et al. 1996), it was suggested that phosphorylation of 4E-BP1 by serum and growth factors is mediated by PI 3-kinase. However, it is not clear whether PI 3-kinase lies directly upstream of 4E-BP1 in a phosphorylation cascade. This is an important question, particularly in light of a recent report demonstrating that wortmannin can inhibit FRAP/mTOR activity directly (Brunn et al. 1996). Here we provide direct evidence that PI 3-kinase and its downstream effector Akt lie in a pathway leading to the in vivo phosphorylation of 4E-BPs. This phosphorylation is sensitive to rapamycin. The rapamycin sensitivity can be overridden by coexpression of a rapamycin-resistant mutant of FRAP/mTOR. Thus, FRAP/mTOR may lie downstream of Akt in the 4E-BP1 phosphorylation cascade.
Results
P110α, the catalytic subunit of PI 3-kinase, and its downstream effector Akt/PKB mediate the phosphorylation of 4E-BP1
To study the role of Akt in the phosphorylation of 4E-BP1, a hemagglutinin-tagged 4E-BP1 (HA–4E-BP1) was generated. We first examined whether the transiently expressed HA–4E-BP1 exhibits a change in electrophoretic mobility after phosphorylation, as was observed for the endogenous 4E-BP1. Human embryonic kidney (HEK) 293 cells were transfected transiently with a HA–4E-BP1 expression vector. After transfection, the cells were deprived of serum for 36 hr and then stimulated with insulin for 30 min. Immunoblot analysis demonstrated a clear shift in mobility of HA–4E-BP1 with insulin stimulation (Fig. 1A, lanes 1,2). The mobility shift was not observed when cells were preincubated with either wortmannin or rapamycin (lanes 3,4), consistent with what has been observed previously for endogenous 4E-BP1 (von Manteuffel et al. 1996).
Figure 1.


PI 3-kinase and Akt elicit phosphorylation of 4E-BP1. (A) Insulin-mediated phosphorylation of 4E-BP1 is both rapamycin and wortmannin sensitive. Human embryonic kidney (HEK) 293 cells were transfected transiently with a hemaglutinin (HA) epitope-tagged 4E-BP1 expression vector. After transfection, cells were deprived of serum for 36 hr, and either mock treated (lane 1) or stimulated with insulin (1 μg/ml) for 30 min (lanes 2–4) in the presence of either wortmannin, [200 nm (Wort.)] (lane 3) or rapamycin [20 ng/ml (Rap.)] (lane 4). Cell extracts were prepared as described in Materials and Methods and HA–4E-BP1 was detected by immunoblot analysis with an anti-HA antibody (12CA5). Molecular size markers (in kD) are indicated. Arrows indicate the different phosphorylated isoforms of HA–4E-BP1. (B) The catalytic subunit of PI 3-kinase p110α elicits phosphorylation of HA–4E-BP1. HEK 293 cells were cotransfected with HA–4E-BP1 expression vector along with one of the following: control vector (lane 1), p110α expression vector (lane 2), or p110αcaax (p110α*) expression vector (lane 3). After transfection, cells were deprived of serum for 36 hr. HA–4E-BP1 was detected as described in A. Small arrows indicate the different phosphorylation forms of 4E-BP1. p110α and p110αcaax were detected as described in Materials and Methods. (C) Akt elicits phosphorylation of HA–4E-BP1. HEK 293 cells were mock transfected (lane 1) or cotransfected with HA–4E-BP1 expression vector and one of the following: control vector (lane 2), HA–c-Akt expression vector (lane 3), or HA–MyrAkt expression vector (lane 4). Cells were deprived of serum for 36 hr. HA–4E-BP1 was detected as described above. Small arrows indicate the different phosphorylation forms of 4E-BP1. (D) A kinase-deficient mutant of Akt inhibits phosphorylation of 4E-BP1 by insulin. HEK 293 cells were cotransfected with a HA–4E-BPI expression vector (100 ng) and the following: control vector (lanes 1,2) or HA–AktK179M expression vector [Akt(kin−)] (lanes 3,4). After transfection, cells were serum-deprived for 36 hr and then stimulated with 100 ng/ml of insulin for 45 min (lanes 2,4). HA–4E-BP1 was detected as described above. HA–AktK179M was detected on the same immunoblot. Small arrows indicate the different phosphorylated forms of 4E-BP1. The results shown are representative of three independent experiments.
Previous studies have indicated a role for PI-3-kinase in the phosphorylation of 4E-BP1 by serum and growth factors (Beretta et al. 1996; von Manteuffel et al. 1996). However, it was also suggested that the effects of extracellular stimuli on 4E-BP1 phosphorylation could be explained by direct activation of FRAP, as the in vitro autokinase activity of FRAP is also inhibited by wortmannin (Brunn et al. 1996). To examine whether PI 3-kinase can affect the phosphorylation state of 4E-BP1, we transiently cotransfected HA-4E-BP1 and PI 3-kinase expression vectors into serum-deprived 293 cells. Cotransfection of HA-4E-BP1 with the catalytic subunit of PI 3-kinase p110α induced phosphorylation of 4E-BP1, as manifested by a shift in its mobility (Fig. 1B, lane 2). This shift in mobility is similar to that observed with insulin stimulation (cf. Fig. 1A, lane 2, with Fig. 1B, lane 2). An activated form of p110α, p110αcaax (p110α*), which is targeted to the plasma membrane by farnesylation (Khwaja et al. 1997) (Fig. 1B, lane 3), also caused this mobility shift (interestingly, overexpression of the wild-type p110α in 293 cells is sufficient to induce 4E-BP1 mobility shift). Thus, PI 3-kinase by itself affects the phosphorylation of 4E-BP1.
Next, we examined whether 4E-BP1 phosphorylation is mediated by Akt. We used two forms of Akt: the wild type c-Akt, and an activated form of Akt, MyrAkt. MyrAkt is comprised of the entire coding sequence of c-Akt fused in-frame to the Src myristoylation signal. This fusion protein is constitutively active, is independent of growth factors, and is wortmannin resistant (Ahmed et al. 1997; Kennedy et al. 1997; see below). Cotransfection of either WTAkt or MyrAkt and HA–4E-BP1 expression vectors caused a mobility shift of 4E-BP1 (Fig. 1C). These results suggest that both PI 3-kinase and its downstream effector Akt are intermediates in the signaling pathway leading to 4E-BP1 phosphorylation. Transient transfections of 293 cells produces a high level of c-Akt expression that was sufficient to elicit the 4E-BP1 mobility shift to the same extent as MyrAkt (Fig. 1C, cf. lanes 3 and 4).
To determine whether the endogenous PI 3-kinase/Akt signaling pathway is involved in mediating the phosphorylation of 4E-BP1 by growth factors, we used a kinase-deficient mutant of Akt containing a point mutation in the ATP-binding domain K179M [Akt(kin−)] (Franke et al. 1995). When expressed at high levels, the kinase dead mutant acted in a dominant negative fashion to abolish the mobility shift of 4E-BP1 normally elicited by insulin treatment (Fig. 1D). Insulin treatment induces the mobility shift of 4E-BP1 (lanes 1,2). Coexpression of Akt(kin−) significantly reduced the insulin-mediated mobility shift (lanes 3,4). These results demonstrate that endogenous Akt is required to transmit the signal leading to 4E-BP1 phosphorylation.
Recent results suggested the possibility that the wortmannin sensitivity of 4E-BP1 phosphorylation is attributable to direct inhibition of FRAP/mTOR activity by wortmannin (Brunn et al. 1996). Therefore, we examined the sensitivity of PI 3-kinase and Akt-mediated phosphorylation of 4B-BP1 to wortmannin and rapamycin. Both wortmannin and rapamycin inhibited the ability of an activated form of p110α to cause the mobility shift of 4E-BP1 (Fig. 2A). However, the mobility shift elicited by the activated Akt was wortmannin resistant but rapamycin sensitive (Fig. 2B). These results indicate that the wortmannin sensitivity of 4E-BP1 phosphorylation by growth factors is attributable to inhibition of PI 3-kinase and not inhibition of FRAP activity.
Figure 2.

Effect of wortmannin and rapamycin on 4E-BP1 phosphorylation by Akt and PI 3-kinase. (A) Phosphorylation of 4E-BP1 by p110αcaax is both wortmannin and rapamycin sensitive. HEK 293 cells were cotransfected with HA–4E-BP1 expression vector and one of the following: control vector (lane 1) or p110αcaax (p110α*) expression vector (lanes 2–4). After transfection, cells were deprived of serum for 36 hr and treated with either wortmannin [200 nm (Wort.); lane 3] or rapamycin [20 ng/ml (Rap.); lane 4]. Cell extracts were prepared and HA–4E–BP1 was detected as described in Fig. 1. Arrows indicate different phosphorylation isoforms of 4E-BP1. (B) Phosphorylation of 4E-BP1 induced by an activated Akt is wortmannin resistant but rapamycin sensitive. HEK 293 cells were cotransfected with HA–4E-BP1 expression vector and with control vector (lane 1), or with HA–MyrAkt expression vector (lanes 2–4). After transfection, cells were deprived of serum for 36 hr. Cells were treated and 4E-BP1 was detected as described in A. HA–MyrAkt was detected on the same immunoblot. Small arrows indicate different phosphorylation forms of 4E-BP1. The figure is representative of two independent experiments.
Taken together these results suggest a linear pathway from growth factor receptors to the activation of PI 3-kinase, which in turn activates Akt and leads to phosphorylation of 4E-BP1.
Akt mediates the phosphorylation of 4E-BP1 and 4E-BP2 in vivo
To confirm that the gel mobility shift observed by cotransfection of Akt with 4E-BP1 is attributable to an increase in 4E-BP1 phosphorylation, we first established a stable 293-cell line overexpressing MyrAkt. 293–MyrAkt or wild-type 293 cells were then labeled metabolically with [32P]orthophosphate, and 4E-BP1 and 4E-BP2 were immunoprecipitated and subjected to SDS-PAGE and autoradiography. In control cells, two phosphorylated isoforms termed β and γ (the fastest migrating isoform, α, is unphosphorylated and hence not detected by 32P labeling) were detected (Fig. 3A, lane 1). After serum or insulin stimulation (lanes 2,3) a 2.5-fold increase in 32P incorporation was observed, and two isoforms of slower mobility (δ and ε) appeared. Rapamycin or wortmannin treatment abrogated the effect of serum and insulin on 4E-BP1 phosphorylation (lanes 4,5). In 293–MyrAkt cells, the four phosphorylated species (β, γ, δ, ε) were present in the absence of stimuli (lane 6, 2.5-fold more total 32P incorporation than in starved 293 cells). The pattern of phosphorylation is very similar to that of 293 cells stimulated with serum or insulin (cf. lane 6 with lanes 2 and 3). In 293–MyrAkt cells, the phosphorylation of 4E-BP1 was sensitive to rapamycin treatment, but completely resistant to wortmannin treatment (lanes 7,8). A similar effect was observed for 4E-BP2. 4E-BP2 was immunoprecipitated from the lysates as 4E-BP1. 4E-BP2 is phosphorylated on fewer residues than 4E-BP1 (A.-C. Gingras and N. Sonenberg, unpubl.), and only one isoform incorporating 32P is detected by SDS-PAGE (Fig. 3B). Total 32P incorporation in 4E-BP2 was increased ∼2- to 2.5-fold in serum- and insulin-stimulated cells (Fig. 3B, cf. lanes 2 and 3 with lane 1). This increase was diminished (1.5-fold) by rapamycin treatment and abolished by wortmannin treatment (lanes 4,5). In MyrAkt-expressing 293 cells, 4E-BP2 phosphorylation was increased approximately twofold, as compared to serum deprived 293 cells (cf. lane 6 with lane 1). As with 4E-BP1, the increase in 4E-BP2 phosphorylation in MyrAkt cells was rapamycin sensitive, but wortmannin insensitive (lanes 7,8). Although the effect of rapamycin on 4E-BP2 phosphorylation was modest (1.5-fold) in the experiment presented here, this inhibitory effect was reproduced several times in 293 cells, with an inhibition varying from 1.5- to 3-fold. This is similar to the inhibition of 4E-BP1 phosphorylation by rapamycin.
Figure 3.


4E-BP1 and 4E-BP2 32P incorporation is increased in 293 MyrAkt cells and is resistant to wortmannin treatment. Cells were labeled with [32P]orthophosphate as described in Materials and Methods and 4E-BP1 (A) and 4E-BP2 (B) were immunoprecipitated successively with polyclonal antibodies, separated by SDS-PAGE, transferred to Immobilon-PSQ and subjected to autoradiography. Different phosphorylated isoforms are indicated for 4E-BP1.
These results further confirm that the differences in the mobility shift observed for 4E-BP1 in Figures 1 and 2 are attributable to changes in the phosphorylation state of 4E-BP1. To determine whether the effects observed on 4E-BP1 phosphorylation are specific to 293 cells, the same experiment was repeated in Rat1a cells stably expressing MyrAkt (Kennedy et al. 1997). Similar results to that observed with 293 cells (two- to threefold increase in phosphorylation) were obtained for both 4E-BP1 and 4E-BP2, and MyrAkt-induced phosphorylation was also sensitive to rapamycin, but resistant to wortmannin (data not shown).
To determine whether Akt-mediated phosphorylation of 4E-BP1 occurs on the in vivo phosphorylation sites, phosphopeptide maps were performed on 32P orthophosphate-labeled 4E-BP1. Ten phosphopeptides were detected in serum-starved 293 cells (Fig. 4A, labeled 1–10 in the order of decreasing intensity). When 293 cells were stimulated with serum, the intensity of some of the spots greatly increased (Fig. 4B, spots 8 and 9), whereas some new spots (11–14) appeared. Peptide 14 was not reproducibly detected in other experiments and will not be discussed further. Rapamycin treatment caused the decrease or disappearance of two serum-dependent spots (11 and 12) and of spots 8 and 9 (Fig. 4C). The effects of wortmannin were even more striking, resulting, for example, in the diminution of spot 7 (Fig. 4D). Serum-starved 293–MyrAkt cells exhibited a phosphopeptide pattern almost identical to that of 293 cells stimulated with serum. Phosphopeptides 11–13 were present in the absence of serum, although the increase in spots 9 and 13 was not as marked as in serum-treated cells (cf. E with A and B). As expected, rapamycin treatment caused a marked decrease in spots 11 and 12 (Fig. 4F). Wortmannin treatment had practically no effect on phosphopeptides 8, 11, 12 (Fig. 4G). Taken together, these data indicate that the increase in phosphorylation of 4E-BP1 in 293–MyrAkt cells occurs on physiologically relevant sites and confirm our data that MyrAkt expression confers resistance to wortmannin, but not to rapamycin on 4E-BP1.
Figure 4.

The phosphopeptide map of 4E-BP1 in 293 MyrAkt cells is identical to that of serum-stimulated 293 cells. 32P-Labeled 4E-BP1 (Fig. 3) was excised from an Immobilon membrane, digested with trypsin–chymotrypsin, and analyzed by two-dimensional phosphopeptide mapping, as described in Materials and Methods. HEK 293 cells (A–D) and HEK 293/ MyrAkt cells (E–G) were deprived of serum for 36 hr. Cells were labeled with 32P as described in Materials and Methods. (A,E) Untreated cells (B–D,F,G) were treated as follows: with 15% FCS for 30 min (B); pretreated with rapamycin (20 ng/ml) for 20 min before addition of FCS (C); pretreated with wortmannin (100 nm) for 20 min before addition of FCS (D); with rapamycin (20 ng/ml) for 20 min (F); with wortmannin (100 nm) for 20 min (G).
To determine whether the changes in 4E-BP1 phosphorylation correlate with 4E-BP1 activity, the interaction of 4E-BP1 with eIF4E was examined, using cap-affinity chromatography. 293 and other human cells contain multiple 4E-BP1 isoforms (Gingras et al. 1996; von Manteuffel et al. 1996). The isoform pattern observed in mouse and rat cells is much simpler, because of a smaller number and better separation of the isoforms. Thus, we performed the analysis in stably transfected Rat1a/MyrAkt and wild-type Rat1a cells. In rodent cells, a total of three 4E-BP1 forms can be detected: (1) a hyperphosphorylated slow migrating isoform (γ), which does not interact with eIF4E; (2) a middle form (β), which is phosphorylated and binds eIF4E with a low affinity; and (3) an unphosphorylated fast migrating species (α), which interacts very strongly with eIF4E (Lin et al. 1995; Beretta et al. 1996; Gingras et al. 1996). Rat1a and Rat1a MyrAkt cells were serum starved and then serum stimulated (Rat1a). A Western blot performed on total extracts indicated that the three isoforms (α, β, and γ) were present in serum-starved cells (Fig. 5A, lane 1, the β form is predominant), only the two slower migrating forms (β and γ) were detected in serum-stimulated Rat1a cells (lane 2, the γ form is predominant). Only the hyperphosphorylated form (γ) was detected in Rat1a MyrAkt cells (lane 3; significantly less 4E-BP1 was present consistently in the MyrAkt cells, for reasons that are not immediately clear), indicating that 4E-BP1 is hyperphosphorylated. A cap-affinity isolation of eIF4E was conducted. In serum-starved Rat1a cells, there was a significant amount of 4E-BP1 (isoforms α and β) that bound eIF4E (Fig. 5B, lane 1). Binding was abolished in serum-stimulated cells (lane 2), and in Rat1a (MyrAkt) cells (lane 3). Taken together, these results suggest that phosphorylation of 4E-BP1 in MyrAkt cells prevents its association with eIF4E and that Akt plays a pivotal role in regulating 4E-BP1 activity in cells.
Figure 5.

Phosphorylation of 4E-BP1 by Akt inhibits interaction with eIF4E. Rat1a and Rat1a/MyrAkt cells were incubated in 0.5% FCS overnight. Rat1a cells were then treated with 20% FCS for 40 min. Cells were lysed by freeze–thaw cycles and extracts were either heat treated (total extract; 100 μg) or incubated (750 μg) with m7GDP–agarose resin, as described in Materials and Methods. Samples were separated by SDS-PAGE and 4E-BP1 protein was analyzed by Western blotting. (A) Total extract (100 μg). (B) Material bound to the m7GDP–agarose resin. Positions of the 4E-BP1 isoforms are indicated.
Akt cannot directly phosphorylate 4E-BP1
To address the question of whether Akt can phosphorylate 4E-BP1 directly, in vitro kinase assay was used. The kinase reaction was performed using histone H2B (as a control substrate) (Kennedy et al. 1997). Histone H2B phosphorylation was increased gradually with time of incubation with immunoprecipitated HA–MyrAkt (Fig. 6, lanes 1–3). After a 45-min incubation, background phosphorylation of H2B was detected even with an immunoprecipitate from mock transfected cells (lane 7). In contrast, only background phosphorylation of GST–4E-BP1 was observed upon incubation with immunoprecipitated HA–MyrAkt (lanes 4–6, and lane 8). Phosphorylation of 4E-BP1 was not observed even after prolonged exposure (data not shown). Therefore, we conclude that Akt cannot phosphorylate 4E-BP1 in vitro and is unlikely to serve as the kinase that directly phosphorylates 4E-BP1 in vivo (see also below).
Figure 6.

Akt does not phosphorylate 4E-BP1 in vitro. HEK 293 cells were transfected transiently with HA–MyrAkt expression vector or with vector alone. Forty-eight hours after transfection, MyrAkt was immunoprecipitated with an anti-HA antibody (HA.11). Immunoprecipitates from mock transfected (lanes 7,8) or from HA–MyrAkt transfected cells (lanes 1–6) were used for kinase reactions, as described in Materials and Methods. GST–4E-BP1 (2 μg) and histone H2B (2 μg) were used as substrates and incubated with the immunoprecipitates for the indicated times. Samples were analyzed by SDS-PAGE. An equal amount of immunoprecipitate was used for each reaction. Equal protein loading was visualized by Coomassie Blue staining. The results shown are representative of two independent experiments.
FRAP/mTOR activity is required for 4E-BP1 phosphorylation by Akt
Previous data and our results suggested that FRAP/mTOR is required for 4E-BP1 phosphorylation. The transfection of wild-type FRAP into serum-deprived wild-type 293 cells did not result in a mobility shift of 4E-BP1 (S.G. Kennedy and N. Hay, unpubl.). To evaluate the role of FRAP/mTOR in our system, we transfected wild-type FRAP and a rapamycin-resistant mutant (S2035T) form of FRAP (Brown et al. 1995) together with HA–4E-BP1 into 293 cells stably expressing MyrAkt. The transiently transfected HA–4E-BP1 exhibited a mobility shift even after 36 hr of serum deprivation (Fig. 7, lane 1). These results are consistent with the increased phosphorylation of 4E-BP1 observed in serum-deprived 293–MyrAkt cells (see Fig. 3). Phosphorylation of 4E-BP1 in 293–MyrAkt cells was unchanged by transfection with wild-type FRAP and FRAP(S2035T) (Fig. 7, lanes 2,5) and was resistant to wortmannin treatment (lanes 3,6). However, phosphorylation of 4E-BP1 in 293–MyrAkt cells transfected with wild-type FRAP was still sensitive to rapamycin treatment (lane 4), whereas transfection of FRAP(S2035T), conferred rapamycin resistance to 4E-BP1 phosphorylation (lane 7). Therefore, we conclude that Akt-induced phosphorylation of 4E-BP1 requires FRAP/mTOR.
Figure 7.

Akt requires FRAP activity for phosphorylation of 4E-BP1. HEK 293 MyrAkt cells were cotransfected with HA–4E-BP1 expression vector and with control vector (lane 1) or with wild-type epitope-tagged FLAG–FRAP expression vector (lanes 2–4), or a rapamycin-resistant mutant FLAG–FRAP S2035T expression vector (lane 5–7). After transfection, cells were serum deprived of for 36 hr and were either left untreated (lanes 1,2,5) or treated with wortmannin, [200 nm (Wort.); lanes 3,6] or rapamycin [20 ng/ml (Rap.); lanes 4,7]. Cell extracts were prepared, and HA–4E-BP1 was detected as described in Fig. 1A. Equal amounts of extract from the same experiment were used for detection of FLAG–FRAP with anti-Flag monoclonal antibodies. FLAG–FRAP is indicated by the large arrow. Small arrows indicate different phosphorylation states of 4E-BP1. The results shown in this figure are representative of three independent experiments.
Discussion
Translation rates are modulated in response to growth factors, hormones, and mitogens. A major target for extracellular stimuli is the translation initiation factor eIF4E. Its activation correlates with the phosphorylation and inactivation of 4E-BPs, as well as with its own phosphorylation on serine 209 (Whalen et al. 1996). Initial studies reported that 4E-BP1 is a direct downstream target of mitogen-activated protein kinase (MAPK) (Lin et al. 1994). Subsequent studies using pharmacological agents such as wortmannin and LY294002, which inhibit PI 3-kinase activity, and mutants of the PDGF receptor that cannot bind PI 3-kinase, have indicated that PI 3-kinase is required for the phosphorylation and inactivation of 4E-BP1, and precluded MAPK as an upstream regulator of 4E-BP1 (Beretta et al. 1996; von Manteuffel et al. 1996). However, a recent report showing that wortmannin and LY294002 inhibit the kinase activity of FRAP/mTOR, which is required for 4E-BP1 phosphorylation, challenged the idea that PI 3-kinase is required for 4E-BP1 phosphorylation (Brunn et al. 1996). In the present study we have provided direct evidence that growth factors mediate phosphorylation of 4E-BP1 with PI 3-kinase, which is the wortmannin-sensitive component in this signaling pathway (Figs. 1B and 2A). Moreover, we have demonstrated that the downstream effector of PI 3-kinase, Akt, is a critical intermediate in the signal transduction pathway leading from growth factors to the phosphorylation of 4E-BP1. Both the wild-type catalytic subunit of PI 3-kinase and the wild-type c-Akt promote the phosphorylation of 4E-BP1 when overexpressed transiently in 293 cells. We found that a constitutively active form of Akt promotes phosphorylation of 4E-BP1 in the absence of growth factors, in a wortmannin-resistant manner, whereas a dominant-interfering mutant of Akt blocks the ability of insulin to induce phosphorylation of 4E-BP1 (Fig. 1D). An activated Akt can also mediate the in vivo phosphorylation of 4E-BP2 (Fig. 3B). The phosphorylation of 4E-BP1 by an activated Akt and in response to growth factors occurred at apparently identical sites, as was demonstrated by phosphopeptide mapping (Fig. 4A,E). An activated Akt inhibits the binding of 4E-BP1 to eIF4E, even in the absence of growth factors, and therefore is presumed to increase eIF4E-dependent translation (Fig. 5).
The fact that identical phosphorylation sites on 4E-BP1 are diminished by rapamycin in both serum-induced and Akt-induced 4E-BP1 phosphorylation (Fig. 4) implies that the rapamycin-sensitive component lies downstream of Akt in this signaling cascade. Indeed, a rapamycin-resistant mutant of FRAP/mTOR confers rapamycin resistance to 4E-BP1 phosphorylation induced by an activated Akt (Fig. 7). Although these results strongly suggest a linear pathway from PI 3-kinase through Akt and to FRAP/mTOR, we cannot exclude a parallel signaling pathway that includes FRAP/mTOR and subsequently, that converges into a common downstream effector leading to phosphorylation of 4E-BP1 (see also Fig. 8). A recent study, which showed that FRAP/mTOR immunoprecipitated from cells is able to phosphorylate 4E-BP1 in vitro (Brunn et al. 1997), is consistent with a linear signaling pathway leading from growth factor receptor to PI 3-kinase, Akt, and FRAP/mTOR. However, the observation that immunoprecipitated FRAP/mTOR can phosphorylate 4E-BP1 does not preclude the possibility that a downstream effector of both FRAP/mTOR and Akt can be coimmunoprecipitated with FRAP/mTOR and consequently, responsible for 4E-BP1 phosphorylation in vitro.
Figure 8.
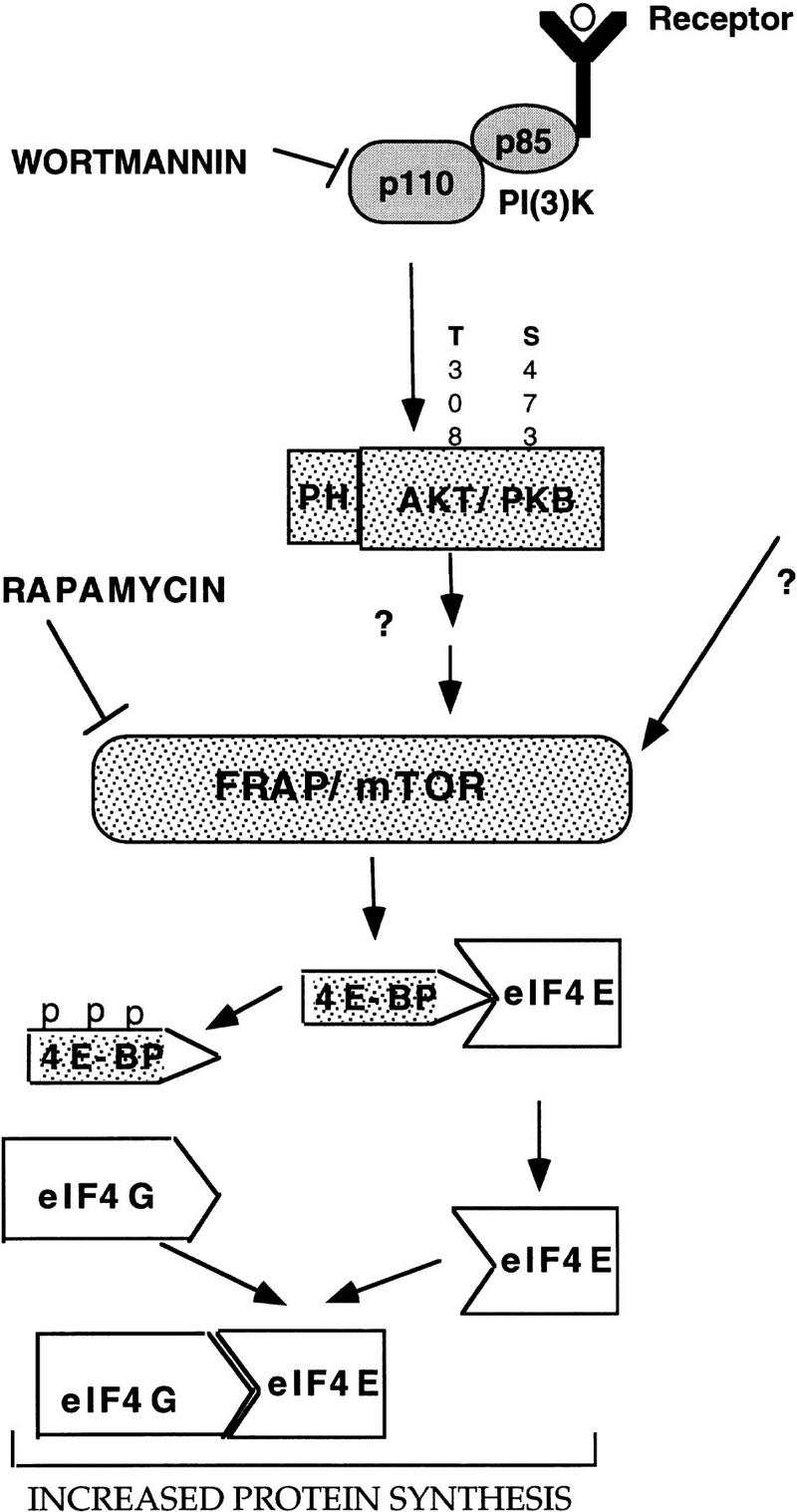
A model illustrating the signaling cascades leading to an increase in protein synthesis. PI 3-kinase, Akt, and FRAP/mTOR are downstream effectors of growth factor receptors that lead to phosphorylation of the 4E-BPs, and subsequent activation of eIF4E. For details see Discussion.
Our studies delineate the signaling pathway leading from growth factor receptor to the phosphorylation of 4E-BP1 and activation of eIF4E and provide direct evidence that PI 3-kinase, Akt, and FRAP/mTOR are key regulators in this pathway (Fig. 8). Akt has been reported to be activated by the lipid products of PI 3-kinase through its PH domain (Franke et al. 1997b). More recent studies showed that at least two upstream kinases are required for the full activation of Akt. One kinase, termed PDK1, is activated directly by the lipid products of PI 3-kinase and is responsible for the phosphorylation of Thr-308 in Akt. The other kinase is required to phosphorylate Ser-473 in Akt (Alessi et al. 1997; Stokoe et al. 1997). We showed that Akt cannot phosphorylate 4E-BP1 directly. It is not clear, however, whether Akt activates FRAP/mTOR directly or modulates the activity of another intermediate such as phosphatase or a kinase.
PI 3-kinase, Akt, and FRAP/mTOR were also implicated in the activation of p70S6k (Brown et al. 1995; Burgering and Coffer 1995; Reif et al. 1997; Fig. 8). Experiments using three different variants of p70S6k showed that overexpression of p70S6k inhibits the phosphorylation of 4E-BP1 by insulin probably by sequestering a common upstream activator. These results also suggest that the 4E-BP1 phosphorylation pathway bifurcates immediately upstream of p70S6k (von Manteuffel et al. 1997). Because the phosphorylation of both p70S6k and 4E-BP1 is inhibited by rapamycin, the common upstream activator could be the target of rapamycin FRAP/mTOR. FRAP/mTOR was shown to mediate the in vivo phosphorylation of both p70S6k and 4E-BP1 (this work; Brown et al. 1995; Brunn et al. 1997). It is unlikely, however, that p70S6k and 4E-BP1 are phosphorylated by the same kinase because the motifs flanking possible 4E-BP1 phosphorylation sites differ from the rapamycin-sensitive p70S6k sites (Moser et al. 1997). The large size of FRAP/mTOR (289 kD) raises the possibility (as depicted in Fig. 8) that it may act as a scaffold protein, which can interact with multiple kinases having different inputs. Thus, one of these kinases could be the p70S6k kinase and another kinase could be the 4E-BP1 kinase.
Akt, apoptosis, cell proliferation, and protein synthesis
Akt appears to have multiple downstream effectors (Marte and Downward 1997). In this study we demonstrated that 4E-BP1 is a new downstream target of Akt. Akt can promote cell survival; it blocks apoptosis accelerated by c-Myc (Kauffmann-Zeh et al. 1997; Kennedy et al. 1997), by UV irradiation (Kulik et al. 1997), by growth factor withdrawal in fibroblasts (Kennedy et al. 1997), in cerebellum neurons (Dudek et al. 1997), and in hemapoietic cells (Ahmed et al. 1997), and by a decrease in cell matrix adhesion (Khwaja et al. 1997). The ability of Akt to promote survival is not dependent on changes in the steady-state levels of Bcl-2 and BclxL (Kennedy et al. 1997). However, Akt inhibits the activity of caspase-3-like proteases that execute the cell death pathway (Kennedy et al. 1997). The ability of Akt to promote cell survival might be dependent on the synergistic effects of its multiple downstream effectors. Interestingly, at least two of its downstream targets, p70S6k and 4E-BP1, are involved in the control of translation. In many cases of apoptosis where cells have already been programmed to die, inhibition of protein synthesis either does not affect the rate of cell death or augments cell death as in tumor necrosis factor (TNF), Fas, or Myc-induced apoptosis (Evan et al. 1992; Polunovsky et al. 1994; Wagner et al. 1994; Natoli et al. 1995; Foote et al. 1996; Karsan et al. 1996; Reinartz et al. 1996). In this regard, it is noteworthy that rapamycin has a pronounced effect on the translation of insulin-like growth factor II (IGF–II) (Nielsen et al. 1995), which acts as a survival factor (Christofori et al. 1994; Ueda and Ganem 1996). However, treatment with rapamycin alone is not sufficient to induce apoptosis (Yao and Cooper 1996; Kauffmann-Zeh et al. 1997), although it was reported that rapamycin can accelerate apoptosis under certain conditions (Shi et al. 1995). It is also intriguing that ectopic expression of eIF4E can rescue cells from c-Myc and growth factor withdrawal-induced apoptosis (Polunovsky et al. 1996), suggesting that at least in part, the pathway uncovered in these studies could be relevant to the ability of Akt to promote survival.
Because both eIF4E and Akt were also shown to play a role in cell proliferation (Lazaris-Karatzas et al. 1990; Cheng et al. 1997), and 4E-BP1 is a negative regulator of cell growth (Rousseau et al. 1996), it is possible that at least in part Akt activity in cell proliferation is dependent on the activation of eIF4E. As was shown for eIF4E, Akt is overexpressed and amplified in certain tumors (Bellacosa et al. 1995; Kerekatte et al. 1995; Cheng et al. 1996; Nathan et al. 1997). Recent studies in Caenorhabditis elegans identified the mammalian equivalents of insulin receptor and PI 3-kinase as modulators of longevity, probably through regulation of metabolism and protein synthesis (Morris et al. 1996; Kimura et al. 1997). Because Akt is conserved in C. elegans (Waterston et al. 1992), it is possible that Akt and its downstream effectors, leading to protein synthesis, are also downstream components of insulin receptor/PI 3-kinase-mediated longevity. Because of its pivotal role in cell survival and proliferation, modulation of Akt activity in vivo might have an impact on therapies of cancer and degenerative diseases. 4E-BPs, the new downstream targets of Akt identified in these studies, can facilitate research on the activity of this multipotent kinase because the mobility-shift assay of 4E-BP1 can provide a simple and attractive read-out for Akt activity in vivo.
Materials and methods
Plasmids and antibodies
The human 4E-BP1 coding sequence was amplified by PCR and introduced in-frame into the cytomegalovirus (CMV)-based vector pACTAG-2 (a kind gift from A. Charest and M. Tremblay, McGill University, Montreal, Canada) to express a fusion protein with three amino-terminal HA tags. HA–c-Akt, HA–c-Akt K179M (Franke et al. 1995), and HA–MyrAkt (Ahmed et al. 1997) expression vectors were generously provided by Philip Tsichlis and Alfonso Bellacosa (Fox Chase Cancer Center, Philadelphia, PA). The BamHI/BglII fragment of HA–c-Akt K179M was introduced into pCDNA3 (Invitrogen) to generate the c-Akt (kin−) expression vector used in this study. FRAP and FRAP(S2035T) (Brown et al. 1995) expression vectors were generously provided by Stuart Schreiber and Eric Brown (Harvard University, Cambridge, MA). Myc–epitope-tagged p110α and p110α* (Khwaja et al. 1997) expression vectors were generously provided by Julian Downward [Imperial Cancer Research Fund (ICRF), London, UK]. Antibody 11208 against human 4E-BP1 was described previously (Gingras et al. 1996). Antibody 11209 is a rabbit polyclonal antibody (Pocono Rabbit Farm, Canadensis, PA) raised against human 4E-BP1 (expressed as a GST fusion protein). Neither antibody cross-reacts with 4E-BP2 in immunoprecipitation studies. Anti-4E-BP2 rabbit polyclonal antibody was raised against a GST–HMK–4E-BP2 fusion protein (described in Pause et al. 1994). The 4E-BP2 crude antisera cross-reacts (as indicated by Western blotting and by immunoprecipitation studies) with 4E-BP1 and, to a lesser extent, with a novel 4E-BP family member, 4E-BP3. The anti-HA antibody 12CA5 (mouse monoclonal) was concentrated from tissue culture supernatant using protein G–Sepharose beads (Pharmacia). The anti-HA mouse monoclonal antibody HA.11 was purchased from BabCO and was used at a dilution of 1:1000. The anti-Flag mouse monoclonal antibody M5 was purchased from Kodak and was used at a dilution of 1:400. The anti-Myc–epitope mouse monoclonal antibody 9E10 was used at a dilution of 1:500 for immunoprecipitation and 1:1000 for Western blotting.
Cell culture and viral infection
Cell culture was performed as described previously (Wagner et al. 1994; Kennedy et al. 1997). Ecotropic MyrAkt retrovirus was made by transient transfection of Bosc23 cells as described previously (Wagner et al. 1994). Virus was used to infect PA317 cells and a stable producer cell line was generated. The resultant amphotropic virus was used to infect HEK 293 cells, and stable clones were selected with G418 (500 μg/ml, Wagner et al. 1994). Clones were pooled and maintained as the stable cell line 293–MyrAkt.
Transient transfections
Transient transfections were conducted using either LipofectAMINE (GIBCO BRL) or calcium phosphate, as indicated in the figure legends. For LipofectAMINE transfection, HEK 293 cells were plated at 1 × 106/6-cm plate and grown overnight in DMEM/10% FCS. Cells were rinsed once with PBS and placed in 600 μl of DMEM. Four hundred nanograms of HA 4E-BP1 and 2.5 μg of the p110α and Akt expression constructs were added to 600 μl of DMEM containing 25 μl of LipofectAMINE and incubated at room temperature for 30 min. This reaction mixture was added to cells and incubated at 37°C for 4 hr. DMEM with 20% FCS (3 ml) was added for 2 hr, then cells were rinsed once with PBS and incubated in DMEM overnight. The medium was changed and cell extracts were prepared after 48 hr. Calcium phosphate transfection was conducted according to (Sambrook et al. 1989). The calcium phosphate/DNA precipitate was incubated with cells overnight in DMEM with 10% FCS. Cells were rinsed once with PBS and placed in DMEM. The following day cells were rinsed again with DMEM.
Extract preparation and Western blotting
Cells were rinsed twice with cold buffer a [20 mm Tris-HCl (pH 7.5), 100 mm KCl, 20 mm β-glycerolphosphate, 1 mm DTT, 0.25 mm Na3VO4, 10 mm NaF, 1 mm EDTA, 1 mm EGTA, 10 nm okadaic acid, 1 mm phenylmethylsulfonyl fluoride] and scraped into a minimal volume of the same buffer. Lysis was performed by three freeze–thaw cycles. Cell debris was pelleted by centrifugation, and the protein concentration in the supernatant was measured using the Bio-Rad assay. For analysis of endogenous 4E-BP1, 75 μg of total cell extract was incubated at 100°C for 7 min to enrich for 4E-BP1, which is heat-stable. Samples were incubated on ice for 5 min, and precipitated material was removed by centrifugation (13,000 rpm, 5 min). When necessary, the extract was TCA precipitated (Pause et al. 1994). Laemmli sample buffer was added to the supernatant, which was then subjected to SDS-15% PAGE. For analysis of transfected HA–4E-BP1, cells were lysed by three freeze–thaw cycles, and 50 μg of protein was analyzed by SDS–10% PAGE. Western blotting, using chemiluminescence detection, was performed as described (Gingras et al. 1996) using either the anti-4E-BP1 11208 antibody (1:1500) or the anti-HA 12CA5 monoclonal antibody (0.5 μg/ml). For analysis of p110α, p110α and p110α* expression vectors were transfected into HEK 293 cells. After 36 hr cells were lysed into 500 μl of RIPA buffer [50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS] containing 1 mm PMSF and subjected to immunoprecipitation for 3 hr with 1 μg of the mouse monoclonal anti-Myc antibody 9E10. Immunoprecipitates were rinsed three times with RIPA buffer and resuspended in Laemmli buffer. Samples were boiled and subjected to SDS–8% PAGE. p110α was detected by incubation with the 9E10 antibody.
Chromatography on m7GDP-agarose
Cell extracts prepared by three freeze–thaw cycles, in buffer containing 20 mm HEPES–KOH (pH 7.5), 75 mm KCl and 1 mm EDTA, were incubated for 1 hr with m7GDP coupled to agarose adipic resin [30 μl of packed beads per reaction; beads were prepared according to Edery et al. (1988)]. Beads were spun down in a microfuge (3000 rpm, 30 sec), washed THREE times with 20 volumes of the same buffer, and resuspended in Laemmli sample buffer. Samples were then analyzed by SDS-PAGE and Western blotting as described above.
Metabolic labeling and immunoprecipitation
HEK 293 cells starved for 30–36 hr or Rat1a cells starved for 16–24 hr were incubated at 37°C for 3 hr in serum-free DMEM containing 0.5 mCi/ml [32P]orthophosphate (DuPont NEN; 3000 mCi/mmole). Rapamycin (20 ng/ml) or wortmannin (100 nm) were added for 20 min, followed by the addition of dialyzed FBS (15%; GIBCO) for 30 min. The medium was removed and the cells were rinsed twice in cold PBS. Cells were lysed in lysis buffer [10% glycerol, 50 mm Tris (pH 7.5), 60 mm KCl, 2 mm CDTA (trans,-1,2-diaminocyclohexane-N,N,N′,N′-tetraacetic acid), 1% Triton X-100, 2 mm DTT, 20 mm β-glycerolphosphate, 10 nm okadaic acid] for 30 min at 4°C. Lysate was harvested by scraping and cell debris was removed by centrifugation. Total radioactivity in the lysate was monitored by spotting 1, 2, 5, and 10 μl of the extract onto a phosphocellulose (P81) paper, which was washed extensively with 75 mm phosphoric acid and dried. Bound radioactivity was measured by scintillation counting (Whalen et al. 1996). The extract (equivalent quantities of radioactivity) was precleared by incubation with protein A beads (50 μl per ml of extract) with end-over-end rotation at 4°C for 1 hr. The supernatant was transferred to a fresh tube, together with 30 μl of 11209 crude antisera per milliliter of extract, and incubated for 3 hr at 4°C. Protein A beads (30 μl packed beads) were added and incubation end-over-end was carried out for 2 hr at 4°C. Beads were spun down (microfuge, 6000 rpm, 2 min) and washed two times in lysis buffer, 2 times in RIPA buffer, and 2 times in LiCl solution (200 mm LiCl, 1 mm DTT). In some experiments, the supernatant (unbound fraction) was further incubated with 11211 anti-4E-BP2 antibody as for 4E-BP1. Immunoprecipitated material was subjected to SDS–15% PAGE and transferred to PVDF membranes (Immobilon-P or Immobilon-PSQ, Millipore), which were dried and autoradiographed. Radioactive bands corresponding to 4E-BPs were excised and Cerenkov counted.
Phosphopeptide maps
Tryptic–chymotryptic digestion of 4E-BP1 immobilized on the PVDF membranes was performed essentially as described (van der Geer and Hunter 1994), with the following modifications. The digest was performed using a 200:1 mixture of TPCK-treated trypsin and chymotrypsin (5 μg, Worthington) for 10 hr, followed by the addition of 5 μg enzyme for 3 hr. The sample was then lyophilized (speed-vac, Savant), resuspended in 200 μl of water, lyophilized again, resuspended in 100 μl of water, lyophilized a third time, resuspended in 100 μl (pH 1.9) buffer (2.5% vol/vol formic acid 88% and 7.8% vol/vol glacial acetic acid) and lyophilized a fourth time. For chromatography, first dimension (electrophoresis) was performed in pH 1.9 buffer using the HTLE 7000 apparatus (CBS Scientific); second dimension was performed in phosphochromatography buffer (37.5% vol/vol n-butanol, 25% vol/vol pyridine, 7.5% vol/vol glacial acetic acid). Plastic-coated cellulose thin-layer chromatography plates (Kodak; 20 cm × 20 cm) were used.
Akt kinase assay
HEK 293 1 × 106 cells were transfected with 3 μg of HA–MyrAkt DNA using LipofectAMINE. Cells were grown in 10% DMEM for 48 hr, then lysed into Akt lysis buffer (Kennedy et al. 1997). Extracts were incubated with 3 μg of monoclonal mouse anti-HA (BabCO, HA.11) for 3 hr. Immunoprecipitations and kinase reactions with histone H2B (2 μg) and GST–4E-BP1 (2 μg) were conducted according to (Franke et al. 1995).
Acknowledgments
We are grateful to S. Schreiber, A. Bellacosa, P. Tsichlis, and J. Downward for plasmids and antibodies. We thank C. Palfrey, B. Raught, and S. Conzen for critical comments and valuable discussions. This work was supported by American Cancer Society (ACS) grant CB-133 and by National Institutes of Health (NIH) grant CA71874 (N.H.), and by the National Cancer Institute of Canada (N.S.). N.S. is a Medical Research Council of Canada Distinguished Scientist and a Howard Hughes Medical Institute International Scholar. A.-C.G. was supported by a Natural Science and Engineering of Canada 1967 Studentship. S.G.K. was supported by training grant GM07151.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL nhay@ben-may.bsd.uchicago.edu; FAX (773) 702-6260.
References
- Ahmed NN, Grimes HL, Bellacosa A, Chan TO, Tsichlis PN. Transduction of interleukin-2 antiapoptotic and proliferative signal via Akt protein kinase. Proc Natl Acad Sci. 1997;94:3627–3632. doi: 10.1073/pnas.94.8.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimoto K, Takahashi R, Moriya S, Nishioka N, Takayanagi J, Kimura K, Fukui Y, Osada S, Mizuno K, Hirai S, Kazlauskas A, Ohno S. EGF and PDGF receptors activate atypical PKC lambda through phosphatidylinositol 3-kinase. EMBO J. 1996;15:788–798. [PMC free article] [PubMed] [Google Scholar]
- Alessi D, James S, Downes CP, Holmes A, Gaffney P, Reese C, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Barbet NC, Schneider U, Helliwell SB, Stansfield I, Tuite MF, Hall MN. Tor controls translation initiation and early G1 progression in yeast. Mol Biol Cell. 1996;7:25–42. doi: 10.1091/mbc.7.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellacosa A, Feo DD, Godwin AK, Bell DW, Cheng JQ, Altomare DA, Wan M, Dubeau L, Scambia G, Masciullo V, Ferrandina G, Panici PB, Mancuso S, Neri G, Testa JR. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Intern J Cancer. 1995;64:280–285. doi: 10.1002/ijc.2910640412. [DOI] [PubMed] [Google Scholar]
- Beretta L, Gingras AC, Svitkin YV, Hall MN, Sonenberg N. Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J. 1996;15:658–664. [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, Schreiber SL. A signaling pathway to translational control. Cell. 1996;86:517–520. doi: 10.1016/s0092-8674(00)80125-7. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Beal PA, Keith CT, Chen J, Shin TB, Schreiber SL. Control of p70 S6 kinase by kinase activity of FRAP in vivo. Nature. 1995;377:441–446. doi: 10.1038/377441a0. [DOI] [PubMed] [Google Scholar]
- Brunn GJ, Williams J, Sabers C, Wiederrecht G, Lawrence JCJ, Abraham RT. Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. EMBO J. 1996;15:5256–5267. [PMC free article] [PubMed] [Google Scholar]
- Brunn GJ, Hudson CC, Sekulic A, Williams JM, Hosoi H, Houghton PJ, Lawrence J, Jr, Abraham RT. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science. 1997;277:99–101. doi: 10.1126/science.277.5322.99. [DOI] [PubMed] [Google Scholar]
- Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- Cheng JQ, Ruggeri B, Klein WM, Sonoda G, Altomare DA, Watson DK, Testa JR. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci. 1996;93:3636–3641. doi: 10.1073/pnas.93.8.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JQ, Altomare DA, Klein MA, Lee WC, Kruh GD, Lissy NA, Testa JR. Transforming activity and mitosis-related expression of the AKT2 oncogene: Evidence suggesting a link between cell cycle regulation and oncogenesis. Oncogene. 1997;14:2793–2801. doi: 10.1038/sj.onc.1201121. [DOI] [PubMed] [Google Scholar]
- Chou MM, Blenis J. The 70 kDa S6 kinase: Regulation of a kinase with multiple roles in mitogenic signaling. Curr Opin Cell Biol. 1995;7:806–814. doi: 10.1016/0955-0674(95)80064-6. [DOI] [PubMed] [Google Scholar]
- Christofori G, Nalik P, Hanahan D. A second signal supplied by insulin-like growth factor II in oncogene-induced tumorigenesis. Nature. 1994;369:414–418. doi: 10.1038/369414a0. [DOI] [PubMed] [Google Scholar]
- Chung J, Kuo CJ, Crabtree GR, Blenis J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell. 1992;69:1227–1236. doi: 10.1016/0092-8674(92)90643-q. [DOI] [PubMed] [Google Scholar]
- Cohen P, Alessi DR, Cross DAE. PDK1, one of the missing links in insulin signal transduction? Fed Eur Biochem Soc Lett. 1997;410:3–10. doi: 10.1016/s0014-5793(97)00490-0. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Edery I, Altmann M, Sonenberg N. High-level synthesis in Escherichia coli of functional cap-binding eukaryotic initiation factor eIE-4E and affinity purification using a simplified cap-analog resin. Gene. 1988;74:517–525. doi: 10.1016/0378-1119(88)90184-9. [DOI] [PubMed] [Google Scholar]
- Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Foote LC, Schneider TJ, Fischer GM, Wang JK, Rasmussen B, Campbell KA, Lynch DH, Ju ST, Marshak-Rothstein A, Rothstein TL. Intracellular signaling for inducible antigen receptor-mediated Fas resistance in B cells. J Immunol. 1996;157:1878–1885. [PubMed] [Google Scholar]
- Franke TF, Yang S, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC. PI3K: Downstream AKTion blocks apoptosis. Cell. 1997a;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC, Toker AT. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997b;275:665–667. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Svitkin Y, Belsham GJ, Pause A, Sonenberg N. Activation of the translational suppressor 4E-BP1 following infection with encephalomyocarditis virus and poliovirus. Proc Natl Acad Sci. 1996;93:5578–5583. doi: 10.1073/pnas.93.11.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MN. The TOR signalling pathway and growth control in yeast. Biochem Soc Transac. 1996;24:234–239. doi: 10.1042/bst0240234. [DOI] [PubMed] [Google Scholar]
- Hawkins PT, Eguinoa A, Qui RG, Stokoe D, Cooke FT, Walter R, Wennstrom S, Claesson WL, Evans T, Symons M, Stephens L. PDGF stimulates an increase in Rac–GTP via the activation of phosphoinositide 3-kinase. Curr Biol. 1995;5:393–403. doi: 10.1016/s0960-9822(95)00080-7. [DOI] [PubMed] [Google Scholar]
- Hemmings BA. Akt signaling: Linking membrane events to life and death decisions. Science. 1997;275:628–630. doi: 10.1126/science.275.5300.628. [DOI] [PubMed] [Google Scholar]
- Hoekstra MF. Responses to DNA damage and regulation of cell cycle checkpoints by the ATM protein kinase family. Curr Opin Genet Dev. 1997;7:170–175. doi: 10.1016/s0959-437x(97)80125-6. [DOI] [PubMed] [Google Scholar]
- Karsan A, Yee E, Harlan JM. Endothelial cell death induced by tumor necrosis factor- alpha is inhibited by the Bcl-2 family member, A1. J Biol Chem. 1996;271:27201–27204. doi: 10.1074/jbc.271.44.27201. [DOI] [PubMed] [Google Scholar]
- Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G I. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes & Dev. 1997;11:701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- Kerekatte V, Smiley K, Hu B, Smith A, Gelder F, De Benedetti A. The proto-oncogene/translation factor eIF4E: A survey of its expression in breast carcinomas. Intern J Cancer. 1995;64:27–31. doi: 10.1002/ijc.2910640107. [DOI] [PubMed] [Google Scholar]
- Khwaja A, Rodriguez-Viciana P, Wenstrom S, Warne PH, Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 1997;16:2783–2793. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Kulik G, Klippel A, Weber MJ. Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol. 1997;17:1595–1606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaris-Karatzas A, Montine KS, Sonenberg N. Malignant transformation by eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature. 1990;345:544–547. doi: 10.1038/345544a0. [DOI] [PubMed] [Google Scholar]
- Lin TA, Kong X, Haystead TA, Pause A, Belsham G, Sonenberg N, Lawrence JCJ. PHAS-I as a link between mitogen-activated protein kinase and translation initiation. Science. 1994;266:653–656. doi: 10.1126/science.7939721. [DOI] [PubMed] [Google Scholar]
- Lin TA, Kong X, Saltiel AR, Blackshear PJ, Lawrence JCJ. Control of PHAS-I by insulin in 3T3-L1 adipocytes. Synthesis, degradation, and phosphorylation by a rapamycin- sensitive and mitogen-activated protein kinase-independent pathway. J Biol Chem. 1995;270:18531–18538. doi: 10.1074/jbc.270.31.18531. [DOI] [PubMed] [Google Scholar]
- Marte BM, Downward J. PKB/Akt: Connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem Sci. 1997;22:355–358. doi: 10.1016/s0968-0004(97)01097-9. [DOI] [PubMed] [Google Scholar]
- Morris JZ, Tissenbaum HA, Ruvkun G. a phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature. 1996;382:536–539. doi: 10.1038/382536a0. [DOI] [PubMed] [Google Scholar]
- Moser BA, Dennis PB, Pullen N, Pearson RB, Williamson NA, Wettenhall RE, Kozma SC, Thomas G. Dual requirement for a newly identified phosphorylation site in p70s6k. Mol Cell Biol. 1997;17:5648–5655. doi: 10.1128/mcb.17.9.5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi H, Brewer KA, Exton JH. Activation of the γ isozyme of protein kinase C by phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1993;268:13–16. [PubMed] [Google Scholar]
- Nathan CA, Liu L, Li BD, Abreo FW, Nandy I, De Benedetti A. Detection of the proto-oncogene eIF4E in surgical margins may predict recurrence in head and neck cancer. Oncogene. 1997;15:579–584. doi: 10.1038/sj.onc.1201216. [DOI] [PubMed] [Google Scholar]
- Natoli G, Ianni A, Costanzo A, De Petrillo G, Ilari I, Chirillo P, Balsano C, Levrero M. Resistance to Fas-mediated apoptosis in human hepatoma cells. Oncogene. 1995;11:1157–1164. [PubMed] [Google Scholar]
- Nielsen FC, Ostergaard L, Nielsen J, Christiansen J. Growth-dependent translation of IGF-II mRNA by a rapamycin-sensitive pathway. Nature. 1995;377:358–362. doi: 10.1038/377358a0. [DOI] [PubMed] [Google Scholar]
- Pause A, Belsham GJ, Gingras AC, Donze O, Lin TA, Lawrence HCJ, Sonenberg N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′cap function. Nature. 1994;371:762–767. doi: 10.1038/371762a0. [DOI] [PubMed] [Google Scholar]
- Polunovsky VA, Rosenwald IB, Tan AT, White J, Chiang L, Sonenberg N, Bitterman PB. Translational control of programmed cell death: Eukaryotic translation initiation factor 4E blocks apoptosis in growth-factor-restricted fibroblasts with physiologically expressed or deregulated Myc. Mol Cell Biol. 1996;16:6573–6581. doi: 10.1128/mcb.16.11.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polunovsky VA, Wendt CH, Ingbar DH, Peterson MS, Bitterman PB. Induction of endothelial cell apoptosis by TNF alpha: Modulation by inhibitors of protein synthesis. Exper Cell Res. 1994;214:584–594. doi: 10.1006/excr.1994.1296. [DOI] [PubMed] [Google Scholar]
- Price DJ, Grove JR, Calvo V, Avruch J, Bierer BE. Rapamycin-induced inhibition of the 70-kilodalton S6 protein kinase. Science. 1992;257:973–977. doi: 10.1126/science.1380182. [DOI] [PubMed] [Google Scholar]
- Proud CG. p70 S6 kinase: An enigma with variations. Trends Biochem Sci. 1996;21:181–185. [PubMed] [Google Scholar]
- Reif K, Burgering B, Cantrell D. Phosphatidylinositol 3-kinase links the interleukin-2 receptor to protein kinase B and p70 S6 kinase. J Biol Chem. 1997;272:14426–14433. doi: 10.1074/jbc.272.22.14426. [DOI] [PubMed] [Google Scholar]
- Reinartz J, Bechtel MJ, Kramer MD. Tumor necrosis factor-alpha-induced apoptosis in a human keratinocyte cell line (HaCaT) is counteracted by transforming growth factor-alpha. Exper Cell Res. 1996;228:334–340. doi: 10.1006/excr.1996.0333. [DOI] [PubMed] [Google Scholar]
- Rousseau D, Gingras AC, Pause A, Sonenberg N. The eIF4E-binding proteins 1 and 2 are negative regulators of cell growth. Oncogene. 1996;13:2415–2420. [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. pp. 16.33–16.35. [Google Scholar]
- Shi Y, Frankel A, Radvanyi LG, Penn LZ, Miller RG, Mills GB. Rapamycin enhances apoptosis and increases sensitivity to cisplatin in vitro. Cancer Res. 1995;55:1982–1988. [PubMed] [Google Scholar]
- Sonenberg N. In: Translational control. Hershey JWB, Mathews MB, Sonenberg N, editors. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1996. pp. 245–269. [Google Scholar]
- Stokoe D, Stephens LR, Copeland T, Gaffney PRJ, Reese CB, Painter GF, Holmes AB, McCormick F, Hawkins PT. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567–570. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- Ueda K, Ganem D. Apoptosis is induced by N-myc expression in hepatocytes, a frequent event in hepadnavirus oncogenesis, and is blocked by insulin-like growth factor II. J Virol. 1996;70:1375–1383. doi: 10.1128/jvi.70.3.1375-1383.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Geer P, Hunter T. Phosphopeptide mapping and phosphoamino acid analysis by electrophoresis and chromatography on thin-layer cellulose plates. Electrophoresis. 1994;15:544–554. doi: 10.1002/elps.1150150173. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield MD. Phosphoinositide 3-kinases: A conserved family of signal transducers. Trends Biochem Sci. 1997;22:267–272. doi: 10.1016/s0968-0004(97)01061-x. [DOI] [PubMed] [Google Scholar]
- von Manteuffel SR, Gingras AC, Ming XF, Sonenberg N, Thomas G. 4EBP-I phosphorylation is mediated by the FRAP-p70S6K pathway and is independent of mitogen-activated protein kinase. Proc Natl Acad Sci. 1996;93:4076–4080. doi: 10.1073/pnas.93.9.4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Manteuffel S, Dennis PB, Pullen N, Gingras AC, Sonenberg N, Thomas G. The insulin-induced signalling pathway leading to S6 and initiation factor 4E binding protein 1 phosphorylation bifurcates at a rapamycin-sensitive point immediately upstream of p70s6k. Mol Cell Biol. 1997;1997:5426–5436. doi: 10.1128/mcb.17.9.5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AJ, Kokontis JM, Hay N. Myc-mediated apoptosis requires wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21waf1/cip1. Genes & Dev. 1994;8:2817–2830. doi: 10.1101/gad.8.23.2817. [DOI] [PubMed] [Google Scholar]
- Waterston R, Martin C, Craxton M, Huynh C, Coulson A, Hillier L, Durbin R, Green P, Shownkeen R, Halloran N, et al. A survey of expressed genes in Caenorhabditis elegans. Nature Genet. 1992;1:114–123. doi: 10.1038/ng0592-114. [DOI] [PubMed] [Google Scholar]
- Whalen SG, Gingras AC, Amankwa L, Mader S, Branton PE, Aebersold R, Sonenberg N. Phosphorylation of eIF-4E on serine 209 by protein kinase C is inhibited by the translational repressors, 4E-binding proteins. J Biol Chem. 1996;271:11831–11837. doi: 10.1074/jbc.271.20.11831. [DOI] [PubMed] [Google Scholar]
- Yao R, Cooper GM. Growth factor-dependent survival of rodent fibroblasts requires phosphatidylinositol 3-kinase but is independent of pp70S6K activity. Oncogene. 1996;13:343–351. [PubMed] [Google Scholar]