

Abstract
Vaccinia virus RNA polymerase terminates transcription in response to a specific signal UUUUUNU in the nascent RNA. Transduction of this signal to the elongating polymerase requires a trans-acting viral termination factor (VTF/capping enzyme), and is coupled to the hydrolysis of ATP. Recent studies suggest that ATP hydrolysis is catalyzed by a novel termination protein (factor X), which is tightly associated with the elongation complex. Here, we identify factor X as NPH-I (nucleoside triphosphate phosphohydrolase-I), a virus-encoded DNA-dependent ATPase of the DExH-box family. We report that NPH-I serves two roles in transcription (1) it acts in concert with VTF/CE to catalyze release of UUUUUNU-containing nascent RNA from the elongation complex, and (2) it acts by itself as a polymerase elongation factor to facilitate readthrough of intrinsic pause sites. A mutation (K61A) in the GxGKT motif of NPH-I abolishes ATP hydrolysis and eliminates the termination and elongation factor activities. Related DExH proteins may have similar roles at postinitiation steps during cellular mRNA synthesis.
Keywords: Vaccina virus, NPH-1 ATPase, transcription termination
Landmark advances in understanding eukaryotic mRNA metabolism have emerged from studies of vaccinia virus mRNA synthesis in vitro. These include the discovery of poly(A) in mRNA (Kates and Beeson 1970), the description of the 5′ cap structure and the elucidation of its synthetic pathway (Ensinger et al. 1975; Wei and Moss 1975), and the observation that initiation of mRNA synthesis is coupled to ATP hydrolysis (Gershowitz et al. 1978). More recently, vaccinia has been in the vanguard with respect to the mechanism of eukaryotic mRNA transcription termination.
Vaccinia encodes its own multisubunit RNA polymerase (a Pol II homolog), transcription initiation and termination factors, and mRNA processing enzymes (Moss 1996). Transcription of the early class of vaccinia genes initiates at virus-specific promoters and terminates at sites downstream of a simple termination signal UUUUUNU in the nascent RNA strand (Shuman and Moss 1988; Moss 1996; Deng and Shuman 1997a). Initiation is specified by a heterodimeric vaccinia protein (ETF; early transcription factor) that binds to the promoter sequence and recruits polymerase to the template (Broyles et al. 1988). Initiation is coupled to ATP hydrolysis by ETF (Li and Broyles 1993). A trans-acting vaccinia termination factor (VTF) is required to transduce the UUUUUNU signal to the elongating polymerase (Shuman et al. 1987). VTF is identical to the vaccinia capping enzyme, a heterodimeric protein that catalyzes three distinct reactions leading to formation of the m7GpppN cap structure (Shuman 1995). Yet, the termination factor activity of this versatile protein is entirely independent of its role in capping and methylating the 5′ end of the nascent RNA chain (Shuman et al. 1987; Hagler et al. 1994; Luo et al. 1995). UV crosslinking studies show that VTF/capping CE enzyme is bound to the nascent RNA during transcription elongation (Hagler et al. 1994). VTF/CE forms a binary complex with vaccinia RNA polymerase in solution in the absence of nucleic acid (Hagler and Shuman 1992b). These findings suggested a model whereby VTF/CE might “read” the sequence of the nascent RNA as it is extruded from the RNA polymerase and elicit an alteration in the properties of the polymerase on encountering the UUUUUNU element.
VTF-mediated termination is an energy-requiring process. The termination event is coupled to the hydrolysis of ATP or dATP (Hagler et al. 1994). Termination site choice and the overall efficiency of termination are determined by a kinetic balance between the rate of signaling and the rate of polymerase movement. Signaling rate is dictated by the concentration of hydrolyzable ATP. For example, lowering the ATP concentration slows the rate of signaling and thereby shifts the distribution of termination sites farther away from the termination signal (Hagler et al. 1994). Elongation rate is influenced by the concentration of NTP precursors and also by DNA template sequences (so-called intrinsic pausing signals) that elicit a transient elongation block even at optimal NTP concentrations. The T-run that encodes the RNA termination signal is one such intrinsic pausing signal (Deng and Shuman 1997b). Limiting the NTP pool (or replacing standard NTPs by certain analogs) slows polymerase elongation rate and thereby shifts the distribution of termination sites nearer to the termination signal (Hagler et al. 1994; Deng and Shuman 1997b). VTF-dependent release of the RNA chain from the template-engaged RNA polymerase occurs in the absence of ongoing elongation, provided that the polymerase has already advanced beyond a threshold distance 3′ of the termination signal. This allows UUUUUNU to be extruded from the nascent RNA-binding pocket of the elongation complex such that it is accessible to VTF/CE (Deng et al. 1996). ATP is required for transcript release.
To begin to address how energy is used during termination, the ATP-hydrolyzing protein must be identified. So far, we have shown that VTF/CE enzyme is not responsible for ATP hydrolysis during transcription termination (Yu et al. 1997) and presented evidence of a novel termination factor tightly associated with the transcription elongation complex. The existence of such a factor (provisionally named factor X) was suggested by the finding that transient exposure of purified elongation complexes to high concentrations of heparin rendered them refractory to VTF-induced termination. Factor X partially purified from virus-infected cells restored VTF-responsiveness to heparin-stripped ternary complexes (Deng and Shuman 1996). Heparin stripping also rendered the elongation complex more susceptible to intrinsic pausing at the 5′ margin of the T-run. This defect, too, was complemented by partially purified factor X, therefore suggesting a second role for factor X during elongation.
Here we show that factor X corresponds to the vaccinia-encoded protein NPH-I (nucleoside triphosphate phosphohydrolase I)—a DNA-dependent ATPase of the DExH-box family. Recombinant NPH-I is necessary and sufficient to restore VTF responsiveness to heparin-stripped elongation complexes and correct their enhanced pausing behavior. Single amino-acid substitutions of NPH-I that inactivate DNA-dependent ATP hydrolysis also abolish termination. We conclude that NPH-I is the energy-coupling factor for mRNA transcription termination. Our studies provide an expanded view of the role of DExH proteins in regulating eukaryotic transcription.
Results
Vaccinia encodes and encapsidates within the virus particle four different nucleic acid-dependent NTPases, one of which, NPH-I, appeared to be a good candidate for termination factor X. Our reasoning was as follows (1) adenine nucleotides are the exclusive energy source for vaccinia transcription termination and NPH-I hydrolyzes dATP and ATP, but is nonreactive with other common NTPs and dNTPs (Paoletti and Moss 1974; Hagler et al. 1994); (2) factor X isolated from virus-infected cells has an associated ATPase function that is activated by single-stranded DNA, which is the obligate cofactor for purified NPH-I (Paoletti and Moss 1974; Deng and Shuman 1996); and (3) factor X and NPH-I have similar native sizes, as gauged by glycerol gradient sedimentation (Paoletti et al. 1974; Deng and Shuman 1996). NPH-I is a 631-amino-acid polypeptide encoded by the vaccinia D11 gene (Broyles and Moss 1987) (Fig. 1). To test the hypothesis that NPH-I is factor X, we expressed NPH-I in bacteria, purified the recombinant NPH-I protein from soluble bacterial lysates, and tested its ability to restore termination competence to heparin-stripped elongation complexes.
Figure 1.

Amino acid sequence of vaccinia NPH-I. The GxGKT and DExH motifs are demarcated by boxes.
Expression and purification of recombinant NPH-I
An inducible T7 RNA polymerase-based system was used to express NPH-I in Escherichia coli. To facilitate purification, the full-length wild type NPH-I protein was fused to an amino-terminal leader peptide containing 10 tandem histidines. NPH-I was purified from a soluble lysate of induced bacteria by Ni-agarose and phosphocellulose column chromatography. The phosphocellulose preparation was nearly homogeneous with respect to the NPH-I polypeptide (Fig. 2A, WT). The phosphocellulose enzyme hydrolyzed ATP in the presence of single-stranded DNA. The release of 32Pi from [γ-32P]ATP was proportional to input enzyme; the reaction proceeded to completion at saturating enzyme concentration (Fig. 2B, WT). We calculated that recombinant NPH-I hydrolyzed 300 molecules of ATP per second per enzyme in the presence of a DNA cofactor. Recombinant NPH-I had no detectable ATPase activity in the absence of added DNA (data not shown). The phosphocellulose preparation of NPH-I was centrifuged through a 15%–35% glycerol gradient. ATPase activity sedimented as a single peak (Fig. 3A), which coincided with the sedimentation profile of the NPH-I polypeptide (data not shown). The apparent sedimentation coefficient of 4.3 S (relative to markers run in parallel) indicated that recombinant NPH-I is a monomer.
Figure 2.

Purification and ATPase activity of recombinant NPH-I. (A) Aliquots (3 μg) of the phosphocellulose preparations of wild-type NPH-I (WT) and the K61A mutant protein were electrophoresed through a 10% polyacrylamide gel containing 0.1% SDS. Polypeptides were visualized by staining with Coomassie brilliant blue dye. The positions and sizes (in kD) of coelectrophoresed marker proteins are indicated on the left. (B) ATPase activity. Assays were performed as described in Materials and Methods. Activity (nmoles Pi release/30 min) is plotted as a function of the input wild-type and K61A phosphocellulose enzyme preparations.
Figure 3.

Sedimentation analysis and DNA binding by recombinant NPH-I. (A) Glycerol gradient sedimentation was performed as described in Materials and Methods. Aliquots (2 μl) of the indicated gradient fractions were assayed for DNA-dependent ATPase activity. The activity profile is shown. The peak positions of marker proteins sedimented in a parallel gradient are indicated by the arrows. (B) DNA-binding assay. Reaction mixtures (20 μl) containing 20 mm Tris-HCl (pH 8.0), 2 mm DTT, 25 fmoles of a 5′ 32P-labeled 40-mer DNA oligonucleotide (5′-CCATGTTTCTGTACTAATCTAGAACGAGTCTATATATTCC), and 25, 5, or 1 ng of wild-type NPH-I of K61A (proceeding from left to right within each titration series) were incubated for 30 min at 30°C. NPH-I was omitted from control reactions (lanes−). The samples were adjusted to 5% glycerol and then applied to a native 8% polyacrylamide gel in 0.25× TBE. Electrophoresis was at 20 mA for 45 min. Free DNA and a protein–DNA complex or retarded electrophoretic mobility (asterisk) were visualized by autoradiographic exposure of the dried gel.
Heparin-stripping of elongation complexes blocks the transcript release step of termination
The experiment in Figure 4 illustrates the effects of heparin-stripping on the transcript release step of the termination reaction. Here, transcription by vaccinia RNA polymerase was programmed by a linear DNA template linked to a streptavidin-coated paramagnetic bead. The G21(TER29)A78 transcription unit consists of a vaccinia early promoter (P) and a 20-nucleotide G-less cassette flanked by a 57-nucleotide A-less cassette. Placed within the A-less cassette is a termination signal, TTTTTTTTT (Fig. 4). Ternary complexes containing radiolabeled 3′OMeGMP-arrested 21-mer RNA were assembled in a reaction mixture containing ATP, UTP, [α-32P]CTP, and 3′OMeGTP, then recovered by centrifugation and concentration of the beads with an externally applied magnet. Heparin-stripped G21 ternary complexes were prepared by adjusting the reaction mixtures to 625 μg/ml of heparin before recovery of the beads. This concentration of heparin strips off several polypeptide components of the elongation complex, including factor X. The pelleted G21 complexes were resuspended in 50 mm Tris, 2 mm DTT and then subjected to three cycles of bead purification and washing to remove the heparin. The control and heparin-stripped G21 ternary complexes were “walked” through the A-less cassette to the first-templated A position (A78), located 50 nucleotides downstream of the first U-residue of the UUUUUUUUU termination signal. Arrest at A78 was achieved by inclusion of cordycepin triphosphate (3′ dATP) in place of ATP during the elongation reaction. The A78 complexes were incubated for 5 min with VTF/CE and the mixture was separated into bead-bound and supernatant fractions. The labeled RNA in each fraction was analyzed by denaturing gel electrophoresis. As shown in Figure 4, control ternary complexes released 62% of the labeled A78 RNA into the supernatant fraction in response to VTF/CE (Fig. 4). We demonstrated previously that RNA release requires VTF/CE, the UUUUUNU signal, and an adenosine nucleotide energy cofactor (Deng et al. 1996; Deng and Shuman 1997a). The heparin-stripped A78 ternary complexes released only 10% of the A78 transcripts in response to VTF/CE. [This value is close to the background levels of 3% to 8% of A78 RNA recovered in the supernatants of control complexes incubated in the absence of VTF/CE (Deng et al. 1996).] Note that both the bound and released A78 RNAs migrate as a doublet, the more slowly migrating species of which corresponds to A78 RNA that has been capped with GMP by VTF/CE (Deng et al. 1996). We surmise from this experiment that heparin stripping blocked the release step of the termination reaction.
Figure 4.

NPH-I restores VTF-responsiveness to heparin-stripped elongation complexes. A schema of the G21(TER29)A78 DNA template is shown. The DNA contains a biotinylated nucleotide incorporated uniquely at the 3′ end of the template DNA strand, which anchors the DNA to a streptavidin-coated magnetic bead. The transcription unit consists of a vaccinia early promoter (P) fused to a 20-nucleotide G-less cassette, which is flanked by a run of three G residues at positions +21 to +23. Situated downstream of the G-less cassette is a 57-nucleotide A-less cassette flanked at its 3′ end by a run of four A residues at positions +78 to + 81. Placed within the A-less cassette is a termination signal, TTTTTTTTT, spanning positions +29 to +37. Ternary complexes containing pulse-labeled OMeG21 RNA were assembled as described in Materials and Methods, then treated for 2 min with 625 μg/ml of heparin (Heparin-Stripped). Control complexes were mock-incubated in the absence of heparin. After bead purification and washing, the ternary complexes were incubated for 30 min on ice in 20 mm Tris-HCl (pH 8.0), 2 mm DTT, with no additional protein, or with 25 ng of the phosphocellulose fraction of wild-type NPH-I or K61A. The G21 complexes were then walked to A78 during a 5-min chase at 30°C in the presence of 6 mm MgCl2 and 1 mm each of 3′ dATP, GTP, CTP, and UTP. The mixtures were then supplemented with 200 fmoles of recombinant VTF/CE (Luo et al. 1995) and incubated for an additional 5 min at 30°C. The bead-bound A78 RNA (lane B) was separated from released A78 RNA (lane F; free) by microcentrifugation of the reaction mixtures. Nucleic acid was then recovered from the pellet and supernatant fractions by phenol extraction and ethanol precipitation. The transcription products were analyzed by electrophoresis through a 17% polyacrylamide gel containing 7 m urea in TBE (90 mm Tris, 90 mm borate, and 2.5 mm EDTA). The labeled A78 transcript was visualized by autoradiography. The percent of RNA released (indicated below the autoradiograph) was quantitated by scanning the gel with a PhosphorImager.
Recombinant NPH-I restores termination competence to heparin-stripped elongation complexes
To test the role of NPH-I in transcript release, the heparin-stripped G21 ternary complexes were incubated with purified recombinant NPH-I in the absence of NTPs and magnesium. The complexes were then walked to A78 and challenged with VTF/CE. Provision of NPH-I fully restored the ability of the heparin-stripped complexes to release their A78 transcripts in response to VTF/CE, that is, 80% of the RNA was recovered in the supernatant fraction (Fig. 4). This result argues that NPH-I is the termination factor X described previously.
ATP hydrolysis by NPH-I is essential for transcript release
We expressed and purified a mutated version of NPH-I in which the lysine residue (K61) of the presumptive ATP-binding site GxGKT was substituted by alanine (Fig. 2A). The K61A protein was unable to hydrolyze ATP under conditions in which wild-type NPH-I catalyzed quantitative conversion of ATP to ADP (Fig. 2B). By extending the protein titration to even higher levels (Fig. 5A), we estimated that the specific activity of K61A was <10−4 of that of wild-type NPH-I. The failure to catalyze ATP hydrolysis was not caused by defective interaction of K61A with the DNA cofactor, insofar as wild-type NPH-I and K61A were both able to bind to a 40-mer DNA oligonucleotide to form a discrete protein–DNA complex that was resolved from free DNA by native gel electrophoresis (Fig. 2B). The protein concentration dependence of protein–DNA formation was essentially identical for wild-type NPH-I and K61A (Fig. 2B). The salient observation was that supplementation of heparin-stripped G21 complexes with K61A protein, followed by walk to A78, failed to restore VTF-responsiveness in transcript release (Fig. 4). Here, K61A-supplemented A78 complexes released only 4% of the RNA, compared with 80% release by complexes receiving wild-type NPH-I. We surmise that transcript release requires ATP hydrolysis by NPH-I.
Figure 5.

Effects of DExH box mutations on ATPase and transcription release. NPH-I mutants D141A and E142A were expressed in bacteria and purified from soluble lysates by Ni-agarose and phosphocellulose chromatography. The purity of the phosphocellulose preparations was equivalent to that of wild-type and K61A (data not shown). (A) ATPase assays were performed as described in Materials and Methods. Activity (nmoles Pi release/30 min) is plotted as a function of the input D141A (○) and E142A (□) phosphocellulose enzyme preparations. The K61A protein (▵) was assayed in parallel. (B) Transcript release. Assays were performed as described in the legend to Fig. 4. Purified heparin-stripped G21 complexes were incubated with 5 ng of the phosphocellulose preparations of wild-type NPH-I, D141A, or E142A, then walked to A78 and challenged with 200 fmoles of recombinant VTF/CE. Bead-bound and released RNAs were separated by centrifugation. The bound and free samples were deproteinized, ethanol-precipitated, and analyzed by denaturing gel electrophoresis. The percent of A78 RNA released is plotted in bar graph format.
This conclusion was underscored by further studies of the effects of alanine substitutions at Asp-141 and Glu-142 within the signature DExH box of NPH-I. The specific ATPase activities of purified recombinant D141A and E142A proteins were 0.1% and 0.02% of the wild-type activity, respectively (Fig. 5A). The catalytically defective D141A and E142A were incapable of complementing the transcript release defect of heparin-stripped elongation complexes (Fig. 5B).
NPH-I is an elongation factor
The G21(TER) transcription unit contains a 20-nucleotide G-less cassette and a run of nine Ts downstream (positions +33 to +41) (Fig. 6). Template-engaged ternary complexes containing pulse-labeled 3′OMeGMP-arrested 21-mer RNA were bead-purified (Fig. 6, lane 1). [Note that the minor 22-, 23-, and 24-mer RNA species labeled during the pulse transcription reaction are 3′ co-terminal nascent transcripts that initiated from template positions upstream of +1 (Hagler and Shuman 1992)]. The G21 complexes were then chased for 10 min in the presence of 1 mm ATP, 1 mm GTP, 1 mm CTP, and 10 μm UTP. With UTP limitation, the elongating polymerase pauses for a sustained interval at the 5′ margin of the T-run before traversing the T-run and elongating to the end of the linear template (Deng and Shuman 1997b). A cluster of paused RNAs (P) and the 195-nucleotide runoff transcript (RT) were observed when control G21 complexes were chased (Fig. 6, lane 2). Heparin-stripped G21 ternary complexes were prepared by adjusting the reaction mixture to 625 μg/ml of heparin after pulse-labeling and before recovery of the beads (Fig. 6, lane 3). This was followed by extensive washing of the beads to remove the heparin. When the heparin-stripped complexes were chased at limiting UTP concentration, intrinsic pausing at the T-run was enhanced and few polymerases reached the end of the template (Fig. 6, lane 4). Addition of purified recombinant NPH-I to the heparin-stripped G21 complexes before the chase restored readthrough of the T-run to the level seen in control reactions and elicited a concomitant increase in the amount of full-length runoff transcripts (Fig. 6, lanes 5–8). The K61A mutant was unable to restore readthrough of the T-run (data not shown), implying that the salutary effects of NPH-I on elongation through intrinsic pause sites requires ATP hydrolysis. This is consistent with earlier findings that intrinsic pausing in the T-run is exacerbated when AMPPNP substitutes for ATP during the chase (Hagler et al. 1994).
Figure 6.
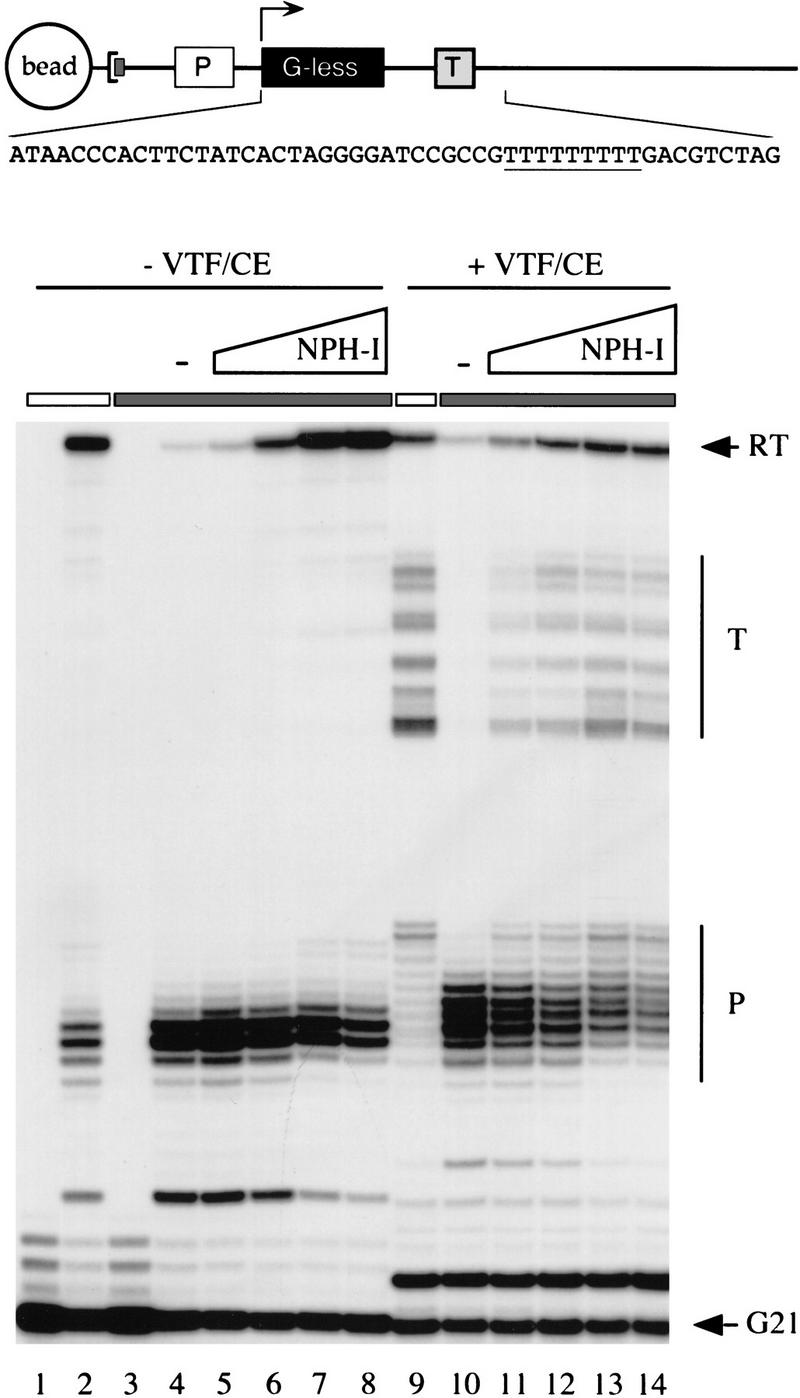
NPH-I suppresses intrinsic pausing and restores termination competence to heparin-stripped complexes. The architecture of the G21(TER) DNA template is shown in cartoon form. The DNA contains a biotinylated nucleotide incorporated uniquely at the 3′ end of the template DNA strand, which anchors the DNA to a streptavidin-coated magnetic bead. A vaccinia virus early promoter element specifies the initiation of transcription at position +1 of a 20-nucleotide G-less cassette. A termination signal (TTTTTTTTT) is situated downstream of the G-less cassette. The sequence of the first 50 nucleotides of the transcribed region (non-template strand) is shown. Pulse-labeled G21 ternary complexes assembled on the G21(TER) template were treated with 625 μg/ml of heparin (lanes 3–8 and 10–14, denoted by the shaded horizontal bar over the lanes) or with buffer (lanes 1, 2, and 9, denoted by an open bar over the lanes) before bead purification and washing. The purified complexes were incubated for 30 min on ice with increasing amounts of the peak glycerol gradient fraction (no. 20) of wild-type NPH-I as follows: 0.4 U (lanes 5,11); 1.6 U (lanes 6,12); 6.3 U (lanes 7,13); 25 U (lanes 8,14). NPH-I was omitted from control reactions (lanes 4,10, indicated by −). Pulse-labeled RNAs were isolated from samples of heparin-stripped (lane 3) and untreated (lane 1) ternary complexes. The remaining samples were chased in 20-μl elongation reaction mixtures containing 20 mm Tris-HCl (pH 8.0), 6 mm MgCl2, 2 mm DTT, and 1 mm ATP, 1 mm GTP, 1 mm CTP, and 10 μm UTP. Where indicated (lanes 9–14), the mixtures were supplemented with 500 fmoles of recombinant VTF/CE just before the addition of NTPs. After incubation for 10 min at 30°C, the elongation reactions were quenched by addition of buffer containing SDS and urea. The samples were extracted with phenol:chloroform and labeled RNA was recovered by ethanol precipitation. Transcription products were analyzed by electrophoresis through a 17% denaturing polyacrylamide gel and radiolabeled transcripts were visualized by autoradiography. The positions of pulse-labeled G21 RNA and the runoff transcript (RT) are indicated by arrows. The terminated (T) and paused (P) RNAs are indicated by vertical bars.
The addition of VTF/CE to the bead-purified control transcription complexes just before the start of the NTP chase resulted in the appearance of a heterogeneous array of transcripts terminated downstream of the termination signal (Fig. 6, lane 9; T). The heparin-stripped complexes formed no terminated RNAs, because most of the polymerases did not elongate through the T-run at low UTP concentration (Fig. 6, lane 10). When the heparin-stripped complexes were supplemented with recombinant NPH-I, however, they responded to VTF/CE and formed a cluster of terminated transcripts during the chase (Fig. 6, lanes 11–14). The size distribution of terminated RNAs made by heparin-stripped complexes reconstituted with NPH-I was identical to that of control complexes that had not been treated with heparin. [Note that some of the pulse-labeled G21 RNAs were not extended during the chase; these correspond to RNA chains that were released from the transcription complexes during the post-isolation incubation (data not shown). The released G21 RNAs are capped by VTF/CE to form a novel labeled species that migrated just above the 21-mer (Fig. 6, lanes 9–14). No such RNA was formed in the absence of added CE (Fig. 6, lanes 4–8).]
When the chase reactions on the G21(TER) template were performed at high UTP concentration (1 mm), the majority of the heparin-stripped complexes traversed the T-run and elongated to the end of the template during the 10-min reaction. Incubation of the stripped G21 complexes with NPH-I alone or VTF/CE alone did not elicit termination; however, termination was restored when both proteins were provided (data not shown). Terminated RNAs were not formed when the stripped G21 complexes were incubated with the K61A mutant and then chased in the presence of VTF/CE (data not shown). We surmise that NPH-I acts in two capacities—as a termination factor in concert with VTF/CE and as an elongation factor per se.
Discussion
The principal findings of this study are (1) transient exposure of ternary complexes to heparin renders vaccinia RNA polymerase unresponsive to VTF/CE-mediated transcript release; (2) heparin-stripped complexes can be restored to VTF/CE responsiveness by addition of purified recombinant NPH-I; (3) NPH-I facilitates elongation through intrinsic pause sites; and (4) ATP hydrolysis by NPH-I is essential for its function in transcription termination and elongation. We conclude that NPH-I is the energy-coupling factor that accounts for the ATP requirement for vaccinia virus transcription termination. The present results, together with previous work on ternary complex structure and function, suggest a plausible model of the macromolecular interactions during transcription termination, which is illustrated in Figure 7. We now know that the VTF-responsive transcription elongation complex consists of the core RNA polymerase and NPH-I. We showed previously by UV cross-linking of purified elongation complexes that the 32P-labeled nascent RNA contacts one or both of the largest polymerase subunits (rpo147, rpo135) and the 95-kD subunit of VTF/CE (Hagler et al. 1994). There was no evidence in that study of RNA cross-linking to a polypeptide the size of NPH-I. Therefore, we posit that the RNA signal recognition step during termination is accomplished by VTF/CE. We suggest that VTF/CE is poised to scan the sequence of the RNA as it is extruded from the RNA-binding pocket of the polymerase. Encounter of VTF/CE with the UUUUUNU signal would activate ATP hydrolysis by NPH-I and effect the release of the nascent RNA from the polymerase. According to this view, NPH-I would be the immediate target of RNA signaling by VTF and the energy of ATP hydrolysis would be used to disrupt contacts between the nascent RNA and the polymerase or interactions between nascent RNA and the template DNA strand. Although the molecular details of this phase are not yet defined, there are mechanistic parallels between the vaccinia termination reaction and ρ-mediated termination by E. coli RNA polymerase (Richardson 1993, 1996).
Figure 7.

A model of the pretermination complex.
Fundamental similarities between the two termination systems include (1) recognition of a cis-acting termination signal in the nascent RNA; (2) 3′ end formation at heterogeneous sites downstream of the signal; (3) recognition of the RNA signal in a context largely devoid of secondary structure (Luo and Shuman 1991); (4) requirement for NTP hydrolysis; (5) kinetic coupling between the rate of polymerase elongation and the rate of action of the termination factor; and (6) the involvement of two protein components in the termination reaction—these being NPH-I and VTF/CE for vaccinia RNA polymerase and ρ and NusG for E. coli RNA polymerase. NusG is required for the function of many ρ-dependent terminators in vivo (Sullivan and Gottesman 1992). Termination in vitro is enhanced by NusG at some ρ-dependent terminators, but is strongly NusG-dependent at others (Burns and Richardson 1995). NusG has been reported to interact physically with RNA polymerase and with ρ (Li et al. 1992, 1993). It has been proposed that NusG binding to the elongation complex is stabilized by RNA-bound ρ and that NusG, in turn, stabilizes ρ interaction with the elongation complex (Nehrke and Platt 1994).
Vaccinia NPH-I is tightly associated with the elongating polymerase in the absence of the second component of the termination system, yet it may, like NusG, serve as a physical or functional bridge between the RNA polymerase and the RNA-bound termination factor. A key distinction between the two systems is that RNA signal recognition and NTP hydrolysis are performed by the same protein in bacteria (ρ), whereas those functions appear to be partitioned between VTF/CE and NPH-I in the vaccinia transcription apparatus. It is remarkable that NPH-I and NusG both have second functions as transcription elongation factors. NusG and NPH-I stimulate transcription elongation by suppressing intrinsic pausing by their cognate RNA polymerases (this study; Burns and Richardson 1995; Burova et al. 1995).
The finding that ρ is an RNA–DNA helicase (Brennan et al. 1987) provides an appealing conceptual framework for how energy use might be coupled to termination signal transduction. It is postulated that ρ binds to its attachment site on nascent RNA, and is activated with respect to NTPase, which then drives translocation or tracking of ρ in a 5′ → 3′ direction along the RNA strand. Termination and transcript release occur when ρ “catches up” with the ternary complex. Whether RNA release occurs because Rho actually disrupts an RNA–DNA hybrid or because it destabilizes contacts between RNA and the polymerase remains unclear.
NPH-I is unlikely to be an RNA-tracking helicase in the manner of ρ, because ATP hydrolysis by NPH-I depends on binding to single-stranded DNA. Single-stranded RNAs, including poly (U), which should mimic a vaccinia termination signal, are incapable of activating its ATPase function (Paoletti and Moss 1974). Fully duplex DNA is also unable to activate NPH-I (Paoletti and Moss 1974). (This makes sense because NPH-I activation by the duplex DNA template would deplete the available ATP pool.) We suggest that the ATPase is activated during elongation and termination through interactions of NPH-I with single-stranded DNA in the transcription bubble. Chemical probing of DNA within the G21 ternary complex shows that the transcription bubble originates 13 nucleotides 5′ of the end of the RNA chain and extends 4 nucleotides 3′ of the catalytic template position (Hagler and Shuman 1992a). We posit that access of NPH-I to the bubble is normally restricted and that activation of the ATPase is triggered by an alteration in the conformation or size of the bubble. This might occur as polymerase encounters the T-run (an intrinsic pause element), which would cause NPH-I to use the energy of ATP hydrolysis to correct the conformational distortion and drive the polymerase forward. VTF/CE, when bound to UUUUUNU, might elicit a unique conformational state that on activation of NPH-I to hydrolyze ATP, results in transcript release. It is tempting to invoke directional tracking by NPH-I along one of the DNA strands of the bubble as a unifying mechanism for its elongation factor and termination factor activities. Were it to track 3′ → 5′ along the template strand, then destabilization of an RNA-DNA hybrid becomes a possibility. Although comparisons with ρ engender enthusiasm for this idea, we have been unable to detect any type of helicase activity by recombinant NPH-I using an array of DNA–DNA duplexes, RNA–RNA duplexes, and RNA–DNA hybrids (C. Gross and S. Shuman, unpubl.). A combination of recombinant NPH-I and recombinant VTF/CE was also devoid of helicase activity. We infer that either the energy of ATP hydrolysis is not used to disrupt a nucleic acid duplex or that a potential helicase function of NPH-I depends on a specific feature of the transcription elongation complex.
Ironically, the nucleic acid-dependent ATPase activities of ρ and NPH-I were reported simultaneously in 1974 (Lowery-Goldhammer and Richardson 1974; Paoletti et al. 1974), but only now can we assign an analogous role to NPH-I in mRNA synthesis. NPH-I and ρ belong to divergent ATPase superfamilies. ρ is structurally related to F1–ATPase (Richardson 1996). NPH-I is one of the two founding members of the DExH family of nucleic acid-dependent NTPases that were purified from vaccinia virions and characterized by Moss and colleagues (Paoletti et al. 1974; Paoletti and Moss 1974; Broyles and Moss 1987). The ever-expanding DExH family includes NTPases involved in DNA repair, recombination, pre-mRNA splicing, and transcription. DExH proteins are components of the transcription factors ETF and TFIIH that are responsible for the requirement for ATP hydrolysis during transcription initiation by vaccinia RNA polymerase and cellular Pol II, respectively (Li and Broyles 1993; Orphanides et al. 1996). TFIIH has been reported recently to facilitate Pol II elongation by suppression of an elongation block at promoter-proximal positions (Dvir et al. 1997). TFIIH contains two distinct DExH-box subunits. Conceivably, the ATP-dependent steps in initiation and early elongation may be catalyzed by different subunits within TFIIH, just as ATP-dependent initiation and elongation steps in vaccinia transcription are catalyzed by ETF and NPH-I. The DExH-box subunit of vaccinia ETF is highly homologous to NPH-I and it appears likely that the two viral proteins diverged after duplication of an ancestral viral gene.
NPH-I is the first DExH protein shown to have a role in transcription termination. We suspect it will not be the last. Xie and Price (1996) have isolated a Drosophila factor that promotes the release of nascent transcripts from Pol II elongation complexes in an ATP-dependent manner.
Henikoff (1993) has proposed that NPH-I is the prototype of a distinct subfamily of DExH proteins that includes Snf2 and its numerous homologs. Snf2-like proteins are implicated in transcriptional activation and repression and in chromatin remodeling (Pazin and Kadonaga 1997). As with NPH-I, most Snf2-like proteins do not score positively when assayed for helicase activity in vitro. This has precipitated suggestions that the “non-helicase” class of DExH-box proteins may use the energy of NTP hydrolysis to disrupt protein–DNA interactions (Pazin and Kadonaga 1997). NPH-I disrupts the RNA polymerase ternary complex in a signal-dependent fashion, but can also stimulate elongation. It may be worthwhile to consider that the positive and negative effects of some Snf2-like proteins on gene expression may be mediated at post-initiation steps of the transcription cycle.
Materials and methods
NPH-I expression and purification
The NPH-I-coding sequence (gene D11) was PCR-amplified from a plasmid containing the vaccinia HindIII D fragment using a primers designed to introduce an NdeI site at the start codon and a BglII site 3′ of the stop codon. The PCR product was digested with NdeI and BglII and then inserted between the NdeI and BamHI sites of plasmid pET16b (Novagen) to generate pET–His–NPH-I. In this context, the NPH-I coding sequence is fused to an amino-terminal leader peptide containing 10 tandem histidines. The entire NPH-I insert was sequenced to confirm that the plasmid encoded a wild-type protein. Alanine substitution mutations K61A, D141A, and E142A were introduced into the NPH-I gene by PCR using the two-stage overlap extension method. The mutated DNA products of the second stage amplification were digested with NdeI and BamHI and then inserted into pET–His–NPH-I in lieu of the corresponding wild-type restriction fragments. This generated plasmids pET–His–K61A, pET–His–D141A, and pET–His–E142A. The presence of the desired mutations was confirmed by DNA sequencing; the segment corresponding to the inserted restriction fragment was sequenced completely to exclude the introduction of unwanted mutations during amplification and cloning. The expression plasmids were transformed into E. coli BL21(DE3). Transformants were inoculated into Luria–Bertani medium containing 0.1 mg/ml of ampicillin and grown at 37°C until the A600 reached ∼0.6. Cultures (200 ml) were then placed on ice for 30 min, adjusted to 2% ethanol, and subsequently incubated at 18°C for 48 hr with continuous shaking. Cells were harvested by centrifugation and the pellets were stored at −80°C. All subsequent procedures were performed at 4°C. The cells were thawed and resuspended in 20 ml of lysis buffer [50 mm Tris-HCl (pH 7.5), 0.15 m NaCl, and 10% sucrose) containing 0.2 mg/ml of lysozyme. After 30 min on ice, the suspensions were adjusted to 0.1% Triton X-100. The lysates were sonicated to reduce viscosity. Insoluble material was removed by centrifugation for 40 min at 18,000 rpm in a Sorvall SS-34 rotor. The supernatants were mixed with 1 ml of Ni-nitrilotriacetic acid-agarose resin (Qiagen) for 1 hr. The slurries were poured into a column and washed with lysis buffer. The columns were eluted serially with IMAC buffer [20 mm Tris-HCl (pH 7.9), 0.5 m NaCl, 1 mm PMSF, and 10% glycerol) containing 5, 25, 50, 200, and 1000 mm imidazole. The polypeptide composition of the column fractions was monitored by SDS-PAGE. Fractions enriched for the expressed NPH-I polypeptide (which eluted at 200 mm imidazole) were pooled and dialyzed against buffer A [50 mm Tris-HCl (pH 8.0), 2 mm EDTA, 2 mm dithiothreitol, 0.1% Triton X-100, and 10% glycerol]). The dialysate was applied to a 1-ml column of phosphocellulose that had been equilibrated with buffer A. The column was eluted stepwise with buffer A containing 50, 100, 200, 400, and 1000 mm NaCl. The NPH-I protein was recovered in the 0.2 m NaCl eluate fraction. The protein concentrations of the phosphocellulose fractions were determined by using the Bio-Rad dye reagent with bovine serum albumin as a standard. An aliquot of the phosphocellulose fraction of wild-type NPH-I was applied to a 4.8-ml 15%–35% glycerol gradient containing 0.2 m NaCl in buffer A. The gradient was centrifuged for 16 hr at 48,000 rpm in a Beckman SW50 rotor. Fractions (0.15 ml) were collected from the bottom of the tube. Marker proteins (catalase, BSA, and cytochrome c) were centrifuged in a parallel gradient.
ATPase assay
ATPase activity was determined by the release of 32Pi from [γ-32P]ATP in the presence of a DNA cofactor. Reaction mixtures (10 μl) containing 40 mm Tris-HCl (pH 8.0), 1 mm MgCl2, 2 mm DTT, 1 mm [γ-32P]ATP, 1 μg of denatured salmon sperm DNA, and NPH-I as specified were incubated for 30 min at 37°C. An aliquot (2 μl) of the reaction mixture was applied to a polyethyleneimine cellulose TLC plate that was developed in 0.5 m LiCl, 1 m formic acid. [γ-32P]ATP and 32Pi were quantitated by scanning the TLC plate with a FUJIX BAS1000 Bio-Imaging Analyzer. One unit (U) of NPH-I hydrolyzed 1 nmole of ATP in 30 min.
Immobilized DNA templates
The G21(TER29)A78 and G21(TER) plasmids contains a vaccinia early promoter fused to a 20-nucleotide G-less cassette (Figs. 4 and 6). G21(TER29)A78 and G21(TER) were linearized with Acc65-1, which cleaved the DNA templates upstream of the vaccinia early promoter region. Biotinylated dAMP was incorporated at the 3′ ends using Klenow DNA polymerase. The biotinylated G21(TER29)A78 and G21(TER) DNAs were then digested with NdeI and PvuII, respectively, and the restriction fragment containing the transcription cassette was isolated by preparative agarose gel electrophoresis. Purified DNA fragments were attached to streptavidin-coated magnetic beads (Dynabeads M280 purchased from Dynal) as described (Hagler et al. 1994).
Transcription in vitro
Ternary transcription complexes were formed as described (Hagler et al. 1994) in reaction mixtures containing (per 20 μl) 20 mm Tris-HCl (pH 8.0), 6 mm MgCl2, 2 mm DTT, 1 mm ATP, 0.1 mm UTP, 1 μm [α32P]CTP (1000 Ci/mmole), 0.1 mm 3′-OMeGTP, vaccinia RNA polymerase (heparin agarose fraction containing ETF), and ∼100 fmoles of bead-linked DNA. Reaction mixtures were incubated at 30°C for 10 min. Heparin (from porcine intestine; molecular weight ∼6000) was added to 625 μg/ml and the samples were incubated for 2 min at 30°C. The beads were concentrated by microcentrifugation for 15 sec and then held in place with a magnet while the supernatant was removed and replaced with 0.1 ml of 20 mm Tris-HCl (pH 8.0), 2 mm DTT. The beads were resuspended and subjected to two further cycles of concentration and washing; after the third wash, the beads were resuspended in a small volume of the wash buffer, and aliquots were distributed into individual reaction tubes to achieve approximately the same concentration of template as that used in the pulse-labeling phase. Elongation reactions were performed by adding nucleoside triphosphates and magnesium as specified in the figure legends. Elongation of the nascent chains beyond the arrest site at G21 depended on removal of the blocking 3′OMeGMP moiety by a hydrolytic activity intrinsic to the vaccinia RNA polymerase elongation complex (Hagler and Shuman 1993).
Acknowledgments
This work was supported by National Institutes of Health grant GM42498.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
#E-MAIL s-shuman@sk11mskcc.org; FAX (212) 717-3623.
References
- Brennan CA, Dombroski AJ, Platt T. Transcription termination factor Rho is an RNA-DNA helicase. Cell. 1987;48:945–952. doi: 10.1016/0092-8674(87)90703-3. [DOI] [PubMed] [Google Scholar]
- Broyles SS, Moss B. Identification of the vaccinia virus gene encoding nucleoside triphosphate phosphohydrolase I, a DNA-dependent ATPase. J Virol. 1987;61:1738–1742. doi: 10.1128/jvi.61.5.1738-1742.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broyles SS, Yuen L, Shuman S, Moss B. Purification of a factor required for transcription of vaccinia virus early genes. J Biol Chem. 1988;263:10754–10760. [PubMed] [Google Scholar]
- Burns CM, Richardson JP. NusG is required to overcome a kinetic limitation to Rho function as an intragenic terminator. Proc Natl Acad Sci. 1995;92:4738–4742. doi: 10.1073/pnas.92.11.4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burova E, Hung SC, Sagitov V, Stitt BL, Gottesman ME. Escherichia coli NusG protein stimulates transcription elongation rates in vivo and in vitro. J Bacteriol. 1995;177:1388–1392. doi: 10.1128/jb.177.5.1388-1392.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Shuman S. An ATPase component of the transcription elongation complex is required for factor-dependent transcription termination by vaccinia RNA polymerase. J Biol Chem. 1996;271:29386–29392. doi: 10.1074/jbc.271.46.29386. [DOI] [PubMed] [Google Scholar]
- ————— Transcription termination by vaccinia RNA polymerase entails recognition of specific phosphates in the nascent RNA. J Biol Chem. 1997a;272:695–698. doi: 10.1074/jbc.272.1.695. [DOI] [PubMed] [Google Scholar]
- ————— Elongation properties of vaccinia virus RNA polymerase: Pausing, slippage, 3′ end addition, and termination site choice. Biochemistry. 1997b;36:15892–15899. doi: 10.1021/bi972037a. [DOI] [PubMed] [Google Scholar]
- Deng L, Hagler J, Shuman S. Factor-dependent release of nascent RNA by ternary complexes of vaccinia RNA polymerase. J Biol Chem. 1996;271:19556–19562. doi: 10.1074/jbc.271.32.19556. [DOI] [PubMed] [Google Scholar]
- Dvir A, Conaway RC, Conaway JW. A role for TFIIH in controlling the activity of early RNA polymerase II elongation complexes. Proc Natl Acad Sci. 1997;94:9006–9010. doi: 10.1073/pnas.94.17.9006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensinger MJ, Martin SA, Paoletti E, Moss B. Modification of the 5′- terminus of mRNA by soluble guanylyl and methyl transferases from vaccinia virus. Proc Natl Acad Sci. 1975;72:2525–2529. doi: 10.1073/pnas.72.7.2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershowitz A, Boone R, Moss B. Multiple roles for ATP in the synthesis and processing of mRNA by vaccinia virus: specific inhibitory effects of adenosine (β,γ imido) triphosphate. J Virol. 1978;27:399–408. doi: 10.1128/jvi.27.2.399-408.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagler J, Shuman S. Structural analysis of ternary complexes of vaccinia RNA polymerase. Proc Natl Acad Sci. 1992a;89:10099–10103. doi: 10.1073/pnas.89.21.10099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— A freeze-frame view of eukaryotic transcription during elongation and capping of nascent mRNA. Science. 1992b;255:983–986. doi: 10.1126/science.1546295. [DOI] [PubMed] [Google Scholar]
- ————— Stability of ternary transcription complexes of vaccinia RNA polymerase at promoter-proximal positions. J Biol Chem. 1992c;267:7644–7654. [PubMed] [Google Scholar]
- ————— Nascent RNA cleavage by purified ternary complexes of vaccinia RNA polymerase. J Biol Chem. 1993;268:2166–2173. [PubMed] [Google Scholar]
- Hagler J, Luo Y, Shuman S. Mechanism of factor-dependent transcription termination by vaccinia RNA polymerase: Kinetic coupling and requirement for ATP hydrolysis. J Biol Chem. 1994;269:10050–10060. [PubMed] [Google Scholar]
- Henikoff S. Transcriptional activator components and poxvirus DNA-dependent ATPases comprise a single family. Trends Biol Sci. 1993;18:291–292. doi: 10.1016/0968-0004(93)90037-n. [DOI] [PubMed] [Google Scholar]
- Kates J, Beeson J. Ribonucleic acid synthesis in vaccinia virus: Synthesis of polyribonucleic acid. J Mol Biol. 1970;50:19–33. doi: 10.1016/0022-2836(70)90101-4. [DOI] [PubMed] [Google Scholar]
- Li J, Broyles SS. The DNA-dependent ATPase activity of vaccinia virus early transcription factor is essential for its transcriptional activation function. J Biol Chem. 1993;268:20016–20021. [PubMed] [Google Scholar]
- Li J, Horwitz R, McCracken S, Greenblatt J. NusG, a new Escherichia coli elongation factor involved in transcriptional antitermination by the N protein of phage λ. J Biol Chem. 1992;267:6012–6019. [PubMed] [Google Scholar]
- Li J, Mason SW, Greenblatt J. Elongation factor NusG interacts with factor ρ to regulate termination and antitermination of transcription. Genes & Dev. 1993;7:161–172. doi: 10.1101/gad.7.1.161. [DOI] [PubMed] [Google Scholar]
- Lowery-Goldhammer C, Richardson JP. An RNA-dependent nucleoside triphosphate phosphohydrolase (ATPase) associated with Rho termination factor. Proc Natl Acad Sci. 1974;71:2003–2007. doi: 10.1073/pnas.71.5.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Shuman S. Antitermination of vaccinia virus early transcription: Possible role of RNA secondary structure. . Virology. 1991;185:432–436. doi: 10.1016/0042-6822(91)90793-b. [DOI] [PubMed] [Google Scholar]
- Luo Y, Mao X, Deng L, Cong P, Shuman S. The D1 and D12 subunits are both essential for the transcription termination factor activity of vaccinia virus capping enzyme. J Virol. 1995;69:3852–3856. doi: 10.1128/jvi.69.6.3852-3856.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B. Poxviridae: The viruses and their replication. In: Fields BN, Knipe DM, Howley PM, et al., editors. Fields virology. Philadelphia, PA: Lippincott-Raven Publishers; 1996. pp. 2637–2671. [Google Scholar]
- Nehrke KW, Platt T. A quaternary transcription termination complex: Reciprocal stabilization of Rho factor and NusG protein. J Mol Biol. 1994;243:830–839. doi: 10.1006/jmbi.1994.1685. [DOI] [PubMed] [Google Scholar]
- Orphanides G, Lagrange T, Reinberg D. The general transcription factors of RNA polymerase II. Genes & Dev. 1996;10:2657–2683. doi: 10.1101/gad.10.21.2657. [DOI] [PubMed] [Google Scholar]
- Paoletti E, Rosemond-Hornbeak H, Moss B. Two nucleic acid-dependent nucleoside triphosphate phosphohydrolases from vaccinia virus: purification and characterization. J Biol Chem. 1974;249:3273–3280. [PubMed] [Google Scholar]
- Paoletti E, Moss B. Two nucleic acid-dependent nucleoside triphosphate phosphohydrolases from vaccinia virus: nucleotide substrate and polynucleotide cofactor specificities. J Biol Chem. 1974;249:3281–3286. [PubMed] [Google Scholar]
- Pazin MJ, Kadonaga JT. SWI2/SNF2 and related proteins: ATP-driven motors that disrupt protein-DNA interactions. Cell. 1997;88:737–740. doi: 10.1016/s0092-8674(00)81918-2. [DOI] [PubMed] [Google Scholar]
- Richardson JP. Transcription termination. Crit Rev Biochem Mol Biol. 1993;28:1–30. doi: 10.3109/10409239309082571. [DOI] [PubMed] [Google Scholar]
- ————— Structural organization of transcription termination factor Rho. J Biol Chem. 1996;271:1251–1254. doi: 10.1074/jbc.271.3.1251. [DOI] [PubMed] [Google Scholar]
- Shuman S. Capping enzyme in eukaryotic mRNA synthesis. Prog Nucleic Acid Res Mol Biol. 1995;50:101–129. doi: 10.1016/s0079-6603(08)60812-0. [DOI] [PubMed] [Google Scholar]
- Shuman S, Moss B. Factor-dependent transcription termination by vaccinia RNA polymerase: Evidence for a cis-acting termination signal in nascent RNA. J Biol Chem. 1988;263:6220–6225. [PubMed] [Google Scholar]
- Shuman S, Broyles S, Moss B. Purification and characterization of a transcription termination factor from vaccinia virions. J Biol Chem. 1987;262:12372–12380. [PubMed] [Google Scholar]
- Sullivan SL, Gottesman ME. Requirement for E. coli NusG protein in factor-dependent transcription termination. Cell. 1992;68:989–994. doi: 10.1016/0092-8674(92)90041-a. [DOI] [PubMed] [Google Scholar]
- Wei C, Moss B. Methylated nucleotides block 5′ terminus of vaccinia virus messenger RNA. Proc Natl Acid Sci. 1975;72:318–322. doi: 10.1073/pnas.72.1.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Price DH. Purification of an RNA polymerase II transcript release factor from Drosophila. J Biol Chem. 1996;271:11043–11046. doi: 10.1074/jbc.271.19.11043. [DOI] [PubMed] [Google Scholar]
- Yu L, Martins A, Deng L, Shuman S. Structure-function analysis of the triphosphatase component of vaccinia virus mRNA capping enzyme. J Virol. 1997;71:9837–9843. doi: 10.1128/jvi.71.12.9837-9843.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]