

Abstract
Msn2p and the partially redundant factor Msn4p are key regulators of stress-responsive gene expression in Saccharomyces cerevisiae. They are required for the transcription of a number of genes coding for proteins with stress-protective functions. Both Msn2p and Msn4p are Cys2His2 zinc finger proteins and bind to the stress response element (STRE). In vivo footprinting studies show that the occupation of STREs is enhanced in stressed cells and dependent on the presence of Msn2p and Msn4p. Both factors accumulate in the nucleus under stress conditions, such as heat shock, osmotic stress, carbon-source starvation, and in the presence of ethanol or sorbate. Stress-induced nuclear localization was found to be rapid, reversible, and independent of protein synthesis. Nuclear localization of Msn2p and Msn4p was shown to be correlated inversely to cAMP levels and protein kinase A (PKA) activity. A region with significant homologies shared between Msn2p and Msn4p is sufficient to confer stress-regulated localization to a SV40–NLS–GFP fusion protein. Serine to alanine or aspartate substitutions in a conserved PKA consensus site abolished cAMP-driven nuclear export and cytoplasmic localization in unstressed cells. We propose stress and cAMP-regulated intracellular localization of Msn2p to be a key step in STRE-dependent transcription and in the general stress response.
Keywords: Stress, yeast, STRE, nuclear localization, protein kinase A, Msn2p, Msn4p
In all living organisms, appropriate reactions to unfavorable environmental conditions (stress factors) are observed. When transcription in eukaryotic cells is controlled by extracellular signals, at least one signaling component has to be translocated from the cytoplasm to the nucleus in a signal-dependent manner. The signaling components may be of low molecular weight (second messengers) or protein members of the signaling cascades. Examples for the latter are MAP kinases, for example, the p42 MAP kinase and p44 ERK1 on mitogenic stimulation (Chen et al. 1992).
Regulated nuclear translocation is also found widely at the level of transcription factors and is achieved by either cytoplasmic anchoring or activation of nuclear localization signals (NLS) by unmasking or modification (Jans 1995; Görlich and Mattaj 1996; Nigg 1997). A prominent example of this type of control is provided by NF-κB. A rapid transcriptional response to a variety of stress stimuli is elicited by phosphorylation of the inhibitory factor IκB followed by its dissociation from the transcription factor and destruction. This leads to unmasking of the NF-κB nuclear localization signal and to the subsequent translocation to the nucleus (Siebenlist, et al. 1995; Baldwin 1996). Another example is NF-ATc, a transcription factor involved in early immune responses that is translocated to the nucleus on dephosphorylation by Ca2+-calcineurin and exported on phosphorylation by GSK-3 (Beals et al. 1997). In Saccharomyces cerevisiae, the transcriptional regulator Yap1p is translocated into the nucleus in response to oxidative stress. This translocation event is regulated by the carboxy-terminal cystein-rich domain (Kuge et al. 1997). Pho4p activity in response to phosphate starvation has been shown to be regulated negatively at the level of cellular localization by phosphorylation through a cyclin-CDK complex (O’Neill et al. 1996). Phosphorylation may also be important for the fast and reversible glucose-regulated nuclear import of the transcriptional repressor Mig1p (DeVit et al. 1997).
In S. cerevisiae, exposure to stress initiates expression of genes encoding proteins with stress-protective functions. Transcriptional control by multiple stress conditions is mediated by the stress reponse element (STRE; core consensus 5′-WAGGGG-3′) (Ruis and Schüller 1995; Siderius and Mager 1997). STRE-driven transcription is regulated by the high-osmolarity glycerol (HOG) pathway (Schüller et al. 1994), a MAP kinase pathway of the yeast S. cerevisiae involved in protection of cells against increases in external osmolarity (Brewster et al. 1993). S. cerevisiae protein kinase A (PKA), which has been implicated in the coordination of several essential cellular events like cell growth (Baroni et al. 1992), entry into cell division (Baroni et al. 1994; Tokiwa et al. 1994), and reprogramming of transcription during the switch from fermentable to nonfermentable carbon sources (Boy-Marcotte et al. 1996) acts as a powerful repressor of STRE-mediated transcription. It appears to provide a link between positive control of cell growth and negative control of stress responses (Ruis 1997).
The transcription factors Msn2p and Msn4p recognize and bind STREs (Estruch and Carlson 1993; Martinez-Pastor et al. 1996; Schmitt and McEntee 1996). The Cys2His2 Zn-finger DNA-binding domains of these proteins are almost identical and both bind specifically to STRE sequences. These proteins appear to be functionally redundant, as double but not single mutants exhibit pleiotropic stress sensitivity. Msn2p seems to have a more pronounced role, as mutants lacking only MSN2 exhibit an already distinct decrease in STRE-mediated transcription (C. Schüller and G. Marchler, unpubl.). Full stress-induced expression of STRE-regulated genes is dependent on the presence of both Msn2p and Msn4p.
A central issue for our understanding of the general stress-response system is the question of how the response to multiple stresses and to PKA activity can be mediated by only one type of transcription factor. We found that Msn2p and Msn4p are translocated reversibly to the nucleus in a stress-dependent manner. This suggests that Msn2p and Msn4p translocation is the key regulatory event in stress induction of transcription. We show that high PKA reverses the nuclear localization of Msn2p under stress conditions. Our results provide a model of how stress and nutrient controls might be integrated in S. cerevisiae.
Results
Msn2p binds to STREs in stressed cells
Msn2p and Msn4p have been shown previously to bind to STREs in vitro (Martinez-Pastor et al. 1996; Schmitt and McEntee 1996). In initial experiments, we observed no significant changes of the STRE-binding activity of myc-tagged Msn2p, comparing extracts prepared from unstressed and stressed cells while levels of Msn2p remained constant under these conditions (data not shown). We concluded from these results that the DNA-binding affinity of Msn2p is independent of stress conditions. Therefore, we analyzed whether changes in the in vivo occupancy of STRE sequences occur due to stress exposure. Wild-type cells and cells lacking Msn2p and Msn4p were grown logarithmically for five generations at 25°C, the culture was split, and an aliquot was exposed to heat stress (37°C) for 10 min and immediately treated with dimethylsulfate (DMS), whereas the other aliquot was DMS-treated at 25°C. Under these conditions, DMS methylates chromosomal DNA at guanine and, to a lesser extent, adenine residues. Methylated chromosomal DNA was prepared and the extent of methylation in promoters containing STREs (CTT1, HSP104) was determined by primer extension (Fig. 1). In both the CTT1 and HSP104 promoter, no significant difference between methylation levels was detectable in wild type and double mutant at 25°C. A distinct increase in protection on STREs (Fig 1A,B, arrows) was apparent in wild-type cells after a 10-min incubation at 37°C. In msn2 msn4 mutant cells, the methylation level of STRE consensus sequences remained the same.
Figure 1.
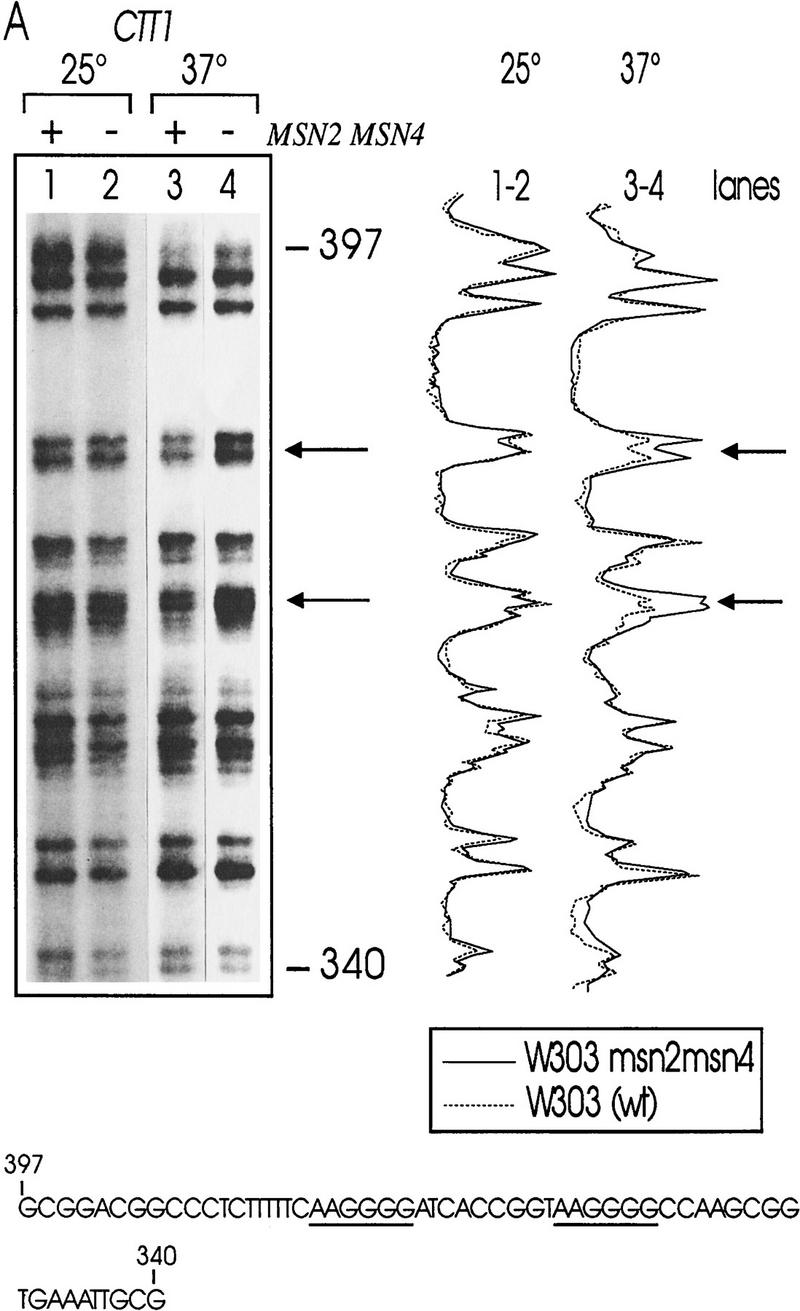
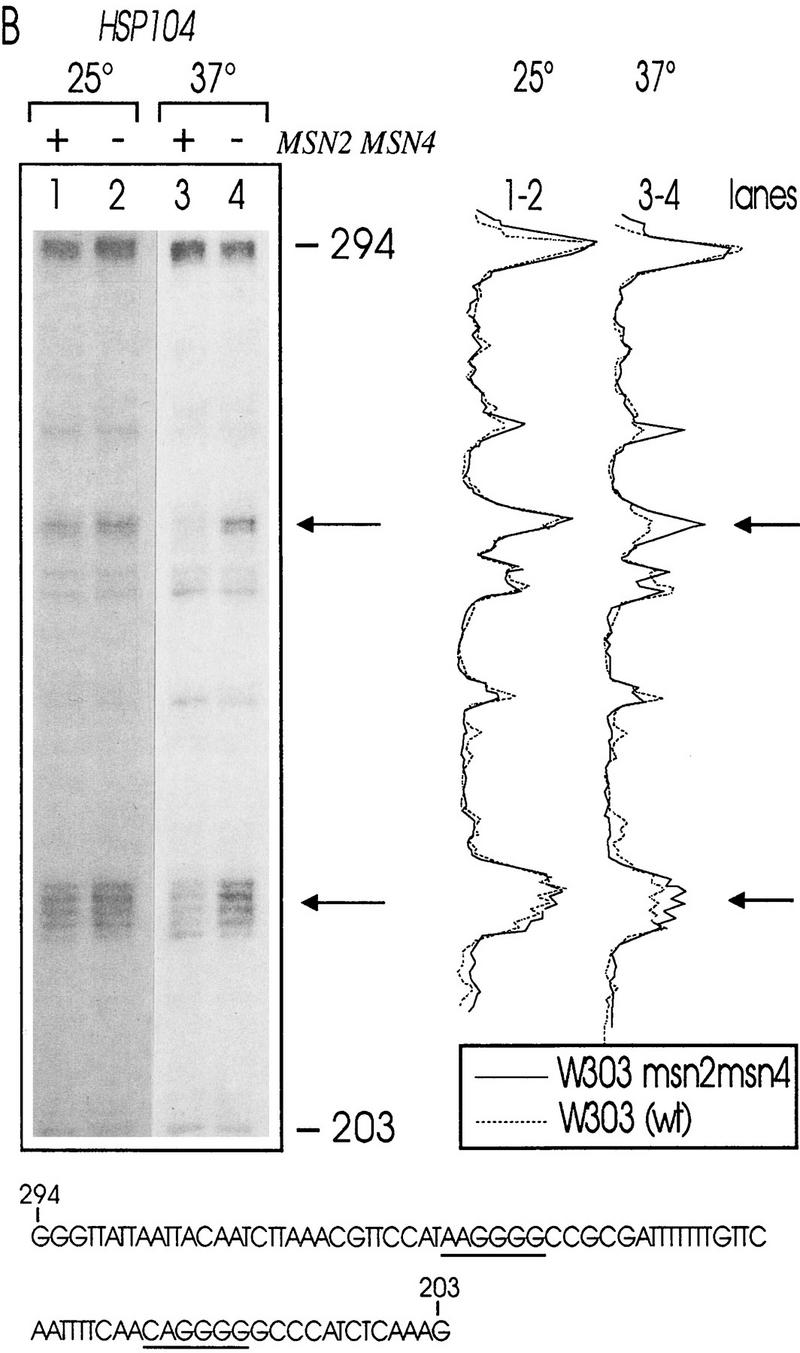
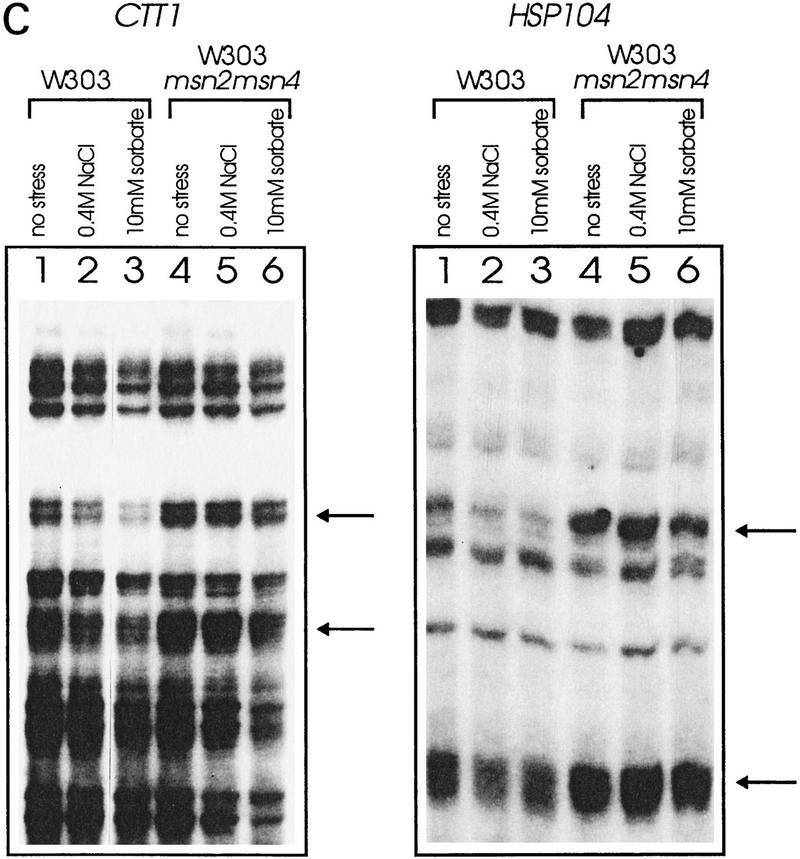
In vivo footprint analysis on the CTT1 (A) and HSP104 (B) promoters during heat shock. DNA of unstressed (25°C) and heat-shocked (37°C) W303 (lanes 1,3) and W303 msn2 msn4 (lanes 2,4) cells was prepared after methylation in vivo with DMS. Primer extension reactions were run on a sequencing gel and autoradiographed. Gels were quantitated with a PhosphorImager. The aligned curves are shown comparing the relative extent of methylation of DNA from mutant (continous lines) and wild-type (broken lines) strain under nonstress and stress conditions. Bands were identified by a conventional sequencing reaction. The STRE core sequence is indicated by arrows or underscoring. (C) In vivo footprint analysis of the CTT1 and HSP104 promoter under osmotic (0.4 m NaCl) and weak organic acid (10 mm sorbic acid) stress. W303 (lanes 1–3) and W303 msn2 msn4 (lanes 4–6) were grown to logarithmic phase and stress-treated for 20 min. Primer extension reactions were run on a sequencing gel and autoradiographed.
To investigate whether this is a common phenomenon, we repeated the experiment with other stress types known to induce STRE-dependent transcription (Schüller et al. 1994; Martinez-Pastor et al. 1996). Increased protection was induced by the food preservative sorbic acid detectable in wild-type (Fig. 1C, cf. lanes 1 and 3), but not in msn2 msn4 mutant cells (Fig. 1C, cf. lanes 4 and 6). High-osmolarity stress (0.4 m NaCl) caused a similar, but less pronounced, increase in protection in wild-type cells only (Fig. 1C, cf. lanes 1 and 2 with 4 and 5). This suggests that STREs are occupied rapidly under stress conditions in a Msn2p- and Msn4p-dependent manner.
Stress-dependent nuclear localization of Msn2p
In vivo DNA binding of Msn2p might be controlled inside the nucleus or by the regulation of nuclear entry. Therefore, we created Msn2p fusions bearing either a myc9 epitope for indirect immunolocalization (Piatti et al. 1996) or, for examination of living cells, a green fluorescent protein (GFP) tag. To verify the functionality of the carboxy-terminally tagged Msn2p derivatives, we assayed the expression of a STRE-driven gene (CTT1) in their presence. In msn2 msn4 mutant cells transformed with empty control vector, only low levels of catalase expression were observed with or without stress (Fig. 2D). Catalase activity was restored to normal and inducible levels by plasmids containing a wild-type MSN2 gene or genes encoding either the myc-tagged version or the Msn2p–GFP fusion. In all of these cases, expression of Msn2p derivatives was driven by the native promoter demonstrating that physiological levels of tagged versions of Msn2p are functional.
Figure 2.
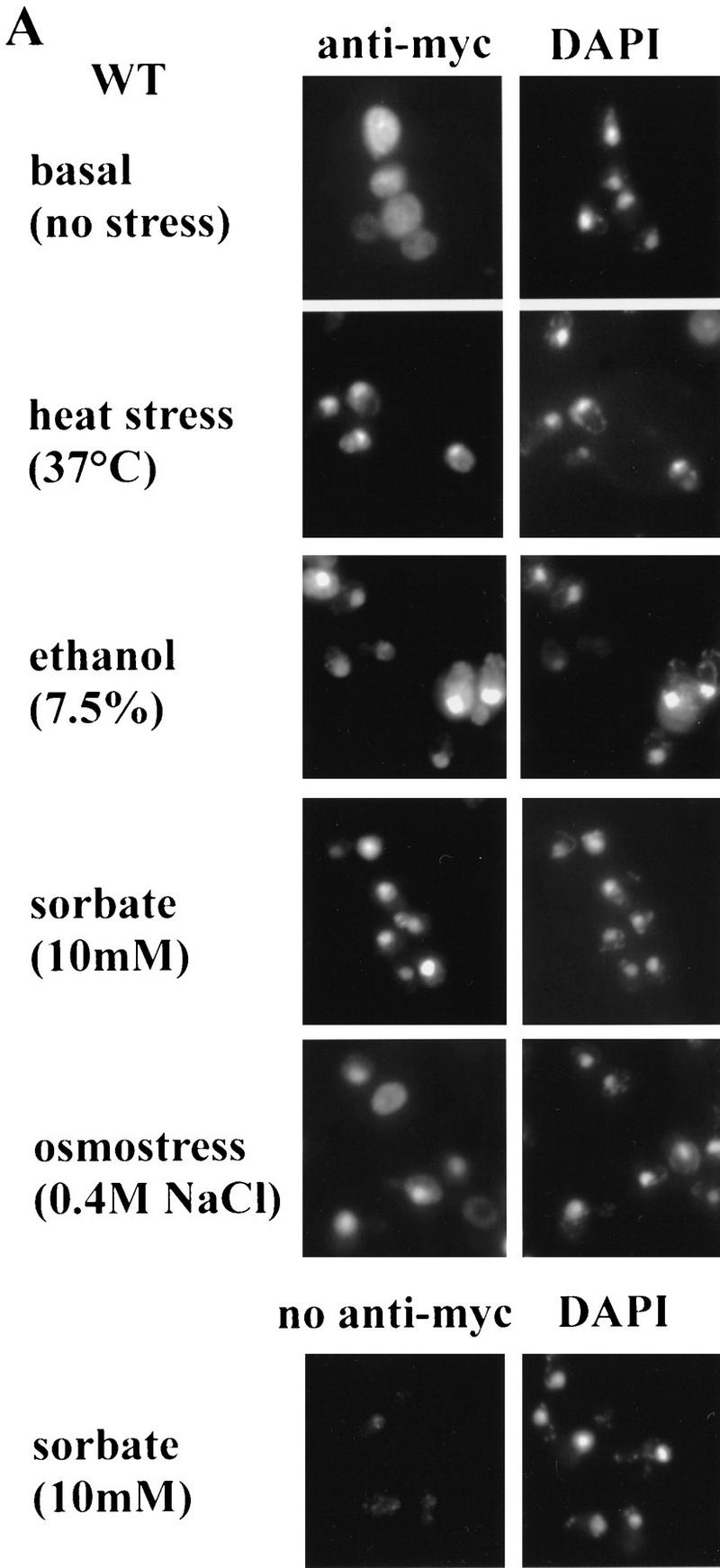


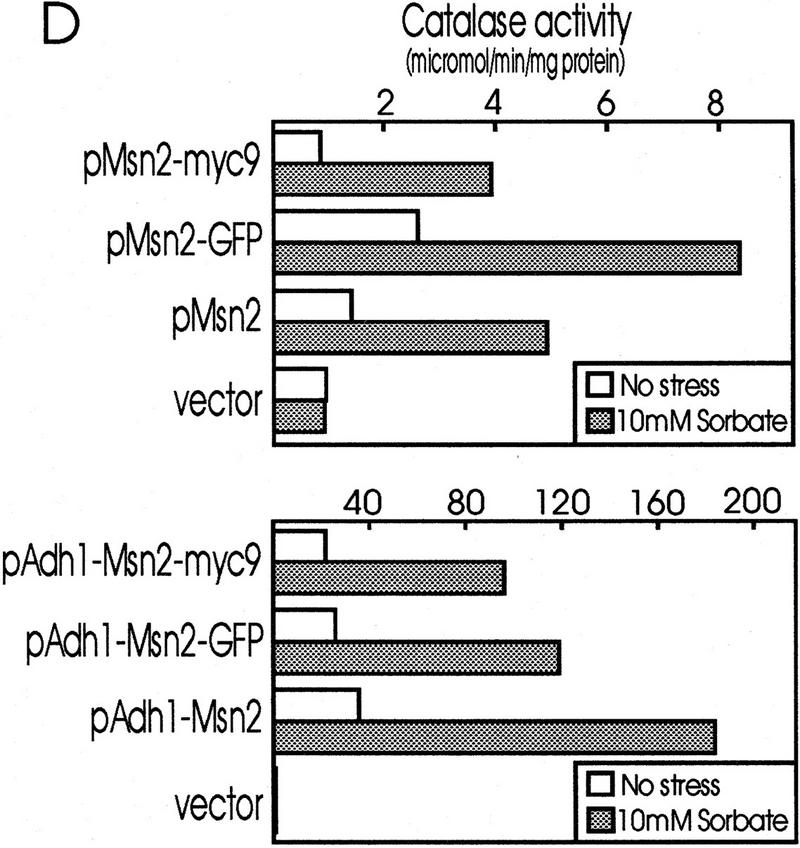
Subcellular localization of Msn2p–myc9 and Msn2p–GFP. (A) Indirect immunofluorescence. W303 msn2 msn4 cells transformed with pAdh1–Msn2 myc9 were grown to logarithmic phase, treated with stress for 20 min and fixed. Indirect immunodetection was performed using monoclonal anti-myc (9E10) antibody and rabbit anti-mouse IgG CY3-conjugated secondary antibody (Sigma). DNA was stained with DAPI. (B) Distribution of Msn2p–GFP fluorescence. Stressed and unstressed W303 msn2 msn4 and W303 hog1 cells transformed with pAdh1–Msn2–GFP were grown to logarithmic phase in synthetic medium. DAPI was added to the cultures 10 min before they were subjected to the indicated stress treatment for 5 min. In living cells, DAPI stains mitochondrial DNA brightly, whereas chromosomal DNA appears as a diffuse spot. (BF) Bright field microscopy. (C) Western blot using a polyclonal anti-Msn2p antiserum showing amounts of Msn2p–GFP under conditions used for fluorescence microscopy; (control) cells without Msn2–GFP plasmid. (D) Biological activity of Msn2p and Msn2p derivatives in W303 msn2 msn4 cells. Mutant cells were transformed with vector (YCplac111), pMsn2, pMsn2–myc9, pMsn2–GFP, pAdh1–Msn2, pAdh1–Msn2–myc9, and pAdh1–Msn2–GFP constructs under control of the native MSN2 or the ADH1 promoter. Cells were grown to logarithmic phase (open bars), one aliquot was treated with sorbic acid (10 mm, 45 min; stippled bars). Catalase activity of extracts was assayed from at least three independent cultures (standard deviation < 20%).
For initial indirect immunofluorescence studies, we used epitope-tagged versions of Msn2p driven by the authentic MSN2 promoter. Although a specific signal is barely detectable under normal growth conditions, a distinct nuclear signal appears after stress treatment. We therefore increased the expression level by replacing the native promoter through the ADH1 promoter. Overexpression of untagged and tagged genes (pAdh1–Msn2 derivatives) resulted in an ∼20-fold increase of both basal and stress-induced expression of the CTT1 gene (Fig. 2D). Indirect immune fluorescence data using monoclonal anti-myc antibody (9E10) are shown in Figure 2A. In unstressed cells, the Msn2p–myc9 fusion protein is distributed diffusely throughout the cytoplasm and is partly excluded from the nucleus. Immunofluorescence of the myc-tagged Msn2p co-stains with DAPI in cells treated for 20 min by either heat shock, ethanol, sorbate, or osmostress (0.4 m NaCl or 0.9 m sorbitol; the latter not shown). Together with the observation that stress causes STRE protection, these data suggest that Msn2p localization controls promoter occupancy.
To confirm this finding in living cells, we used a Msn2p–GFP fusion protein. Logarithmic cells carrying the Msn2p–GFP fusion showed uniform cytoplasmic staining in the absence of stress (Fig. 2B). After stress treatment, we observed that the nuclei of almost all cells were stained within a few minutes. In addition to the stress conditions documented in Figure 2B, we found a similar nuclear concentration of Msn2p–GFP in cells exposed to sudden glucose starvation (see Fig. 3), low pH (2.8) or methanol (10%) (data not shown). Msn2p–GFP fluorescence colocalizes with DAPI-stained DNA in stressed cells. No influence of a hog1 mutation on Msn2p translocation by osmostress (Fig. 2B) or by other stresses (data not shown) was observed. Western control experiments demonstrated that the levels of Msn2p–GFP remain unchanged under these conditions (Fig. 2C).
Figure 3.
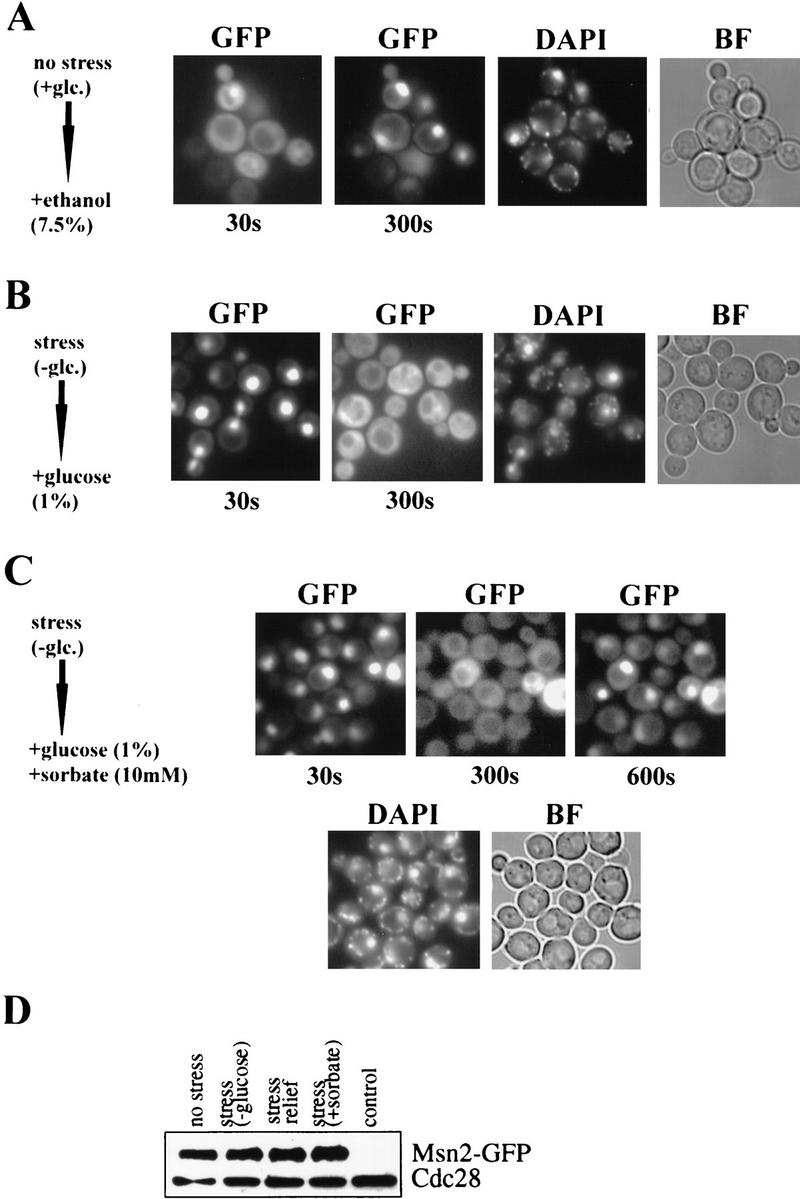
Dynamic distribution of Msn2p–GFP in W303 msn2 msn4 cells transformed with pAdh1–Msn2–GFP. Import (A): Cells were grown to early logarithmic phase, treated with 7.5% ethanol and pictures were taken after 30 and 300 sec. Export (B): Cells were starved for carbon source and glucose (1%) was added. Pictures were taken after 30 and 300 sec. Import–export (C): Msn2p–GFP distribution in a group of carbon-starved cells after simultaneous addition of 1% glucose and 10 mm sorbic acid (30, 300, and 600 sec). In all cases, DAPI fluorescence was recorded immediately after the final GFP recording. (D) Western blot showing amounts of Msn2p–GFP in extracts from cells grown under conditions described in B and C.
Msn2p nuclear import and export is fast and protein synthesis-independent
To study the kinetics of Msn2p translocation (Fig. 3), logarithmic W303 msn2 msn4 cells transformed with a Msn2p–GFP fusion plasmid were treated with 7.5% ethanol and fluorescence of a group of cells was recorded after 30 and 300 sec, when nuclear import was completed (Fig. 3A). De novo protein synthesis was not required for stress-induced nuclear import, as preincubation with 100 μg cycloheximide/ml did not prevent translocation (data not shown). These data demonstrate that stress-induced translocation of Msn2p is a direct and rapid process.
Because the activation of stress genes is normally transient and tightly controlled, one can assume that the relief of stress might, by analogy, cause a rapid shutdown of stress gene transcription. In the case of nuclear-localized Msn2p, the shutdown could either be achieved by modification, degradation, or export of the factor. Western blots show that the levels of Msn2p do not exhibit large variations before stress, after stress, or after stress relief (Fig. 3D). When import was induced by a short heat shock (36°C), relocation of Msn2p–GFP to the cytoplasm was observed at room temperature within several minutes (data not shown). A consecutive heat shock again caused nuclear concentration of the GFP-tagged protein. Similarly, export of Msn2p–GFP from the nucleus was observed when stress agents (sorbic acid, ethanol, NaCl) were removed by transferring the cells to fresh medium. Activation of transcription by glucose starvation has been shown to be mediated by Msn2p and Msn4p (Martinez-Pastor et al. 1996). Carbon-source starvation is achieved by transferring the cells to medium lacking glucose and results in rapid nuclear concentration of the Msn2p–GFP fusion protein. Relief of stress can be accomplished by adding glucose to cells in carbon-source starvation medium. Figure 3B shows glucose-starved cells to which 1% glucose was added. Immediately after addition of glucose (30 sec), Msn2p–GFP is still concentrated in the nuclei. Within 300 sec, nuclear fluorescence faded with concomitant increase of cytoplasmic staining. The constance of the overall cellular concentration of Msn2p–GFP under these conditions was demonstrated by a control Western blot (Fig. 3D), suggesting that no significant degradation occurred. Therefore, Msn2p–GFP is exported from the nucleus to the cytoplasm on stress relief. Analogous to stress-induced import, no significant dependence of the export on de novo protein synthesis was evident. To demonstrate the reversibility of Msn2p import and export on individual cells, stress relief and stress agent were applied simultaneously. Carbon-starved cells were treated with 1% glucose and 10 mm sorbate. The time course of Msn2p–GFP localization under these conditions is shown in Figure 3C (30, 300, and 600 sec). Apparently, glucose refeeding (relief of stress) and sorbate stress compete under such conditions so that Msn2p–GFP, which is concentrated initially in the nucleus at time point 30 sec, is exported after 300 sec, but partly re-imported into nuclei after 600 sec. This implies that Msn2p localization is dynamically controlled by stress and nonstress conditions, perhaps by two competing mechanisms.
PKA regulates Msn2p localization
The PKA pathway is known to negatively regulate STRE-dependent transcription (Marchler et al. 1993). To determine how Msn2p function is affected by PKA, we studied the localization of Msn2p in cells with different levels of kinase activity. Constitutively low PKA activity is exhibited in strain RS58-5A-1 (bcy1 tpk1w tpk2 tpk3) lacking the gene for the regulatory subunit (BCY1) and two of the three genes for catalytic subunits of PKA (TPK2, TPK3), while carrying a functionally impaired allele of the third (tpk1w) (Cameron et al. 1988). Such mutants have abnormally high levels of STRE-regulated transcripts. Consistent with a major role for Msn2p, deletion of MSN2 in this strain abolished the high level of catalase T produced in this genetic background in the absence of stress (Fig. 4B). In vivo footprinting analysis shows enhanced protection of STREs (indicated by arrows) in the CTT1 promoter in unstressed logarithmic RS58-5A-1 cells in comparison with the isogenic wild-type strain SP1 (Fig. 4A). Consistently, Msn2p–GFP displayed constitutive nuclear localization in RS58-5A-1 cells, in contrast to cytoplasmic staining observed in wild-type cells (Fig. 4C). Notably, levels of Msn2p–GFP are significantly lower in strain RS58-5A-1 compared with wild type (data not shown). This is probably an indirect effect attributable to the mutant background as Msn2p–GFP was found to be stable when PKA activity was impaired in a cdc25-1 strain (see below). These results implied that PKA is negatively regulating STRE-dependent transcription through control of Msn2p localization.
Figure 4.
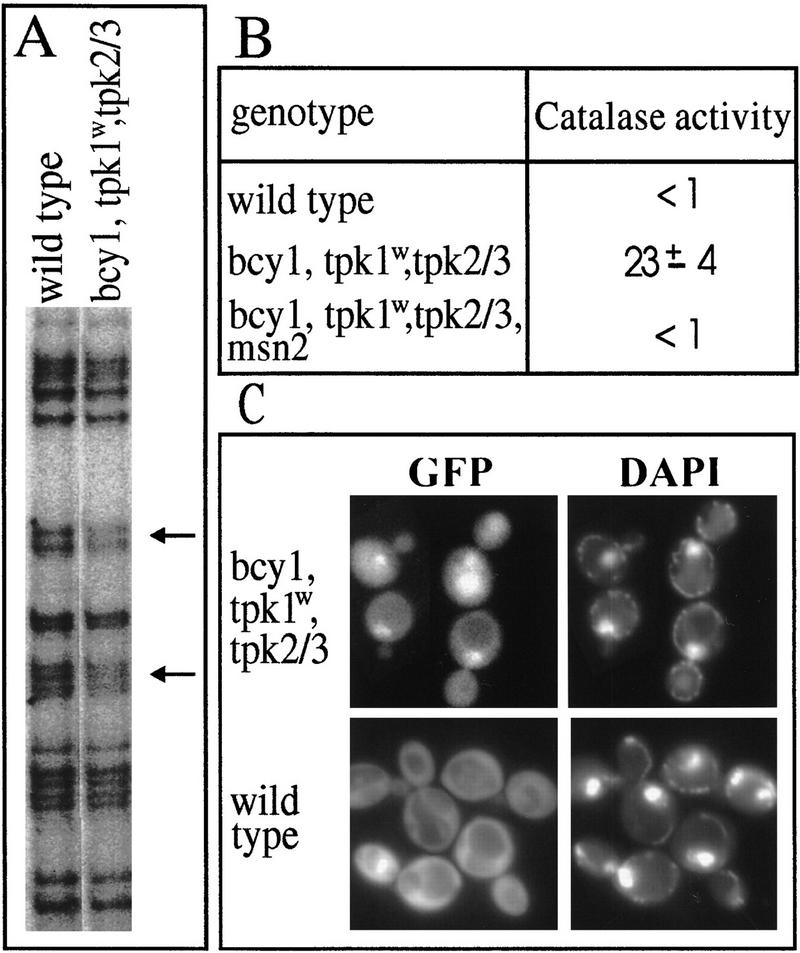
Msn2p in a low PKA mutant. (A) The strains SP1 (wild type) and RS58-5A-1 (bcy1 tpk1w tpk2 tpk3) were grown logarithmically and in vivo footprinting was performed on the CTT1 promoter. Products of primer extension with the CTT1-specific oligonucleotide were separated and autoradiographed. The STRE core sequences are indicated by arrows. (B) Catalase activity (micromoles H2O2 decomposed/min × mg of protein) in extracts of SP1, RS58-5A-1, and RS58-5A-1 msn2 cells was assayed under nonstress conditions. (C) Msn2p–GFP localization in RS58-5A-1 and SP1 under nonstress conditions. DNA was visualized with DAPI.
PKA activity can also be manipulated by exogenous cAMP in cells lacking the high-affinity phosphodiesterase gene (pde2). Strain OL556 carries a temperature sensitive allele of the CDC25 gene (cdc25-5) and a pde2 mutation that allows uptake of externally supplied cAMP (Boy-Marcotte et al. 1996). At the nonpermissive temperature (37°C) endogenous cAMP production ceases, attributable to the cdc25-5 allele whose product fails to activate the yeast Ras proteins and thereby prevents activation of adenylate cyclase. At the permissive temperature (26°C) we observed no difference in the regulation and levels of expression of CTT1 and of a STRE–LacZ fusion in this strain compared with wild-type strains (data not shown). To assay the effect of cAMP on Msn2p, OL556 cells were grown to logarithmic phase at 26°C and cAMP was added (3 mm). After 1 hr, the culture was shifted to the nonpermissive temperature of 37°C. To recover cells from heat shock, the culture was kept at 37°C for at least 1 hr. During this time span, heat shock-induced CTT1 mRNA levels decrease again to the basal level in wild-type cells (Martinez-Pastor et al. 1996). In the OL556 strain, the heat shock induction of catalase activity was suppressed by 3 mm cAMP indicating inhibition of stress-induced transcription by high PKA activity (data not shown). The culture was divided and cAMP was removed by washing one aliquot in 37°C medium. The cultures were then incubated further at 37°C. CTT1 mRNA rose to a detectable level within 15 min in the absence of cAMP, but not in its presence, in a cycloheximide-insensitive manner (Fig. 5B). OL556 cells treated in the same way were subjected to in vivo footprinting analysis. Protection of CTT1 STREs was increased significantly in the cAMP-free cells compared with cAMP-containing cells (Fig. 5A). In absence of cAMP, but not its presence, Msn2p–GFP accumulated in the nucleus at the nonpermissive temperature (Fig. 5C). To investigate whether cAMP depletion is sufficient to trigger nuclear accumulation of Msn2p at room temperature, we used a strain (TC41-1; Hubler et al. 1993) that is unable to sythesize cAMP because of a deletion of the adenylate cyclase gene (CDC35). TC41-1 cells expressing Msn2p–GFP were grown in the presence of cAMP (2 mm). cAMP was removed by washing the cells in medium and nuclear localization of Msn2p–GFP was observed within 20 min (data not shown). Taken together, these data suggest that high PKA activity inactivates transcription of STRE-regulated genes by lowering the nuclear localization of Msn2p. To establish whether an increase of PKA activity causes nuclear export, we followed the fate of already nuclear localized Msn2p–GFP. Nuclear localization of Msn2p–GFP was triggered by treating OL556 cells with 10 mm sorbate at the permissive temperature (Fig. 5D). Activation of PKA by subsequent addition of cAMP (25 mm) caused fading of the nuclear GFP signal with a concomitant increase of fluorescence activity in the cytoplasm within 10 min (Fig. 5D). According to a control Western blot, no significant degradation of Msn2p–GFP occurs during this type of experiment (Fig. 5E). These results demonstrate that translocation of Msn2p to nuclei, even under sustained stress conditions, is reversed by cAMP, a factor activating PKA activity.
Figure 5.
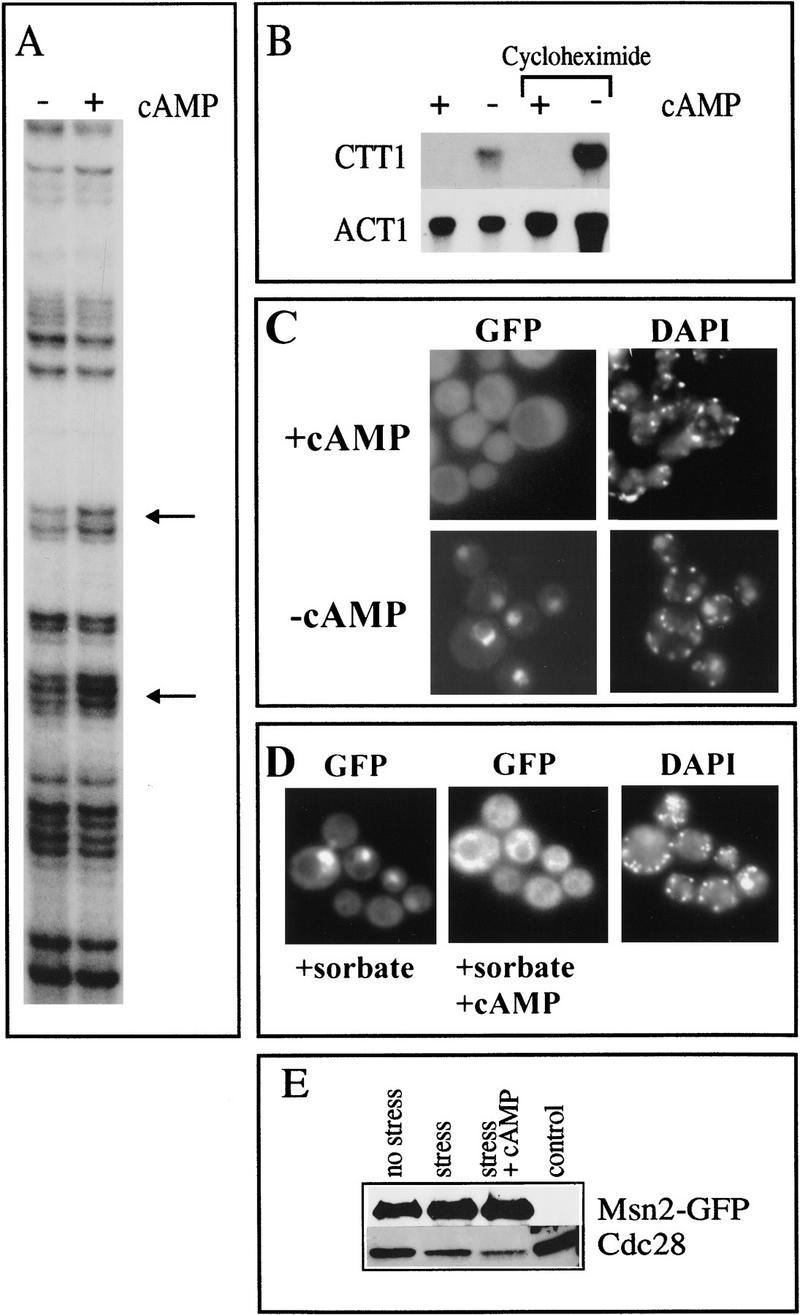
cAMP levels influence Msn2p localization. (A) In vivo footprint analysis of the CTT1 STREs. OL556 (cdc25-5 pde2) cells were grown and shifted to nonpermissive temperature (37°C) in the presence of 3mM cAMP and released by dividing the culture and removing cAMP from one aliquot. STRE protection in the CTT1 promoter was determined after 20 min in cells with (+cAMP) and without cAMP (−cAMP). (B) CTT1 transcript levels. Logarithmic OL556 cells, were shifted for 1 hr to 37°C in presence of 3 mm cAMP (+cAMP). The culture was divided and cAMP removed from aliquots (−cAMP). The procedure was done in the presence and absence of cycloheximide. Cells from all aliquots were harvested 15 min after cAMP had been removed from −cAMP aliquots, RNA was prepared, and probed with CTT1 and ACT1 after electrophoresis and blotting. (C) Msn2p–GFP localization in strain OL556 at the nonpermissive temperature with and without cAMP. OL556 transformed with pAdh1–Msn2p–GFP was grown and shifted as in A. Fluorescence signals were recorded at the same time points in presence (+cAMP) and absence (−cAMP) of cAMP. (D) Msn2p–GFP distribution in stressed OL556 cells after addition of cAMP. OL556 carrying pAdh1–Msn2–GFP was grown at 26°C to logarithmic phase. Sorbate (10 mm) was added to induce nuclear localization. Msn2p–GFP fluorescence was observed in a group of cells 0.5 min (+sorbate) and 10 min after addition of 25 mm cAMP (+sorbate +cAMP). (E) Control Western blot showing amounts of Msn2–GFP in extracts from cells grown under conditions described in D.
Stress and cAMP influence the localization of the Msn2p homolog Msn4p
Msn4p has been reported to bind to STREs and to have functions overlapping with Msn2p (Martinez-Pastor et al. 1996). We investigated the intracellular localization of Msn4p using a GFP fusion protein (Fig. 6). Msn4p–GFP is distributed diffusely in the cytoplasm of unstressed cells, whereas, very much like Msn2p, it is rapidly concentrated in nuclei on sorbate stress. Other stresses had comparable effects (data not shown). cAMP responsiveness was tested in strain OL556. The protein was found to be nuclear in the absence of cAMP, but localized in the cytoplasm in its presence. Therefore, Msn2p and Msn4p may share common control mechanisms regulating intracellular localization by stress and cAMP.
Figure 6.
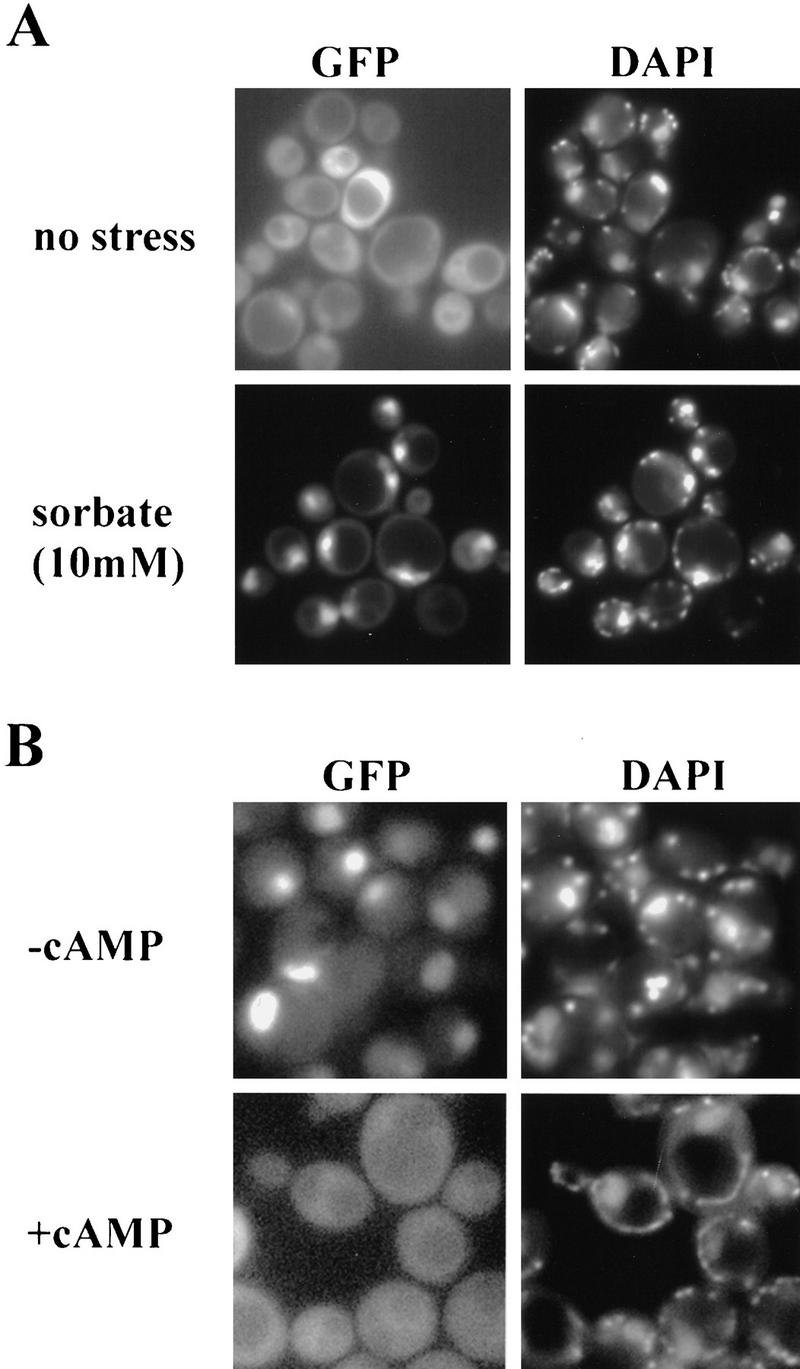
Msn4p–GFP distribution in stressed and cAMP-treated cells. (A) W303 msn2 msn4 tranformed with pAdh1–Msn4–GFP was grown logarithmically and treated with 10 mm sorbate. (B) OL556 cells transformed with pAdh1–Msn4–GFP were shifted to 37°C with and without 3 mm cAMP. DAPI was added 10 min before treatments.
Analysis of Msn2p regions required for regulation of localization
To identify the domains involved in regulation of Msn2p, we carried out a deletion and mutagenesis analysis (Fig. 7A). Whereas a truncated protein lacking the DNA-binding domain behaved like wild type, regulation was lost with deletion of the carboxy-terminal 136 amino acids (data not shown). The carboxy-terminal region of the protein (pAMG3) is sufficient for nuclear localization of a GFP fusion and may therefore contain a NLS that can be replaced by the SV40–NLS (pASMG1). Deletion of the amino-terminal 264 amino acids (pAMG4) did not impair regulated localization. Further deletion to position 287 (pAMG2) resulted in constitutive nuclear localization. To determine whether potential PKA modification sites (RRXS) are important for Msn2p regulation, we created a series of serine to alanine and aspartate substitutions. S288A and S288D mutations resulted in a GFP fusion protein (pAMG5/pAMG6) with nuclear localization under nonstress conditions, which, however, translocated to the cytoplasm in presence of high exogenous cAMP. Export was abolished completely in combination with the exchange of S620, S625, and S633 to aspartate (pAMG7) or S582, S620, S625, and S633 to alanine (pAMG8). Therefore, the PKA sites are important but partially redundant for nuclear export driven by high exogenous cAMP.
Figure 7.
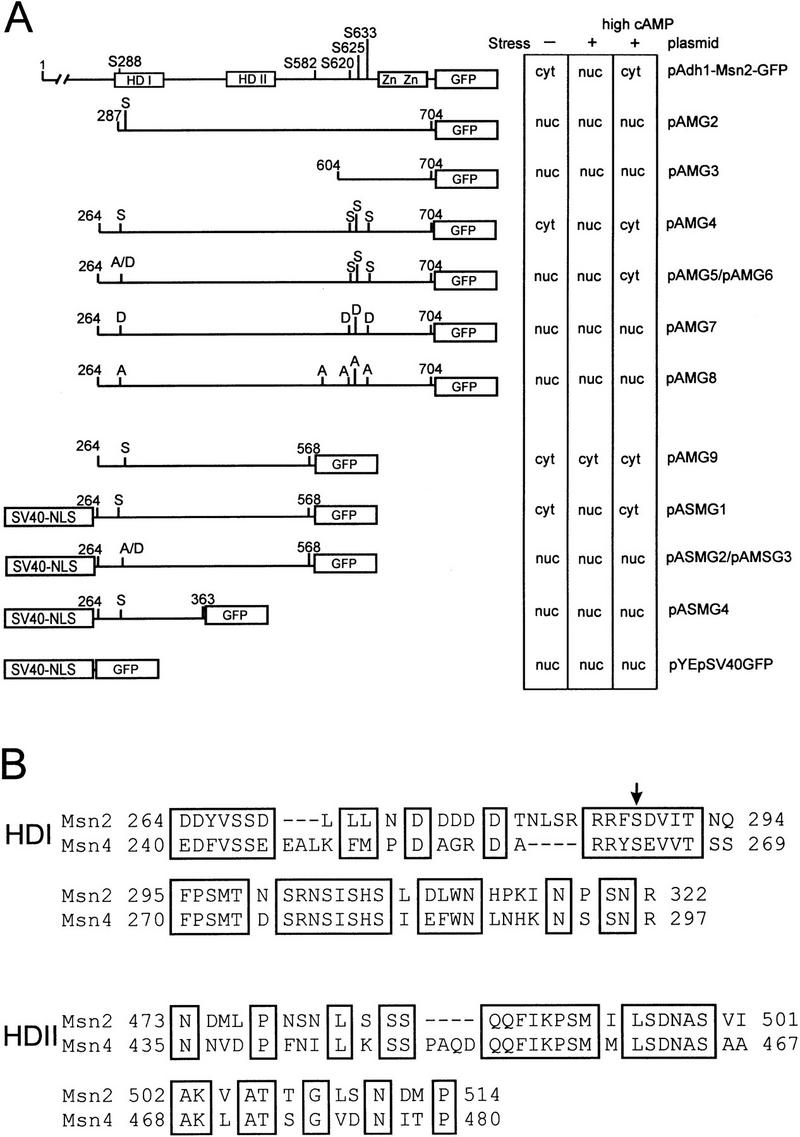
Functional regions of Msn2p. Deletion constructs were assayed for stress-induced import and cAMP-driven export in OL556 cells. Microcolonies of transformants were suspended in fresh medium (− stress), treated with 10 mm sorbate (+stress), and subsequently incubated with 25 mm cAMP (+high cAMP). The effects of stress and cAMP treatment were scored after 10 min (cyt: cytoplasmic localization due to indeterminable nuclear staining; nuc: clearly identifiable nuclear staining). Serine to alanine or aspartate substitutions are indicated. All SV40–NLS-directed constructs exhibit a certain background of cytoplasmic fluorescence. (HD I) Homology domain I; (HD II) homology domain II; (Zn) zinc finger. (B) Alignment of HD I and HD II (pileup algorithm, Wisconsin GCG package).
To determine the specific function of S288, a fragment of Msn2p (264 to 568; pAMG9) in which all carboxy-terminal PKA sites are eliminated was fused to the SV40–NLS. The fusion protein (pASMG1) showed wild type-like behavior in response to stress and cAMP. Mutagenesis of S288 abolished both stress responsiveness and cAMP-driven nuclear export (pASMG2, pASMG3). Further carboxy-terminal truncation of pASMG1 destroyed any kind of regulation (pASMG4, amino acids 264–363). In summary, nuclear export requires the region containing the PKA consensus site around S288 and the region between amino acids 363 and 568. Within this region (264–568), two stretches with high similarity to the Msn4p sequence (Fig. 7B) are apparent, both of which are included in the domains necessary for Msn2p nuclear export. These homology domains are indicated in Figure 7A as HD I and HD II. The PKA consensus site in HD I (containing S288 of Msn2p) is conserved between both proteins (Fig. 7B, arrow). These data suggest that functional PKA consensus sites are important for the regulation of the intracellular localization of Msn proteins.
Discussion
In the budding yeast S. cerevisiae, many different stress situations activate a common response system whose central parts are two transcription factors, Msn2p and Msn4p. These two factors have partly overlapping functions—being able to bind and activate STRE dependent promoters. Double mutants (msn2 msn4) are normally unable to survive severe stress probably because they are defective in mounting proper countermeasures. Any attempt to explain the generality of the stress response to various individual challenges might have to deal with the question of how these transcription factors function. The results presented here strongly support the notion that diverse stress signals converge on mechanisms that determine the nuclear concentration of Msn2p. Nuclear transport phenomena might therefore be the key to understand how different stress conditions are integrated into one cellular response.
Nuclear localization of Msn2p is a general response to stress
Our conclusion that Msn2p and Msn4p localization must be at the heart of the system is mostly based on cytological observations. Both in situ immunodetection of an epitope-tagged Msn2p as well as GFP fusions reveal the same situation. Under favorable environmental conditions, the amount of nuclear Msn2p/Msn4p appears low in most cells. After cells encounter any stressful condition, however, the pool of existing proteins is quickly redistributed toward high nuclear concentrations. This effect can be seen in virtually all cells. A causal relationship between the translocation event and gene activity can be inferred by the fact that an increase in nuclear Msn2p concentration correlates well with the occupation of STREs, as suggested by in vivo footprinting experiments. STRE-dependent transcriptional induction is normally occurring rapidly, that is, within minutes after exposing cells to a variety of stress conditions. According to the observed kinetics of this process, Msn2p/Msn4p nuclear translocation appears to be the rate-determining step in the activation process.
In many STRE containing promoters, transcriptional induction occurs after sudden temperature upshift, lowering of the pH, osmotic shock, glucose starvation, and treatment with alcohols or weak organic acids (sorbic acid). In almost every case, the induction is dependent completely on the presence of Msn2p and Msn4p (Martinez-Pastor et al. 1996). One notable exception is the response to osmotic stress, which is still discernible in the msn2 msn4 double mutant even if the absolute levels of transcription are affected severely. In this case, the wild-type response requires a functional Hog1p MAP kinase whose activity is induced specifically by high external osmolarity (Brewster et al. 1993). Our observations on Msn2p nuclear import correlate very well with the STRE expression data. In all stress situations, including osmotic challenge, the nuclear levels of Msn2p become elevated. This increase in nuclear concentration can be observed in hog1 null mutants under osmotic stress. The data suggest that a separate, specialized HOG pathway-dependent mechanism must enhance the osmotic response (Schüller et al. 1994; Martinez-Pastor et al. 1996). After initial perception, the osmostress signal may bifurcate at one point to converge again on the level of the STRE employing different and perhaps interacting transcription factors with Msn2p being one of them. Another explanation could be that high osmolarity normally causes enough collateral damage to induce the general stress response system. Whatever the primary cause of Msn2p and Msn4p translocation, we suggest that this translocation is an integrating response to activate a large set of stress-related genes. Therefore, Msn2p localization may be regarded as a measure of a yeast cell’s well-being.
How might nuclear localization of Msn2p be regulated?
Two different modes of regulation could impinge on Msn2p to achieve the observed differences in spatial distribution—regulated nuclear import and regulated export. With regard to import control, two major themes have emerged over the last few years. One involves the obscuring of a nuclear import signal by phosphorylation. The other mechanism involves the retention of a transcription factor by a cytoplasmic anchor. Our dissection of Msn2p clearly indicates that position and nature of the NLS is apparently not so important for regulated translocation. Instead, the integrity of a ∼300-amino-acid domain seems crucial to cause cytoplasmic accumulation under nonstress conditions. This observation may point to a retention mechanism that depends on a cytoplasmic partner. The interaction of NF-κB with IκB is perhaps the prototypical example in this regard. Curiously, like Msn2p in yeast, NF-κB seems to guide a large variety of stress responses in higher eukaryotic systems. Here, stress signal-activated kinases seem to influence the phosphorylation-induced instability of IκB. Unfortunately, there is no structural similarity between the two transcription factors to allow any meaningful extrapolation from NFκB on Msn2p control. Compared with these two factors, a more specialized situation has been studied in the case of the S. cerevisiae transcription factor Yap1p (Kuge et al. 1997). Localization of Yap1p involves a carboxy-terminal cystein-rich domain sufficient to cause cytoplasmic retention in unstressed cells. As some of these cystein residues are required for proper regulation they have been proposed to act as sensors for oxidative damaging conditions. The mechanism of retention, however, is not yet understood but might also require a cytoplasmic anchor. In any case, translocation of this factor appears to be controlled specifically by oxidative stress, whereas a broad variety of stresses affect cellular localization in the case of Msn2p.
What is the role of PKA in the regulation of Msn2p
With the exception of the HOG pathway, which seems to bypass Msn2p, little is known about signal generation and transduction for any of the Msn2p-activating stresses. Nevertheless, one might ask whether modification of Msn2p could be important for releasing or maintaining cytoplasmic anchorage. For example, Pho4p, a factor inducing the response to low phosphate, was shown to be modified by a cyclin-CDK complex, and this phosphorylation may provide the information for anchorage-dependent retention (O’Neill et al. 1996). Glucose-regulated Mig1p nuclear localization is correlated dynamically and reversibly with its phosphorylation status, which is probably regulated by the Snf1 protein kinase. Again phosphorylation may prevent nuclear import (DeVit et al. 1997). We presented here an intriguing connection between Msn2p localization and the activity of protein kinase A. Similar to Pho4p, there is an inverse correlation between PKA activity and nuclear localization. The finding that substitutions at the consensus PKA sites, particularly at position 288, lead to constitutive nuclear residence of Msn2p is highly suggestive for a role of direct modification.
Despite this evidence, we have no clear indication where and how PKA interferes mechanistically with Msn2p and Msn4p distribution. For example, one cannot completely disregard the possibility that PKA regulates the activity of stress signal transduction systems before they converge on Msn proteins. Similarly, one cannot yet exclude that stress might regulate the intracellular distribution of the transcription factors through modulating PKA activity. What we have made clear, however, is the fact that stress conditions generally lead to higher nuclear concentration, whereas increases in PKA activity induce cytoplasmic accumulation. Changes in the distribution between nucleus and cytoplasm happen rapidly and without noticable changes in the overall protein concentration. Therefore we find considerable attraction in a model in which stress-induced import and PKA induced export are mediated by independent mechanisms. In a dynamic equilibrium, either stress factors or PKA activity might dominate leading to a steady-state situation where the major part of Msn2p and Msn4p is located either in the nucleus or in the cytoplasm depending on the extent of stress as well on PKA activity.
What is the physiological role of dynamic Msn2p localization?
The influence of PKA activity on Msn2p provides a model explaining how growth conditions determine stress resistance in yeast. Previous observations indicated that strains with low PKA activity are more stress resistant than cells with high PKA activity (Shin et al. 1987). We show here that in mutants with constitutive low PKA activity (bcy1 tpk1w tpk2 tpk3), Msn2p is localized predominantly in the nucleus, which causes high levels of expression of stress-inducible genes and increased stress resistance (Schüller et al. 1994; Ruis 1997). In contrast, high PKA activity would shut down expression of stress genes by preventing nuclear accumulation of Msn2p. This would explain why mutants with high PKA activity (e.g., bcy1) are stress-sensitive (Thevelein 1994; de Winde et al. 1997). Our results suggest that PKA sets the threshold by preventing the nuclear accumulation of Msn2p/Msn4p for how much stress signal is required to mount the stress defense program.
For yeast, it might be advantageous to adjust stress protein synthesis according to growth conditions, thereby avoiding any unnecessary burden detrimental to optimal growth. A high level of expression of Msn2p or Msn4p reduces the growth rate, perhaps by the constitutive activation of STRE-regulated genes (Estruch and Carlson 1993; Martinez-Pastor et al. 1996; C. Schüller, unpubl.). In a competitive environment, a return to fast growth after stress relief and adaptation could be of vital importance.
Materials and methods
Yeast strains, media, and growth conditions
In strain W303–STRE–lacZ (MATa ura3-1 leu2-3 his3-11 ura3, trp1-1 ade2-1 can1-1000 URA3::STRE–lacZ) the MSN2 and MSN4 genes were disrupted using the msn2::HIS3 and msn4::TRP1 (Estruch and Carlson 1993), HOG1 was deleted with TRP1 (Brewster et al. 1993). OL556 (MATa/α cdc25-5/cdc25-5 pde2/pde2 his3/his3 leu2/leu2 ura3/URA3::STRE–lacZ; Boy-Marcotte et al. 1996) was obtained from E. Boy-Marcotte. TC41-1 (MATa leu2-3 leu2-112 his3-532 his4 ura3 cdc35::ura3 cam) was provided by W. Heidemann. SP1 (MATa leu2 his3 trp1 ade8 can1 ura3) and the derivative RS58-5A-1 (MATa leu2 his3 trp1 ade8 can1 ura3 tpk1w tpk2::HIS3 tpk3::TRP1 bcy1::LEU2) have been described previously (Cameron et al. 1988). RS58-5A-1 msn2::URA3 was created by transformation. Cells were generally grown to logarithmic growth phase from OD600 0.2 to OD600 1-2 by dilution of overnight precultures in rich medium (YPD) or in synthetic medium. Logarithmic cultures were used to visualize GFP fusions. GFP fusions were also visualized in medium suspensions of microcolonies (diameter up to ∼1 mm). Stress was caused by addition of concentrated solutions to logarithmic cultures or suspensions to the desired final concentration followed by incubation with shaking for the indicated times. For heat stress treatment cultures were grown at least four generations at 26°C before the growth temperature was raised to 37°C within 3–4 min. Glucose starvation was achieved by washing cells two times in glucose free medium.
Plasmids
Plasmid pMsn2::Ura3 was created from pt32-deltaXB::His3 (Estruch and Carlson 1993) by replacing the HIS3 sequences with URA3. Plasmid pMsn2 was generated by cloning a 3.5-kb KspI–HindIII fragment containing the MSN2 gene into a modified EcoRI site (changed to a KspI site with oligonucleotide 5′-AATTGCCGCGGC-3′) of Ycplac111 centromere vector (Gietz and Sugino 1988). A NotI site was created by introducing a linker into the SalI site (position +7) of the MSN2 gene. A NotI site at the 3′ end of MSN2 was introduced before the stop codon by PCR using 5′ oligonucleotide 5′-TCGACTCCGTCTGGGATCCTCTTCGATCTTTCCATC-3′ and 3′ oligonucleotide 5′-TTTAAGCTTAAATTATGGGCGGCCGCCAATGTCTCCATGTTTTTTATGAGT-3′ (NotI site, italicized; stop codon, underscored; HindIII site, bold). The 0.3-kb NdeI–HindIII fragment of pMsn2 was replaced by the NdeI–HindIII-cut PCR fragment generating pMsn2–NotI. pMsn2–myc9 was generated by ligating a myc9 NotI cassette (Piatti et al. 1996) into the NotI site. For construction of the Msn2p–GFP fusion protein, a GFP NotI cassette was generated by PCR, using oligonucleotides G1 5′-AAGCGGCCGCACCATGGTGAGCAAGGGCG-3′ (NcoI site, underscored) and G2 5′-TTTGCGGCCGCTCTTGTACAGCTCGTCCAT-3′ (NotI sites, italicized); vector pEGFP–N3 (Clonetech) served as template. Plasmid pMsn2–GFP was generated by ligation of a GFP NotI cassette into pMsn2–NotI. pAdh1–Msn2, was created by replacing the KspI–SalI fragment of pMsn2–NotI with a PCR-generated KspI–SalI fragment of the yeast ADH1 promoter using 3′ oligonucleotide Adh1–SalI 5′-ATGGTCGACCGTCATTGTATATGAGATGATAGTTGATTGTATGCTTGG-3′ (SalI, italicized) and 5′ oligonucleotide Adh1–KspI 5′-CTAAACCGCGGAATATTTCGGGATATCC-3′ (KspI, italicized). Replacement of the KspI–SalI fragment of plasmids pMsn2–myc9 and pMsn2–GFP with the ADH1 promoter generated pAdh1–Msn2–myc9 and pAdh1–Msn2–GFP. For studies in ura3 background, the KspI–HindIII fragment of pAdh1–Msn2–GFP was cloned into vector pRS316 (Sikorski and Hieter 1989). Expression of fusion proteins was verified by Western blotting using monoclonal antibody to the Myc epitope (9E10) or polyclonal rabbit antiserum against Msn2p.
Deletions and mutations of Msn2p were generated by reinserting PCR fragments into appropriately cut pAdh1–Msn2–GFP. pAMG2 was constructed by insertion of the SalI–NcoI-cut PCR fragment obtained using the oligonucleotides X12B 5′-CACGCCGAAGATTTGTCGACGTTATAACAAAC-3′ and G3 5′-TGGCCGTTTACGTCGCCGTCCAGC-3′, pAMG3 by insertion of the SalI–NcoI cut PCR fragment obtained by oligonucleotides X8 5′-GTCGACTCAACTGGCAATGGTGCTGG-3′ and G3. pAMG4 by insertion of the SalI–NcoI cut PCR fragment obtained by oligonucleotides X12 5′-GTCGACGTGTTCTAGTGATCTCGTATTG-3′ and G3 were used. Mutagenesis of S288 to alanine or aspartate was carried out by PCR with oligonucleotides X12–A288 5′-CCATGCTAGATGTCGACGTTTCTAGTGATCTCTTATTGAATGATGATGATGATGACACTAATTTATCACGCCGAAGATTTGCCGACGTTATAAC-3′ (pAMG5) and X12–D288 5′-AATGTCGACGTTTCTAGTGATCTCTTATTGAATGATGATGATGATGACACTAATTTATCACGCCGAAGATTTGATGACGTTATAACAAACC-3′ (pAMG6) both times using 5′ primer G3 and inserting the SalI–NcoI fragment back into pAdh1–Msn2–GFP. For pAMG7, the XhoI–SalI-cut PCR fragment obtained with oligonucleotides X12–D288 and Msn2–D3 5′-TTTCTCCTCGAGTTCCTTTGTTGATTCTATTACGACATCTGATCTTCTGGACGGTGTCATATCTTTTCTCCTGTAATCTGGCCTTCTTTCC-3′ and pAMG8, the XhoI-SalI-cut PCR fragment obtained with oligonucleotides X12–A288 and Msn2–A3 5′-CTCCTCGAGTTCCTTTGTTGATTCTATTACGACAGCTGATCTTCTGGACGGTGTCATTGCTTTCTCCTGTAAGCTGGCCTTCTTTCCTT-3′, were inserted into XhoI–SalI-cut pAdh1–Msn2–GFP. For pAMG8, a template containing a serine 581 to alanine substituion derived from site-directed mutagenesis was used. pAMG9 was generated by insertion of the SalI–NcoI-cut fragment obtained by PCR with oligonucleotides X12 and X5 5′-AGCCATTGTAGTGTGACCCTGCTGTGG-3′ into pAdh1–Msn2–GFP. For the construction of pASMG1, an ADH1 promoter fused to SV40–NLS, was generated by cloning the annealed oligonucleotides SV40-1 5′-TCGAAGCACCCAAGAAGAAGCGGAAGGTGGGGATCC-3′ and SV40-2 5′-TCGAGGATCCCCACCTTCCGCTTCTTCTTGGGTGCA3′ into SalI-cut pAdh1–Msn2–GFP. An adjacent XhoI site was generated by amplification with SV40-3 5′-TCCATGGCATGCTCGAGGATCCCCACC-3′ and the Adh1–KspI primer. pASMG1 was generated by joining the Adh1–SV40–NLS KspI–XhoI fragment with the SalI–NcoI cut product of PCR using oligonucleotides X12 and X5 into KspI–NcoI cut pAdh1–Msn2–GFP pASMG2/3 were generated in a similar way but using fragments obtained by PCR with primers X12-A and X12-D respectively using 5′ primer X5 in both cases. pASMG4 was generated in a similar way from a PCR fragment obtained with oligonucleotides X12 and X5D 5′-TGTCGACATTGTCCATGGTAGCGTCATTG-3′. PCR reactions were performed with Vent polymerase (NEB).
Indirect in situ immunofluorescence and GFP fluorescence microscopy
Fixation and staining of cells was carried out as described by Nasmyth et al. (1990). The method was modified by omitting methanol/acetone treatment of fixed spheroplasts. Myc9-tagged Msn2p was detected by anti-Myc monoclonal antibodies (9E10) and indirect immunofluorescence detection was achieved by using goat anti-mouse IgG CY3 conjugated secondary antibodies (Sigma). GFP was visualized without fixation. Nuclei were stained by addition of 2 μg/ml of 4,6 diamidino-2-phenylindol (DAPI) DNA dye to the cultures 10 min before microscopy. Aliquots (2 μl) of the cultures were put on microscope slides and covered with 18 × 18 mm coverslips. All fixed and living cells were viewed using a Zeiss Axioskop fluorescence microscope. Images were scanned with a Quantix CCD camera using IP LAB software under Windows95, processed in Adobe Photoshop 4.0, and printed on a Kodak videoprinter.
In vivo footprinting
In vivo footprinting was carried out by the method described by Koch et al. (1996). Briefly, cultures were grown logarithmically from precultures for five generations to OD600 1.2 to 1.5 in YPD at 30°C (or at 26°C in the case of heat shock). Sorbic acid (final concentration, 10 mm) or NaCl (0.4 m) was added and cells were harvested after 20 min, resuspended in 2 ml YPD plus the respective agent and exposed to DMS (5 μl) for 5 min at room temperature. For heat shock, the cultures were concentrated in 10 ml of fresh YPD, shifted to 37°C for 10 min and incubated with 10 μl of DMS for 5 min at 37°C. The reaction was stopped by washing in cold TNEβ (10 mm Tris-HCl, pH 7.5, 50 mm NaCl, 1 mm EDTA, 0.1 m β-mercaptoethanol), and DNA was prepared after spheroplasting in SCEβ (1 m Sorbitol, 0.1 m Tri-Na-citrate.2H2O, 10 mm EDTA, 0.1 m β-mercaptoethanol, pH 7–7.5), and subsequent lysis (2% SDS, 0.1 m Tris-HCl, pH 9, 50 mm EDTA, at 65°C). Protein was removed by precipitation in 0.63 m potassium acetate at 4°C overnight. DNA from the supernatant was purifed by several ethanol precipitation/solvation cycles. Primer extension was carried out with 32P end-labeled oligonucleotides for 40 cycles (1 min at 94°C; 2 min at 55°C, 3 min at 72°C), the products were precipitated twice and dissolved in loading buffer (50 mm NaOH, 0.5 mm EDTA, 4 m urea, 0.1% bromo-phenol-blue/xylene cyanol) and resolved on an 8% sequencing gel. Gels were autoradiographed and scanned with a PhosphorImager and quantitated. Oligonucleotides used in the primer extension reaction were HSP104 promoter, 5′-TGAAGGAATTGAGGCAAGATTACAATGCC-3′, and CTT1 promoter, 5′-TTCAGCATACGAATAGTACTGGCAATGAC-3′.
Extract preparation for EMSA and Western blot analysis
Extracts for EMSA were prepared from cells grown at 30°C at OD600 1–2. Pellets were suspended in cold buffer A [50 mm HEPES, pH 8.0, 0.4 m (NH4)2SO4, 1 mm EDTA, and 5% glycerol], broken with glass beads, and clarified by centrifugation. Extracts were precipitated with (NH4)2SO4 to 50% saturation and resuspended in buffer B (75 mm KCl, 25 mm HEPES, pH 8.0, 1 mm EDTA, and 12% glycerol).
For Western analysis, cells from OD600 = 1.0 cultures were washed once, resuspended in buffer A, and then broken up by vortexing with glass beads for 3 × 4 min. Glass beads and debris were spun out by two 20-min centrifugation steps. Extracts (100 μl) were boiled with 50 μl 4× sample buffer. For Western blot analysis, 50 μg of protein were loaded on to 8% SDS-PAGE and separated using the Mini Protean system (Biorad). Proteins were transferred to Nitrocellulose membranes (Schleicher & Schuell) by Semi-dry Blotting Device (Biorad). Immunodetection of proteins was carried out as described in the ECL manual (Amersham) using primary polyclonal affinity-purified αMsn2p and αCdc28p antibodies. Complete protease inhibitor mix tablets (Boehringer) were used in all buffers.
Electrophoretic mobility shift assays, Northern analysis, and enzyme assays
EMSA was carried out as described by Martinez-Pastor et al. (1996). CTT1-20 and CTT1-23 are described by Marchler et al. (1993). A supershift was performed by addition of 9E10 monoclonal anitbody to the binding mix 2 hr before the addition of the labeled oligonucleotide. Northern analysis and measurement of β-galactosidase and catalase enzyme activities were carried out as described by Martinez-Pastor et al. (1996). To test whether the processes studied require protein synthesis, cycloheximide (100 μg/ml) was added 30 min before harvesting.
Acknowledgments
We thank W. Zachariae and T. Dechat for help with immunofluorescence; E. Waigmann for providing her microscope; E. Boy-Marcotte, W. Heidemann, and J. Aitchison for materials; A. Hartig, G. Griffioen, C. Brochard, and H.P. Rottensteiner for fruitful discussions; and H. Wrba for technical assistance. This work was supported by grant P11303 of the Fonds zur Förderung der wissenschaftlichen Forschung, Vienna, Austria (to H.R.) and grant PB94-0994 of the Direccion General de Investigation Scientifica y Tecnica (DGICYT), Spain (to F.E.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
This work is dedicated to our late friend and colleague Manuel M. Simon.
E-MAIL cs@abc.univie.ac.at; FAX 43-1-7995272.
References
- Baldwin AS., Jr The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Baroni MD, Monti P, Marconi G, Alberghina L. cAMP-mediated increase in the critical cell size required for the G1 to S transition in Saccharomyces cerevisiae. Exp Cell Res. 1992;201:299–306. doi: 10.1016/0014-4827(92)90277-f. [DOI] [PubMed] [Google Scholar]
- Baroni MD, Monti P, Alberghina L. Repression of growth-regulated G1 cyclin expression by cyclic AMP in budding yeast. Nature. 1994;371:339–342. doi: 10.1038/371339a0. [DOI] [PubMed] [Google Scholar]
- Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 1997;275:1930–1933. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- Brewster JL, de Valoir T, Dwyer ND, Winter E, Gustin MC. An osmosensing signal transduction pathway in yeast. Science. 1993;259:1760–1763. doi: 10.1126/science.7681220. [DOI] [PubMed] [Google Scholar]
- Boy-Marcotte E, Tadi D, Perrot M, Boucherie H, Jacquet M. High cAMP levels antagonize the reprogramming of gene expression that occurs at the diauxic shift in Saccharomyces cerevisiae. Microbiology. 1996;142:459–467. doi: 10.1099/13500872-142-3-459. [DOI] [PubMed] [Google Scholar]
- Cameron S, Levin L, Zoller M, Wigler M. cAMP-independent control of sporulation, glycogen metabolism, and heat shock resistance in S. cerevisiae. Cell. 1988;53:555–566. doi: 10.1016/0092-8674(88)90572-7. [DOI] [PubMed] [Google Scholar]
- Chen R-H, Sarniecki C, Blenis J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol Cell Biol. 1992;12:915–927. doi: 10.1128/mcb.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVit MJ, Waddle JA, Johnston M. Regulated nuclear translocation of the Mig1 glucose repressor. Mol Biol Cell. 1997;8:1603–1618. doi: 10.1091/mbc.8.8.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Winde JH, Thevelein JM, Winderickx J. From feast to famine: Adaptation to nutrient depletion in yeast. In: Hohmann S, Mager WH, editors. Yeast stress responses. Austin, TX: Landes; 1997. pp. 7–52. [Google Scholar]
- Estruch F, Carlson M. Two homologous zinc finger genes identified by multicopy suppression in a SNF1 protein kinase mutant of Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:3872–3881. doi: 10.1128/mcb.13.7.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Sugino A. New yeast—Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- Görlich D, Mattaj IW. Nucleocytoplasmic transport. Science. 1996;271:1513–1518. doi: 10.1126/science.271.5255.1513. [DOI] [PubMed] [Google Scholar]
- Hubler L, Bradshaw-Rouse J, Heideman W. Connections between the Ras-cyclic AMP pathway and G1 cyclin expression in the budding yeast Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:6274–6282. doi: 10.1128/mcb.13.10.6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jans DA. The regulation of protein transport to the nucleus by phosphorylation. Biochem J. 1995;311:705–716. doi: 10.1042/bj3110705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch C, Schleiffer A, Ammerer G, Nasmyth K. Switching transcription on and off during the yeast cell cycle: Cln/Cdc28 kinases activate bound transcription factor SBF (Swi4/Swi6) at start, whereas Clb/Cdc28 kinases displace it from the promoter in G2. Genes & Dev. 1996;10:129–141. doi: 10.1101/gad.10.2.129. [DOI] [PubMed] [Google Scholar]
- Kuge S, Jones N, Nomoto A. Regulation of yAP-1 nuclear localization in response to oxidative stress. EMBO J. 1997;16:1710–1720. doi: 10.1093/emboj/16.7.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler G, Schüller C, Adam G, Ruis H. A Saccharomyces cerevisiae UAS element controlled by protein kinase A activates transcription in response to a variety of stress conditions. EMBO J. 1993;12:1997–2003. doi: 10.1002/j.1460-2075.1993.tb05849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Pastor MT, Marchler G, Schüller C, Marchler-Bauer A, Ruis H, Estruch F. The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE) EMBO J. 1996;15:2227–2235. [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K, Adolf G, Lydall D, Seddon A. The identification of a second cell cycle control on the HO promoter in yeast: cell cycle regulation of SWI5 nuclear entry. Cell. 1990;62:631–647. doi: 10.1016/0092-8674(90)90110-z. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Nucleocytoplasmic transport: Signals, mechanisms and regulation. Nature. 1997;386:779–783. doi: 10.1038/386779a0. [DOI] [PubMed] [Google Scholar]
- O’Neill EM, Kaffman A, Jolly RJ, O’Shea EK. Regulation of PHO4 nuclear localization by the PHO80-PHO85 cyclin-CDK complex. Science. 1996;271:209–212. doi: 10.1126/science.271.5246.209. [DOI] [PubMed] [Google Scholar]
- Piatti S, Böhm T, Cocker JH, Diffley JFX, Nasmyth K. Activation of S-phase-promoting CDKs in late G1 defines a “point of no return” after which Cdc6 synthesis cannot promote DNA replication in yeast. Genes & Dev. 1996;10:1516–1531. doi: 10.1101/gad.10.12.1516. [DOI] [PubMed] [Google Scholar]
- Ruis H. Yeast stress responses: Achievements, goals and a look beyond yeast. In: Hohmann S, Mager WH, editors. Yeast stress responses. Austin, TX: Landes; 1997. pp. 231–247. [Google Scholar]
- Ruis H, Schüller C. Stress signaling in yeast. BioEssays. 1995;17:959–965. doi: 10.1002/bies.950171109. [DOI] [PubMed] [Google Scholar]
- Schmitt AP, McEntee K. Msn2p, a zinc finger DNA-binding protein, is the transcriptional activator of the multistress response in Saccharomyces cerevisiae. Proc Natl Acad Sci. 1996;93:5777–5782. doi: 10.1073/pnas.93.12.5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüller C, Brewster JL, Alexander MR, Gustin MC, Ruis H. The HOG pathway controls osmotic regulation of transcription via the stress response element (STRE) of the Saccharomyces cerevisiae CTT1 gene. EMBO J. 1994;13:4382–4389. doi: 10.1002/j.1460-2075.1994.tb06758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D-Y, Matsumoto K, Iida H, Uno I, Ishikawa T. Heat shock response of Saccharomyces cerevisiae mutants altered in cAMP-dependent protein phosphorylation. Mol Cell Biol. 1987;7:244–250. doi: 10.1128/mcb.7.1.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebenlist U, Brown K, Franzoso G. NF-κB: A mediator of pathogen and stress responses. In: Baeuerle P, editor. Inducible gene expression, Vol.1, Environmental stresses and nutrients. Boston, MA: Birkhäuser; 1995. pp. 93–142. [Google Scholar]
- Siderius M, Mager WH. General stress response: In search of a common denominator. In: Hohmann S, Mager WH, editors. Yeast stress responses. Austin, TX: Landes; 1997. pp. 213–230. [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevelein JM. Signal transduction in yeast. Yeast. 1994;10:1753–1790. doi: 10.1002/yea.320101308. [DOI] [PubMed] [Google Scholar]
- Tokiwa G, Tyers M, Volpe T, Futcher B. Inhibition of G1 cyclin activity by the Ras/cAMP pathway in yeast. Nature. 1994;371:342–345. doi: 10.1038/371342a0. [DOI] [PubMed] [Google Scholar]