

Abstract
Bacterial biofilm formation causes significant industrial economic loss and high morbidity and mortality in medical settings. Biofilms are defined as multicellular communities of bacteria encased in a matrix of protective extracellular polymers. Because biofilms have a high tolerance for treatment with antimicrobials, protect bacteria from immune defense, and resist clearance with standard sanitation protocols, it is critical to develop new approaches to prevent biofilm formation. Here, a novel benzimidazole molecule, named antibiofilm compound 1 (ABC-1), identified in a small-molecule screen, was found to prevent bacterial biofilm formation in multiple Gram-negative and Gram-positive bacterial pathogens, including Pseudomonas aeruginosa and Staphylococcus aureus, on a variety of different surface types. Importantly, ABC-1 itself does not inhibit the growth of bacteria, and it is effective at nanomolar concentrations. Also, coating a polystyrene surface with ABC-1 reduces biofilm formation. These data suggest ABC-1 is a new chemical scaffold for the development of antibiofilm compounds.
INTRODUCTION
Biofilm formation by bacterial pathogens is an increasing cause of morbidity and mortality associated with chronic and nosocomial infections. Biofilms are defined as conglomerations of bacterial cells protected by a self-synthesized extrapolymeric substance (EPS). Biofilms are notoriously difficult to eradicate and cause recalcitrant infections (11, 44). Bacterial biofilms are responsible for many chronic infections, including dental caries, osteomyelitis, and cystic fibrosis (14, 20, 26). Furthermore, biofilms are associated with 65% of nosocomial infections (30). Biofilm-based infections have been estimated to be responsible for upwards of 500,000 deaths and billions of dollars in treatment costs annually in the United States alone (44). Therefore, it is imperative to identify new strategies to combat biofilm-based infection.
Biofilm infections are particularly challenging to treat. Bacteria within a biofilm have increased tolerance for antibiotic treatment compared to planktonic cells (17, 30). This tolerance can allow biofilm-based bacteria to survive 1,000 times the dose of antibiotics that their planktonic counterparts can survive (9). Furthermore, biofilms are able to resist clearance by the host immune system due to exclusion of host cell antibodies and phagocytic cells (28). Biofilms interfere with clinical therapy for chronic, persistent, and wound-related infections on various indwelling medical devices (10). Biofilms also trigger inflammation and impair the wound-healing process (44). Frequently, the only effective treatment option for biofilm-based chronic wound infections is to amputate the infected limb (21).
Bacterial biofilm formation also causes significant economic loss in industrial settings (43). Any moist, nonsterile environment provides a fruitful ground for the development of biofilms. In industrial settings, equipment such as processing plants and cooling towers can harbor bacterial biofilms (7). These bacteria lead to corrosion of equipment, contamination of products, and loss of energy efficiency. Furthermore, biofilm formation on the hulls of ships, a process known as biofouling, significantly increases drag and reduces fuel efficiency (45).
Traditional antibiotics and disinfectants were developed for their ability to kill or inhibit the growth of planktonic bacteria, and they are often ineffective against biofilms. Bacteria also quickly develop resistance to such treatments. Therefore, there is an urgent need to identify new strategies that specifically target bacteria in the biofilm state. To this end, we performed a high-throughput screen to identify molecules that would prevent bacterial biofilm formation. From this screen, we determined that the molecule 5-methoxy-2-[(4-methylbenzyl)sulfanyl]-1H-benzimidazole, here named antibiofilm compound 1 (ABC-1), can efficiently inhibit biofilm formation by multiple bacterial pathogens, including Vibrio cholerae, Klebsiella pneumoniae, Shigella boydii, Erwinia amylovora, a cystic fibrosis (CF) isolate of Pseudomonas aeruginosa, and a methicillin-resistant Staphylococcus aureus (MRSA) isolate under both static and flow conditions. Biofilm formation was inhibited on a variety of surfaces, including polystyrene, glass, and silicone catheters. Importantly, ABC-1 does not significantly inhibit the growth of Gram-negative bacteria and impacts the growth of Gram-positive bacteria only at relatively high concentrations, leading to minimal selective pressure for resistance (15). While ABC-1 does not disperse preformed biofilms under the conditions examined here, ABC-1 embedded in a polymeric multilayer matrix effectively inhibits biofilm formation on a surface.
MATERIALS AND METHODS
Bacterial strains and media.
The bacterial strains and plasmids used in this study are listed in Table 1. V. cholerae, P. aeruginosa, K. pneumoniae, and S. boydii cells were grown at 37°C with constant aeration in Luria-Bertani (LB) broth. The lone exception was that V. cholerae was grown at 30°C for the chemical screen. S. aureus cells were grown at 37°C with constant aeration in tryptic soy broth (TSB). E. amylovora was grown at 28°C in LB broth. For biofilm growth, LB medium was used for all strains except S. aureus, for which TSB medium supplemented with 0.5% glucose and 3% NaCl was used. For expression studies, isopropyl β-d-1-thiogalactopyranoside (IPTG) was used at a concentration of 100 μM. When necessary, antibiotics were used at concentrations of 50 μM (kanamycin) and 5 μM (chloramphenicol).
Table 1.
Strains used in this study
| Species and/or strain | Relevant genotypea | Comment and/or parent strain | Reference or source |
|---|---|---|---|
| Vibrio cholerae | ΔhapR | C6706str2 | 32 |
| Pseudomonas aeruginosa | WT | Cystic fibrosis isolate CF 145 | Martha Mulks |
| Klebsiella pneumoniae | WT | Clinical isolate | 18 |
| Erwinia amylovora | WT | Ea1189 | 3 |
| Shigella boydii | WT | 3408-67 | STEC Center at Michigan State University |
| Staphylococcus aureus Newman | WT | Blood isolate | ATCC 25904 |
| Staphylococcus aureus USA300 | WT | CA-MRSA | 27 |
WT, wild type.
High-throughput screen.
We screened approximately 20,000 compounds from the ChemDiv diversity set, 16,000 compounds from the Maybrige HF library, 13,000 compounds from the Chembridge build block set, 2000 biologically active compounds from the MS2000 library, 446 compounds in the Biofocus-NCC, and 25,000 natural-product extracts from the laboratory of David Sherman at the University of Michigan Center for Chemical Genomics. A total of 0.2 μl compound (final concentration, 7.5 to 10 μM) was added to 20 μl of LB containing kanamycin, chloramphenicol, and IPTG in a 384-well plate. Twenty microliters of LB containing a 1:150 dilution of an overnight culture of the reporter strain was added to each well. The reporter strain consists of V. cholerae strain C6706str2 carrying two compatible plasmids. The first plasmid is an IPTG-inducible vector whereby the GGDEF enzyme VC1216 was placed under the control of the Ptac promoter, and the second vector encodes a transcriptional fusion of a cyclic di-GMP (c-di-GMP)-inducible promoter located near the gene VC1673 (unpublished data). This culture was incubated overnight at 30°C, and the absorbance (optical density at 600 nm [OD600]) and luminescence were determined using a Pherastar plate reader. Each plate contained one row of a negative control (no compound addition) and a positive control (no IPTG addition). Preliminary experiments revealed that dimethyl sulfoxide (DMSO) addition at the percentage used did not affect induction of the transcriptional reporter. Approximately 66,000 compounds were screened once, and 1,039 small molecules and 357 natural-product extracts exhibiting greater than 3 standard deviations difference from the negative control were rescreened in triplicate. The top 331 compounds from this rescreen were selected, and the 50% inhibitory concentration (IC50) was determined in duplicate.
Preparation of ABC-1.
To a mixture of 5-methoxy-2-mercaptobenzimidazole (1.673 g; 9.285 mmol) and p-xylyl bromide (1.718 g; 9.285 mmol) in dimethylformamide (DMF; 33.5 ml) at 0°C, Cs2CO3 (4.53 g, 13.93 mmol) was added, and the suspension was stirred for 24 h while being allowed to warm from 0°C to room temperature (RT). Distilled water (150 ml) was added with vigorous stirring. The murky white suspension turned into a clearer solution with white granules. After being stirred for 20 min at RT, the mixture was filtered, and the residue was rinsed with water (3 × 20 ml) and hexanes (3 × 20 ml). The crude product was >95% pure by 1H nuclear magnetic resonance (NMR) analysis and was not further purified. The yield was 2.437 g (92%). 1H NMR (500 MHz, CDCl3) δ 2.25 (s, 3H), 3.76/3.78 (apparent d, 3H), 4.38/4.42 (apparent d, 2H), 6.74 to 7.17 (m, 6H), 7.52 (d, 1H, J = 7.5 Hz), 8.87 (broad signal, 1H). 13C NMR (125 MHz, CDCl3) δ 21.30, 37.39, 37.63, 55.89, 55.96, 94.15, 101.18, 110.39, 111.10, 112.45, 119.07, 128.98, 129.58, 133.94, 156.61. (The apparent doublets may arise from the 1,3-proton shift between the nitrogen atoms in the heterocycle.)
Biofilm assays.
Biofilm formation was measured under both static and flow conditions. For the static condition, we used a quantitative crystal violet assay on polystyrene 96-well and minimum biofilm eradication concentration (MBEC) plates (Biosurface Technologies, Bozeman, MT) as described previously (13, 38). The MBEC technology is a 96-well plate cover containing 96 polystyrene pegs that sit in the 96 wells of a conventional plate. Briefly, cultures grown overnight were standardized to an OD595 of 0.1, 200 μl was transferred to the wells of a 96-well polystyrene microtiter plate (Corning, Inc., Corning, NY), and the MBEC lid was placed on top of the wells. Biofilms were grown on the pegs of the lid under shaking conditions for 24 h. The lid was removed, and the pegs were gently washed twice with 200 μl of phosphate-buffered saline to remove nonadherent cells. Adherent biofilms on the pegs were fixed with 200 μl of 100% ethanol prior to staining them for 2 min with 200 μl of 0.41% (wt/vol) crystal violet in 12% ethanol (Protocol crystal violet; Biochemical Sciences, Swedesboro, NJ). The pegs were washed several times with phosphate-buffered saline to remove excess stain. Quantitative assessment of biofilm formation was obtained by immersing the pegs in a sterile polystyrene microtiter plate containing 200 μl of 100% ethanol and incubating it at room temperature for 10 min (35), and the absorbance at 595 nm was determined using a SpectraMax M5 microplate spectrophotometer system (Molecular Devices, Sunnyvale, CA). Three independent experiments were performed for each of these assays, with each experiment representing three wells.
For biofilm experiments under flow conditions, biofilms were grown in disposable flow cells (Stovall Life Science, Greensboro, NC) as previously described (38). Each flow cell channel measures 1 mm deep, 4 mm wide, and 40 mm long. In brief, the inlet side of the flow cell was connected to a sterile reservoir filled with the appropriate growth medium. The outlet side was connected to a waste reservoir to create a “once-through” flow cell system. Tubing upstream of each individual cell was injected with 0.5 ml of standardized overnight culture of the test strain, and the chamber was incubated in upright position at 37°C for 1 h. The flow was then resumed, with a flow rate of 0.2 ml/min for V. cholerae, P. aeruginosa, S. boydii, S. aureus, and K. pneumoniae and a flow rate of 0.15 ml/min for E. amylovora. For flow cell experiments, S. aureus was grown in TSB supplemented with 0.5% glucose and 3% NaCl. For the Gram-negative bacteria except E. amylovora, the bacteria were grown in LB broth. E. amylovora was grown in 0.5× LB broth with 3% sucrose. The nonadherent bacteria were eventually flushed by the flow of the medium, thereby replacing the volume of the flow cell once every minute. Adherent cells were stained with the Live/Dead cell viability assay stain (Invitrogen). This assay stains live cells green and dead cells red. Very few dead cells (less than 1%) were visualized in any of the experiments. Biofilm formation on the flow cell was imaged both macroscopically and microscopically at 24 h and 48 h. Three sections of the flow cell chosen randomly were imaged, and representative images are shown. Each section represents dimensions of 250 μm by 250 μm with a resolution of 512 by 512 pixels and shows the same depth. Cross sections of each section were performed at 0.5 to 1 μm for different pathogens. Heat intensity plots were generated from the z-stack images by the Zeiss LSM 5 PASCAL Software, which translates the fluorescence intensity of the stained cells into a relative intensity output. Catheter experiments were performed by replacing the three-chambered flow cell with a 14 French 5-ml all silicone catheter (Bard, Inc., Covington, GA). For biofilm dispersal studies, biofilms were allowed to develop in the flow cells for 24 h. This was followed by the addition of 100 μM ABC-1 (stock ABC-1 was dissolved in DMSO) to the growth medium, and biofilm dispersal was monitored at regular intervals by confocal laser scanning microscopy (CLSM).
Microscopy.
For CLSM analysis of biofilms, the medium flow was stopped, and the fluorescent dyes Syto-9 and propidium iodide (Molecular Probes, Eugene, OR) were injected into the flow cell chamber and incubated for 30 min in the dark. Confocal microscopic images were acquired using either an Olympus FluoView 1000 Laser Scanning Confocal Microscope (Olympus America Inc., Melville, NY) or a Carl Zeiss Pascal Laser Scanning Microscope (Carl Zeiss, Jena, Germany) equipped with a 63×/1.4-numerical-aperture Plan-Apochromat objective. The Syto-9 and propidium iodide fluorophores were excited with an argon laser at 488 nm; the emission band-pass filters used for Syto-9 and propidium iodide were 515 ± 15 nm and 630 ± 15 nm, respectively. CLSM z-stack image analysis and processing were performed using either Carl Zeiss LSM 5 PASCAL software (version 3.5) or Olympus Fluoview software, respectively. Image stacks of biofilms were acquired from at least three distinct regions on the flow cell. The thickness of the biofilm was measured starting from the z-section at the flow-cell/biofilm interface to the z-section at the top of the biofilm surface containing <5% of the total biomass. Three-dimensional (3D) images of the biofilm were rendered using the Zeiss LSM image application.
Preparation of antimicrobial-compound polyelectrolyte multilayer thin films.
Unless otherwise indicated, all solutions were prepared using deionized (DI) water supplied by a Barnstead Nanopure-UV 4-stage purifier (Barnstead International, Dubuque, IA) equipped with a UV source and a final 0.2-μm filter with resistivity greater than 18 MΩ · cm. The polymers used in this study were branched polyethylenimine (BPEI), polyacrylic acid (PAA), and polyethylene glycol (PEG). BPEI (mass, approximately 25,000 Da), PAA sodium salt (35% [wt/vol] solution in water; mass, approximately 15,000 Da), and PEG (mass, approximately 10,000 Da) were purchased from Sigma-Aldrich, St. Louis, MO. The concentrations of BPEI, PAA, and PEG were maintained at 1 mg/ml. Deposition of the multilayers was performed as described previously. In brief, an automated Carl Zeiss slide stainer with a custom-designed ultrasonic bath was used to deposit multilayers as described previously (23) on the polystyrene surface of the MBEC assay plate. The robotic arm of the slide stainer to which the MBEC assay plate is attached can be programmed to carry out the immersion, washing, and sonication steps for the specified period. Before depositing the multilayers, the surface of the polystyrene plate was extensively cleaned using a Harrick plasma cleaner (Harrick Scientific Corporation, Broading Ossining, NY) for 15 min. The pressure and oxygen flow rate in the plasma chamber were maintained at 0.15 torr and 50 standard cm3 per min, respectively, as previously described (23).
The multilayers incorporating antibiofilm compound were deposited using an approach similar to that described elsewhere (31). Briefly, the O2-plasma-cleaned plate was dipped in BPEI solution prepared as described above (the pH was adjusted to approximately 10.5) for 30 min, followed by dipping it in DI water (pH = 10.5) twice for 10 min each time to remove the loosely bound polymer. Thereafter, the plate was dipped in PAA and PEG solutions for 20 min each (the pH was adjusted to 2.0) to deposit one bilayer. After every polymer dipping cycle, the plate was washed twice with DI water (pH 2.0). Five such bilayers of PAA/PEG were deposited. PEG formed the final layer.
Ten milligrams of ABC-1 dissolved in 2 ml DMSO was subsequently diluted to 100 μM by adding DI water (pH 2.0). Five and one-half bilayers of compound/PEG were deposited on the PAA/PEG-coated plate using the same immersion and washing steps described above. ABC-1 formed the final layer. The plate was cured for 30 min using a UV sterilizer once the multilayer deposition was complete.
RESULTS
ABC-1 and 2-[(4-chlorobenzyl)thio]-5-methoxy-1H-benzimidazole (ABC-2) inhibit biofilm formation in V. cholerae.
We performed a screen of approximately 66,000 compounds and natural-product extracts at the Center for Chemical Genomics at the University of Michigan to identify novel compounds that inhibit biofilm formation. The premise behind the screen was to identify small molecules that negatively impacted induction of a V. cholerae c-di-GMP-inducible transcriptional fusion. c-di-GMP is a second-messenger signal that induces biofilm formation in the majority of bacteria (19). Therefore, we hypothesized that compounds that could reduce the expression of a c-di-GMP-responsive transcriptional reporter might have antibiofilm properties, either through a reduction in the level of c-di-GMP or via other mechanisms.
The transcription fusion used in the screen encoded a c-di-GMP-induced promoter we have recently identified located in the VC1673 gene fused to a luciferase reporter (unpublished data). This fusion was inserted into a V. cholerae strain in which the levels of c-di-GMP were artificially induced through overexpression of the c-di-GMP synthesis enzyme, VC1216, encoded on a second plasmid. The compounds and natural-product extracts were screened in a 384-well format. In addition to measuring luciferase expression, cell growth was simultaneously quantified by measuring the OD600 of each well. Compounds that inhibited luciferase expression at levels greater than 3 standard deviations from the negative-control wells without significantly impacting growth were considered hits and screened further in triplicate. Dose-response curves were performed in duplicate on the top 331 hits to determine the concentration of each compound that inhibits luciferase expression by 50% (IC50). Importantly, the screen was performed using intact, growing bacteria. Therefore, any hits that showed activity must have been able to cross the lipid bilayer of bacteria (or signal from across the cytoplasmic membrane). Moreover, we excluded from further analysis any compounds that significantly inhibited the growth of bacteria because it was hypothesized that anti-infective strategies that reduced virulence without blocking or inhibiting growth would lead to less selection for resistant strains (6).
From the screen, we identified 188 compounds that inhibited VC1673-lux with an IC50 lower than 10 μM. Here, we report the activities of two structurally related benzimidazole compounds from this group, ABC-1 and ABC-2, that possess antibiofilm activity (Fig. 1). To examine the antibiofilm properties of ABC-1 and ABC-2, V. cholerae was grown in the presence of 10 μM ABC-1 or ABC-2, and biofilm formation was determined in a static culture using the MBEC crystal violet biofilm assay (see Materials and Methods). Each of these compounds significantly inhibited V. cholerae biofilm development (Fig. 2). We chose to more fully characterize the antibiofilm activity of ABC-1, as it more strongly inhibited biofilms in V. cholerae.
Fig. 1.
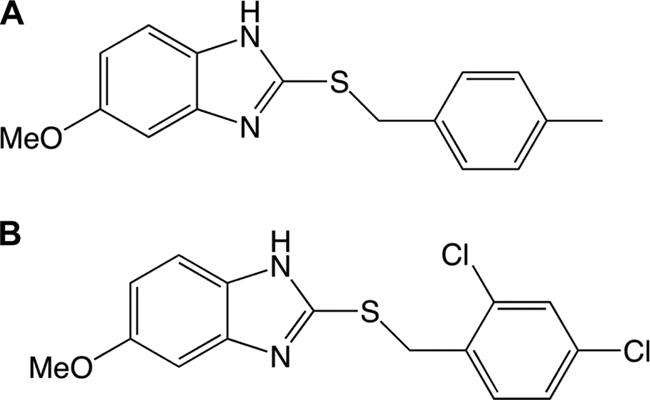
Chemical structures of ABC-1, 5-methoxy-2-[(4-methylbenzyl)sulfanyl]-1H-benzimidazole (A), and ABC-2, 2-[(4-chlorobenzyl)thio]-5-methoxy-1H-benzimidazole (B).
Fig. 2.

ABC-1 and ABC-2 inhibit biofilm formation by V. cholerae. V. cholerae was grown in LB medium for 8 h in the wells of MBEC plates with 10 μM ABC-1 or ABC-2 or an equivalent amount of DMSO. Biofilm was quantified by staining with crystal violet and elution with ethanol as described in the text. The results represent the means plus standard errors of the mean (SEM). Student's paired t test was used to determine the statistical significance (*, P < 0.05).
ABC-1 is a broad-spectrum inhibitor of biofilm formation.
ABC-1 possessed potent antibiofilm activity toward V. cholerae; however, the most useful compounds possess antibiofilm properties against a broad spectrum of different bacterial pathogens. We therefore examined the activity of ABC-1 to inhibit biofilm formation by five human pathogens and one plant pathogen in the MBEC static biofilm assay. The bacteria we chose to examine were a CF clinical isolate of P. aeruginosa (CF-145), K. pneumoniae, E. amylovora, S. boydii, MRSA strain USA300, and S. aureus strain Newman. The rationale for examining each of these species of bacteria is discussed below. ABC-1 was used at a concentration of 100 μM for the Gram-negative bacteria and 25 μM for S. aureus. Similar to our results for V. cholerae, ABC-1 significantly inhibited biofilms in each of these bacteria in the MBEC static biofilm assay (Fig. 3). It should be noted that ABC-1 did not significantly affect the growth of any of these pathogens at the concentrations used. From these data, we conclude that ABC-1 possesses broad-spectrum antibiofilm activity against multiple bacterial pathogens.
Fig. 3.

ABC-1 exhibits broad-spectrum antibiofilm activity. Cultures were incubated in the wells of MBEC plates, and biofilm formation was quantitated by staining with crystal violet and elution with ethanol as described in the text. ABC-1 was used at a concentration of 100 μM for the Gram-negative bacteria and 25 μM for S. aureus. The results represent the means plus SEM. Student's paired t test was used to determine the statistical significance of the treated versus untreated conditions (*, P < 0.05).
ABC-1 is active against V. cholerae and P. aeruginosa at nanomolar concentrations.
The IC50 to inhibit biofilm formation for both V. cholerae and P. aeruginosa CF-145 was determined using the MBEC assay by measuring biofilm formation across a range of ABC-1 concentrations in duplicate. The data were fitted to a log (inhibitor) versus response curve using nonlinear regression with variable-slope analysis (Graph Pad Prism). The IC50 of ABC-1 to inhibit biofilm formation of V. cholerae was 32.3 nM, with a 95% confidence interval from 14.1 to 74.4 nM. Similarly, the calculated IC50 of ABC-1 to inhibit biofilm formation of P. aeruginosa was 45.9 nM, with a 95% confidence interval of 44.3 to 47.5 nM (Fig. 4). Thus, ABC-1 possesses antibiofilm activities at nanomolar concentrations and inhibits biofilm formation at similar concentrations in both V. cholerae and P. aeruginosa.
Fig. 4.

ABC-1 inhibits biofilm formation by V. cholerae and P. aeruginosa at nanomolar concentrations. A concentration response curve measuring the antibiofilm activity of ABC-1 was performed in duplicate. P. aeruginosa and V. cholerae are represented as circles and squares, respectively. The calculated IC50 for P. aeruginosa is 45.9 nM with a 95% confidence interval (error bars) of 44.3 to 47.5. The IC50 for V. cholerae is 32.3 nM with a 95% confidence interval of 14.1 to 74.4.
ABC-1 inhibits biofilm formation of V. cholerae under flow conditions.
Our results thus far showed that ABC-1 possessed broad-spectrum antibiofilm activity for inhibition of biofilm formation under static conditions. However, natural biofilms and biofilms associated with disease typically form in environments exposed to fluid flow. Therefore, we analyzed the impact of ABC-1 on biofilm formation using flow cells exposed to a continuous flow of fresh medium to better approximate physiologically relevant biofilm formation. We first measured V. cholerae biofilm formation using a flow cell apparatus that enabled noninvasive observation of biofilm formation under a continuous flow in the presence and absence of 10 μM ABC-1. After 24 h, the biofilms were stained with a Live/Dead cell viability assay stain to differentiate between intact and compromised cells, and biofilms were visualized using CLSM. This analysis allowed us to generate a three-dimensional view of the biofilm through the measurement of signal intensity. The biofilm structure can be observed as an intensity map in Fig. 5A and B, where the intensity of the signal represents the density of cells in a particular location. These results show that 10 μM ABC-1 dramatically reduced biofilm formation under shear conditions.
Fig. 5.

ABC-1 inhibits biofilm formation by multiple Gram-negative pathogens under flow conditions. Shown is a three-dimensional view of biofilm in flow cells through intensity mapping after 24 to 48 h of growth for V. cholerae (A and B), P. aeruginosa CF-145 (C and D), K. pneumoniae (E and F), E. amylovora (G and H), and S. boydii (I and J). (A, C, E, G, and I) Biofilm growth in the absence of ABC-1. (B, D, F, H, and J) Biofilm growth in the presence of ABC-1. The intensity mapping had the maximum value set at 250, and the maps were developed with five color layers. Additional layers of color indicate greater intensity of the signal.
ABC-1 inhibits biofilm formation in multiple Gram-negative pathogens.
V. cholerae persists as biofilms in environmental reservoirs, but the importance of biofilm formation during infection remains unclear (22). Therefore, we determined the ability of ABC-1 to inhibit biofilm formation by several pathogens for which biofilm formation is an important virulence determinant. One such pathogen is P. aeruginosa, the most prominent pathogen in lung infections of CF individuals. Biofilm formation by P. aeruginosa during a CF infection is hypothesized to be an important survival strategy in the colonized lung (reviewed in reference 20). Over time, P. aeruginosa colonizing the lung mutates into a mucoid biotype, which is associated with higher levels of cyclic di-GMP and robust biofilm formation. The mucoid biotype leads to chronic colonization that cannot be cleared with antibiotic treatment (15). Although aggressive antibiotic therapy can reduce the rate of lung damage, these chronic infections eventually lead to lung failure, necessitating a lung transplant (12).
The CF-145 P. aeruginosa strain that we examined in the MBEC assay is a mucoid isolate (Fig. 3). Similar to our results under static conditions, ABC-1 possessed potent antibiofilm activity against the CF strain under flow conditions. In the untreated flow chamber, biofilm growth was observed throughout the chamber, attached to the surface and exhibiting vertical growth (Fig. 5C). In contrast, P. aeruginosa treated with 100 μM ABC-1 had greatly reduced biofilm formation (Fig. 5D). ABC-1 treatment led to a low signal intensity of fluorescence and no signs of aggregation after 24 h. Also, similar results were obtained when a laboratory isolate of P. aeruginosa (strain PA01) was treated with ABC-1 (data not shown).
We also determined the antibiofilm activity of ABC-1 against K. pneumoniae under flow conditions. K. pneumoniae is an important opportunistic pathogen commonly associated with nosocomial urinary tract infections (UTIs), including catheter-associated UTIs (CAUTIs), as well as sepsis and pneumonia (36). ABC-1 treatment at 100 μM K. pneumoniae inhibited biofilm formation under flow conditions at 24 h both in static MBEC assays (Fig. 3) and under continuous-flow conditions (Fig. 5, compare panels E and F). In the untreated flow cell, thick cellular aggregates were present throughout the flow cell chamber (Fig. 5E), while this characteristic feature was absent in ABC-1 treatment cells accompanied by less surface coverage (Fig. 5F). In addition, the biofilms that did form in the presence of ABC-1 began to disperse after 30 h, while the untreated K. pneumoniae bacteria continued to make uniform biofilms (data not shown).
E. amylovora, the causative agent of fire-blight infections, leads to wilting of infected plants by blocking xylem function through the formation of biofilms (25). Therefore, we determined the antibiofilm activity of ABC-1 on E. amylovora. Identical to our results described above, addition of 100 μM ABC-1 produced a pronounced decrease in E. amylovora biofilm development under both static (Fig. 3) and continuous-flow (Fig. 5G and H) conditions. Untreated E. amylovora developed thick biofilms, whereas ABC-1-treated E. amylovora showed sparser distribution of biofilms in the flow chamber (Fig. 5H).
We next examined the antibiofilm activity of ABC-1 against S. boydii, a pathogen known to cause dysentery in humans through fecal-oral contamination. S. boydii is a prominent pathogen in the food production sector, where biofilm formation in food-processing equipment has been implicated in food-borne outbreaks (4). Furthermore, it was previously demonstrated that S. boydii can persist and form biofilms on the surfaces of parsley plants, which may be another mechanism of product contamination (2). This feature contributes to the ineffectiveness of aqueous sanitizers in inactivating human pathogens found on plant tissues. Therefore, antibiofilm strategies targeting S. boydii that can be used alone or in combination with sanitizers are needed. As seen with the other bacteria we examined, development of S. boydii biofilms was significantly impacted upon treatment with 100 μM ABC-1 under both static (Fig. 3) and flow (Fig. 5I and J) conditions. S. boydii produced less robust biofilm formation under the conditions examined than the other pathogens studied. Nevertheless, at 48 h, biofilm formation by S. boydii in the flow chamber was observed (Fig. 5I). Treatment with ABC-1 greatly reduced biofilm formation by S. boydii (Fig. 5 J).
ABC-1 inhibits biofilm formation by S. aureus.
The above-mentioned results show that ABC-1 possesses broad-spectrum antibiofilm activity against multiple Gram-negative bacterial pathogens. However, Gram-positive bacteria, like S. aureus, Staphylococcus epidermidis, and Streptococcus, are problematic biofilm-forming pathogens. S. aureus is one of the most important pathogens that cause infections associated with indwelling medical devices, such as prosthetic joints, prosthetic heart valves, and intravascular catheters, thereby creating an enormous health care problem (47).
To examine if ABC-1 exhibits antibiofilm properties against Gram-positive bacteria, we examined the activity of ABC-1 against two strains of the Gram-positive pathogen S. aureus: (i) S. aureus Newman, a blood isolate, and (ii) the USA300 community-acquired (CA)-MRSA strain. Before examining the antibiofilm properties of ABC-1, we initially determined the impact of ABC-1 on the growth of S. aureus. Interestingly, we found that ABC-1 concentrations of 50 μM or greater inhibited the growth of S. aureus (data not shown). This finding contrasts with our results with Gram-negative bacteria, in which ABC-1 did not inhibit growth at these concentrations. Because we are interested in the antibiofilm activity of ABC-1 rather than its growth inhibition properties, we examined the impact of 25 μM ABC-1 on S. aureus biofilm formation to avoid activity due to toxicity effects of the compound. Inhibition of biofilm formation by both S. aureus strains under static conditions was observed to be statistically significant (P < 0.05) but less robust than that observed for most Gram-negative pathogens (Fig. 3). However, greater inhibition of biofilms by ABC-1 was observed under flow conditions. When subjected to shear conditions, S. aureus strain Newman in the absence of ABC-1 formed significant biofilm on the surface of the flow cell (Fig. 6A). The biofilm was patchy and consisted of numerous mushroom-like structures. In contrast, ABC-1-treated S. aureus exhibited less surface area coverage, and biofilms were present as small patches (Fig. 6B). Similarly, biofilm formation by CA-MRSA in the presence of ABC-1 was reduced compared to the untreated condition (Fig. 6C and D). These continuous-flow condition experiments on S. aureus revealed that ABC-1 treatment significantly decreased biofilm formation by S. aureus Newman and CA-MRSA.
Fig. 6.

ABC-1 inhibits biofilm formation by S. aureus under flow conditions. Shown is a three-dimensional view of biofilm in flow cells through intensity mapping of S. aureus Newman (A and B) and S. aureus USA300 (C and D). (A and C) Biofilm growth in the absence of ABC-1. (B and D) Biofilm growth in the presence of ABC-1.
ABC-1 inhibits biofilm formation on a silicone catheter.
Thus far, we have shown that ABC-1 inhibits biofilm formation on a polystyrene surface under static conditions and a glass surface exposed to fluid flow. To explore the activity of ABC-1 on a medically relevant surface, we examined biofilm formation by P. aeruginosa strain PA01 on the surface of a medical silicone catheter under flow conditions in the absence and presence of 100 μM ABC-1. Untreated P. aeruginosa was able to form robust biofilms on the inside surface of the catheter, as could be observed macroscopically (Fig. 7A). CLSM analysis of these biofilms revealed thick, even biofilms covering the entire surface (Fig. 7B). Treatment with ABC-1, however, virtually abolished biofilm of P. aeruginosa on the catheter. No obvious macroscopic biofilm was observed on the catheter (Fig. 7A), and very few cells were attached, as measured by CLSM (Fig. 7B).
Fig. 7.
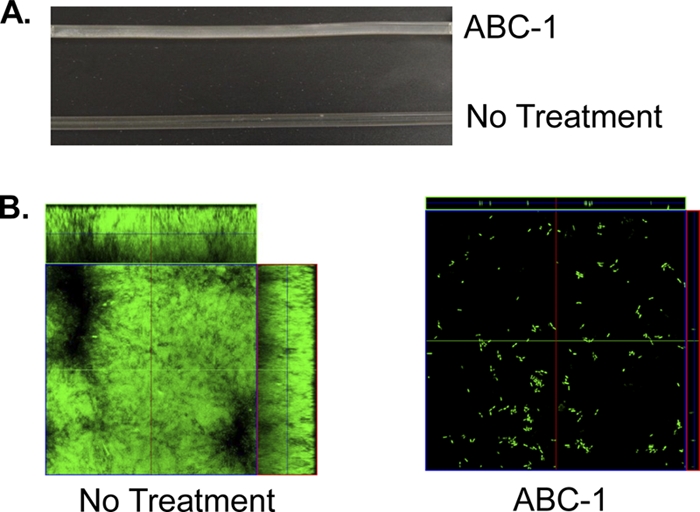
ABC-1 inhibits biofilm formation on catheters. Biofilm formation by P. aeruginosa PA01 was determined macroscopically (A) and using CLSM (B) on a silicone catheter in the absence and presence of 100 μM ABC-1. The biofilms were grown for 48 h.
Impact of ABC-1 on biofilm dispersion.
In the experiments described thus far, ABC-1 was added concurrently with bacterial inoculation. We wondered if ABC-1 was capable of dispersing preformed biofilms. For these experiments, biofilms of K. pneumoniae and P. aeruginosa strain CF-145 grown to 24 h were exposed to a continuous flow of medium with 100 μM ABC-1. After an additional 3, 6, and 24 hours of incubation, the treated and untreated biofilms were visualized by CLSM to determine if dispersal had occurred. ABC-1 did not cause significant architectural changes in preformed biofilms (Fig. 8A to D).
Fig. 8.

ABC-1 does not disperse preformed biofilms. Shown is a three-dimensional view of biofilm formation in flow cells through intensity mapping of K. pneumoniae (A and B) and P. aeruginosa CF-145 (C and D). (A and C) Untreated conditions. (B and D) ABC-1 was added after the biofilms had been allowed to form for 24 h. The images shown are 3 h after addition of ABC-1.
Coating surfaces with ABC-1 reduces biofilm formation.
Biofilm-based bacterial disease is often associated with biofilm development on indwelling medical devices, such as catheters and prosthetic joints (8). One strategy for reducing these infections is to treat the device surfaces with antimicrobials or antibiofilm compounds to prevent the formation of biofilms. To determine if ABC-1 can be used in this manner, the polystyrene surfaces of MBEC pegs were coated with ABC-1 using pH-sensitive polyelectrolyte multilayers (see Discussion). Biofilm formation by V. cholerae and P. aeruginosa CF-145 was then measured on regular MBEC pegs, MBEC pegs coated with the polymer only, and MBEC pegs coated with the polymer containing ABC-1. Our results showed that biofilm formation by both pathogens was reduced at least 50% compared to the untreated or the polyelectrolyte control surfaces (Fig. 9). Thus, ABC-1 inhibited biofilm formation on a coated surface.
Fig. 9.

Coating of a surface with ABC-1 reduces biofilm formation. Cultures of V. cholerae and P. aeruginosa were allowed to form biofilms on the pegs of an MBEC plate that were coated with polymer and ABC-1 or polymer only or that were uncoated. Biofilm was quantitated by staining the pegs with crystal violet and elution with ethanol as described in the text. The results represent the means and SEM from at least three independent experiments. Student's paired t test was used to compare the biofilm growth on an uncoated surface to that on an ABC-1-coated surface (*, P < 0.05).
ABC-1 does not impact c-di-GMP signaling.
We hypothesized that ABC-1 could inhibit biofilms through inhibition of c-di-GMP signaling. To test this, the levels of c-di-GMP were examined in V. cholerae and P. aeruginosa PA01 treated with 35 μM ABC-1 and untreated, using HPLC tandem mass spectrometry. In comparison to untreated controls, growth of V. cholerae in ABC-1 led to a modest reduction in intracellular c-di-GMP, but ABC-1 had no effect on the levels of c-di-GMP in P. aeruginosa (data not shown). Furthermore, we established an in vitro assay to measure the activity of the diguanylate cyclase (DGC) enzyme VC2370, which synthesizes c-di-GMP, and the phosphodiesterase (PDE) enzyme VieA, which degrades c-di-GMP (42). ABC-1 did not significantly affect the in vitro activities of these enzymes (data not shown). Consistent with these results, S. aureus is not predicted to encode enzymes that synthesize or degrade c-di-GMP, yet ABC-1 was able to inhibit biofilm formation in the species. Therefore, we conclude that ABC-1 inhibits biofilm formation through an unknown mechanism that is independent of c-di-GMP signaling. The mechanism by which ABC-1 exerts antibiofilm activity is under investigation.
DISCUSSION
Here, we describe a novel benzimidazole antibiofilm compound, ABC-1, that inhibits biofilm formation in several prominent biofilm-forming pathogens. ABC-1 is a benzimidazole that possesses strong structural similarity to the drugs omeprazole and lansoprazole, which are used extensively as proton pump inhibitors to control stomach acid secretion. Interestingly, these compounds also have antimicrobial actions against Helicobacter pylori (34). In recent years, benzimidazoles have been shown to inhibit the growth of oral bacteria in acid environments, such as progressing caries lesions (33). Importantly, benzimidazoles and their derivatives possess anti-inflammatory activity (1). Because ABC-1 has strong structural similarity to these molecules, it is likely that the presence of ABC-1 in medical devices will not cause an inflammatory response. Furthermore, the potential anti-inflammatory activity of ABC-1 could be beneficial in treating chronic infections, such as those found in the CF lung, where it is thought that host inflammation is responsible for much of the host cell damage observed (20). In addition, benzimidazoles have low toxicity (16, 39). We have determined that omeprazole does not possess significant antibiofilm activity under the conditions we examined (data not shown), suggesting ABC-1 has novel properties not found in the benzimadazoles used clinically.
Recently, a number of 2-aminobenzimidazoles were found to have antibiofilm activity by using a rational design strategy (37). These compounds contain the bicyclic benzimidazole structure found in both ABC-1 and ABC-2 but are otherwise structurally quite different. Active analogs have a 2-NH2 group and a functionalized amino substituent at C-6, in contrast to ABC-1 and -2, which bear a functionalized thioether unit at C-2. Furthermore, the 2-aminobenzimidazoles showed no antibiofilm activity toward P. aeruginosa or Acinetobacter baumannii, leading to the conclusion that these compounds are not effective against Gram-negative pathogens (37). Again in contrast, ABC-1 exhibits broad-spectrum activity against multiple Gram-negative bacteria, including P. aeruginosa. We hypothesize that this difference might be due to increased diffusion of ABC-1 across the outer membrane of Gram-negative bacteria compared with the 2-aminobenzimidazoles. Nevertheless, based on the work of Rogers et al. (37) and our findings here, the benzimidazole functional group is a promising scaffold for the development of new antibiofilm compounds.
The increased prevalence of antibiotic-resistant bacteria heralds a need for new drugs and novel strategies to identify better drug targets. Several screening strategies have been employed to identify small-molecule inhibitors of biofilm development. The quest for such small-molecule biofilm inhibitors using either target- or structure-based screening has led to the identification of a significant number of biofilm inhibitors (reviewed in reference 40). Coadministration of antibiotics and antibiofilm molecules could form the basis of future clinical protocols against biofilm-based infections.
Biofilm encroachment on biomaterial is an important concern postsurgery. In the past, several studies of modifying surfaces have shown that pH-sensitive polyelectrolyte multilayers can be effectively used to release therapeutics at a specific pH (24, 46). Drugs are incorporated within weak polyelectrolyte assemblies at low pH due to hydrogen bonding of the small molecule with the matrix to form thin multilayers. This hydrogen bonding is disrupted at higher pH, contributing to release of the drugs incorporated in these multilayers (5, 41). This approach of using pH-sensitive erasable multilayers to release lysozyme, a protein similar to brain-derived neurotrophic factor (BDNF), was recently extended to polyacrylic acid/polyethylene glycol assembly (31). In the present work, we incorporated the antibacterial compound ABC-1 in the multilayer assembly at low pH. The compound was released due to disruption in H bonding when the films were immersed in solutions with neutral pH. Using this approach, we showed that surfaces modified with ABC-1 are able to significantly inhibit biofilm formation by V. cholerae and a cystic fibrosis isolate of P. aeruginosa.
ABC-1 possesses a number of properties that are attractive for drug development. ABC-1 is a low-molecular-weight compound that is structurally related to pharmaceuticals that are currently in widespread use, and ABC-1 inhibits biofilm formation at nanomolar concentrations. The chemical properties of ABC-1 fit those described by Lipinski et al. as important for compounds that are effective as therapeutics in humans (29). Furthermore, ABC-1 is easily synthesized in significant quantities using a one-step synthesis procedure. In preliminary studies, we have found that ABC-1 has no overt toxicity to mice at high concentrations (unpublished results), which is not surprising based on its similarity to omeprazole and lansoprazole. ABC-1 inhibits biofilm formation on multiple surface types in a broad-spectrum fashion. Moreover, ABC-1 is able to prevent biofilm formation when it is added in solution or used to coat a surface. Our results open the possibility of using ABC-1-modified materials for the construction of biofilm-resistant catheters and implantable biomaterial, given its broad-spectrum activity against both Gram-positive and Gram-negative bacterial strains. Therefore, ABC-1 exhibits potential as a new therapeutic strategy to inhibit biofilm formation.
ACKNOWLEDGMENTS
This work was supported by NIH grant 1K22AI080937-01 and support from the Region V “Great Lakes” RCE (NIH award 2-U54-AI-057153) to C.M.W. K.S. was supported by a postdoctoral research fellowship from the Michigan State University Center for Waters Sciences.
We thank Martha Mulks and Shannon Manning for sharing strains and Eric Kim for preparing Fig. 1. We thank Martha Larsen and Steve vander Roest at the Center for Chemical Genomics at the University of Michigan for help with the small-molecule screen.
Footnotes
Published ahead of print on 27 June 2011.
REFERENCES
- 1. Achar K. C., Hosamani K. M., Seetharamareddy H. R. 2010. In-vivo analgesic and anti-inflammatory activities of newly synthesized benzimidazole derivatives. Eur. J. Med. Chem. 45:2048–2054 [DOI] [PubMed] [Google Scholar]
- 2. Agle M. E., Martin S. E., Blaschek H. P. 2005. Survival of Shigella boydii 18 in bean salad. J. Food Prot. 68:838–840 [DOI] [PubMed] [Google Scholar]
- 3. Burse A., Weingar H., Ullrich M. S. 2004. NorM, an Erwinia amylovora multidrug efflux pump involved in vitro competition with other epiphytic bacteria. Appl. Environ. Microbiol. 70:693–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chan Y. C., Blaschek H. P. 2005. Comparative analysis of Shigella boydii 18 foodborne outbreak isolate and related enteric bacteria: role of rpoS and adiA in acid stress response. J. Food Prot. 68:521–527 [DOI] [PubMed] [Google Scholar]
- 5. Chung A. J., Rubner M. F. 2002. Methods of loading and releasing low molecular weight cationic molecules in weak polyelectrolyte multilayer films. Langmuir 18:1176–1183 [Google Scholar]
- 6. Clatworthy A. E., Pierson E., Hung D. T. 2007. Targeting virulence: a new paradigm for antimicrobial therapy. Nat. Chem. Biol. 3:541–548 [DOI] [PubMed] [Google Scholar]
- 7. Coetser S. E., Cloete T. E. 2005. Biofouling and biocorrosion in industrial water systems. Crit. Rev. Microbiol. 31:213–232 [DOI] [PubMed] [Google Scholar]
- 8. Costerton W., et al. 2003. The application of biofilm science to the study and control of chronic bacterial infections. J. Clin. Invest. 112:1466–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Evans R. C., Holmes C. J. 1987. Effect of vancomycin hydrochloride on Staphylococcus epidermidis biofilm associated with silicone elastomer. Antimicrob. Agents Chemother. 31:889–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fux C. A., Costerton J. W., Stewart P. S., Stoodley P. 2005. Survival strategies of infectious biofilms. Trends Microbiol. 13:34–40 [DOI] [PubMed] [Google Scholar]
- 11. Hall-Stoodley L., Costerton J. W., Stoodley P. 2004. Bacterial biofilms: from the natural environment to infectious diseases. Nat. Rev. Microbiol. 2:95–108 [DOI] [PubMed] [Google Scholar]
- 12. Hansen C. R., Pressler T., Hoiby N. 2008. Early aggressive eradication therapy for intermittent Pseudomonas aeruginosa airway colonization in cystic fibrosis patients: 15 years experience. J. Cyst. Fibros. 7:523–530 [DOI] [PubMed] [Google Scholar]
- 13. Harrison J. J., et al. 2010. Microtiter susceptibility testing of microbes growing on peg lids: a miniaturized biofilm model for high-throughput screening. Nat. Protoc. 5:1236–1254 [DOI] [PubMed] [Google Scholar]
- 14. Hassett D. J., et al. 2009. Pseudomonas aeruginosa hypoxic or anaerobic biofilm infections within cystic fibrosis airways. Trends Microbiol. 17:130–138 [DOI] [PubMed] [Google Scholar]
- 15. Hay I. D., Remminghorst U., Rehm B. H. 2009. MucR, a novel membrane-associated regulator of alginate biosynthesis in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 75:1110–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hennessy D. R., Ali D. N., Tremain S. A. 1994. The partition and fate of soluble and digesta particulate associated oxfendazole and its metabolites in the gastrointestinal tract of sheep. Int. J. Parasitol. 24:327–333 [DOI] [PubMed] [Google Scholar]
- 17. Hoyle B. D., Costerton J. W. 1991. Bacterial resistance to antibiotics: the role of biofilms. Prog. Drug Res. 37:91–105 [DOI] [PubMed] [Google Scholar]
- 18. Jagnow J., Clegg S. 2003. Klebsiella pneumoniae MrkD-mediated biofilm formation on extracellular matrix- and collagen-coated surfaces. Microbiology 149:2397–2405 [DOI] [PubMed] [Google Scholar]
- 19. Jenal U., Dorman C. J. 2009. Small molecule signaling. Curr. Opin. Microbiol. 12:125–128 [DOI] [PubMed] [Google Scholar]
- 20. Jensen P. O., Givskov M., Bjarnsholt T., Moser C. 2010. The immune system vs. Pseudomonas aeruginosa biofilms. FEMS Immunol. Med. Microbiol. 59:292–305 [DOI] [PubMed] [Google Scholar]
- 21. Jeys L., Grimer R. 2009. The long-term risks of infection and amputation with limb salvage surgery using endoprostheses. Recent Results Cancer Res. 179:75–84 [DOI] [PubMed] [Google Scholar]
- 22. Kamruzzaman M., et al. 2010. Quorum-regulated biofilms enhance the development of conditionally viable, environmental Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 107:1588–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kidambi S., Chan C., Lee I. S. 2004. Selective depositions on polyelectrolyte multilayers: Self-assembled monolayers of m-dPEG acid as molecular template. J. Am. Chem. Soc. 126:4697–4703 [DOI] [PubMed] [Google Scholar]
- 24. Kim B. S., Lee H., Min Y. H., Poon Z., Hammond P. T. 2009. Hydrogen-bonded multilayer of pH-responsive polymeric micelles with tannic acid for surface drug delivery. Chem. Commun. 4194–4196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koczan J. M., McGrath M. J., Zhao Y., Sundin G. W. 2009. Contribution of Erwinia amylovora exopolysaccharides amylovoran and levan to biofilm formation: implications in pathogenicity. Phytopathology 99:1237–1244 [DOI] [PubMed] [Google Scholar]
- 26. Kolenbrander P. E., Palmer R. J., Jr., Periasamy S., Jakubovics N. S. 2010. Oral multispecies biofilm development and the key role of cell-cell distance. Nat. Rev. Microbiol. 8:471–480 [DOI] [PubMed] [Google Scholar]
- 27. Lauderdale K. J., Malone C. L., Boles B. R., Morcuende J., Horswill A. R. 2010. Biofilm dispersal of community-associated methicillin-resistant Staphylococcus aureus on orthopedic implant material. J. Orthop. Res. 28:55–61 [DOI] [PubMed] [Google Scholar]
- 28. Leid J. G., et al. 2005. The exopolysaccharide alginate protects Pseudomonas aeruginosa biofilm bacteria from IFN-gamma-mediated macrophage killing. J. Immunol. 175:7512–7518 [DOI] [PubMed] [Google Scholar]
- 29. Lipinski C. A., Lombardo F., Dominy B. W., Freeney P. J. 1997. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 23:3–25 [DOI] [PubMed] [Google Scholar]
- 30. Mah T. F., O'Toole G. A. 2001. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 9:34–39 [DOI] [PubMed] [Google Scholar]
- 31. Mehrotra S., et al. 2010. Time controlled protein release from layer-by-layer assembled multilayer functionalized agarose hydrogels. Adv. Funct. Mater. 20:247–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miller M. B., Skorupski K., Lenz D. H., Taylor R. K., Bassler B. L. 2002. Parallel quorum sensing systems converge to regulate virulence in Vibrio cholerae. Cell 110:303–314 [DOI] [PubMed] [Google Scholar]
- 33. Nguyen P. T., Baldeck J. D., Olsson J., Marquis R. E. 2005. Antimicrobial actions of benzimidazoles against oral streptococci. Oral Microbiol. Immunol. 20:93–100 [DOI] [PubMed] [Google Scholar]
- 34. Olbe L., Carlsson E., Lindberg P. 2003. A proton-pump inhibitor expedition: the case histories of omeprazole and esomeprazole. Nat. Rev. Drug Discov. 2:132–139 [DOI] [PubMed] [Google Scholar]
- 35. O'Toole G. A., et al. 1999. Genetic approaches to study of biofilms. Methods Enzymol. 310:91–109 [DOI] [PubMed] [Google Scholar]
- 36. Podschun R., Ullmann U. 1998. Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin. Microbiol. Rev. 11:589–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rogers S. A., Huigens R. W., III, Melander C. 2009. A 2-aminobenzimidazole that inhibits and disperses gram-positive biofilms through a zinc-dependent mechanism. J. Am. Chem. Soc. 131:9868–9869 [DOI] [PubMed] [Google Scholar]
- 38. Sambanthamoorthy K., Schwartz A., Nagarajan V., Elasri M. O. 2008. The role of msa in Staphylococcus aureus biofilm formation. BMC Microbiol. 8:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sanyal P. K. 2002. Worm control in ruminants in India: prospects of biological control for integrated nematode parasite management. FAO animal production and health paper.Food and Agriculture Organization of the United Nations, Rome, Italy [Google Scholar]
- 40. Shunmugaperumal T. Microbial colonization of medical devices and novel preventive strategies. Recent Pat. Drug Deliv. Formul. 4:153–173 [DOI] [PubMed] [Google Scholar]
- 41. Sukhishvili S. A. 2005. Responsive polymer films and capsules via layer-by-layer assembly. Curr. Opin. Colloid Interface Sci. 10:37–44 [Google Scholar]
- 42. Tamayo R., Tischler A. D., Camilli A. 2005. The EAL domain protein VieA is a cyclic diguanylate phosphodiesterase. J. Biol. Chem. 280:33324–33330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Van Houdt R., Michiels C. W. 2010. Biofilm formation and the food industry, a focus on the bacterial outer surface. J. Appl. Microbiol. 109:1117–1131 [DOI] [PubMed] [Google Scholar]
- 44. Wolcott R. D., et al. 2010. Chronic wounds and the medical biofilm paradigm. J. Wound Care 19:45–46, 48,–50, 52–53 [DOI] [PubMed] [Google Scholar]
- 45. Yebra D. M., Kill S., Dam-Johansen K. 2004. Antifouling technology—past, present and future steps towards efficient and environmentally friendly antifouling coatings. Progr. Organic Coatings 50:75–104 [Google Scholar]
- 46. Zhu Y. F., Shi J. L. 2007. A mesoporous core-shell structure for pH-controlled storage and release of water-soluble drug. Microporous Mesoporous Mater. 103:243–249 [Google Scholar]
- 47. Ziebuhr W. 2001. Staphylococcus aureus and Staphylococcus epidermidis: emerging pathogens in nosocomial infections. Contrib. Microbiol. 8:102–107 [DOI] [PubMed] [Google Scholar]