

Abstract
The activation of MAPKs is controlled by the balance between MAPK kinase and MAPK phosphatase activities. The latter is mediated by a subset of phosphatases with dual specificity (VH-1 family). Here, we describe a new member of this family encoded by the puckered gene of Drosophila. Mutations in this gene lead to cytoskeletal defects that result in a failure in dorsal closure related to those associated with mutations in basket, the Drosophila JNK homolog. We show that puckered mutations result in the hyperactivation of DJNK, and that overexpression of puc mimics basket mutant phenotypes. We also show that puckered expression is itself a consequence of the activity of the JNK pathway and that during dorsal closure, JNK signaling has a dual role: to activate an effector, encoded by decapentaplegic, and an element of negative feedback regulation encoded by puckered.
Keywords: puckered, JNK, Drosophila, dpp, Phosphatase, Signal transduction, dorsal closure
In many cases, cell differentiation represents a transition between two states of cellular activity—one in which cells proliferate and acquire information about their fates and identities, and another in which cells stop dividing and manifest the information gathered earlier. Many of the signaling pathways leading to cell differentiation depend on phosphorylation cascades. Mounting evidence points to signaling through MAP kinase (MAPK) pathways as a key component in this transition. Three distinct types of MAPK pathways have been identified: p42–p44 ERKs (extracellular signal-related kinases), p38 kinases, and p46–p54 JNKs (Jun N (amino)-terminal kinases). These major subfamilies transduce signals from different stimuli. The ERKs respond predominantly to growth factors and hormones and are activated in a Ras-dependent manner. The p38 and JNKs respond to different environmental stresses and are activated preferentially downstream of Rac1 and Cdc42 small G proteins (for review, see Canman and Kastan 1996). In most cases, MAPK activation is a transient event, even in the continuing presence of the stimulus that leads to its activation. MAPK activity is controlled by the balance of MAPK kinase and MAPK phosphatase activities.
The dorsal closure of the Drosophila embryo provides an example of cell differentiation and how this is usually coupled to morphogenetic events and movements that shape late stages in development. Half way through embryogenesis, the dorsal surface of the embryo is covered by an extraembryonic membrane, the amnioserosa, which contacts the epidermis. After proliferation stops, the epidermis stretches dorsally and, as it encroaches the amnioserosa, closes the existing gap. Three phases lead to the successful completion of this event. The dorsal-ward movement of the epidermal cells, an anteroposterior stretching of the embryo and the seaming of the dorsal epidermis (Martinez-Arias 1993). The completion of this process takes several hours and is associated with specialized behavior of the dorsal-most epidermal cells. These cells display planar polarity reflected in the arrangement of the cytoskeleton, which is essential for the normal process of dorsal closure (Ring and Martinez-Arias 1993; Young et al. 1993).
There are several mutations that disrupt the process of dorsal closure. In basket (bsk) and hemipterous (hep) mutants, dorsal closure fails and the embryo exhibits a hole in the dorsal cuticle. hep encodes a Drosophila homolog of MKK7, a kinase that regulates JNK MAPKs (Holland et al. 1997; Tournier et al. 1997) and bsk encodes a Drosophila homolog of JNK (Riesgo-Escovar et al. 1996; Sluss et al. 1996). The involvement of the JNK pathway in dorsal closure is further emphasized by the observation that mutants for Djun, a target of DJNK signaling, fail to close dorsally (Hou et al. 1997; Kockel et al. 1997; Riesgo-Escovar and Hafen 1997), and that ectopic expression of a dominant-negative form of Drac1 (DN–Drac1), the Drosophila homolog of Rac1, also leads to the same dorsal closure defects (Harden et al. 1995).
Here, we describe the molecular characterization of the puckered (puc) gene. In puc mutants, dorsal closure takes place, but an abnormal organization of the cells at the leading edge of the epidermis results in a defective process and puckering, which provides the name of the mutant (Ring and Martinez-Arias 1993). We show that puc encodes a member of the MAPK subfamily (MKPs) of the VH1 like dual specificity phosphatases. Our results indicate that puc regulates signaling through the JNK pathway and participates in a negative feedback loop leading to a transient activity of the JNK signal during dorsal closure.
Results
Molecular characterization of puc
The puc gene was identified through a P(lacZ) insertional mutation that highlights the most dorsal epidermal cells as they finish proliferation, and causes defects during dorsal closure (Ring and Martinez-Arias 1993). The pucE69 allele is caused by a single insertion at 84E. Genomic DNA from the region around the insertion point was isolated with a probe for lacZ against a λ library constructed with pucE69 genomic DNA. A fragment of the resultant λ clone, containing only genomic DNA sequences, was then used as a probe to screen a wild-type genomic library to isolate DNA from a larger genomic region. The isolated clones were aligned and mapped by restriction analysis (Fig. 1A; Materials and Methods).
Figure 1.


Characterization of the puc gene and predicted protein. (A) Genomic organization of the puc locus. Structure of cDNA exons (shaded boxes) and introns (broken lines) is shown below the genomic map. Exon/intron boundaries are approximate to within the restriction fragment indicated. The P-element integration site of pucB48 is located in the first intron of cDNA12, pucE69, pucA251.1, and puc320 are located within the second intron of cDNA12. (E) EcoRI; (H) HindIII; (B) BamHI; (S) SalI; (X) XhoI. (B) DNA sequence of puc cDNA12 and predicted amino acid sequence. Identified motifs are the signature sequence for PTPases (boxed) and potential MAPK phosphorylation sites (P/L-X-S/T-P; circled letters).
A 3.7-kb genomic DNA fragment close to the P-element insertion site in pucE69 was used to screen a 12–24 hr embryonic cDNA library (Brown and Kafatos 1988). Three cDNA clones were isolated and mapped onto the genomic region (Fig. 1A; Material and Methods). The longest, cDNA12, is 2.3 kb long and contains an ORF capable of encoding a protein of 496 amino acids with a predicted molecular mass of 57.6 kD (Fig. 1B). Northern blot analysis of embryonic RNA detects a 2.9-kb RNA present throughout embryogenesis (see Fig. 4A, below).
Figure 4.
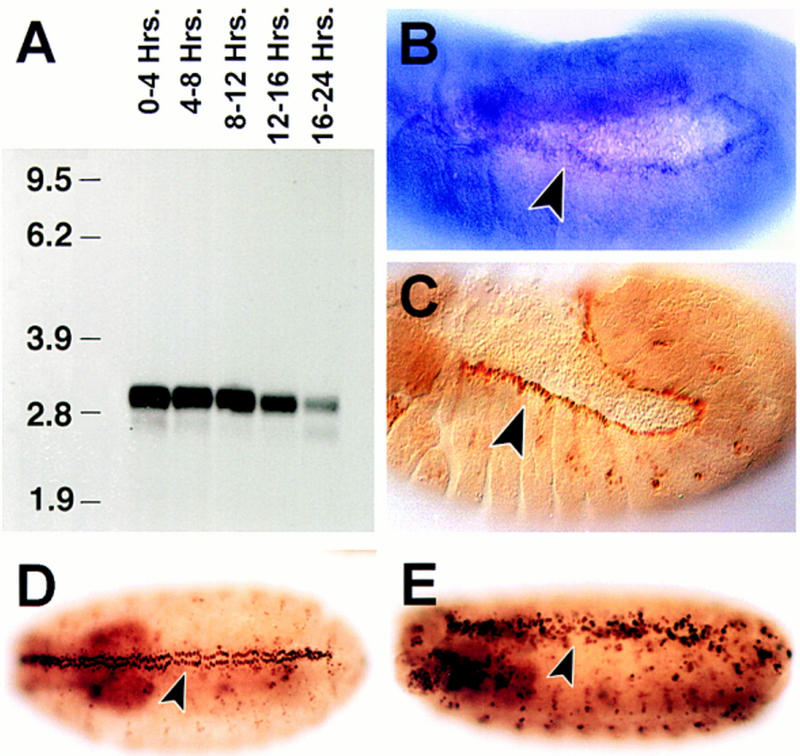
puc expression: Its modulation by puc activity. (A) Northern analysis of puc RNA expression in embryos at various times during development. (A) 0–4 hr; (B) 4–8 hr; (C) 8–12 hr; (D) 12–16 hr; and (E) 16–20 hr. The 2.9-kb puc transcript is apparent. (B) puc RNA detected in stage 13 by whole mount in situ hybridization. The expression in the dorsal-most epidermal cells is indicated. (C) stage 13 pucE69 heterozygous embryos stained with an antibody against β-galactosidase. The arrowhead points to the cells of the leading edge of the epidermis expressing β-gal. Notice that these are the same cells as in B. At early stages, evident puc expression is present in amnioserosa cells. (D) β-galactosidase expression of puc320 heterozygous embryos (stage 14). (F) A considerably higher number of cells, and at higher levels, express β-galactosidase in puc320/puc320 stage 15 embryos (arrowhead).
The pucE69 insertion and three additional lethal P-elements insertions, puc320, pucA251.1, and pucB48, which do not complement pucE69, have been identified. puc320, pucA251.1, and pucE69 map to the second intron of the cDNA (Fig. 1A). The mutations caused by pucE69 and puc320 insertions have been reverted to wild type with loss of the resident P element, which suggests that these P elements are the cause of the puc mutation (Ring 1993). The pucB48 P-element insertion site has been sequenced and maps to the first intron of puc. RT–PCR of single embryos detects puc transcripts in wild type, but not in extracts from homozygous pucB48 or pucB48R23, a lethal revertant of pucB48 that removes the genomic region adjacent to the insertion (data not shown).
The Puc ORF encodes a protein tyrosine phosphatase, with a catalytic domain between amino acids 214 and 226 that includes the invariant cysteine known to be required for phosphatase activity (Guan et al. 1991). The protein contains eight putative sites for phosphorylation by MAPK, distributed throughout the carboxy-terminal part of the protein (Fig. 1B). The predicted protein contains no clear hydrophobic sequences indicative of either signal sequence or transmembrane domain, which suggests that the Puc protein is neither a secreted nor an integral membrane protein (Fig. 1B).
The phosphatase encoded by puc has high similarity to nonreceptor dual specificity phosphatases of the VH-1 subfamily (Fig. 2A). Phylogenetic analysis (see Materials and Methods), indicates that its closest relative is the protein encoded by the Caenorhabditis elegans CEL-F08B1 ORF (Wilson et al. 1994) that has 38% identical residues over 158 amino acid overlap. When conservative residues are taken into account, the comparison yields 59.5% similarity between the two sequences. Very high similarities with other proteins of this family (see Fig. 2) highlight the conservation of their catalytic sites, which are identical at 9–11 of 13 amino acids. Interestingly, Puc, like yeast MSG-5, lacks the amino-terminal domains with homology to the cdc25 proteins that are present in all the mammalian MKPs (Keyse and Ginsburg 1993). A single copy of an internally repeated domain of unknown function (amino acids 238–312; amino acids 386–460) (Fig. 2B) is present in all VH-1 family phosphatases.
Figure 2.

Homology of Puc to VH-1 family phosphatases. (A) Sequence alignment (ClustalV) of Puc and other VH-1 family phosphatases [Drosophila Puc; human CL100 (Keyse and Emslie 1992); human Pac1 (Rohan et al. 1993); human HVH-2 (Guan and Butch 1995); human HVH-3 (Ishibashi et al. 1994; Kwak and Dixon 1995); human HVH-5 (Martell et al. 1995); human Pyst1 (Groom et al. 1996); human Pyst2 (Groom et al. 1996); human MKP-4 (Muda et al. 1997); C. elegans CEL-F08B1 (Wilson et al. 1994) and Saccharomyces cerevisiae MSG5 (Doi et al. 1994)]. Identical residues are in black, conservative changes in blue. CEL-F08B1, Pyst1 and homologs, Pyst2, and HVH-5 gave the highest homology scores to Puc in BLAST/BEAUTY searches (BCM Launcher). The other enzymes complete the whole series of distinct human dual phosphatases isolated so far. Yeast MSG-5, which share some characteristics with Puc, is also included. Phylogenetic trees (DNAstar program) point to the C. elegans CEL-F08B1 as the closest relative of Puc in the databases. CEL-F08B1 has been identified recently in the C. elegans Genome Project, but its function is unknown. Residues in the alignment highlighted in red represent putative MAPK phosphorylation sites. Interestingly, they seem to cluster for almost every protein in a low homology region at the carboxy-terminal end of the catalytic domain (double underlined), which suggest a possible functional homology. (B) Matrix alignment of Puc with itself shows the existence of an internal repeat in the protein. These domains correspond to the putative MAPK phosphorylated region and a further sequence close to the carboxy-terminal end of the molecule. Again, in the second repeat, several tentative phosphorylation sites can be identified.
Puckered encodes a Drosophila JNK phosphatase
To establish whether Puc is a phosphatase, we first determined its enzymatic activity towards p-nitrophenyl phosphate (PNPP), a chromogenic substrate structurally related to phosphotyrosine (Keyse and Emslie 1992). The first 424 amino acids of Puc, including the phosphatase domain, were fused in-frame to GST. Variable amounts of the GST–Puc protein were added to reactions containing PNPP and cleavage was analyzed spectrophotometrically (Fig. 3A). PNPP cleavage was dependent on the addition of the GST–Puc fusion protein and increased linearly with the added protein. These results indicate that Puc functions as a protein phosphatase.
Figure 3.

puc encodes a JNK phosphatase. (A) In vitro phosphatase activity of a GST–Puc fusion protein. Results of PNPP assay in which cleavage of PNPP was measured by change in absorbance at 410 nm as a function of added protein. (•) Purified GST–Puc protein; (○) control points from extracts obtained from untransformed bacteria following similar protocols. (Bottom) Schematic representation of the fusion protein. The phosphatase catalytic domain is highlighted in black (residues 214–222). (B) Endogenous JNK and ERK activity of wild-type (wt), pucE69/pucE69 (puc) and hep1/hep1 (hep) embryos. (Top) JNK assays were performed with normalized amounts of embryo extracts (1 embryo/μl) prepared in the presence of phosphatase inhibitors (as indicated in Materials and Methods). Kinase activity is measured in arbitrary units from imaging analysis. (Solid circles) Wild-type (wt) extracts; (shaded triangles) puc embryo extracts; (shaded squares) hep embryos. JNK activity increases twofold in puc mutants and reduced threefold in hep. (Bottom) ERK assays were performed by in-gel kinase assay with a normalized amount of extract, in the linear range for JNK, equivalent to five embryos. Histograms represent quantitation of kinase activity (arbitrary units). Wild-type, puc, and hep extracts have equivalent levels of ERK activity. (C) Puc phosphatase activity on heterologous JNK and ERK. (Top) JNK activity induced in HeLa cells was measured in the absence of any extract to deduce the basal level of activity (100% JNK activity—broken line). Equivalent amounts were incubated with normalized embryo extracts (1 embryo/μl) prepared in the absence of phosphatase inhibitors. The results are expressed in percentage of JNK activity. (Solid circles) Wild-type extracts; (shaded triangles) puc embryo extracts; (shaded squares) hep embryos. Wild-type embryos have high levels of JNK phosphatase activity (HeLa JNK activity is reduced fivefold). Puc extracts do not show JNK phosphatase activity, indeed HeLa JNK activity gets increased because of the high levels of JNK activity of puc extracts (it can be brought back to basal levels by previous heat inactivation; see also Discussion). In hep extracts, JNK phosphatase activity is reduced to 50% of that of wild-type embryos. (Bottom) ERK activity of preactivated human ERK (hERK*) was assayed as indicated in Materials and Methods. Extracts (5 embryos) from wild-type, puc, and hep embryos display the same level of ERK phosphatase activity, reducing hERK* activity by 40%. Histograms represent quantitation of kinase activity (arbitrary units). Positive controls were performed with purified CL100 phosphatase (50 μg/ml) (data not shown).
Members of the VH-1 family of phosphatases have been implicated in the down regulation of MAPK activity (for review, see Keyse 1995). To study if Puc could have such function, we measured the endogenous MAPK activity of embryo extracts, prepared in the presence of phosphatase inhibitors (see Materials and Methods). Extracts prepared from puc mutant embryos showed a twofold increase in JNK activity relative to wild type by use of (1–86) cJun–GST captured on glutathione–Sepharose beads, whereas ERK activity on myelin basic protein (MBP) was unaffected (Fig. 3B). We also examined the ability of extracts without phosphatase inhibitors to inhibit activated JNK that was affinity purified from anysomicin-treated HeLa cells (see Materials and Methods). Extracts derived from wild-type Drosophila embryos were able to inhibit HeLa JNK activity up to 80%. In contrast, extracts from puc mutants were unable to repress exogenous JNK and a gain of kinase activity was observed (Fig. 3C), probably caused by a feedback loop between JNK and puc (see below). ERK phosphatase activity measured in these extracts was unaffected in puc mutants (Fig. 3C). These results strongly suggest that puc encodes a protein capable of regulating JNK, but not ERK, activity through dephosphorylation.
If puc encodes a JNK phosphatase activity, then it should inactivate JNK in vivo. To test if this is the case, we placed the puc cDNA under the control of Gal4/UAS to target its expression during development (see Materials and Methods). Ubiquitous expression of Puc results in a dorsal hole during embryogenesis (see Fig. 5, below) reminiscent of the phenotype of mutations in hep and bsk that encode a JNKK and a JNK, respectively.
Figure 5.
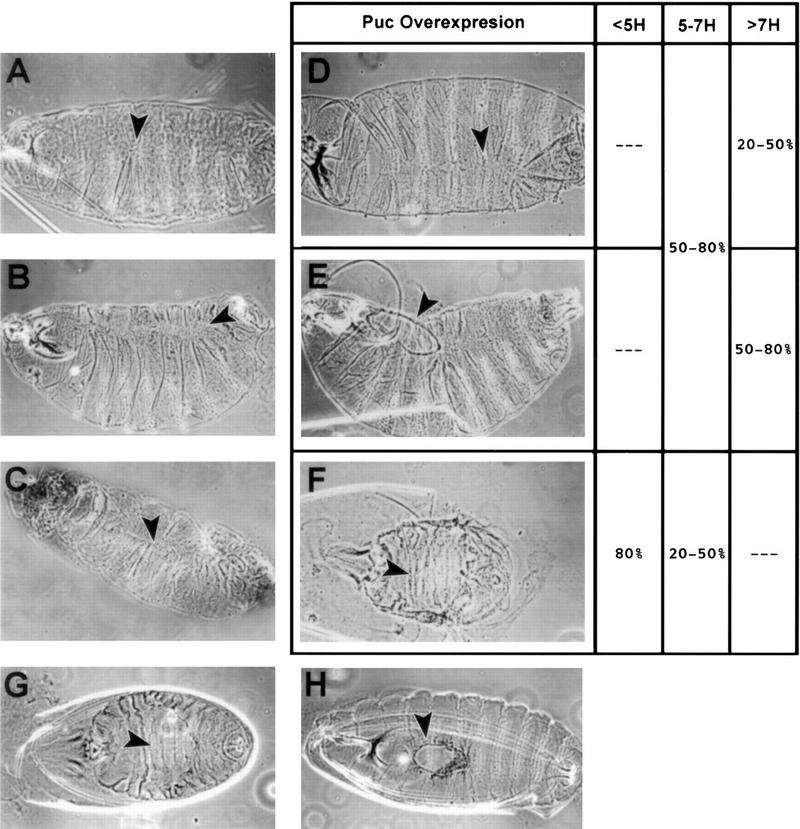
Cuticle phenotypes of puc mutants and those generated by ectopic expression of puc and dpp. Rescue of hep phenotype by puc in heterozygous condition. (A) Dorsal view of pucEh/pucEh embryo. (B) Dorsolateral view of pucE69/pucE69 mutant embryos. (C) Dorsolateral view of ArmGal4/UASdpp embryo. Arrowheads point to defects, puckering, and naked cuticle, along the dorsal midline. (D–E) Embryos from a cross of flies carrying the UASpuc with flies carrying a hsGal4 insert that were exposed to a 30 min heat shock at various times during development. Cuticle preparations revealed three classes of phenotype depending on the age of the embryo at the time of heat shock. (D) puc loss of function-like; (E) dorsal hole; (F) dorsal open. Arrowheads indicate dorsal cuticle defects. Dorsal open embryos are observed more frequently after early heat shocks. Dorsal hole phenotypes appear at intermediate times and puc loss-of-function-like embryos are present mainly after late heat shocks (see columns at the right). (G) hep1 hemizygous embryo, note the dorsal open phenotype. (H) hep1 hemizygous, pucE69 heterozygous embryo. A partial rescue of the dorsal open phenotype leads to small dorsal holes (arrowhead).
These results suggest that the phosphatase encoded by Puc act as a JNK phosphatase in vivo and might play a role in the regulation of the activity of JNK signaling during dorsal closure.
The expression and activity of Puc during embryonic development are regulated by the Drosophila JNK pathway
The temporal pattern of puc expression during embryogenesis was determined by use of a puc cDNA probe. Northern analysis detects a 2.9-kb RNA present throughout embryogenesis (Fig. 4A). This RNA was apparent in early embryos (0–4 hr AEL) suggesting the presence of maternal transcripts. Minor transcripts of 2.6 and 2.4 kb are also detected.
The spatial distribution of puc was determined by whole mount in situ hybridization. puc expression was detected, by use of a variety of genomic and cDNA derived probes, only in a small number of the experiments, which suggests a very low level of expression or transcript instability. We were able to observe expression of puc in the dorsal-most cells at the leading edge of the epidermis (Fig. 4B). This pattern was identical to that described previously for the early expression of β-galactosidase in the insertional alleles (cf. Fig. 4B and C). After stage 11, puc mRNA expression slowly decays on the leading edge, whereas β-galactosidase is found up to completion of dorsal closure (Ring and Martinez-Arias 1993).
It has been shown previously that β-galactosidase expression by the pucE69 insertion is abolished in hep and bsk mutants (Glise et al. 1995; Riesgo-Escovar et al. 1996). Furthermore, β-galactosidase activity is enhanced after overexpression of activated forms of Drac1 (DRac1V12) and Dcdc42 (Dcdc42V12) (Glise and Noselli 1997) that activate JNK signaling. As puc encodes a JNK phosphatase that appears to down-regulate the JNK pathway, we studied the expression of puc (β-galactosidase) in puc mutants, in which we have shown that JNK activity is enhanced. The misregulation of pucE69 LacZ activity had been reported previously (Ring and Martinez-Arias 1993; Glise and Noselli 1997). We found that for all puc insertional alleles, mutant embryos show higher levels and more cells expressing β-galactosidase than wild type, for example, in the leading edge of the epidermis, the amnioserosa, the ectoderm, and the nervous system (puc320; Fig. 4D,E).
The requirement for the DJNK pathway to activate puc expression was also mirrored in the reduction of JNK phosphatase activity in mutants for hep: JNK activity is reduced threefold in hep1 mutants extracts, as expected for a loss of function in a JNK activator (Fig. 3B). In addition, extracts from hep1 mutants show less phosphatase activity on exogenous JNK (a 50% reduction), but identical levels of ERK phosphatase function, compared with wild-type embryo extracts (Fig. 3C).
Altogether, these results support a role for the Drosophila JNK pathway in the control of puc expression.
Loss and overexpression of puc have different effects in dorsal closure
Embryos mutant for puc develop defects along the dorsal midline of the larval cuticle during dorsal closure. These defects manifest as misaligned segments in the weakest allele, pucEh (Ring and Martinez-Arias 1993) (Fig. 5A), and become more pronounced in stronger alleles. For example, in embryos carrying the pucE69 allele (Fig. 5B), dorsal hairs are absent along the midline, leaving a strip of naked cuticle along most of the midline; these embryos display strong puckering of the epidermis (Ring and Martinez-Arias 1993).
To study if puc restrains JNK activity during the different steps of dorsal closure, we analyzed the phenotype of embryos in which we had expressed puc ectopically using hsGal4 and a UASpuc line (Fig. 5). Embryos heat-shocked very early [between 4 and 5 hr after egg laying (AEL)] failed to achieve dorsal closure and exhibit a large dorsal opening (Fig. 5F). Most of the embryos heat shocked between 5 and 7 hr AEL displayed dorsal holes (Fig. 5E) or phenotypes similar to puc loss of function alleles (Fig. 5D). Heat-shocking embryos after 7 hr AEL produced a puc loss-of-function phenotype, and occasionally a dorsal hole that, on average, was smaller than that of embryos heat-shocked earlier.
In these experiments, early overexpression of Puc mimics the phenotype of bsk mutants and the complete inactivation of DJNK signaling. Late expression of Puc affects the ability of the dorsal-most cells to differentiate properly and induces the same defects as puc loss-of-function alleles. Furthermore, puc in heterozygous condition can rescue the dorsal open characteristic of low JNK signaling: Hemizygous hep1 embryos develop a dorsal open phenotype (Fig. 5G), which is partially rescued by pucE69 (Fig. 5H) or pucA251.1 (data not shown) in heterozygous condition.
These results support a role of puc in limiting DJNK activity during dorsal closure and suggest the existence of a feedback loop through which JNK dependent expression of puc regulates JNK activity (see Discussion).
puckered alters actin and nonmuscle myosin localization and affects epidermal morphogenesis
During dorsal closure, the shape of the epidermal cells in the dorsal region of the embryo changes dramatically. Beginning with cells immediately flanking the amnioserosa, there is an elongation along the dorsoventral axis that gradually spreads ventrally through the epidermis. These cell shape changes stretch the opposing sides of the lateral epidermis until they meet along the dorsal midline.
Dorsal cuticle puckering and dorsal holes are indicative of a defect in dorsal closure, probably dependent on cell shape changes (for review, see Martinez-Arias 1993). In bsk, hep, and Djun mutants, cell shape changes are disrupted (Glise et al. 1995; Riesgo-Escovar et al. 1996; Riesgo-Escovar and Hafen 1997; Hou et al. 1997; Kockel et al. 1997). It is interesting that expression of DN–Drac1 displays phenotypes that are similar to that of hep and bsk mutants, suggesting that Drac1 might initiate signaling through this cascade. Drac1 seems to be involved in the control of the cell cytoskeleton. In wild-type embryos, the onset of dorsal closure coincides with a specific subplasmalemmal accumulation of nonmuscle myosin at the leading edge of the dorsal-most epidermal cells (Fig. 6A,B; Young et al. 1993). It is likely that nonmuscle myosin (NMM) probably contributes to the elongation of these cells by participating with actin in forming a dorsal constriction. In DN–Drac1 embryos, NMM and actin in epidermal cells are strongly reduced (Harden et al. 1995). These changes in the cytoskeleton are also evident in Djun mutant embryos (Hou et al. 1997).
Figure 6.
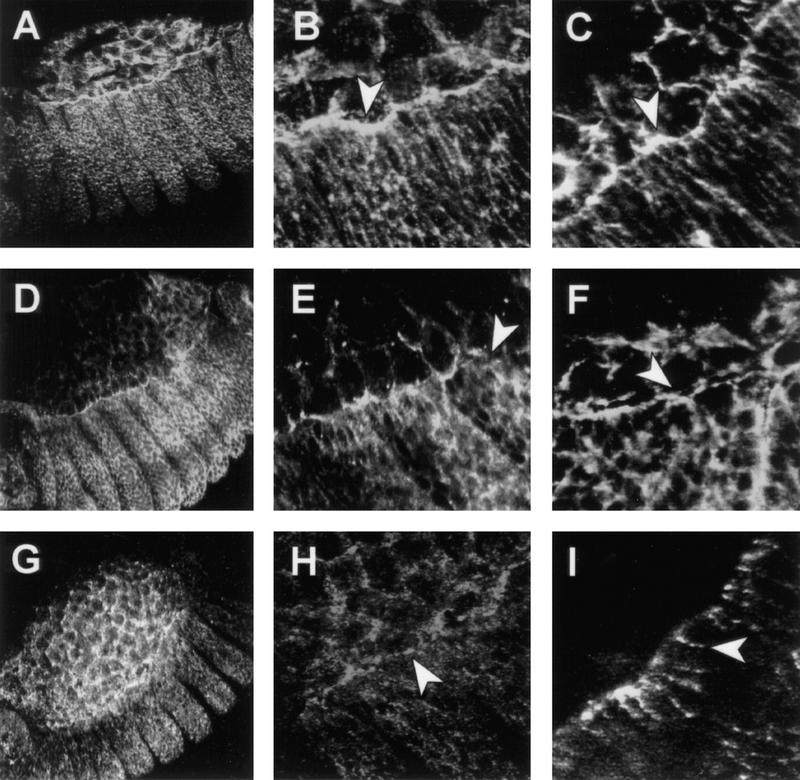
puc controls the accumulation of actin and myosin in the leading edge of the epidermis during dorsal closure. Confocal fluorescent micrographs of the boundary between the amnioserosa and the epidermis in stage 13 embryos. The distribution of nonmuscle myosin (A,B,D,E,G,H) and filamentous actin (C,F,I) are shown in wild-type embryos (A–C), pucE69 embryos (D–F) and ArmGal4/UASpuc embryos (G–I). Embryos were stained for filamentous actin with phalloidin or for nonmuscle myosin with antibodies. Whereas actin and NMM are accumulated along the leading edge in wild-type embryos (arrowheads in B and C), in puc mutants their level decreases and it is possible to observe gaps (arrowheads in E and F) between the amnioserosa and the epidermis. After Puc overexpression, NMM is maintained in the amnioserosa (arrowhead in H) but the level of expression in the epidermis is severely reduced. Actin ceases to be expressed in the amnioserosa and it appears on patchy spots in the epidermis (arrowhead in I).
We compared the effects of the absence of puc (pucE69) and its overexpression (ArmGal4/UASpuc) in the levels and organization of the actin cytoskeleton and NMM. We find that in pucE69 both myosin and actin do not change dramatically their expression in the periphery of the cells in lateral regions of the embryo, but fail to accumulate along the leading edge of the epidermis (Fig. 6D–F). Cell shape changes proceed almost normal. In contrast, epidermal cells of ArmGal4/UASpuc embryos fail to change their shapes and accumulate low levels of spatially disorganized myosin at the leading edge (Fig. 6G,H). In these embryos, actin fails to be expressed in the amnioserosa and its levels are reduced in the epidermis (Fig. 6I). Actin and myosin tend to form clumps in these epidermal cells.
Our data indicate that puc is an essential component in the control of the different steps of dorsal closure progression by modulating the apical accumulation of actin and myosin at the leading edge and directing cell shape changes. These results correlate with those of the effects of overexpression of DN–Drac1 and Djun mutants, and further suggest a role for Puc in the control of JNK activity over the cytoskeleton.
dpp in the leading edge is regulated by puc and its overexpression disrupts completion of dorsal closure
The gene dpp encodes a member of the TGFβ superfamily and has been identified as a secreted signaling molecule that mediates inductive interactions during Drosophila development (Wharton et al. 1993). During the second half of embryogenesis, dpp is expressed in specific regions in the embryo, particularly in the dorsal-most epidermal cells that express puc (Fig. 7B). It has been shown recently that the maintenance of dpp expression along the leading edge of the epidermis depends on the activity of the DJNK pathway (Glise and Noselli 1997; Hou et al. 1997; Riesgo-Escovar and Hafen 1997).
Figure 7.
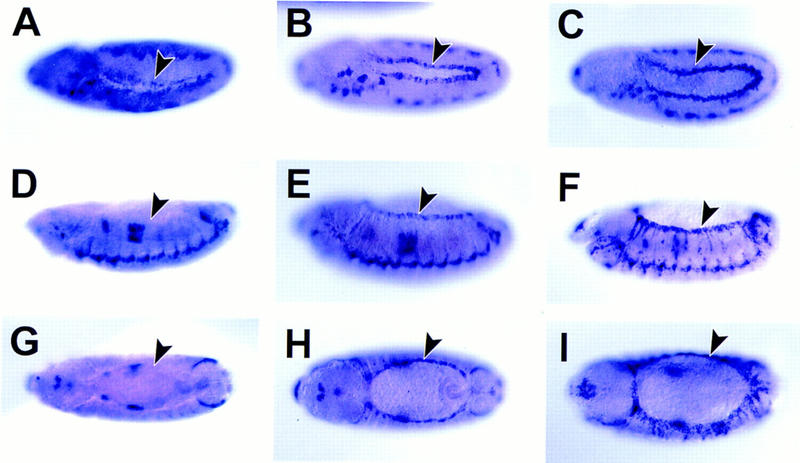
Effects of wild-type, mutant pucE69 and Puc expressed under the control of the Armadillo promoter on dpp transcription. (A,D,G) dpp RNA expression in ArmGal4/UASpuc embryos. (B,E,H) dpp RNA expression in wild-type embryos. (C,F,I) dpp RNA expression in pucE69 embryos. (A–C) lateral views of stage 11 embryos. Anterior is to the left; dorsal is up. At this early stage, it is possible to observe a reduction in dpp expression after ectopic expression of Puc and new cells expressing dpp along the epidermal border in puc mutants (arrowheads). (D–F) lateral views of stage 13 embryos. (G–I) dorsal views of stage 13 embryos. At this stage, dpp disappears from the dorsal-most epidermal cells after ectopic Puc expression and it is present in at least two rows of cells at the leading edge of each lateral hemisegment in puc mutants (arrowheads). The expression of dpp in the visceral mesoderm is unaffected.
In pucE69 mutant embryos, the expression of dpp in the dorsal-most epidermal cells is enhanced from stage 11 (cf. Fig. 7B with C; see also Glise and Noselli 1997). Furthermore, after germ-band shortening there are more epidermal cells expressing dpp than in wild type, but this expression is still mainly limited to dorsal cells (cf. Fig. 7E and H with F and I). In contrast, ubiquitous expression of Puc leads to a decrease in the expression of dpp in those cells during stage 11 (Fig. 7A) and the complete absence at stages 12 and 13 (Fig. 7D,G). Puc overexpresion does not affect the levels of dpp on the visceral mesoderm or the ventral epidermis. These results suggest a role for puc in the control of dpp expression, which could be mediated by Puc activity on JNK signaling (See Discussion).
The null phenotype for dpp, a completely ventralized embryo, reflects its initial role over the dorsal epidermis and might obscure the function of later patterns of expression (Wharton et al. 1993). Mutants for thick veins, however, which encodes a dpp receptor, display dorsal holes similar to those of hep or bsk mutants (Affolter et al. 1994). To test if the higher levels of dpp present in puc mutants could be involved in puckering phenotypes, we overexpressed dpp using ArmGal4 as the driving system. ArmGal4/UAS dpp embryos undergo an extreme dorsalization of the epidermis, but they still have a dorsal midline, in which, in many cases, we observed phenotypes (Fig. 5C) very similar to those observed in pucE69 mutants (cf. Fig. 5C with B). This indicates that, besides an early function of dpp in epidermal cell stretching, the downregulation of dpp in the dorsal-most epidermal cells is necessary for completion of dorsal closure.
Discussion
Two different MAPKs, encoded by the bsk and rolled (rl) genes, have been identified so far in Drosophila. The phenotype of mutations in these genes suggests that they play different roles during development. The MAPK encoded by rl has been implicated downstream of multiple RTK signal transduction pathways during cell fate determination (Diaz-Benjumea and Hafen 1994). On the other hand, the MAPK encoded by bsk behaves as a homolog of mammalian JNK and is involved in the process of dorsal closure (Riesgo-Escovar et al. 1996; Sluss et al. 1996).
Down-regulation of the activity of MAPK proteins through a feedback mechanism that is dependent on their own signaling ability has been suggested as a way of regulating the activity of MAPK pathways (Sun et al. 1993). Here, we have identified a novel member of the MKP subfamily of VH-1 protein tyrosine phosphatase (PTPs), which is involved in the regulation of JNK activities during dorsal closure in Drosophila. This phosphatase is associated with insertional mutations in the puc gene (Ring and Martinez-Arias 1993).
Three arguments lead us to the conclusion that the PTP that we have cloned is encoded by the puc gene. First, four independent P-element insertions cause puc mutant phenotypes and map to two different introns of the gene that we have cloned. Two of these mutations have been reverted to wild type with loss of the resident P element. Also, transcripts for this gene are not present in embryos of one of these mutants and one lethal revertant. Second, extracts from puc mutants have more JNK activity than wild type. And third, whereas loss of function of puc leads to ectopic expression of dpp in the dorsal epidermis, ubiquitous expression of the Puc protein leads to the loss of dpp expression.
We noticed the presence of maternally contributed RNA in early embryos. Consistent with a function of this RNA, the phenotype of pucEh/Df(3R)dsx10 embryos derived from Df(3R)dsx10 females was stronger than that of embryos derived from pucEh females (Ring 1993). Similar differences were also observed in reciprocal crosses between strong and weak alleles. The function of this maternal RNA during oogenesis and early embryogenesis is currently under study.
Evidence that Puc is a JNK-specific phosphatase is provided by biochemical assays. When extracts from wild-type and puc mutant embryos are assayed for their endogenous JNK activity, puc mutants show a significant increase in JNK activity compared with wild type. When similar extracts are tested for their ability to inactivate preactivated JNK, up to 50% inhibition was obtained after 30 min from wild-type embryos compared with abolition of activity in extracts from puc mutants. In similar assays, we never observed changes in ERK and ERK phosphatase activities.
A feedback loop between the Puc phosphatase and JNK signaling
The expression of most members of the VH-1 family of PTPs is subject to tight transcriptional regulation (i.e., Charles et al. 1993). The same is likely to be true for puc because it displays dynamic patterns of expression in the embryo (Ring and Martinez-Arias 1993) and the adult (E. Martin-Blanco, unpubl.).
During and after germ band shortening, puc is expressed in the dorsal-most epidermal cells that play a leading role in the process of dorsal closure. In embryos mutant for the JNKK encoded by hep or for the JNK encoded by bsk, there is no puc expression in these cells and dorsal closure fails in a manner similar to that produced by overexpression of puc (Glise et al. 1995; Riesgo-Escovar et al. 1996). These results suggest a model in which signaling through Hep and Bsk leads to the expression of effectors of dorsal closure and a regulator encoded by puc. The function of the latter is to exert a negative feedback on the signaling cascade of hep and bsk. Interestingly, in mutants for Djun, a likely target of JNK activity, puc expression is absent at the leading edge of the epidermis (N. Perrimon, pers. comm.), suggesting a transcriptional link between the activity of the JNK encoded by bsk and the expression of puc. The phenotypes of gain and loss of function of puc, together with the levels of JNK and JNK phosphatase activities of extracts from puc embryos support this model.
Expression of a dominant-negative Drac1 protein results in defects in the dorsal epidermis that are similar to those of hep and bsk mutants and, occasionally, produces defects that resemble weak puc mutant phenotypes (Harden et al. 1995). A similar situation can be observed when Puc is overexpressed at different times in embryogenesis: Whereas early overexpression leads to a hep/bsk mutant phenotype, overexpression during dorsal closure leads to phenotypes that resemble puc. The function of puc in a negative feedback loop can account for these observations. Early overexpression of Puc or dominant-negative Drac1 would inactivate signaling through JNK and thus inhibit the expression not only of puc, but also of the effectors of the pathway. On the other hand, overexpression of puc later would have allowed for some expression of the effectors but would result in the late inhibition of the feedback loop by suppression of puc expression. Therefore, the phenotype of puc mutants represents a failure in the negative regulation of signaling after the process has been initiated.
A similar feedback has been proposed as a mechanism by which a rapid and transient response to extracellular signals may occur. Erp, another member of the VH-1 phosphatase family, is expressed as an early event in the proliferative response of fibroblasts to serum and yet, constitutive expression of Erp in fibroblasts suppresses proliferation, suggesting this same negative feedback mechanism (Noguchi et al. 1993).
puc, signaling, and dorsal closure
During the first half of embryogenesis, the dorsal side of the Drosophila embryo is covered by an extraembryonic membrane, the amnioserosa, which bridges the two edges of the open epidermal sheet. The process of dorsal closure closes the gap by bringing these two epidermal edges together and intruding the amnioserosa into the embryo (Ring and Martinez-Arias 1993; Young et al. 1993).
The cell shape changes that accompany this process suggest that cytoskeletal rearrangements play an important role. Consistent with this, zipper mutant embryos (Nüsslein-Volhard et al. 1984), which lack the cytoplasmic myosin heavy chain (Young et al. 1993), display defective cell shape changes that lead to an abnormal dorsal closure. On this basis, a model for dorsal closure has been suggested in which myosin plays an active role in producing force for cell shape change and as a mechanically contractile band at the leading edge that powers the epidermal movements during dorsal closure (Young et al. 1993).
In puc mutants, the dorsal-most epidermal cells retain nonmuscle myosin and actin expression, albeit at reduced levels. In contrast, after Puc overexpression, the polarized localization of nonmuscle myosin is lost and a strong reduction in both myosin and actin levels is observed, correlating with an extreme lack of cell shape change. These defects are very reminiscent of the effects of dominant-negative Drac and Djun mutations on these cytoskeletal elements (Harden et al. 1995; Hou et al. 1997). In all these cases, actin became excluded from the amnioserosa. This disruption of the cytoskeleton is probably the cause of the absence of dorsoventral elongation of the epidermal cells associated with dorsal holes.
It is possible that independently of its effects on hep and bsk, DRac1 could be controlling the process of dorsal closure by direct effects on the cytoskeleton. The fact that mutations in hep supress the effects of the expression of an activated form of Drac1 (Drac1V12) (Glise and Noselli 1997), and that the inhibition of DJNK activity by puc seems to produce equivalent cytoskeletal and morphogenetic defects as the expression of DN-Drac, however, suggest that during dorsal closure, the outcome of Drac1 signaling is the activation of DJNK.
puc control of Dpp expression and cellular morphogenesis
The control of DJNK activity by Puc affects the maintenance and modulation of dpp, which might mediate many of the requirements for dorsal closure (Fig. 8). In hep (Glise and Noselli 1997) and Djun (Hou et al. 1997; Riesgo-Escovar and Hafen 1997) mutants, the expression of dpp is abolished in the dorsal epidermal cells and dorsal closure is never initiated. On the other hand, dpp expression along the leading edge is augmented in puc mutants and is abolished after Puc overexpression.
Figure 8.

A model for the role of puc in JNK signaling during dorsal closure. Halfway during embryogenesis, in the cells at the leading edge of the epidermis, the hep/bsk pathway becomes activated, probably by Drac. As a consequence, DJun is itself activated and gets involved in the maintenance of dpp and puc expression. Puc will drive its own down regulation through inactivation of bsk, and it will control the level of expression of dorsal closure effectors as dpp. dpp might have two different roles: to induce the cellular events required for dorsal closure in the lateral cells and to participate in the specializations in the dorsal-most cells required for the last steps of closure. In puc mutants, JNK signaling becomes hyperactivated in the leading edge, the dorsal-ward stretching of the lateral cells proceeds normally, but the excess of dpp interferes with proper cell differentiation and affects midline alignment. When puc is ectopically expressed early throughout the epidermis, it blocks signaling through the bsk pathway leading to the disappearance of dpp from the dorsal-most cells and to a failure in dorsal closure. Late Puc overexpression it affects only the cellular differentiation of the leading edge cells.
Mutants for the dpp receptor thick veins have a prominent dorsal hole similar to that of hep and bsk mutants (Affolter et al. 1994). It is therefore interesting that, as we have shown here, overexpression of Dpp leads to similar problems during dorsal closure as does loss of puc function. dpp would affect first the morphogenetic changes of the lateral epidermal cells during closure progression (Riesgo-Escovar and Hafen 1997), and second, the proper recognition and adhesion of the leading edge cells at closure completion.
Taken together, the existing results suggest that the outcome of JNK signaling during dorsal closure is the expression via DJun of puc and dpp in the dorsal-most cells of the epidermis. Whereas dpp provides an effector of dorsal closure, puc encodes the regulatory element that controls the amount of signaling through the pathway. Experiments in tissue culture in vertebrates have suggested the possibility of such feedback loops on signaling cascades, and we have shown here one case in which it occurs in vivo. The existence of this feedback mechanism provides a sensitive way of regulating and controlling signaling through the pathway, something that might be important during morphogenetic events like the one of dorsal closure that requires coordinated fine tuning of cellular behavior.
Materials and methods
Drosophila strains and culture
All flies were maintained at 25°C on standard medium. The P[ry+, lacZ]E69 line (renamed pucE69) was generated from the starter strain that carried the ry506 P[ry+, lacZ]C49 chromosome, kindly provided by C. O’Kane, as described in Ring and Martinez-Arias (1993). P[w+, lacZ]320c (renamed puc320) was a gift from J. Campos-Ortega (University of Koln, Germany). l(3)84EhK19 (renamed pucEh) was supplied by B. Baker (Stanford University, CA). P[w+, lacZ]A251.1 (renamed pucA251.1) was a gift from W. Gehring (Biozentrum, University of Basel, Switzerland). The P[w+, lacZ]B48 (renamed pucB48) and the revertant pucB48R23 were a gift from C. Ruhslow and A. Pai (New York University, NY). The hep1 mutant stock was a gift of S. Noselli (Centre de Biologie du Developpement, CNRS, Toulouse, France). The UASdpp line was a generous gift of F. M. Hoffmann (University of Wisconsin, Madison). The HsGal4 line was from A. Brand (Wellcome/CRC Institute, University of Cambridge, UK). The ArmGal4 is expressed ubiquitously during embryonic development and is a gift of J.P. Vincent (NIMR, Medical Research Council, London, UK).
Embryo cuticle preparations were done according to Wieschaus and Nüsslein-Volhard (1986) except that embryos were not fixed before mounting.
Immunocytochemistry and whole mount in situ hybridization
For all procedures, embryos were dechorionated, fixed for 20 min at room temperature in heptane/4% paraformaldehyde, and devitellinized with methanol. mRNAs were detected by in situ hybridizations with DIG-labeled DNA probes following standard protocols (Tautz and Pfeiffle 1989). Two puc probes were used, a probe generated by PCR with primers flanking the second exon of cDNA12 and a probe generated from a HindII–BglII fragment of cDNA12 that includes 73 nucleotides from the pNB40 plasmid and 114 nucleotides from the 5′ end of cDNA12 located within the 5′ untranslated region. The dpp probe was generated from a 2-kb EcoRI fragment from a dpp cDNA clone (St. Johnston et al. 1990).
Antibody staining was performed by standard techniques (Ashburner 1989). The primary antibodies were commercial rabbit anti-β-galactosidase and rabbit anti-non muscle myosin antibody (Kiehart and Feghali 1986). Biotinylated secondary antibodies and streptavidin conjugated to horseradish peroxidase or Texas Red (Jackson laboratories) were used. F-actin staining was performed with rhodamine–phalloidin on embryos devitellinized with 80% ethanol as described previously (Harden et al. 1995).
Analysis of genomic DNA at the puc locus
Genomic DNA was isolated from pucE69/TM3sb flies, partially digested with Sau3A and size selected on 0.75% low melting point agarose following the protocol described by Kaiser and Murray (1985). Fragments were subcloned into λEMBL3 with the Stratagene cloning kit. Recombinants were packaged into phage particles by use of the Stratagene Gigapack II Plus Packaging Extract and transformed into Escherichia coli strain P2392 (Stratagene). The pucE69 genomic library was screened with a probe against lacZ sequences. Genomic DNA from the resultant 12-kb clone was then used to probe a wild-type Drosophila genomic library (gift from J. Tamkin, University of Colorado, Boulder) to isolate wild-type genomic DNA from the puc locus. Library screening was done by standard techniques (Sambrook et al. 1989). DNA probes were radioactively labeled with [α-32P]ATP (Amersham) by random priming with hexadeoxynucleotides (Pharmacia) according to the protocol described by Feinberg and Vogelstein (1984). The genomic map shown in Figure 2 was generated by restriction mapping of overlapping genomic clones. Resident P elements in pucE69, puc320, and pucA251.1 (Ring 1993) were localized by Southern hybridization by standard techniques (Sambrook et al. 1989). pucB48 was mapped by sequencing of a rescued plasmid.
Cloning and sequencing of puc cDNA
A 12- to 24-hr embryonic cDNA library in pNB40, kindly provided by N. Brown (Brown and Kafatos 1988) was screened by use of the 3.7-kb EcoRI puc genomic fragment. Filters for hybridization were prepared with the added step of washing nitrocellulose filters in CHCl3 for 2 min before baking. Hybridization and probe preparation were performed by standard technique (Sambrook et al. 1989). Restriction fragments of cDNAs were subcloned and sequenced in both directions by automatic sequencing.
Northern hybridization and RT–PCR
RNA purification from Drosophila embryos was performed by the hot phenol/chloroform method (Jowett 1986). Northern blot analysis was done by standard techniques (Sambrook et al. 1989). The Northern was probed with the 1037-bp BglII–EagI fragment of puc cDNA12 that had been labeled with 32P by random priming.
RT–PCR was performed from single embryos as follows: Extracts from embryos (stage 13–14) were made in the cold in 10 μl of PCR buffer [10 mm Tris (pH 8.2), 1.2 mm EDTA, 25 mm NaCl, 0.1% Tween 20, 0.1% gelatin] supplemented with 5 units of RNAsin/ml. The extracts were mixed with 10 μl of proteinase K (50 μg/ml) in RNAsin–PCR buffer and incubated for 1 hr at 50°C, followed by denaturation at 94°C for 10 min. Five microliters was taken into a 20-μl reverse transcriptase reaction [1 unit of RT, 0.5 μm dNTPs, 5 units of RNAsin, 0.1 μg of a reverse primer from the third exon of puc (3.3REV; GAGGTCAATCTGGATGAGCAG)] at 42°C, for 30 min. After heat inactivation, the transcription reactions were subjected to PCR amplification after the addition of a forward primer from the first exon of puc (1.1ATG) (GTGCATATGTGTGTGAATCGAG) and Taq polymerase.
Construction of recombinant expression plasmids
The Puc fusion construct (pGST–PucT1) contains the NdeI (blunt-ended)–EcoRI fragment of pNB40/cDNA12 ligated to the expression plasmid PGEX-2T (Smith and Johnson 1988) digested with SmaI and EcoRI. This recombinant plasmid encodes the first 424 amino acids of Puc, including the phosphatase catalytic domain, fused in frame to the carboxy-terminal end of GST.
The UASpuc construct was made by directional cloning of the BglII–NotI fragment of PNB40/cDNA12 in the vector pUAST (Brand and Perrimon 1993). Embryo injection and selection of recombinants were performed by standard procedures.
Protein purification and in vitro phosphatase activity assay
One-liter cultures grown to mid-log phase from freshly transformed cells were induced by IPTG (1 mm) and harvested after 3 hr. Harvested cells were frozen in dry ice for at least 30 min and thawed at 0°C. Two microliters of HKEDN buffer [25 mm HEPES (pH 7.6), 0.1 m KCl, 0.1 mm EDTA, 0.5 mm DTT, 0.1% NP-40, 10 mg/ml of leupeptin, 0.1 mm benzamidin, 10 mg/ml of pepstatin A, 1 mm PMSF, 10 mg/ml of aprotinin, 1 mg/ml of phenanthroline] was added, and aliquots of 1 ml were sonicated. Recombinant protein was insoluble and went to the pellet by centrifugation (TL100 rotor, 68,000 rpm for 45 min at 4°C). The pellet fraction was frozen in dry ice. The fusion protein was recovered in a denatured form, dissolved in 6 m guanidine-HCl in HKEDN and refolded by gradual removal of the denaturant by dialysis (Claasen et al. 1991).
Hydrolysis of PNPP was in a reaction volume of 200 μl containing 50 mm imidazole at pH 7.5, 0.1% β-mercaptoethanol, 20 mm PNPP, and different concentrations of recombinant protein or control extracts at 30°C for 30 min. The reaction was stopped by addition of 800 μl of 0.25 m NaOH, and the absorbance at 410 nm (A410) was measured (Keyse and Emslie 1992).
Kinase assays and measurement of JNK/ERK–phosphatase activity
For assessment of endogenous ERK and JNK activity, 50 dechorionated embryos at stage 13, identified under the microscope by their mutant phenotypes, were homogenized in 50 μl of ice-cold extraction buffer (25 mm HEPES at pH 7.7, 0.3 m NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 0.1% Triton X-100, 0.5 mm DTT, 20 mm β-glycerophosphate, 0.1 mm Na3VO4, 10 μg/ml each of leupeptin and aprotinin). The extracts were clarified by centrifugation (12,000g, 4°C for 20 min).
For measurement of endogenous ERK activities, an in-gel kinase assay was performed according to the procedure of Kameshita and Fujisawa (1989) by use of myelin basic protein (MBP) as the substrate. Extracts prepared in the manner described above were fractionated on a SDS/10% polyacrylamide gel containing 500 μg/ml of MBP. The gel was then washed twice in Tris-HCl buffer (50 mm at pH 8) containing 20% (vol/vol) isopropanol, followed by washes in Tris-HCl buffer supplemented with 5 mm β-mercaptoethanol (buffer A). The proteins were then denatured for 1 hr with two changes of buffer A containing 6 m guanidine-HCl and finally renatured for 16–20 hr at 4°C by incubating the gel in buffer A containing 0.04% (vol/vol) Tween 40. Renatured MBP kinase activity was detected by incubating the gel for 30–45 min at 30°C in kinase buffer (50 mm Tris-HCl at pH 8, 5 mm DTT, 5 mm MgCl2, 1 mm EGTA, 50 μm ATP, 20 μCi/ml [α-32P]ATP). Unincorporated radioactivity was removed by extensive washing of the gel in 5% (wt/vol) TCA solution containing 1% (wt/vol) tetrasodium pyrophosphate, dried, and analyzed by phosphorimaging. Intensity of bands was measured by the ImageQuant (Molecular Dynamics, Kent, UK) software.
For measurement of endogenous JNK activity, an aliquot of the clarified total extract was diluted so that the final concentration of the buffer was modified to 20 mm HEPES (pH 7.7), 75 mm NaCl, 2.5 mm MgCl2, 0.1 mm EDTA, 0.05% Triton X-100, 0.5 mm DTT, 20 mm β-glycerophosphate, 0.1 mm Na3VO4, and 10 μg/ml each of leupeptin and aprotinin. The diluted extracts were then precleared by mixing with 10 μl of glutathione (GSH)–agarose for 3–4 hr at 4°C. After this time, GSH–agarose was pelleted and the supernatants were transferred into new tubes and tumbled overnight at 4°C in the presence of GST–cJun (1–86) coupled to glutathione–agarose beads. The beads were finally pelleted and washed extensively (5 × 1 ml) in ice-cold HEPES-binding buffer (20 mm HEPES at pH 7.7, 50 mm NaCl. 2.5 mm MgCl2, 0.1 mm EDTA, 0.05% Triton X-100). The beads were then resuspended in 35 μl of kinase buffer (20 mm HEPES at pH 7.6, 20 mm MgCl2, 20 mm β-glycerophosphate, 0.1 mm Na3VO4, 2 mm DTT, 20 μm ATP, 10 μCi [α-32P]ATP), and the reaction was allowed to continue for 1 hr at 30°C. The reactions were terminated by washing the beads twice with ice-cold HEPES-binding buffer and the phosphorylated proteins were eluted by the addition of Laemmli buffer. The proteins were then resolved on a SDS/10% polyacrylamide gel. The gel was Coomassie stained, dried, and exposed to PhosphorImager screens.
In experiments designed to measure ERK- and JNK-specific phosphatase activity, embryonic extracts were prepared in the manner described above but with the exclusion of phosphatase inhibitors in all buffers. ERK phosphatase activity was measured by the ability of clarified extracts to inhibit the phosphotransferase activity of activated human ERK toward myelin basic protein. Clarified extracts (10 μl) were mixed with kinase buffer containing activated human ERK and MBP (10 μg) in a final volume of 50 μl. Kinase reactions were initiated by the addition of [α-32P]ATP (2.5 μCi/reaction) and the reaction allowed to proceed at 35°C for 15 min before termination by EDTA (20 mm). The entire reaction volume was then spotted to P81 phosphocellulose paper. After several washes in phosphoric acid (1%) and a rinse in acetone, the P81 paper was air dried and exposed to PhosphorImager screens for quantitation.
To measure JNK–phosphatase activity, clarified extracts were preincubated (30 min at 35°C) with GST–cJun (1–86) coupled to glutathione–agarose beads with prebound activated JNK derived from anisomycin-treated HeLa cells. Kinase reactions were initiated by the addition of kinase buffer (20 mm HEPES at pH 7.6, 20 mm MgCl2, 2 mm DTT, 20 mm ATP, 10 μCi [γ-32P]ATP) and allowed to proceed for 1 hr at 35°C. Control reactions were performed in kinase buffer (lacking phosphatase inhibitors) either in the absence of any extract or kinase buffer containing CL100 (kindly provided by S. Keyse, ICRF, Ninewells Hospital, Dundee, UK). Reactions were terminated as described above and fractionated on a SDS/10% polyacrylamide gel. The intensity of phosphorylated GST–cJun (1–86) protein was quantitated by PhosphorImaging.
Loading controls for all the extracts (in the presence or absence of phosphatase inhibitors) were performed by Western blotting. Western blots were tested with an anti-Drosophila ERK (Rolled) antibody kindly provided by L. Zipursky (HHMI, University of California, Los Angeles). This antibody recognizes a single 45-kD band. The results were quantified by densitometric scanning of ECL films, and the results of kinase and phosphatase assays were normalized to these values.
Acknowledgments
We thank C. O’Kane, W. Gehring, B. Baker, J. Campos-Ortega, J.P. Vincent, F.M. Hoffman, J. Pradel, A. Brand, C. Ruhslow, A. Pia, and the Bloomington Stock center for generously providing Drosophila stocks; L. Zipursky for the anti-Rolled antibody; S. Keyse for the CL100 protein; C.M. Marshall for the activated human ERK; and E. Hafen, S. Noselli, and N. Perrimon for helpful discussions and communication of data prior to publication. We are grateful to S. Keyse, M. Freeman, and A. Prokop for comments on the manuscript, and S. Rolfe for technical assistance. E.M.-B. was supported by The Wellcome Trust and the European Union. A.G. and J.R. were supported by a Jane Coffin Child fellowship and a Commonwealth studentship respectively. K.V. and A.M.T. are supported by the Wellcome Trust. A.M.-A. is a Senior Fellow of The Wellcome Trust.
Note
The cDNA sequence data reported in this paper have been submitted to the GenBank/EMBL library under accession no. AJ223360.
Footnotes
E-MAIL ama11@cus.cam.ac.uk; FAX 44-1223-336676.
References
- Affolter M, Nellen D, Nussbaumer U, Basler K. Multiple requirements for the receptor serine/threonine kinase thickveins reveal novel functions of TGFβ homologs during Drosophila embryogenesis. Development. 1994;120:3105–3117. doi: 10.1242/dev.120.11.3105. [DOI] [PubMed] [Google Scholar]
- Ashburner M. Drosophila, A laboratory handbook. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Brown NH, Kafatos FC. Functional cDNA libraries from Drosophila embryos. J Mol Biol. 1988;203:425–437. doi: 10.1016/0022-2836(88)90010-1. [DOI] [PubMed] [Google Scholar]
- Canman CE, Kastan MB. Signal transduction—3 paths to stress relief. Nature. 1996;384:213–214. doi: 10.1038/384213a0. [DOI] [PubMed] [Google Scholar]
- Charles CH, Sun H, Lau LF, Tonks NK. The growth factor-inducible immediate-early gene 3CH134 encodes a protein-tyrosine-phosphatase. Proc Natl Acad Sci. 1993;90:5292–5296. doi: 10.1073/pnas.90.11.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claasen LA, Ahn B, Koo HS, Grossman L. Construction of deletion mutants of the Escherichia coli UvrA protein and their purification from inclusion bodies. J Biol Chem. 1991;266:11380–11387. [PubMed] [Google Scholar]
- Diaz-Benjumea FJ, Hafen E. The sevenless signaling cassette mediates Drosophila EGF receptor function during epidermal development. Development. 1994;120:569–578. doi: 10.1242/dev.120.3.569. [DOI] [PubMed] [Google Scholar]
- Doi K, Gartner A, Ammerer G, Errede B, Shinkawa H, Sugimoto K, Matsumoto K. MSG5, A novel protein phosphatase promotes adaptation to pheromone response in Saccharomyces cerevisiae. EMBO J. 1994;13:61–70. doi: 10.1002/j.1460-2075.1994.tb06235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem. 1984;137:266–267. doi: 10.1016/0003-2697(84)90381-6. [DOI] [PubMed] [Google Scholar]
- Glise B, Noselli S. Coupling of Jun amino-terminal kinase and Decapentaplegic signaling pathways in Drosophila morphogenesis. Genes & Dev. 1997;11:1738–1747. doi: 10.1101/gad.11.13.1738. [DOI] [PubMed] [Google Scholar]
- Glise B, Bourbon H, Noselli S. hemipterous encodes a novel Drosophila MAP kinase kinase, required for epithelial cell sheet movement. Cell. 1995;83:451–461. doi: 10.1016/0092-8674(95)90123-x. [DOI] [PubMed] [Google Scholar]
- Groom LA, Sneddon AA, Alessi DR, Dowd S, Keyse SM. Differential regulation of the MAP, SAP and RK/P38 kinases by Pyst1, a novel cytosolic dual-specificity phosphatase. EMBO J. 1996;15:3621–3632. [PMC free article] [PubMed] [Google Scholar]
- Guan KL, Butch E. Isolation and characterization of a novel dual specific phosphatase, HVH2, which selectively dephosphorylates the mitogen-activated protein-kinase. J Biol Chem. 1995;270:7197–7203. doi: 10.1074/jbc.270.13.7197. [DOI] [PubMed] [Google Scholar]
- Guan K, Broyles SS, Dixon JE. A Tyr/Ser protein phosphatase encoded by vaccinia virus. Nature. 1991;350:359–362. doi: 10.1038/350359a0. [DOI] [PubMed] [Google Scholar]
- Harden N, Loh HY, Chia W, Lim L. A dominant inhibitory version of the small GTP-binding protein Rac disrupts cytoskeletal structures and inhibits developmental cell shape changes in Drosophila. Development. 1995;121:903–914. doi: 10.1242/dev.121.3.903. [DOI] [PubMed] [Google Scholar]
- Holland PM, Suzanne M, Campbell JS, Noselli S, Cooper JA. MKK7 is a stress-activated mitogen-activated protein kinase kinase functionally related to hemipterous. J Biol Chem. 1997;272:24994–24998. doi: 10.1074/jbc.272.40.24994. [DOI] [PubMed] [Google Scholar]
- Hou XS, Goldstein ES, Perrimon N. Drosophila Jun relays the Jun amino-terminal kinase signal transduction pathway to the Decapentaplegic signal transduction pathway in regulating epithelial cell sheet movement. Genes & Dev. 1997;11:1728–1737. doi: 10.1101/gad.11.13.1728. [DOI] [PubMed] [Google Scholar]
- Ishibashi T, Bottaro DP, Michieli P, Kelley CA, Aaronson SA. A novel dual-specificity phosphatase induced by serum stimulation and heat-shock. J Biol Chem. 1994;269:29897–29902. [PubMed] [Google Scholar]
- Jowett T. Preparation of nucleic acids. In: Roberts DB, editor. Drosophila: A practical approach. Oxford, UK: IRL Press; 1996. pp. 275–286. [Google Scholar]
- Kaiser K, Murray NE. The use of phage λ replacement vectors in the construction of representative genomic DNA libraries. In: Glover DM, editor. DNA cloning: A practical approach. Oxford, UK: IRL Press; 1985. pp. 1–47. [Google Scholar]
- Kameshita I, Fujisawa H. A sensitive method for detection of Calmodulin-dependent Protein Kinase-II activity in sodium dodecyl sulfate-polyacrylamide gel. Anal Biochem. 1989;183:139–143. doi: 10.1016/0003-2697(89)90181-4. [DOI] [PubMed] [Google Scholar]
- Keyse SM. An emerging family of dual-specificity MAP kinase phosphatases. Biochim Biophys Acta. 1995;1265:152–160. doi: 10.1016/0167-4889(94)00211-v. [DOI] [PubMed] [Google Scholar]
- Keyse SM, Emslie EA. Oxidative stress and heat-shock induce a human gene encoding a protein-tyrosine phosphatase. Nature. 1992;359:644–647. doi: 10.1038/359644a0. [DOI] [PubMed] [Google Scholar]
- Keyse SM, Ginsburg M. Amino-acid-sequence similarity between CL100, a dual-specificity MAP kinase phosphatase and cdc25. Trends Biochem Sci. 1993;18:377–378. doi: 10.1016/0968-0004(93)90092-2. [DOI] [PubMed] [Google Scholar]
- Kiehart DP, Feghali R. Drosophila cytoplasmic myosin. Biophys J. 1986;49:A186. doi: 10.1083/jcb.103.4.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockel S, Zeitlinger J, Staszewski LM, Mlodzik M, Bohmann D. Jun in Drosophila development: Redundant and non redundant functions and regulation by two MAPK signal transduction pathways. Genes & Dev. 1997;11:1748–1758. doi: 10.1101/gad.11.13.1748. [DOI] [PubMed] [Google Scholar]
- Kwak SP, Dixon JE. Multiple dual-specificity protein-tyrosine phosphatases are expressed and regulated differentially in liver-cell lines. J Biol Chem. 1995;270:1156–1160. doi: 10.1074/jbc.270.3.1156. [DOI] [PubMed] [Google Scholar]
- Martell KJ, Seasholtz AF, Kwak SP, Clemens KK, Dixon JE. HVH-5—A protein-tyrosine-phosphatase abundant in brain that inactivates Mitogen-Activated Protein-kinase. J Neurochem. 1995;65:1823–1833. doi: 10.1046/j.1471-4159.1995.65041823.x. [DOI] [PubMed] [Google Scholar]
- Martinez-Arias A. Development and patterning of the larval epidermis of Drosophila. In: Bate CM, Martinez-Arias A, editors. The development of Drosophila melanogaster. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 517–608. [Google Scholar]
- Muda M, Boschert U, Smith A, Antonsson B, Gillieron C, Chabert C, Camps M, Martinou I, Ashworth A, Arkinstall S. Molecular cloning and functional characterization of a novel Mitogen-Activated Protein kinase phosphatase, MKP-4. J Biol Chem. 1997;272:5141–5151. doi: 10.1074/jbc.272.8.5141. [DOI] [PubMed] [Google Scholar]
- Noguchi T, Metz R, Chen L, Mattei MG, Bravo R. Structure, mapping and expression of erp, a growth factor-inducible gene encoding a nontransmembrane protein tyrosine phosphatase, and effect of ERP on cell growth. Mol Cell Biol. 1993;13:5195–5205. doi: 10.1128/mcb.13.9.5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusslein-Volhard C, Wieschaus E, Kluding H. Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster. 1. Zygotic loci on the 2nd chromosome. Roux’s Arch Dev Biol. 1984;193:267–282. doi: 10.1007/BF00848156. [DOI] [PubMed] [Google Scholar]
- Riesgo-Escovar JR, Hafen E. Drosophila Jun kinase regulates expression of decapentaplegic via the ETS-domain protein Aop and the AP-1 transcription factor DJun during dorsal closure. Genes & Dev. 1997;11:1717–1727. doi: 10.1101/gad.11.13.1717. [DOI] [PubMed] [Google Scholar]
- Riesgo-Escovar JR, Jenni M, Fritz A, Hafen E. The Drosophila Jun-N-terminal kinase is required for cell morphogenesis but not for Djun-dependent cell fate specification in the eye. Genes & Dev. 1996;10:2759–2768. doi: 10.1101/gad.10.21.2759. [DOI] [PubMed] [Google Scholar]
- Ring JM. Identification and characterization of the velcro locus of Drosophila melanogaster. Ph.D. thesis. Cambridge, UK: University of Cambridge; 1993. [Google Scholar]
- Ring, J.M. and A. Martinez-Arias. 1993. puckered, a gene involved in position-specific cell differentiation in the dorsal epidermis of the Drosophila larva. Development (Suppl.) 121: 251–259. [PubMed]
- Rohan PJ, Davis P, Moskaluk CA, Kearns M, Krutzsch H, Siebenlist U, Kelly K. Pac-1—a mitogen-induced nuclear protein tyrosine phosphatase. Science. 1993;259:1763–1766. doi: 10.1126/science.7681221. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Gritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sluss HK, Han ZK, Barrett T, Goberdhan DCI, Wilson C, Davis RJ, Ip YT. A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes & Dev. 1996;10:2745–2758. doi: 10.1101/gad.10.21.2745. [DOI] [PubMed] [Google Scholar]
- Smith DB, Johnson KS. The single-step purification of polypeptides expressed in Escherichia coli as fusion proteins with Glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- St. Johnston RD, Hoffmann FM, Blackman RK, Segal D, Grimaila R, Padgett RW, Irick HA, Gelbart WM. Molecular organization of the decapentaplegic gene in Drosophila melanogaster. Genes & Dev. 1990;4:1114–1127. doi: 10.1101/gad.4.7.1114. [DOI] [PubMed] [Google Scholar]
- Sun H, Charles CH, Lau LF, Tonks NK. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell. 1993;75:487–493. doi: 10.1016/0092-8674(93)90383-2. [DOI] [PubMed] [Google Scholar]
- Tautz D, Pfeifle C. A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma. 1989;98:81–85. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- Tournier C, Whitmarsh AJ, Cavanagh J, Barret T, Davis RJ. Mitogen-Activated Protein Kinase Kinase 7 is an activator of the c-Jun NH2-terminal Kinase. Proc Natl Acad Sci. 1997;94:7337–7342. doi: 10.1073/pnas.94.14.7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton KA, Ray RP, Gelbart WM. An activity gradient of decapentaplegic is necessary for the specification of dorsal pattern elements in the Drosophila embryo. Development. 1993;117:807–822. doi: 10.1242/dev.117.2.807. [DOI] [PubMed] [Google Scholar]
- Wieschaus E, Nusslein-Volhard C. Looking at embryos. In: Roberts DB, editor. Drosophila, a practical approach. Oxford, Washington, D.C: IRL Press; 1986. [Google Scholar]
- Wilson R, Ainscough R, Anderson K, Baynes C, Berks M, et al. 2.2 Mb of contiguous nucleotide-sequence from chromosome-III of C. elegans. Nature. 1994;368:32–38. doi: 10.1038/368032a0. [DOI] [PubMed] [Google Scholar]
- Young PE, Richman AM, Ketchum AS, Kiehart DP. Morphogenesis in Drosophila requires nonmuscle myosin heavy chain function. Genes & Dev. 1993;7:29–41. doi: 10.1101/gad.7.1.29. [DOI] [PubMed] [Google Scholar]