

Abstract
eIF2B is a heteropentameric guanine-nucleotide exchange factor essential for protein synthesis initiation in eukaryotes. Its activity is inhibited in response to starvation or stress by phosphorylation of the α subunit of its substrate, translation initiation factor eIF2, resulting in reduced rates of translation and cell growth. We have used an in vitro nucleotide-exchange assay to show that wild-type yeast eIF2B is inhibited by phosphorylated eIF2 [eIF2(αP)] and to characterize eIF2B regulatory mutations that render translation initiation insensitive to eIF2 phosphorylation in vivo. Unlike wild-type eIF2B, eIF2B complexes with mutated GCN3 or GCD7 subunits efficiently catalyzed GDP exchange using eIF2(αP) as a substrate. Using an affinity-binding assay, we show that an eIF2B subcomplex of the GCN3, GCD7, and GCD2 subunits binds to eIF2 and has a higher affinity for eIF2(αP), but it lacks nucleotide-exchange activity. In contrast, the GCD1 and GCD6 subunits form an eIF2B subcomplex that binds equally to eIF2 and eIF2(αP). Remarkably, this second subcomplex has higher nucleotide-exchange activity than wild-type eIF2B that is not inhibited by eIF2(αP). The identification of regulatory and catalytic eIF2B subcomplexes leads us to propose that binding of eIF2(αP) to the regulatory subcomplex prevents a productive interaction with the catalytic subcomplex, thereby inhibiting nucleotide exchange.
Keywords: Phosphorylation, eIF2, GEF, translation initiation
Guanine-nucleotide exchange factors regulate the activity of G proteins by controlling their rate of conversion from the inactive GDP-bound form to the active GTP-bound state (Bourne et al. 1991; Boguski and McCormick 1993). One regulatory mechanism controlling protein synthesis initiation in eukaryotes involves inhibition of the guanine-nucleotide exchange factor eIF2B in response to starvation or stress conditions. eIF2B converts its substrate, eukaryotic translation initiation factor 2 (eIF2), from an inactive eIF2 ⋅ GDP binary complex to eIF2 ⋅ GTP. This active complex binds charged initiator tRNAMet (Met-tRNAMeti), forming a ternary complex, which interacts with the 40S ribosomal subunit. Following addition of mRNA and the 60S ribosomal subunit, the G-protein cycle is completed by hydrolysis of eIF2-bound GTP and the release of eIF2 ⋅ GDP (for review, see Merrick 1992; Trachsel 1996). Thus, inhibition of eIF2B activity prevents eIF2 recycling, thereby reducing rates of translation initiation and cell growth.
Although functionally similar to the small GTPases and exchange factors of the Ras superfamily, eIF2 and eIF2B are complex proteins of three (α–γ) and five (α–ε) nonidentical subunits, respectively. We considered it likely that the subunit complexity of eIF2B is attributable, at least in part, to its novel mechanism of regulation. eIF2B is inhibited indirectly by phosphorylation of its substrate, eIF2, on the α subunit (eIF2α) at residue serine 51. Three protein kinases, called PKR, HCR, and GCN2, specifically phosphorylate Ser-51 of eIF2α under different stress conditions (Clemens 1996). PKR (previously known as p68 kinase or DAI) is part of the antiviral response and is activated by double-stranded RNA, whereas HCR (also called HRI) is activated in mammalian reticulocytes in response to heme deprivation. Both kinases completely inhibit eIF2B to shut off total protein synthesis. In the yeast Saccharomyces cerevisiae, the protein kinase GCN2 phosphorylates eIF2α in response to amino-acid or purine starvation to induce translation of GCN4 mRNA, encoding a transcriptional activator of amino acid biosynthetic genes (Hinnebusch 1996). General translation and cell growth are inhibited in yeast cells expressing mutant hyperactive forms of GCN2 (termed GCN2c) (Wek et al. 1990), human PKR, or rabbit HCR (Dever et al. 1993) just as occurs in mammalian cells when eIF2 is highly phosphorylated.
In the one published report demonstrating the catalytic activity of yeast eIF2B in vitro, its regulation by eIF2(αP) was not examined (Cigan et al. 1993). The results of extensive genetic analysis, however, support the conclusion that yeast eIF2B is inhibited by eIF2(αP) just as it is in mammalian cells. In addition, these studies have provided evidence for specific functions of eIF2B subunits. All five subunits (encoded by GCN3, GCD7, GCD1, GCD2, and GCD6) show extensive sequence identity with the corresponding mammalian polypeptides (called eIF2Bα–ε, respectively) (Bushman et al. 1993a; Price et al. 1996a,b; Pavitt et al. 1997). The four subunits encoded by GCD genes are essential, whereas the GCN3 subunit appears to be dispensable for eIF2B catalytic activity. GCN3 is required for inhibition of translation initiation by eIF2(αP) in vivo (Hinnebusch and Fink 1983; Hannig and Hinnebusch 1988), however, implying that it mediates the inhibitory effects of eIF2(αP) on eIF2B catalytic activity. A recent study using recombinant eIF2B expressed in insect cells showed that rat eIF2Bα has a function similar to GCN3, as it was not required for eIF2B catalytic activity, but was needed for inhibition by eIF2(αP) (Fabian et al. 1997).
We previously obtained molecular and genetic evidence that the GCN3, GCD7, and GCD2 subunits all play roles in the inhibition of eIF2B by eIF2(αP). When co-overexpressed, these subunits formed a stable trimeric subcomplex in vivo that could partially suppress the inhibitory effects of eIF2(αP) (Table 1; Yang and Hinnebusch 1996). This effect implied that GCD2 and GCD7 participate directly with GCN3 in the inhibition of eIF2B by eIF2(αP). GCN3, GCD7, and the carboxy-terminal half of GCD2 all share sequence similarity (Paddon et al. 1989; Bushman et al. 1993a), suggesting that their homologous regions might be devoted to this regulatory mechanism. This hypothesis was confirmed by the isolation of regulatory mutations in each of the three subunits that were clustered within the shared homologous regions (Vazquez de Aldana and Hinnebusch 1994; Pavitt et al. 1997). Surprisingly, given the apparent redundancy of function, a single missense mutation in any one gene was sufficient to completely eliminate the regulation of eIF2B by eIF2(αP).
Table 1.
Suppression of eIF2α hyperphosphorylation toxicity in vivo by mutation or overexpression of eIF2B subunits
| Relevant genotype
|
Growth rate of cells expressing the eIF2α kinase GCN2c-513
|
|---|---|
| eIF2B mutationsa | |
| none (wild type) | 1+ |
| gcn3Δ | 5+ |
| GCD7–S119P (M1) | 5+ |
| GCD7–I118T, D178Y (M2) | 5+ |
| Overexpressed subunitsb | |
| none (vectors only) | 1+ |
| GCN3, GCD7, GCD1, GCD2, GCD6 | 4+ |
| GCD7, GCD1, GCD2, GCD6 | 5+ |
| GCN3, GCD7, GCD2 | 3+ |
| GCD1, GCD6 | 1+ |
| GCD6 | 1+ |
Data from Pavitt et al. (1997); growth scored on a scale of 0 for no growth to 5+ for wild-type growth, based on colony sizes.
Data taken from Yang and Hinnebusch (1996); growth scored on a scale of 0 for no growth to 6+ for wild-type growth, based on colony sizes.
Here we describe a biochemical analysis of the mechanism of inhibition of yeast eIF2B by eIF2(αP). We have used simple assays to demonstrate that eIF2B complexes with regulatory mutations in the GCN3 or GCD7 subunits overcome inhibition by eIF2(αP) by accepting it as a substrate for guanine-nucleotide exchange. In addition, we show that the five eIF2B subunits can be divided into two distinct subcomplexes, each of which binds to eIF2 in vitro. One subcomplex, consisting of GCN3, GCD7, and GCD2, binds more tightly to the phosphorylated form of eIF2 and has no exchange activity. The second subcomplex, composed of GCD1 and GCD6, binds equally to eIF2 and eIF2(αP) and has high-level exchange activity that is insensitive to eIF2 phosphorylation. These results suggest to us that eIF2B has distinct regulatory and catalytic binding surfaces for eIF2 that functionally interact to control eIF2B activity.
Results
An in vitro assay for yeast eIF2B activity and its inhibition by eIF2(αP)
To develop an in vitro assay for yeast eIF2B-catalyzed guanine-nucleotide exchange we purified eIF2 from a yeast strain overexpressing all three eIF2 subunits in which eIF2γ was modified by addition of six amino-terminal histidine residues (see Materials and Methods). This polyhistidine tag did not affect the function of eIF2γ in vivo as determined from growth rates in wild-type and GCN2c mutant cells; the latter being a sensitive test for the inhibitory effects of eIF2α phosphorylation on eIF2 activity (data not shown). The purified yeast eIF2 formed eIF2 ⋅ [3H]GDP binary complexes that were very stable at 10°C when challenged with a 100-fold excess of nonradiolabeled GDP (Fig. 1A). As a source of eIF2B to stimulate nucleotide exchange on eIF2 ⋅ [3H]GDP, we used extracts from yeast cells in which all five subunits were overexpressed by ∼10-fold. We showed previously that the overexpressed subunits form intact eIF2B complexes, as the majority of each subunit could be co-immunoprecipitated with an antibody against the GCD6 subunit (Dever et al. 1995). Addition of 150 μg of cell extract to the eIF2 ⋅ [3H]GDP complexes led to rapid dissociation of the bound radionucleotide, whereas an equal amount of an extract from a strain expressing wild-type levels of eIF2B gave a slow rate of nucleotide exchange, only slightly above the spontaneous rate with no extract (Fig. 1A). These findings showed that the rapid dissociation of eIF2 ⋅ [3H]GDP binary complexes stimulated by the first extract could be attributed to the exchange activity of overexpressed eIF2B.
Figure 1.

Guanine–nucleotide exchange catalyzed by wild-type and mutant eIF2B, and its inhibition by phosphorylation of eIF2. eIF2 ⋅ [3H]GDP binary complexes were preformed, with or without prior phosphorylation of eIF2 by HCR, and challenged with nonradiolabeled GDP (see Materials and Methods). (A–D) Level of eIF2 ⋅ 3H]GDP (filled symbols connected with solid lines) and eIF2(αP) ⋅ [3H]GDP (open symbols connected with broken lines) binary complexes remaining with time, following incubation with cell extracts (150 μg) from yeast strains bearing high-copy plasmids encoding the form of eIF2B indicated in each inset: (A) Wild-type five-subunit eIF2B (h.c. eIF2B); (B) eIF2B*4s, regulatory mutant eIF2B containing the four essential eIF2B subunits, but lacking GCN3; (C) eIF2B*5s-M1, eIF2B containing five eIF2B subunits with a regulatory mutation in GCD7 (GCD7-S119P); and (D) eIF2B*5s-M2, eIF2B containing five eIF2B subunits with a regulatory mutation in GCD7 (GCD7-I118T,D178Y). Control reactions with cell extracts from yeast strains containing empty vectors (filled circles linked with solid lines) are shown in A–D, and a reaction with extract buffer substituted for yeast cell extract (crosses linked by dotted lines) is shown in A.
To examine the regulation of eIF2B activity, we phosphorylated eIF2 in vitro using purified rabbit HCR. Isoelectric-focusing (IEF) PAGE analysis showed that eIF2α was rapidly and completely phosphorylated following the addition of HCR (Fig. 2A). eIF2(αP) ⋅ [3H]GDP binary complexes formed with the same efficiency and stability as binary complexes with unphosphorylated eIF2. Addition of the same extract overexpressing all five wild-type eIF2B subunits, however, failed to stimulate release of the bound nucleotide (Fig. 1A). This result demonstrates for the first time that eIF2(αP) is not a substrate for yeast eIF2B in vitro.
Figure 2.

Phosphorylation of purified eIF2 in vitro. (A) Purified eIF2 was phosphorylated for the indicated times (lanes 2–4), after which samples were resolved by IEF PAGE and eIF2α detected by Western blotting. (Lane 1) Unphosphorylated purified eIF2. (B) Guanine–nucleotide exchange reactions were performed exactly as described in Fig. 1, except that unlabeled GDP was used throughout. Samples were taken at the indicated times following the addition of cell extracts (150 μg) from yeast strains bearing high-copy plasmids encoding the indicated form of eIF2B (as in Fig. 1). eIF2α was resolved and detected as in A. (Lanes 1,2) Samples from reactions with unphosphorylated eIF2 ⋅ GDP binary complexes; (lanes 3–14) binary complexes prephosphorylated with HCR kinase; (lane 15) buffer and wild-type eIF2B extract only with no added eIF2 ⋅ GDP binary complexes. The positions of phosphorylated and unphosphorylated eIF2α are indicated.
eIF2B regulatory mutants can catalyze guanine-nucleotide exchange on eIF2(αP)
Having established an in vitro assay for eIF2B activity that mimics the regulation observed in vivo, we wished to examine guanine-nucleotide exchange using mutant forms of eIF2B to determine their mechanism of action. As indicated above, we showed previously that the reduced cell growth rate resulting from high levels of eIF2(αP) caused by expression of hyperactive GCN2c kinases in yeast can be alleviated by deletion of GCN3 (Dever et al. 1993) or completely suppressed by certain missense mutations in GCN3, GCD7, or GCD2 (Vazquez de Aldana and Hinnebusch 1994; Pavitt et al. 1997). The growth phenotypes of the gcn3Δ and gcd7 regulatory mutants relevant to this study are summarized in Table 1. We also showed previously by immunoblot analysis that the regulatory mutations did not affect the expression levels of any eIF2B subunits (except, of course, GCN3 in the gcn3Δ mutant). In addition, a combination of genetic tests and co-immunoprecipitation experiments demonstrated that the GCD7 mutations did not reduce the association of GCN3 with eIF2B, eliminating this possible mode of action (Pavitt et al. 1997). Therefore, it seemed likely to us that these regulatory mutations act by one of two mechanisms: (1) by allowing the inhibitor to act as a substrate for guanine-nucleotide exchange; or (2) by altering the relative binding affinities for phosphorylated and unphosphorylated eIF2 so that eIF2(αP) is rendered an ineffective inhibitor of mutant eIF2B. These models predict different fates for eIF2(αP) ⋅ GDP binary complexes. The first model predicts eIF2(αP) ⋅ GDP is a substrate for nucleotide exchange, while in the second model it is not. We used our exchange assay to distinguish between these two mechanisms.
To examine the regulatory defect caused by the mutations, we analyzed extracts from cells overexpressing the four essential subunits of eIF2B [i.e., lacking GCN3 to mimic the effect of a deletion of GCN3 (termed eIF2B*4s)], or overexpressing all five subunits of eIF2B with one of two missense mutations in GCD7 [(GCD7-S119P (termed eIF2B*M1) or GCD7-I118T,D178Y (termed eIF2B*M2)]. We found that all three mutant protein complexes promoted dissociation of [3H]GDP from eIF2 binary complexes independently of eIF2 phosphorylation (Fig. 1B–D). Interestingly, eIF2B*4s catalyzed guanine-nucleotide exchange at a faster rate than did wild-type eIF2B, apparently increasing the rate of exchange by twofold at early time points (Fig. 1A,B).
It was important to demonstrate that eIF2(αP) was not being dephosphorylated during the exchange reactions. Accordingly, we used IEF PAGE analysis to examine the level of eIF2α phosphorylation at three time points during the reaction. The data in Figure 2B revealed that 97%–98% of the eIF2α was phosphorylated at time=0 and that no dephosphorylation occurred over the course of the GDP exchange reactions. These results demonstrate that the regulatory alterations in the mutant eIF2B complexes allow both eIF2(αP) and eIF2 to be used as substrates for GDP exchange with similar efficiencies. These findings can account for the fact that strains bearing these eIF2B mutations maintain protein synthesis and continue growing when a majority of the eIF2 in the cell is phosphorylated (Vazquez de Aldana and Hinnebusch 1994; Dever et al. 1995; Pavitt et al. 1997), as eIF2(αP) no longer inhibits the guanine-nucleotide exchange activity of eIF2B.
GCD6 has nucleotide-exchange activity in vitro that is enhanced by co-overexpression of GCD1
To overexpress eIF2B in the experiments just described, we used a two-plasmid system, one to overexpress GCD1 and GCD6 and one of a series of second plasmids overexpressing wild-type or mutant versions of GCD2, GCD7, and GCN3. This system allowed us to investigate whether any eIF2B partial complexes have guanine-nucleotide exchange activity. We performed GDP-exchange assays using cell extracts co-overexpressing wild-type GCN3, GCD2, and GCD7 (h.c. GCN3/GCD2/GCD7), the mutants (h.c. GCN3/GCD2/GCD7*M1 or h.c. GCN3/GCD2/GCD7*M2) or the combination of GCD2 and GCD7 (h.c. GCD2/GCD7) (Fig. 3A–D). In each case, no significant nucleotide exchange was observed over background levels, showing that these overexpressed subunits do not possess eIF2B catalytic activity.
Figure 3.
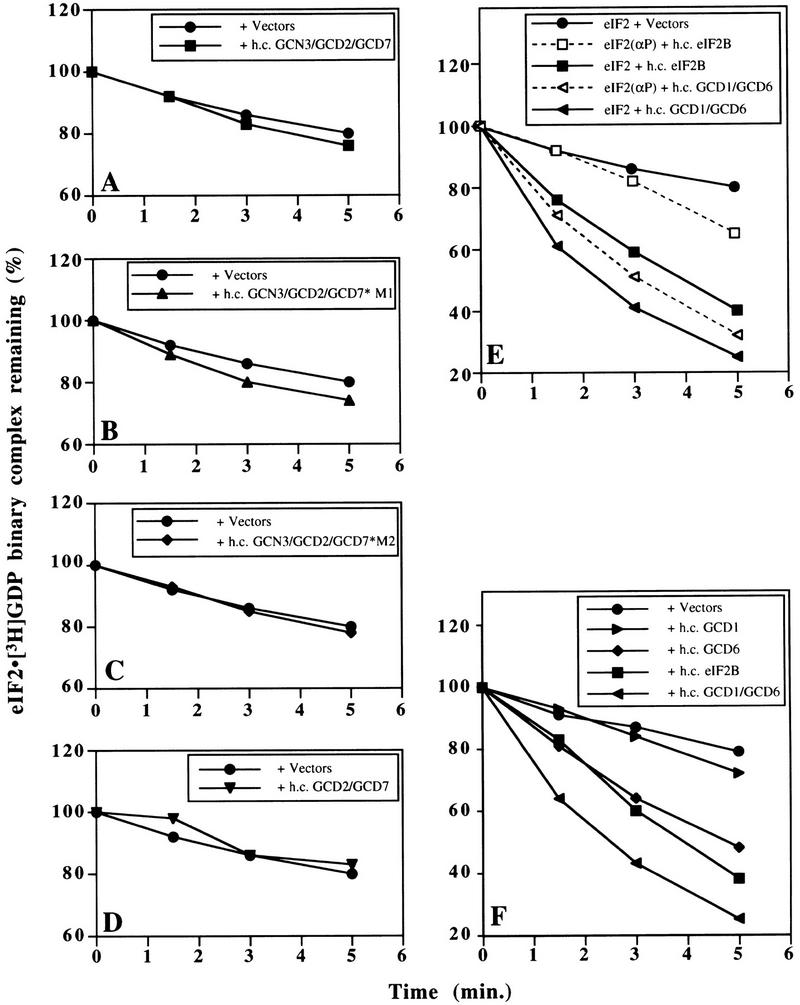
Guanine–nucleotide exchange catalyzed by eIF2B subcomplexes. Experiments were performed exactly as in Fig. 1 except with yeast cell extracts (150 μg) containing the overexpressed eIF2B subunits indicated in each inset. (A) Wild-type GCN3, GCD7, and GCD2 (h.c. GCN3/GCD2/GCD7); (B) wild-type GCN3 and GCD2 with regulatory mutant GCD7-S119P (GCD7*M1); (C) wild-type GCN3 and GCD2 with regulatory mutant GCD7-I118T,D178Y (GCD7*M2); (D) wild-type GCD7 and GCD2 only; (E) wild-type GCD1 and GCD6 (h.c. GCD1/GCD6) and all five subunits of wild-type eIF2B (h.c. eIF2B); (F) wild-type GCD1 alone (h.c. GCD1) and wild-type GCD6 alone (h.c. GCD6) in addition to the extracts shown in E. Control reactions with cell extracts from yeast cells containing empty vectors (filled circles linked with solid lines) are shown in all panels.
In sharp contrast to these results, we found that the overexpressed GCD1 and GCD6 subunits (h.c. GCD1/GCD6) showed full eIF2B catalytic activity (Fig. 3E), identical to the highest activity seen with the extract overexpressing eIF2B*4s (Fig. 1B). This result was unexpected because, unlike overexpressed wild-type eIF2B or eIF2B*4s, co-overexpression of GCD1 and GCD6 alone did not overcome the inhibitory effects of eIF2 hyperphosphorylation in yeast cells (Table 1). To characterize this exchange activity further, we asked whether it was inhibited by prephosphorylation of eIF2α. Similar to our findings on the eIF2B*4s and mutant five-subunit complexes, nucleotide-exchange activity in the h.c. GCD1/GCD6 extracts was unaffected by phosphorylation of eIF2α (Fig. 3E). Importantly, immunoblotting experiments determined that the levels of GCD1 and GCD6 were the same in extracts overexpressing GCD1 and GCD6 as in extracts overexpressing wild-type five subunit eIF2B (data not shown).
We next tested whether GCD1 or GCD6 alone possesses guanine-nucleotide exchange activity. A recent study using recombinant rat eIF2B expressed in insect cells found that eIF2Bε alone (the homolog of GCD6) possessed very low-level exchange activity, ∼30-fold below that of wild-type eIF2B (Fabian et al. 1997). Activity this low would be undetectable in our assay. We found, however, that overexpressed GCD6 alone possessed eIF2B activity only slightly lower than that observed with wild-type five-subunit eIF2B (Fig. 3F). GCD1 and GCD6 show 47% sequence similarity over the entire length of GCD1 (Bushman et al. 1993a). Despite this sequence similarity, we found that GCD1 alone had no exchange activity. Surprisingly, immunoblotting experiments showed that the level of GCD6 was approximately fourfold higher when overexpressed singly compared with co-overexpression with GCD1 (data not shown). When the relative protein levels are accounted for, these results show that GCD6 is the catalytic subunit of eIF2B and that GCD1 enhances its nucleotide-exchange activity ∼8- to 10-fold. In addition, they imply that GCD1 and GCD6 form a stable subcomplex that binds to eIF2 in the absence of the other three subunits. We provide additional evidence to support the latter assertion below.
Wild-type and mutant eIF2B bind with higher affinity to eIF2(αP) than to eIF2
By use of enzyme kinetic methods, it was demonstrated that mammalian eIF2(αP) was not a substrate for eIF2B and that eIF2B has a higher affinity for the inhibitor, eIF2(αP), than for the substrate, eIF2 (Goss et al. 1984; Rowlands et al. 1988). Rowlands et al. (1988) proposed that eIF2(αP) is a competitive inhibitor of nucleotide exchange that acts through repeated noncatalytic binding and release of the eIF2B. Using our exchange assay, we showed above that yeast eIF2(αP) was not a substrate for yeast eIF2B. Next, we used the fact that the purified polyhistidine-tagged eIF2 binds to Ni-NTA–agarose affinity resin to devise a pull-down assay to measure stable binding between eIF2B overexpressed in yeast extracts and purified eIF2. The eIF2 ⋅ eIF2B complexes formed in solution were captured on the affinity resin, washed, and eluted with imidazole. The fraction of eIF2B subunits bound to eIF2 was assessed by SDS-PAGE and Western blotting. Using this assay, we could show that binding of all five eIF2B subunits to eIF2(αP) was reproducibly about 2-fold higher than to unphosphorylated eIF2 and 10-fold higher than the background level of binding seen with no eIF2 added to the reactions (Fig. 4A,B, lanes 2–4). Except GCD2, none of the individual eIF2B subunits present in extracts overexpressing single eIF2B subunits bound to eIF2 or eIF2(αP) significantly above background levels (Fig. 5A,B, lanes 5–16 and Fig. 7A,B, lanes 5–12, below). [GCD2 bound to eIF2 but not eIF2(αP) (Fig. 5A,B, lanes 10,11); we are currently investigating further this result.] These findings indicate that, like the mammalian enzyme, yeast eIF2B has a higher binding affinity for eIF2(αP) than for eIF2, consistent with the idea that eIF2(αP) is a competitive inhibitor of eIF2B. Similar results were obtained when we examined the binding of the four-subunit form of eIF2B (eIF2B*4s; Fig. 4A,B, lanes 6–8).
Figure 4.

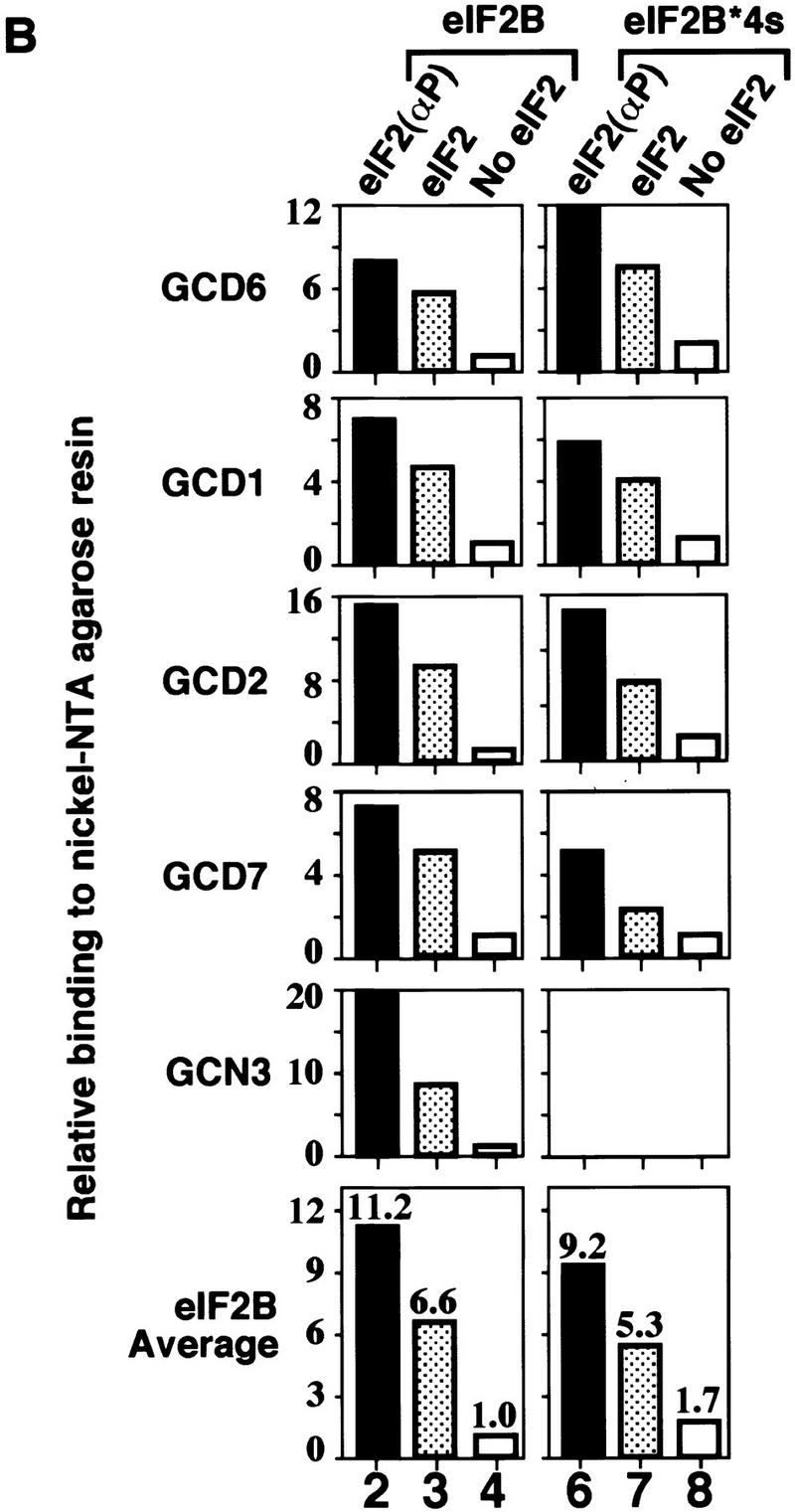
Binding of wild-type eIF2B and mutant eIF2B lacking the GCN3 subunit to His-tagged eIF2. Yeast whole cell extracts (100 μg) from strains overexpressing wild-type eIF2B (lanes 1–4) or eIF2B lacking the GCN3 subunit (eIF2B*4s, lanes 5–8) were incubated with 2.5 μg of prephosphorylated purified His-tagged eIF2 (lanes 2,6), unphosphorylated His-tagged eIF2 (lanes 3,7), or no eIF2 (lanes 4,8); proteins bound to eIF2 were recovered by Ni-NTA affinity chromatography. One third of each reaction was separated by SDS-PAGE and proteins identified by Western blotting. (A) Western blot analysis with specific polyclonal antisera to eIF2α and eIF2B subunits. (Input) Ten micrograms of each cell extract used (lanes 1,5). For GCN3, only the lower band represents the GCN3 signal, the upper diffuse band in each lane is a nonspecific cross-reacting band (Pavitt et al. 1997). (B) Histograms showing densitometry of signals for each eIF2B antiserum used in pellet lanes (lanes 2–4,6–8) from A relative to the density of the signal in lane 4, which was assigned an arbitrary value of 1. Mean densitometry for all subunits is shown.
Figure 5.
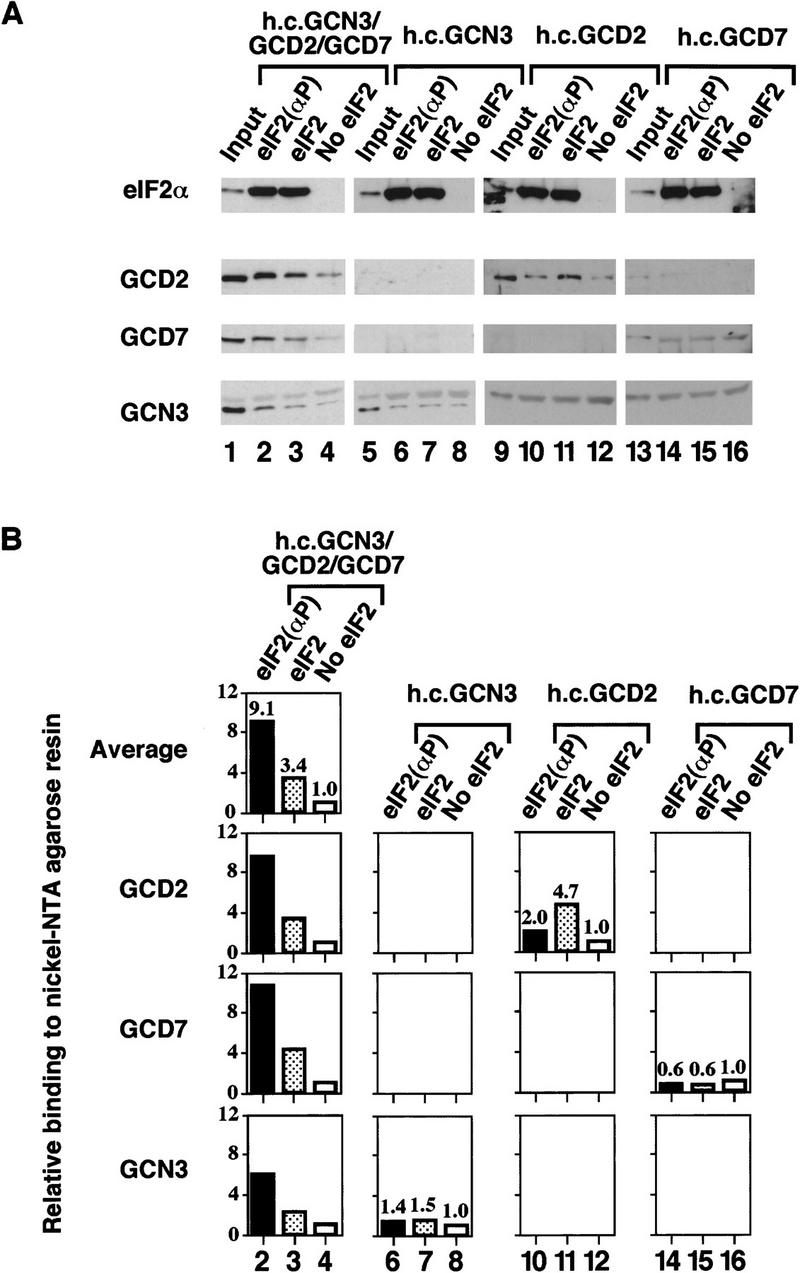
Binding of the regulatory GCN3/GCD7/GCD2 subcomplex to His-tagged eIF2. Overexpressed trimeric GCN3/GCD7/GCD2 subcomplex (lanes 1–4) or overexpressed single eIF2B subunits GCN3 (lanes 5–8), GCD2 (lanes 9–12), or GCD7 (lanes 13–16) were bound to eIF2 as in Fig. 4. (A) Western blot analysis of the binding eIF2B subunits to Ni–NTA–agarose in the presence of prephosphorylated purified His-tagged eIF2 (lanes 2,6,10,14), unphosphorylated purified His-tagged eIF2 (lanes 3,7,11,15), or without purified eIF2 (lanes 4,8,12,16). (Input) 10 μg of each cell extract used (lanes 1,5,9,13). (B) Histograms showing densitometry of signals for eIF2B antisera shown in pellet lanes (lanes 2–4,6–8,10–12,14–16) from A relative to the density of the signal in lanes 4,8,12, and 16, which were assigned an arbitrary value of 1. Mean densitometry for the three subunits of the trimeric regulatory complex is shown.
Figure 7.
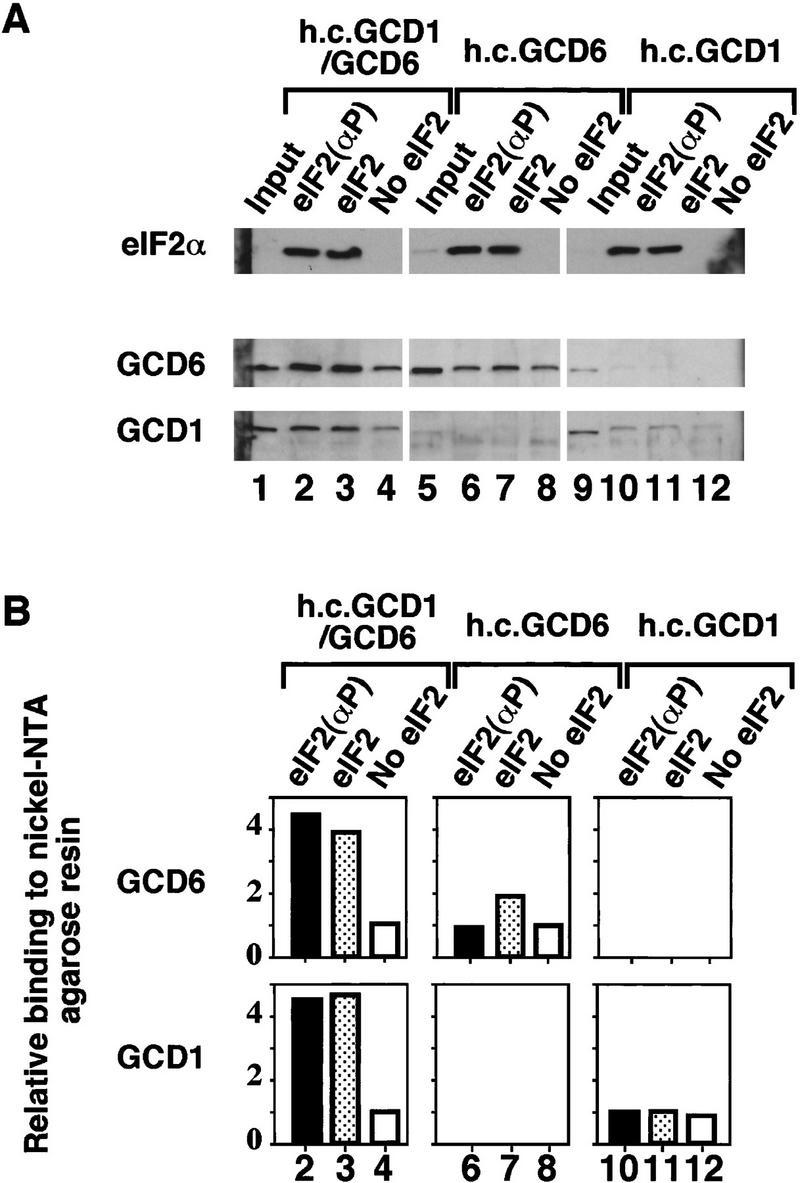
Binding of the catalytic GCD1/GCD6 subcomplex to His-tagged eIF2. As described for Fig. 4 except that the cell extracts used contained the overexpressed catalytic GCD6/GCD1 subcomplex (lanes 1–4) or overexpressed single subunits GCD6 (lanes 5–8) or GCD1 (lanes 9–12). (A) Western blot analysis of eIF2α and eIF2B subunits. The binding of 33 μg of cell extracts to Ni-NTA–agarose beads in the presence of purified His-tagged eIF2 prephosphorylated with HCR kinase (lanes 2,6,10), unphosphorylated purified His-tagged eIF2 (lanes 3,7,11), or without added purified His-tagged eIF2 (lanes 4,8,12). (Input) 10 μg of each cell extract used (lanes 1,5,9). (B) Histograms showing densitometry of signals for each eIF2B antibody shown in pellet lanes (lanes 2–4,6–8,10–12,14–16) from A relative to the density of the signal in lanes 4, 8, and 12, which were assigned an arbitrary value of 1.
We showed previously that overexpressed GCN3, GCD2, and GCD7 form a stable trimeric complex that can suppress the toxic effects of eIF2 phosphorylation in vivo (Yang and Hinnebusch 1996). To explain this finding we suggested that this trimeric subcomplex binds to eIF2(αP) independently of GCD1 and GCD6. In agreement with this prediction, co-overexpressed GCN3, GCD2, and GCD7 bound to eIF2(αP) at levels approximately 10-fold above background and 3-fold higher than to eIF2 (Fig. 5A,B, lanes 2–4). The implications of these results for the mechanism of action of eIF2B and the trimeric GCN3/GCD2/GCD7 subcomplex are discussed later.
GCD1 forms a subcomplex with GCD6 to stabilize the interaction of GCD6 with eIF2
As the catalytic activity of overexpressed GCD6 was greatly stimulated by the co-overexpression of GCD1, we surmised that GCD1 and GCD6 can form a stable subcomplex in the absence of the other three subunits. To provide direct evidence for this conclusion, we modified GCD1 by adding a carboxy-terminal polyhistidine tag and verified by growth assays that the His-tagged GCD1 gene on a single-copy-number plasmid was fully capable of substituting for wild-type GCD1 in vivo (data not shown, see Materials and Methods). When co-overexpressed with GCD6, His-tagged GCD1 specifically bound a large fraction of the excess GCD6 to Ni–NTA silica resin, while only a small fraction of overexpressed GCD6 was bound to the resin in a control reaction where GCD1 was untagged (Fig. 6A,B, lanes 4,5). These data demonstrated that GCD1 and GCD6 could form a stable subcomplex in vivo. Next we asked if GCD1 could stabilize the binding between GCD6 and eIF2. In a pull-down experiment with extracts overexpressing untagged GCD1 and GCD6 and purified His-tagged eIF2, we found that, when co-overexpressed, GCD1 and GCD6 bound to eIF2 at four- to fivefold higher levels than when each protein was overexpressed singly (Fig. 7, lanes 2–4 vs. lanes 6–8 and 10–12). Interestingly, unlike the GCN3/GCD2/GCD7 trimeric subcomplex, the level of binding of the GCD1/GCD6 subcomplex to eIF2 was not affected by eIF2α phosphorylation. Together with the results of the guanine-nucleotide exchange assays, these data indicate that GCD6 is the eIF2B catalytic subunit and that GCD1 stabilizes the binding of GCD6 to eIF2, thereby enhancing its catalytic activity.
Figure 6.
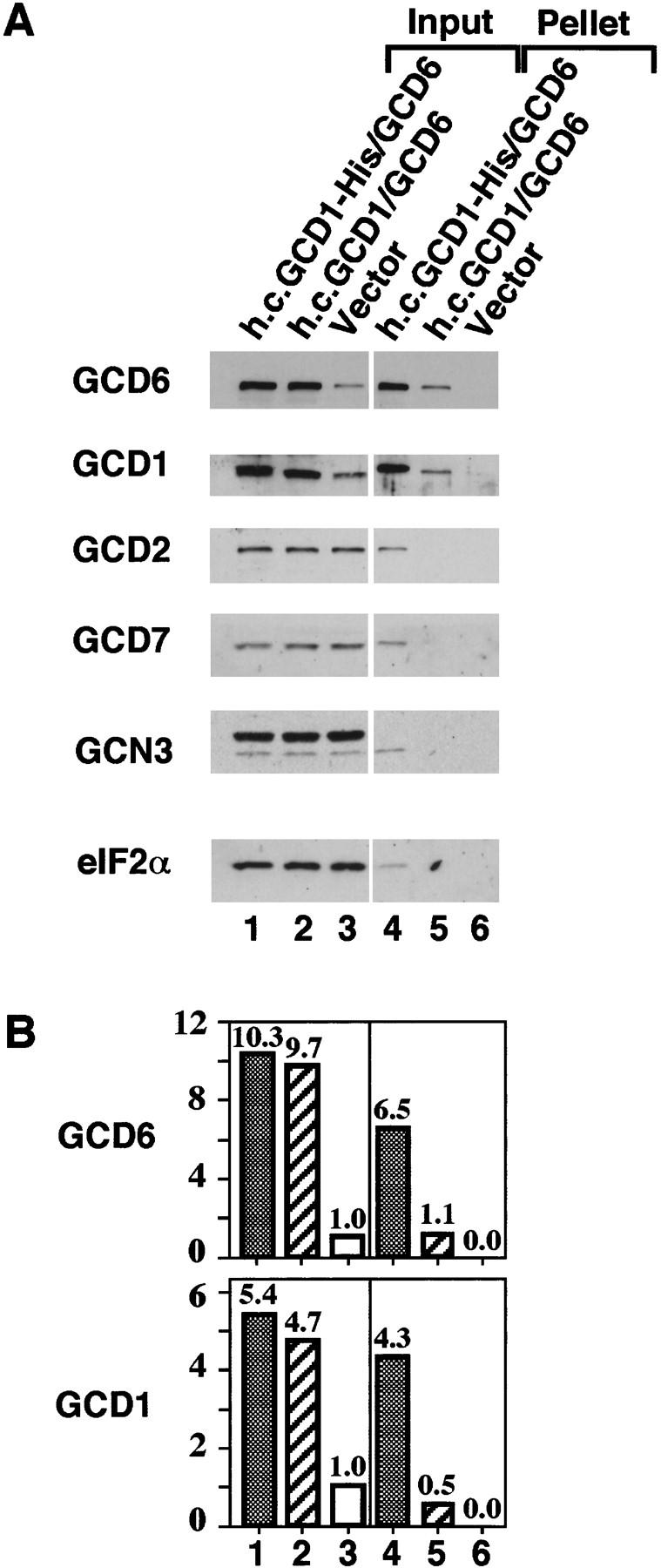
Subcomplex formation between His-tagged GCD1 and GCD6. Ni-NTA–silica affinity chromatography of whole-cell extracts from strains co-overexpressing His-tagged GCD1 and wild-type GCD6 (lanes 1,4), co-overexpressing wild-type GCD1 and GCD6 (lanes 2,5), or carrying only the plasmid vector (lanes 3,6). (A) Western blot analysis of the binding of eIF2B subunits from 120 μg of cell extracts to Ni–NTA–silica resin (lanes 4–6) and from 10 μg of each cell extract input (lanes 1–3). (B) Histograms showing densitometry of signals for GCD1 and GCD6 antiserum from A relative to the density of the signal in lane 3, which was assigned an arbitrary value of 1.
Discussion
Mechanism of action of eIF2B regulatory mutations
Regulatory mutations isolated in GCN3, GCD2, and GCD7 overcome the inhibitory effects of eIF2 phosphorylation on cell growth and activation of GCN4 expression (Vazquez de Aldana and Hinnebusch 1994; Pavitt et al. 1997). These mutations alter residues within homologous regions of each subunit. A single missense substitution in any one subunit is sufficient to disrupt this regulatory function, suggesting that all three subunits act together to mediate the negative regulation of eIF2B by eIF2(αP). Fabian et al. (1997) recently demonstrated that rat eIF2B expressed in insect cells catalyzed guanine-nucleotide exchange. They also showed that a four-subunit form of eIF2B lacking the α subunit (the homolog of GCN3) was not sensitive to regulation by eIF2(αP), thereby mimicking in vitro the effects of deleting GCN3 in yeast cells. By use of a competition assay in which nucleotide exchange on eIF2 ⋅ [3H]GDP is inhibited by the addition of eIF2(αP), however, they could not elucidate the mechanism by which inhibition was overcome.
In our in vitro assays for guanine-nucleotide exchange and eIF2 binding by eIF2B, all of the eIF2 in the preformed eIF2 ⋅ [3H]GDP binary complexes was either fully phosphorylated (at least 98% phosphorylated) or unphosphorylated. We began by showing that eIF2(αP) was completely inactive as a substrate for guanine-nucleotide exchange by wild-type eIF2B (Fig. 1). The eIF2 binding experiments demonstrated that eIF2B bound to eIF2(αP) with a higher affinity than to unphosphorylated eIF2 (Fig. 4). Together, these experiments established that wild-type yeast eIF2B has a higher affinity for the inhibitor than the substrate, but cannot catalyze GDP exchange with the bound inhibitor.
To explain the action of our regulatory mutations, we set out to distinguish between two possible mechanisms for how these mutations eliminate the inhibitory effect of eIF2(αP) on eIF2B activity: (1) the mutations simply reduce the binding affinity of eIF2B for the inhibitor eIF2(αP) versus the substrate (Hinnebusch 1994) and (2) eIF2(αP) is accepted as a substrate for guanine-nucleotide exchange by mutant eIF2B (Pavitt et al. 1997). The major difference between these models is the fate of eIF2(αP) ⋅ GDP in the nucleotide-exchange assay. In the first model, eIF2(αP) is still an inhibitor (just an ineffective one) so no GDP exchange should occur with eIF2(αP) ⋅ GDP as a substrate. In contrast, we observed nucleotide exchange with mutant eIF2B extracts (eIF2B*) at equal rates with both eIF2(αP) and eIF2, as predicted by the second model (Fig. 1B–D). These results imply that the regulatory mutations we analyzed overcome a block in a step of the catalytic process that occurs after binding eIF2(αP). In addition, binding of the eIF2B*4s mutant complex to eIF2 and eIF2(αP) was examined with the result that no change in binding preference for eIF2 versus eIF2(αP) was seen relative to wild-type eIF2B (Fig. 4). Because the first model requires a substantial change in the relative binding affinities of eIF2B for eIF2 and eIF2(αP) and none was found, these results corroborate the conclusions drawn from the nucleotide-exchange assays.
Separate catalytic and regulatory subcomplexes of eIF2B subunits
Our second major finding is the formation of a subcomplex between the GCD1 and GCD6 subunits (Fig. 6), which binds with equal affinity to eIF2(αP) or eIF2 (Fig. 7) and has the full nucleotide exchange activity of eIF2B in vitro (Fig. 3). It was shown recently that rat eIF2Bε alone (the homolog of GCD6) has very low catalytic activity in a recombinant baculovirus expression system (Fabian et al. 1997). Similarly, yeast GCD6 alone has catalytic activity (Fig. 3). In addition, we find that GCD1 alone has no catalytic activity, but stabilizes the binding of eIF2 to GCD6 (Fig. 7), stimulating catalytic activity approximately ninefold. These findings demonstrate that GCD6 is the catalytic subunit of eIF2B and imply that GCD1 serves to stabilize the eIF2 ⋅ eIF2B interaction. Consistent with the latter conclusion, we found that several conditional-lethal (temperature-sensitive) mutations in GCD1 were suppressed in vivo by overexpression of all three subunits of eIF2, implying that an increase in eIF2 concentration restores the eIF2 ⋅ eIF2B interaction weakened by these gcd1 mutations (Dever et al. 1995; G.D. Pavitt and A.G. Hinnebusch, unpubl.).
There is a striking parallel to this proposed role for GCD1 in the binding of the nucleotide-exchange factor GrpE to the ATPase domain of its substrate DnaK. In the crystal structure of this complex, a GrpE homodimer binds to DnaK, but only one GrpE molecule (GrpE1) makes contacts with DnaK. These interactions promote the structural rearrangements in DnaK required to mediate nucleotide exchange. The second GrpE molecule binds only to GrpE1, thereby stabilizing the productive interaction of GrpE1 with DnaK (Harrison et al. 1997). As GCD1 is similar along its entire length to GCD6 (Bushman et al. 1993a) and as shown here, stabilizes the binding of eIF2 to GCD6 and enhances GCD6 exchange activity, the functional relationship between the GrpE subunits is easy to imagine for GCD1 and GCD6.
We showed previously that GCD2, GCD7, and GCN3 can form a stable subcomplex in vivo that, when overexpressed, mimicked the effect of eIF2B regulatory mutations by suppressing the growth inhibition caused by eIF2 hyperphosphorylation (Yang and Hinnebusch 1996; see Table 1). Using genetic data, we argued that this subcomplex does not possess guanine-nucleotide exchange activity on its own, but can sequester eIF2(αP) and allow native eIF2B to catalyze guanine-nucleotide exchange on the pool of unphosphorylated eIF2 in the cell. In support of this model, we found that the GCN3/GCD7/GCD2 subcomplex does not possess guanine-nucleotide exchange activity (Fig. 3) and that it can interact stably with eIF2 with a binding preference for eIF2(αP) similar to that of native eIF2B (Figs. 4,5). Therefore, when overexpressed, the GCN3, GCD7, and GCD2 form a regulatory subcomplex that could act in vivo to titrate eIF2(αP) away from native eIF2B.
A second function for eIF2B?
The finding that the GCD1/GCD6 subcomplex has high-level nucleotide-exchange activity in cell extracts but is not sufficient to provide the essential function of eIF2B in vivo is interesting. GCD2 and GCD7 are essential genes. In addition, as shown in Table 1, overexpression of the four essential subunits of eIF2B (eIF2B*4s) is sufficient to overcome the growth-inhibitory effects of eIF2α hyperphosphorylation (a sensitive, but indirect measurement of eIF2B activity), whereas co-overexpression of just GCD1 and GCD6 has no such effect. In cell extracts, however, both complexes show identical high-level eIF2B activity that is insensitive to eIF2 phosphorylation. How can we explain this difference between cell extracts and intact cells?
According to the currently accepted view of protein synthesis initiation, eIF2 is spontaneously released from the 40S ribosome following hydrolysis of eIF2-bound GTP. This allows 60S subunit joining, to form the 80S initiation complex, and the commencement of translation elongation. Subsequently, eIF2B catalyzed guanine-nucleotide exchange on eIF2 occurs free from the ribosome (Merrick 1992). Several reports, however, suggest that, with phosphorylation of mammalian eIF2, eIF2(αP) ⋅ GDP remains associated with ribosomes, and exogenously added eIF2B can stimulate the release of eIF2(αP) ⋅ GDP (De Benedetti and Baglioni 1983; Thomas et al. 1985; Gross et al. 1987; Ramaiah et al. 1992). This finding suggests that eIF2B functions to release eIF2 ⋅ GDP from ribosomal subunits during the initiation process and that this second activity is also inhibited by eIF2 phosphorylation. Consistent with this idea, 48S initiation complexes bearing eIF2 were observed in gcd1 and gcd2 mutants (Cigan et al. 1991; Foiani et al. 1991). Therefore, we speculated that yeast eIF2B similarly performs a second function to release eIF2 ⋅ GDP from ribosomes. Although the GCD1/GCD6 subcomplex is competent for nucleotide exchange on free eIF2 ⋅ GDP in vitro, it is possible that GCD2 and GCD7 are additionally required in vivo to localize eIF2B to the ribosome or to release eIF2 ⋅ GDP from ribosomal subunits.
Separate regulatory and catalytic binding surfaces for eIF2 mediate inhibition of nucleotide exchange
Our results demonstrate that the catalytic and regulatory subcomplexes in eIF2B can both bind eIF2 independently in vitro. The stability of the interaction between eIF2 and the regulatory subcomplex is significantly increased by phosphorylation of eIF2, whereas the catalytic subcomplex binds equally to phosphorylated and unphosphorylated eIF2.
As eIF2γ possesses the GDP/GTP-binding domain (Hannig et al. 1993), and eIF2α bears the phosphorylation site, it is tempting to speculate that the eIF2B catalytic subcomplex interacts with the γ subunit of eIF2, whereas the regulatory subcomplex binds eIF2α. We propose that the presence of the regulatory subcomplex in eIF2B provides a high-affinity interaction with the α subunit of eIF2 that is incompatible with the proper binding of eIF2 to the catalytic subcomplex required for nucleotide exchange. Thus eIF2 would initially bind to eIF2B in a nonproductive mode dominated by interactions between eIF2α and the regulatory subcomplex. When eIF2 is unphosphorylated, as depicted in Figure 8A, an isomerization in eIF2B would then shift binding into a productive mode involving interactions between eIF2γ and the catalytic subcomplex. When phosphorylated eIF2 binds, however, the isomerization would be inhibited, preventing nucleotide exchange (Fig. 8B). The catalytic subcomplex alone is capable of high-affinity binding to eIF2, but cannot distinguish between eIF2 and eIF2(αP) because it does not interact with the α subunit of eIF2 (Fig. 8C). The eIF2B complexes with regulatory mutations in GCN3 and GCD7 analyzed here fail to distinguish between eIF2 and eIF2(αP) in the nucleotide exchange assays, but at least the eIF2B*4s complex showed the higher affinity binding of eIF2(αP) versus eIF2 characteristic of wild-type eIF2B. These data suggest that the mutant eIF2B complexes catalyze nucleotide exchange with eIF2(αP) because the isomerization from nonproductive to productive binding is no longer blocked (Fig. 8D).
Figure 8.

A sequential binding model for eIF2B catalyzed guanine-nucleotide exchange and its inhibition by eIF2(αP). (A) Proposed two-binding surface scheme for GDP/GTP exchange with unphosphorylated eIF2. The eIF2 (unfilled oval labeled α, β, γ) in a binary complex with GDP (light-grey filled circle with a light center) makes initial contact with eIF2B via a surface on the regulatory GCN3/GCD7/GCD2 subcomplex (stippled shape labeled 2 3 7). A hypothetical conformational change in eIF2B (movement indicated by grey arrows) occurs to promote correct contact between eIF2γ and the eIF2B catalytic subcomplex (filled black with subunits labeled 6 1), permitting exchange of GDP for GTP (grey filled circle with dark center). (B) Inhibition of nucleotide exchange by phosphorylated eIF2. eIF2(αP) (as in A with an added filled circle, labeled ∼P) binds to the eIF2B regulatory subcomplex with high affinity (broad arrow), this inhibits the conformational change in eIF2B, preventing nucleotide exchange. (C) In the absence of the eIF2B regulatory subcomplex, direct binding of eIF2γ to the catalytic subcomplex allows nucleotide exchange even with eIF2(αP). (D) Regulatory mutant eIF2B can perform nucleotide exchange with eIF2(αP). eIF2(αP) binds to mutant eIF2B (eIF2B*), making contact with the altered regulatory subcomplex (altered shaped stippled box labeled 2 3 7). The regulatory defect allows the conformational change needed for productive interaction between eIF2γ and the catalytic subcomplex even when eIF2 is phosphorylated.
Equilibrium binding assays will be needed to determine the dissociation constants for interactions between eIF2 and the different eIF2B complexes and subcomplexes to establish the validity of these interpretations. In addition, a clearer picture of how eIF2(αP) inhibits eIF2B will require a deeper understanding of the steps in the guanine-nucleotide exchange reaction. With the assays developed here and identification of the yeast GCD1/GCD6 subcomplex as a highly efficient exchange factor, it should be possible to analyze the mechanism of the exchange reaction in greater detail. It may also be feasible to obtain structural information on the eIF2B catalytic subunit, GCD6, or the stable complex formed between eIF2, GCD1, and GCD6.
Materials and methods
Yeast strains
Standard methods were used for transformation (Ito et al. 1983) and genetic manipulation of yeast strains (Sherman et al. 1974). For making extracts containing overexpressed eIF2B subunits, plasmid transformants of strain BJ1995 (MATα leu2 trp1 ura3-52 gal2 pep4-3 prb1-1122) (Jones 1991) were employed. Strain GP3511 (MATα leu2-3 leu2-112 ura3-52 ino1 gcn2Δ pep4::LEU2 sui2Δ HIS4-lacZ bearing plasmid pAV1089[SUI2 SUI3 His-tagged GCD11 URA3]) was used for overexpression and purification of polyhistidine-tagged eIF2. GP3511 was constructed as follows: (1) GCN2 was deleted with p1144 (Dever et al. 1992) in strain H1648 (MATα leu2-3 leu2-112 ura3-52 ino1 sui2Δ HIS4–lacZ bearing plasmid p918[SUI2 LEU2]) (a gift from Tom Dever), to generate strain H2573; (2) plasmid shuffling (Boeke et al. 1984) was used to introduce pAV1089, generating GP3507; and (3) PEP4 was disrupted by use of the pep4::LEU2 plasmid pT517 (a gift from Tom Stevens, University of Oregon, Eugene) to generate GP3511.
Plasmids
Standard methods were used for the manipulation of DNA (Sambrook et al. 1989). pRS425 and pRS426 are LEU2- and URA3-marked 2μ plasmids, respectively (Christianson et al. 1992). Plasmid p1873 is a derivative of pRS425 for overexpressing GCD1 and GCD6 (Dever et al. 1995). Derivatives of pRS426 for overexpressing different combinations of eIF2B subunits were: p1871 (GCD2, GCD7, GCN3) and p1872 (GCD2, GCD7) (Dever et al. 1995), and p2297 (GCD2), p2305 (GCD7), p2304 (GCN3), p2301 (GCD1), p2300 (GCD6), and p2302 (GCD1, GCD6) (Yang and Hinnebusch 1996).
pAV1089 is a 2μ, URA3 plasmid used to overexpress all three subunits of eIF2 [SUI2 (α), SUI3 (β), and GCD11 (γ)] with polyhistidine-tagged GCD11 that was constructed in three steps. (1) Site-directed mutagenesis (Altered Sites mutagenesis kit, Promega) introduced the polyhistidine tag SGHHHHHHTG (single-letter code) between the first and second codons of GCD11 in plasmid Ep517 (Dorris et al. 1995), generating plasmid pAV1027 (CEN LEU2 GCD11–6×His). This plasmid complemented the gcd11Δ allele in strain EY551 (MATa leu2-3 leu2-112 ura3-52 gcd11::hisG (ΔHpaI–HpaI) HIS4–lacZ bearing plasmid Ep293[YCp50 GCD11]) (Dorris et al. 1995) when Ep293 was eliminated by plasmid shuffling. (2) The GCD11 fragment used in this plasmid failed to complement fully a deletion of GCD11 in some strains when present on a low-copy plasmid (T. Dever pers. comm.). To overcome this defect, an additional 0.8 kb of GCD11 5′ noncoding DNA was subcloned upstream of the GCD11–6×His allele to generate pAV1043. (3) Subcloning the GCD11–6×His allele from pAV1043 on an NheI–XbaI fragment into p1778, the 2μ plasmid overexpressing eIF2α and eIF2β [YEp24 SUI2 SUI3] (Dever et al. 1995), linearized with NheI1 to generate pAV1089. When transformed into yeast this plasmid overexpressed the three subunits of eIF2 to the same levels as the control plasmid p1780 overexpressing eIF2α, eIF2β, and untagged eIF2γ (∼10-fold) (Dever et al. 1995).
p2337 is a 2 μ URA3 plasmid for overexpressing polyhistidine-tagged GCD1 and wild-type GCD6, and was constructed as follows. (1) Site-directed mutagenesis (as above) introduced the polyhistidine tag (residues SGHHHHHHTG in single-letter code) immediately before the nonsense codon of GCD1 in plasmid pBM10[CEN4 TRP1 GCD1] (Cigan et al. 1993), generating plasmid pAV1013 [CEN4 TRP1 GCD1–6×His]. This plasmid complemented the gcd1::LEU2 disruption in strain MC1061(MATa ura3-52 leu2-3 leu2-112 trp1- Δ63 gcd1::LEU2 HIS4–lacZ bearing plasmid pBM10[GCD1 TRP1]) (Cigan et al. 1993) following the loss of plasmid pBM10 on medium containing tryptophan. (2) p2337 was constructed by replacing wild-type GCD1 in p1873[2 μ URA3 GCD1 GCD6] with the GCD1–6×His allele in pAV1013 by ligation of the 9.95-kb XhoI–KpnI fragment of p1873 to the 2.4-kb SalI—KpnI GCD1–6×His-containing fragment of pAV1013.
pAV1139 (M1) and pAV1140 (M2) are high-copy plasmids for overexpressing wild-type GCN3 and GCD2 together with GCD7 regulatory mutant proteins with missense substitutions GCD7-S119P and GCD7-I118T,D178Y, respectively. They were constructed by ligation of three DNA fragments: a 2.6-kb ClaI–XbaI GCD2 fragment from pCP46 (Paddon and Hinnebusch 1989), the 2.1-kb ClaI—Bsp120 I fragment containing either GCD7-S119P (M1) from pAV1079 (Pavitt et al. 1997) or GCD7-I118T,D178Y (M2) from p1563 (Vazquez de Aldana and Hinnebusch 1994), and the high-copy GCN3 URA3 plasmid p2304, cut with SpeI and NotI. These plasmids contain restriction fragments equivalent to those present in p1871, from which wild-type GCD7, GCD2, and GCN3 were co-overexpressed (Dever et al. 1995).
General protein methods
Protein concentrations were determined by the Bradford assay (BioRad) using BSA as a standard. eIF2 and eIF2B proteins were resolved by 12% SDS-PAGE (Sambrook et al. 1989) and detected by Western blotting as described (Dever et al. 1995; Yang and Hinnebusch 1996) with the appropriate rabbit polyclonal primary antisera (Cigan et al. 1989, 1991; Bushman et al. 1993b), an HRP-conjugated anti-rabbit secondary antibody, and the enhanced chemiluminescence system from Amersham.
Purification of His-tagged eIF2
His-tagged eIF2 was purified by use of Ni-NTA–agarose (Qiagen) and heparin–Sepharose (Pharmacia) chromatography as described by Erickson and Hannig (1996) with the following modifications. We used the plasmid pAV1089 to overexpress His-tagged eIF2 in the protease deficient (pep4), gcn2Δ strain GP3511 described above. We added phosphatase inhibitors NaF (50 mm) and Na3VO4 (100 μm) to breaking buffer and Ni-column-loading buffer, and NaF only (5 mm) to all subsequent buffers. Cell lysis was done in a bead-beater (Biospec Products) with ice-cold acid-washed glass beads with 5×45-sec beatings and 1-min intervals for cooling. Column fractions containing eIF2 were identified by 12% SDS-PAGE and by assaying for ternary complex formation activity (Hannig et al. 1993) with yeast [3H]Met-tRNAMeti (Donahue et al. 1988). Active fractions were pooled, desalted and concentrated in storage buffer (20 mm Tris-HCl at pH 7.5, 100 mm KCl, 0.1 mm MgCl2, 25% vol/vol glycerol, 1 mm DTT, 0.1 mm PMSF, 5 mm NaF) by use of Centriprep 30 spin concentrators (Amicon), and stored under liquid nitrogen in 50-μl aliquots. Subunit integrity of the final preparation was confirmed by 12% SDS-PAGE, Western blotting with antisera to the α, β, and γ subunits, IEF PAGE to determine the phosphorylation state of eIF2α (see below), and assaying for ternary complex formation. The yield was ∼5 mg of eIF2 from 100 grams (wet weight) of yeast cells, at 75%–80% purity.
Guanine–nucleotide exchange assays
Total yeast cell protein extracts (10–15 μg/μl) containing the indicated overexpressed subunits of eIF2B were prepared from cells grown in synthetic complete media. Cells were grown to A600 = 0.5–1.0, harvested by centrifugation, washed with sterile deionized water, resuspended in breaking buffer (20 mm Tris-HCl at pH 7.5, 100 mm KCl, 10 mm 2-aminopurine (Sigma), 3 mm MgCl2, 5 mm NaF, 1 mm DTT, 10% vol/vol glycerol, 1 mm PMSF, 1 μg/ml of leupeptin, 0.7 μg/ml of pepstatin, and 1 μg/ml of aprotinin) and broken with acid-washed glass beads (0.4- to 0.52-mm diameter) in a Braun homogenizer (B. Braun) as described (Moehle and Hinnebusch 1991).
Binary complexes of eIF2 ⋅ [3H]GDP (50 μl) were formed in 12 × 75 mm glass tubes containing binary complex buffer [20 mm Tris-HCl at pH 7.5, 100 mm KCl, 2 mg/ml of creatine phosphokinase, 3 mm MgCl2, 5 mm NaF, 1 mm DTT, 0.1 mm ATP, 0.1 mm EDTA, and 15% (vol/vol) glycerol] with the addition of 8–10 μg (∼50 pmole) of purified yeast eIF2 and 20 pmole of [3H]GDP (11.6 Ci/mmole) (Amersham). After 12 min at 23°C, reactions were held on ice. Under these conditions, ∼7 pmole of eIF2 ⋅ [3H]GDP was formed. In experiments where the effect of eIF2α phosphorylation was assessed, eIF2 in binary complex buffer was first incubated with or without 1 μl (∼0.1 μg) of purified rabbit reticulocyte HCR kinase (Jackson and Hunt 1985) for 10 min at 23°C. The presence of HCR did not affect binary complex formation.
To measure dissociation of binary complexes, reactions (from above) and 150 μg of cell extracts overexpressing eIF2B subunits, as indicated in Figures 1 and 3, were held at 10°C for 4 min, a 100-fold excess of unlabeled GDP was added to the binary complex, and, 1 min later, the cell extract (or extract buffer as a control) was added to start the reaction. Ten-microliter samples were removed immediately (assay time=0) and at the indicated times, added to 2 ml of ice-cold wash buffer (20 mm Tris-HCl at pH 7.5, 100 mm KCl, 5 mm MgCl2, 0.1 mm EDTA), filtered through nitrocellulose filters (Whatman) using a vacuum manifold (Millipore), and washed twice with 4 ml each of ice-cold wash buffer. Filters were dried and counted by liquid scintillation in Econofluor 2 (Packard). Counting efficiency was 56%. Assays were done in duplicate or triplicate and in at least three separate experiments. Mean results of typical experiments are shown. Variation in values was ±0%–6% per time point with a mean variation for all time points of ±2.1% (t = 0 excluded).
IEF PAGE
Samples of purified eIF2 (∼1 μg in 5 μl) in binary complex buffer (as above) were diluted with 150 μl of IEF sample buffer (Dever 1997). Fifty microliters of each sample was resolved on a vertical IEF polyacrylamide slab gel (Dever 1997).
Ni pull-down assays with His-tagged eIF2
In these assays purified His-tagged eIF2 is kept at a constant concentration and is in large molar excess over eIF2B and eIF2 present in cell extracts. Thus the fraction of total eIF2B bound is a function only of the dissociation constant (Phizicky and Fields 1995). Purified yeast eIF2 (2.5 μg) was incubated with or without HCR (1 μl) at 30°C for 10 min in 25 μl of pull-down buffer [20 mm Tris-HCl at pH 7.5, 100 mm KCl, 2 mm magnesium acetate, 0.4 mg/ml of creatine phosphokinase, 5 mm NaF, 5 mm 2-mercaptoethanol, 0.1 mm ATP, 0.1 mm EDTA, and 7% (vol/vol) glycerol]. Control reactions without eIF2 contained pull-down buffer only. Reactions were added to 100 μg of whole-cell extracts (prepared as described for nucleotide exchange assays) from yeast strains overexpressing different eIF2B subunits in 35 μl of PD buffer (20 mm Tris-HCl at pH 7.5, 100 mm KCl, 2 mm magnesium acetate, 5 mm NaF, 5 mm 2-mercaptoethanol, 1 mm PMSF, 1 μg/ml of leupeptin, 0.7 μg/ml of pepstatin, and 1 μg/ml of aprotinin). eIF2 and cell extracts were incubated together at 10°C for 10 min then added to 50 μl of Ni–NTA–agarose beads (Qiagen) prewashed in PD buffer. Reactions were mixed by rotating on a Nutator (Becton-Dickinson) at 4°C for 30 min. The agarose beads were collected by low-speed centrifugation and washed 3× with 500 μl of buffer PD-I-5 (PD buffer with 5 mm imidazole added) by rotation on a Labquake shaker (Barnstead-Thermolyne). Finally, bound material was eluted with 60 μl of buffer PD-I-250 by rotation for 30 min at 4°C and collection of the beads by centrifugation. The eluted material was mixed with 4× Laemmli sample buffer and heated to 37°C for 10 min prior to fractionation by 12% SDS-PAGE.
Ni pull-down assays with His-tagged GCD1
Assays were performed as described above with the following changes: No purified eIF2 was added, 400 μg of cell extract was used, and 10 μl of Ni–NTA–silica resin (Qiagen) was used in place of Ni–NTA–agarose, as this gave lower nonspecific binding of yeast proteins.
Acknowledgments
S.R.K. was supported by National Institutes of Health grants DK15658 and DK13499 awarded to L.S. Jefferson. We are grateful to Lynne Hugendubler for technical assistance. We thank Ernie Hannig for communicating eIF2 purification methods to us prior to publication, and Tom Dever and Tom Stevens for yeast strains and plasmids. In addition, we thank Tom Dever and Krishnamurthy Natarajan for comments on the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
Corresponding author.
E-MAIL ahinnebusch@nih.gov; FAX (301) 496-6828.
References
- Boeke JD, LaCroute F, Fink GR. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-Fluoro-orotic acid resistance. Mol Gen Genet. 1984;197:345–346. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]
- Boguski MS, McCormick F. Proteins regulating Ras and its relatives. Nature. 1993;366:643–654. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature. 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- Bushman JL, Asuru AI, Matts RL, Hinnebusch AG. Evidence that GCD6 and GCD7, translational regulators of GCN4, are subunits of the guanine nucleotide exchange factor for eIF-2 in Saccharomyces cerevisiae. Mol Cell Biol. 1993a;13:1920–1932. doi: 10.1128/mcb.13.3.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman JL, Foiani M, Cigan AM, Paddon CJ, Hinnebusch AG. Guanine nucleotide exchange factor for eIF-2 in yeast: Genetic and biochemical analysis of interactions between essential subunits GCD2, GCD6 and GCD7 and regulatory subunit GCN3. Mol Cell Biol. 1993b;13:4618–4631. doi: 10.1128/mcb.13.8.4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P. Multifunctional yeast high-copy-number shuttle vectors. Gene. 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- Cigan AM, Pabich EK, Feng L, Donahue TF. Yeast translation initiation suppressor sui2 encodes the alpha subunit of eukaryotic initiation factor 2 and shares identity with the human alpha subunit. Proc Natl Acad Sci. 1989;86:2784–2788. doi: 10.1073/pnas.86.8.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cigan AM, Foiani M, Hannig EM, Hinnebusch AG. Complex formation by positive and negative translational regulators of GCN4. Mol Cell Biol. 1991;11:3217–3228. doi: 10.1128/mcb.11.6.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cigan AM, Bushman JL, Boal TR, Hinnebusch AG. A protein complex of translational regulators of GCN4 is the guanine nucleotide exchange factor for eIF-2 in yeast. Proc Natl Acad Sci. 1993;90:5350–5354. doi: 10.1073/pnas.90.11.5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens MJ. Protein kinases that phosphorylate eIF2 and eIF2B, and their role in eukaryotic cell translational control. In: Hershey JWB, Mathews MB, Sonenberg N, editors. Translational control. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1996. pp. 139–172. [Google Scholar]
- De Benedetti A, Baglioni C. Phosphorylation of initiation factor eIF2α, binding of mRNA to 48S complexes, and its reutilization in initiation of protein synthesis. J Biol Chem. 1983;258:14556–14562. [PubMed] [Google Scholar]
- Dever TE. Using GCN4 as a reporter of eIF2α phosphorylation and translational regulation in yeast. Methods. 1997;11:403–417. doi: 10.1006/meth.1996.0437. [DOI] [PubMed] [Google Scholar]
- Dever TE, Feng L, Wek RC, Cigan AM, Donahue TD, Hinnebusch AG. Phosphorylation of initiation factor 2α by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell. 1992;68:585–596. doi: 10.1016/0092-8674(92)90193-g. [DOI] [PubMed] [Google Scholar]
- Dever TE, Chen JJ, Barber GN, Cigan AM, Feng L, Donahue TF, London IM, Katze MG, Hinnebusch AG. Mammalian eukaryotic initiation factor 2α kinases functionally substitute for GCN2 in the GCN4 translational control mechanism of yeast. Proc Natl Acad Sci. 1993;90:4616–4620. doi: 10.1073/pnas.90.10.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Yang W, Astrom S, Bystrom AS, Hinnebusch AG. Modulation of tRNAMeti, eIF-2 and eIF-2B expression shows that GCN4 translation is inversely coupled to the level of eIF-2 ⋅ GTP ⋅ Met-tRNAMeti ternary complexes. Mol Cell Biol. 1995;15:6351–6363. doi: 10.1128/mcb.15.11.6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue TF, Cigan AM, Pabich EK, Castilho-Valavicius B. Mutations at a Zn(ll) finger motif in the yeast elF-2β gene alter ribosomal start-site selection during the scanning process. Cell. 1988;54:621–632. doi: 10.1016/s0092-8674(88)80006-0. [DOI] [PubMed] [Google Scholar]
- Dorris DR, Erickson FL, Hannig EM. Mutations in GCD11, the structural gene for eIF-2γ in yeast, alter translational regulation of GCN4 and the selection of the start site for protein synthesis. EMBO J. 1995;14:2239–2239. doi: 10.1002/j.1460-2075.1995.tb07218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson FL, Hannig EM. Ligand interactions with eukaryotic translation initiation factor 2: Role of the γ-subunit. EMBO J. 1996;15:6311–6320. [PMC free article] [PubMed] [Google Scholar]
- Fabian JR, Kimball SR, Heinzinger NK, Jefferson LS. Subunit assembly and guanine nucleotide exchange activity of eukaryotic initiation factor-2B expressed in Sf9 cells. J Biol Chem. 1997;272:12359–12365. doi: 10.1074/jbc.272.19.12359. [DOI] [PubMed] [Google Scholar]
- Foiani M, Cigan AM, Paddon CJ, Harashima S, Hinnebusch AG. GCD2, a translational repressor of the GCN4 gene, has a general function in the initiation of protein synthesis in Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:3203–3216. doi: 10.1128/mcb.11.6.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss DJ, Parkhurst LJ, Mehta HB, Woodley CL, Wahba AJ. Studies on the role of eukaryotic nucleotide exchange factor in polypeptide chain initiation. J Biol Chem. 1984;259:7374–7377. [PubMed] [Google Scholar]
- Gross M, Wing M, Rundquist C, Rubino MS. Evidence that phosphorylation of the eIF-2α prevents the eIF-2B-mediated dissociation of eIF-2 ⋅ GDP from the 60S subunit of complete initiation complexes. J Biol Chem. 1987;262:6899–6907. [PubMed] [Google Scholar]
- Hannig EM, Cigan AM, Freeman BA, Kinzy TG. GCD11, a negative regulator of GCN4 expression, encodes the gamma subunit of eIF-2 in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:506–520. doi: 10.1128/mcb.13.1.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannig EM, Hinnebusch AG. Molecular analysis of GCN3, a translational activator of GCN4: Evidence for posttranslational control of GCN3 regulatory function. Mol Cell Biol. 1988;8:4808–4820. doi: 10.1128/mcb.8.11.4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison CJ, Hayer-Hartl M, Di Liberto M, Hartl FU, Kuriyan J. Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science. 1997;276:431–435. doi: 10.1126/science.276.5311.431. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG. Translational control of GCN4: An in vivo barometer of initiation factor activity. Trends Biochem Sci. 1994;19:409–414. doi: 10.1016/0968-0004(94)90089-2. [DOI] [PubMed] [Google Scholar]
- ————— . Translational control of GCN4: Gene-specific regulation by phosphorylation of eIF2. In: Hershey JWB, Mathews MB, Sonenberg N, editors. Translational control. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1996. pp. 199–244. [Google Scholar]
- Hinnebusch AG, Fink GR. Positive regulation in the general amino acid control of Saccharomyces cerevisiae. Proc Natl Acad Sci. 1983;80:5374–5378. doi: 10.1073/pnas.80.17.5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Fukada Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hunt T. A novel approach to the isolation of rabbit reticulocyte heam-controlled eIF-2α protein kinase. Biochem Biophys Acta. 1985;826:224–228. doi: 10.1016/0167-4781(85)90010-7. [DOI] [PubMed] [Google Scholar]
- Jones EW. Tackling the protease problem in Saccharomyces cerevisiae. Methods Enzymol. 1991;194:428–453. doi: 10.1016/0076-6879(91)94034-a. [DOI] [PubMed] [Google Scholar]
- Merrick WC. Mechanism and regulation of eukaryotic protein synthesis. Microbiol Rev. 1992;56:291–315. doi: 10.1128/mr.56.2.291-315.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehle CM, Hinnebusch AG. Association of RAP1 binding sites with stringent control of ribosomal protein gene transcription in Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:2723–2735. doi: 10.1128/mcb.11.5.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddon CJ, Hinnebusch AG. gcd12 mutations are gcn3-dependent alleles of GCD2, a negative regulator of GCN4 in the general amino acid control of Saccharomcyes cerevisiae. Genetics. 1989;122:543–550. doi: 10.1093/genetics/122.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddon CJ, Hannig EM, Hinnebusch AG. Amino acid sequence similarity between GCN3 and GCD2, positive and negative translational regulators of GCN4: Evidence for antagonism by competition. Genetics. 1989;122:551–559. doi: 10.1093/genetics/122.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavitt GD, Yang W, Hinnebusch AG. Homologous segments in three subunits of the guanine nucleotide exchange factor eIF2B mediate translational regulation by phosphorylation of eIF2. Mol Cell Biol. 1997;17:1298–1313. doi: 10.1128/mcb.17.3.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky EM, Fields S. Protein-protein interactions. Methods for detection and analysis. Microbiol Rev. 1995;59:94–123. doi: 10.1128/mr.59.1.94-123.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NT, Kimball SR, Jefferson LS, Proud CG. Cloning of cDNA for the γ-subunit of mammalian translation initiation factor 2B, the guanine nucleotide-exchange factor for eukaryotic initiation factor 2. Biochem J. 1996a;318:631–636. doi: 10.1042/bj3180631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NT, Mellor H, Craddock BL, Flowers KM, Kimball SR, Wilmer T, Jefferson LS, Proud CG. eIF2B, the guanine nucleotide-exchange factor for eukaryotic initiation factor 2. Biochem J. 1996b;318:637–643. doi: 10.1042/bj3180637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaiah KVA, Dhindsa RS, Chen J-J, London IM, Levin D. Recycling and phosphorylation of eukaryotic initiation factor 2 on 60S subunits of 80S initiation complexes and polysomes. Proc Natl Acad Sci. 1992;89:12063–12067. doi: 10.1073/pnas.89.24.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowlands AG, Panniers R, Henshaw EC. The catalytic mechanism of guanine nucleotide exchange factor action and competitive inhibition by phosphorylated eukaryotic initiation factor 2. J Biol Chem. 1988;263:5526–5533. [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sherman F, Fink GR, Lawrence CW. Methods of yeast genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1974. [Google Scholar]
- Thomas NSB, Matts RL, Levin DH, London IM. The 60S ribosomal subunit as a carrier of eukaryotic initiation factor 2 and the site of reversing factor activity during protein synthesis. J Biol Chem. 1985;260:9860–9866. [PubMed] [Google Scholar]
- Trachsel H. Binding of initiator methionyl-tRNA to ribosomes. In: Hershey JWB, Mathews MB, Sonenberg N, editors. Translational control. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1996. pp. 113–138. [Google Scholar]
- Vazquez de Aldana CR, Hinnebusch AG. Mutations in the GCD7 subunit of yeast guanine nucleotide exchange factor eIF-2B overcome the inhibitory effects of phosphorylated eIF-2 on translation initiation. Mol Cell Biol. 1994;14:3208–3222. doi: 10.1128/mcb.14.5.3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek RC, Ramirez M, Jackson BM, Hinnebusch AG. Identification of positive-acting domains in GCN2 protein kinase required for translational activation of GCN4 expression. Mol Cell Biol. 1990;10:2820–2831. doi: 10.1128/mcb.10.6.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Hinnebusch AG. Identification of a regulatory subcomplex in the guanine nucleotide exchange factor eIF2B that mediates inhibition by phosphorylated eIF2. Mol Cell Biol. 1996;16:6603–6616. doi: 10.1128/mcb.16.11.6603. [DOI] [PMC free article] [PubMed] [Google Scholar]