

Abstract
Oligonucleotide microarrays were employed to quantitate mRNA levels from a large number of genes regulated by the p53 transcription factor. Responses to DNA damage and to zinc-inducible p53 were compared for their transcription patterns in cell culture. A cluster analysis of these data demonstrates that genes induced by γ radiation, UV radiation, and the zinc-induced p53 form distinct sets and subsets with a few genes in common to all these treatments. Cell type- or cell line-specific p53 responses were detected. When p53 proteins were induced with zinc, the kinetics of induction or repression of mRNAs from p53-responsive genes fell into eight distinct classes, five different kinetics of induction, and three different kinetics of repression. In addition, low levels of p53 in a cell induced or repressed only a subset of genes observed at higher p53 levels. The results of this study demonstrate that the nature of the p53 response in diverse mRNA species depends on the levels of p53 protein in a cell, the type of inducing agent or event, and the cell type employed. Of 6000 genes examined for p53 regulatory responses, 107 induced and 54 repressed genes fell into categories of apoptosis and growth arrest, cytoskeletal functions, growth factors and their inhibitors, extracellular matrix, and adhesion genes.
Keywords: p53-regulated genes, oligonucleotide microarrays, EB-1 cells
Mutations in the p53 tumor suppressor gene occur with unusually high frequency in a broad spectrum of human cancers (Hollstein et al. 1991; Bennett et al. 1992). Germ-line mutations of p53 gene, the Li–Fraumeni syndrome, predispose to diverse types of cancers (Malkin et al. 1990). Wild-type p53 has been shown to block the transformation by activated oncogenes and inhibit tumor cell growth in vitro (Eliyahu et al. 1989; Finlay et al. 1989; Baker et al. 1990; Michalovitz et al. 1990). Additionally, p53's function as a tumor suppressor is further supported by the observation that p53 null mice, generated by homologous targeting, are susceptible to spontaneous development of tumors at a young age (Donehower et al. 1992; Lozano and Liu 1998). A normal cell has a low level of p53 protein, because of its short half-life, and it is likely in a latent form. The levels and activity of p53 increase in response to cellular stress, such as DNA damage by irradiation or chemotherapeutic agents, activation of oncogenes or viral infection, hypoxia, or very low levels of ribonucleoside triphosphate pools. Subsequently, activated p53 mediates cell cycle arrest or programmed cell death (apoptosis), depending on the cell type or the presence of activated oncogenes. This results in the elimination of clones of cells that contain mutations or prevents a high mutation rate by blocking the duplication of damaged DNA (Levine 1997; Prives and Hall 1999).
The mechanisms leading to p53-induced cell cycle arrest and apoptosis have been extensively investigated. Wild-type p53 is a sequence-specific transcriptional activator (Fields and Jang 1990; Raycroft et al. 1990; Farmer et al. 1992). The consensus binding site for p53 is defined as two copies of the 10-bp motif, 5′-PuPuPuC(A/T)(T/A)GPyPyPy-3′, separated by 0–13 bp (Kern et al. 1991; el-Deiry et al. 1992). The DNA-binding activity has been mapped to the core portion of p53 (residues 102–292) (Bargonetti et al. 1993; Pavletich et al. 1993; Wang et al. 1993), which harbors most of the missense mutations identified in human tumors. The interaction between p53 and its target sequence has been illustrated by crystal structures (Cho et al. 1994). The amino-terminal 42 amino acids of p53 are essential for its transcriptional activation (Fields and Jang 1990; Unger et al. 1992; Lin et al. 1994). The carboxy-terminal domain contains a tetramer-ization motif (Iwabuchi et al. 1993; Wang et al. 1994).
Sequence-specific DNA binding is essential for p53 to function as a tumor suppressor (Pietenpol et al. 1994). Identification of transcriptional targets of p53 is critical in understanding pathways by which p53 affects cellular outcomes such as growth arrest and apoptosis. Many target genes have been identified that contain p53-responsive elements and are induced on p53 activation. Among them, the p21/Waf1/Cip1 gene is most well studied. As an inhibitor for cyclin-dependent kinases, particularly those that function during the G1 phase of the cell cycle, p21/Waf1/Cip1 is believed to mediate, at least in part, p53-induced G1 arrest (el-Deiry et al. 1993; Harper et al. 1993; Xiong et al. 1993). A group of genes that are involved in cellular responses to oxidative stress have been shown to be p53-regulated by SAGE analysis, suggesting a role for reactive oxygen species (ROS) in p53-mediated apoptosis (Polyak et al. 1997). In addition to its sequence-specific transactivation, p53 is also capable of repressing gene expression from a broad spectrum of promoters that do not appear to contain p53-responsive elements (Mack et al. 1993; Murphy et al. 1996). This general repressive effect on transcription is thought to be a consequence of p53 inhibiting transcription activators or components of the basal transcription machinery and may be important in mediating apoptosis (Seto et al. 1992; Murphy et al. 1996).
Recent development of DNA microarray technology permits one to monitor a large number of cellular transcripts in a parallel fashion (Schena et al. 1995; DeRisi et al. 1997; Wodicka et al. 1997; Iyer et al. 1999). We report an investigation of p53-regulated gene expression using oligonucleotide microarrays. EB-1 is a human colon carcinoma cell line containing a stably tranfected p53 cDNA driven by the metallothionein promoter (Shaw et al. 1992). Induction of p53 in these cells results in apoptosis. More than 6000 human genes were initially screened for p53-regulated genes in EB-1 cells, and subsequently 400 genes of interest were studied in detail for their expression kinetics following p53 induction. A classification of these genes can now be made on the basis of their expression profiles. Finally, the expression of these 400 genes was also monitored and compared in different cancer-derived cell lines following UV or γ irradiation. From these studies it is clear that the p53 response is quite heterogeneous, depending upon several variables.
Results
A human colon cancer cell line with inducible p53
EB-1 is a colon tumor-derived cell line carrying a wild-type p53 gene under the control of an inducible metallothionein (MT) promoter. This cell line was chosen because the MT promoter has been shown to exhibit little or no detectable basal activity and to give sustained high levels of p53 expression by stimulation with 100 μm zinc chloride. Induction of p53 in EB-1 cells results in progressive cell death within a 4-day period with clear characteristics of apoptosis. Higher concentration of zinc (200 μm) induces apoptosis within 24 hr. The parental EB (deficient for endogenous p53) cells continue to grow normally in the presence of zinc chloride at all concentrations (Shaw et al. 1992). The steady-state p53 level increased as early as 2 hr after stimulation with 100 μm zinc chloride, peaked between 4 and 8 hr, and the high level of p53 protein was maintained throughout the 24-hr experimental period (Fig. 1A). Human MDM2 was also induced in these cells, but the induction was delayed compared with that of p53, indicating that p53 expressed from the transgene is indeed active (Fig. 1A). To examine the dependence of p53 induction on the concentration of zinc, we performed additional control experiments (Fig. 1B). Lower amounts of p53 could be induced by 50 μm after 8 hr, whereas 100 μm and 200 μm zinc chloride gave rise to much higher levels of p53 in a dose-dependent manner (Fig. 1B). No significant cell death was observed with 50 or 100 μm zinc during the first 24 hr of incubation; however, massive cell death occurred with 200μm zinc within 24 hr. Thus, for the initial screening and the time course of expression we used samples from 100 μm zinc-induced EB-1 cells, whereas the kinetics of a subset of genes were compared to those in 50 μm zinc-induced cells.
Figure 1.
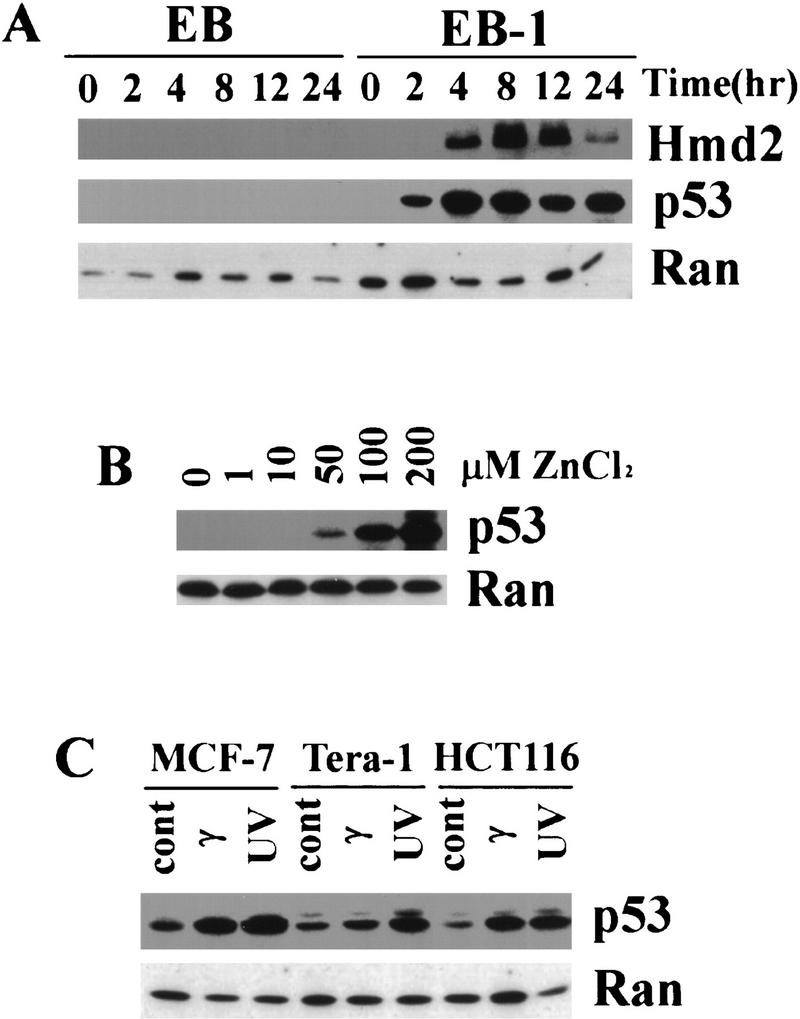
Inducible p53 in colon carcimona cell line EB-1. p53 and human MDM2 were detected by Western blots using monoclonal antibodies 1801 and 2A9, respectively. A small GTPase Ran was used as control. (A) EB-1 cells were stimulated with 100 μm zinc chloride starting at time 0 hr, and the level of p53 protein was elevated as early as 2 hr. Maximum induction of p53 was seen between 4 and 8 hr. Human MDM2 protein was also induced in these cells with a delay compared with p53 induction. No induction of p53 or hMDM2 in parental EB cells was observed following the same stimulation. The amount of 10 μg of total protein was loaded in each lane, and the same blot was probed by anti-p53, anti-hMDM2, and anti-Ran antibodies. (B) The levels of induced p53 protein in EB-1 cells depend on the zinc concentration in the culture media. EB-1 cells were cultured in presence of 0, 1, 10, 50, 100, and 200 μm of zinc chloride for 8 hr. A minimum of 50 μm of zinc was required to induce p53 proteins in these cells, and 100 and 200 μm zinc induced p53 to much higher levels in a dose-dependent manner. The amount of 20 μg of total protein was loaded in each lane. (C) Induction of p53 by UV and γ irradiation in different cancer-derived cell lines. Each cell line was treated with radiation as indicated, and cell lysates were prepared after 3–6 hr (see Materials and Methods). p53 was induced following both UV and γ irradiation in all the cell lines tested. The amount of 65 μg of total protein was loaded in each lane.
Initial screening for p53-regulated genes
Both EB-1 and parental EB cells were stimulated with 100 μm zinc chloride for 0 (control) and 12 hr. Poly(A) RNAs were isolated from both cell lines before and after p53 induction, labeled, and hybridized to the oligonucleotide microarrays (Human GeneChip 6500, Affymetrix), which contains probes for 7464 genes representing 6003 unique accession numbers at time of design (Lockhart et al. 1996). Induction by p53 was evaluated for each gene by calculating the ratio of gene intensity in EB-1 cells to that in parental EB cells following 12 hr in zinc (EB-1/EB ratio) (Alon et al. 1999). The reciprocal of this ratio was employed as an index for p53-mediated repression (EB/EB-1 ratio). Data from two independent experiments were averaged. To compile a list of p53-responsive genes, only those genes with greater than threefold induction or repression in the presence of zinc and with an induced intensity of 100 units or greater were selected, because of large experimental variations for genes with intensity values <100 and expression ratio <3. This list, containing 107 induced genes ranging from 3- to 64-fold and 54 repressed genes ranging from 3- to 20-fold, can be obtained from our web site, http://www.molbio.princeton.edu/levinelb/p53genes. Thus, 1.8% of the genes on the chip were induced and 0.9% of the genes were repressed.
Many known p53-responsive genes were identified in the experiment, including p21/Waf1/Cip1, IGFBP-3, GADD45, and TGF-α. In addition, a number of previously unknown genes were also shown to be regulated by p53 (see the list of p53-responsive genes on the web site). Many of these results were confirmed by either Northern blot analysis (data not shown) or reverse transcription–polymerase chain reaction (RT–PCR).
To classify p53-regulated genes, each induced gene (n = 107) was placed into a broad functional category. This group was then compared with 100 randomly selected genes from the Affymetrix GeneChip array. Interestingly, genes classified as coding for cytoskeletal features, growth factors, and extracellular matrix or cell adhesion proteins were significantly (P < 0.02) more frequent in the list of p53-induced genes (Fig. 2). As anticipated, genes associated with apoptosis or growth arrest were also more abundant in the p53-induced gene list (Fig. 2). In contrast, genes that appear to be involved in cellular metabolism or have unknown functions were less prevalent compared with the randomly selected genes.
Figure 2.

Functional grouping of p53-induced genes (open bars) compared with the same grouping of 100 randomly selected genes (solid bars). Significant differences (P < 0.02) are indicated by an asterisk. p53-induced genes were enriched for genes involved in apoptosis, growth arrest, the cytoskeleton, growth factors, and cell adhesion. Genes involved in cell metabolism were less abundant compared with a randomly selected gene pool.
Eight clusters of temporal expression profiles
From the initial screening, 161 genes appeared to be p53 responsive. To study in detail the expression kinetics of these genes in response to p53 induction, a spotted oligonucleotide microarray representing 400 genes was developed from our initial list of p53-regulated genes, together with genes reported to be potential p53 targets and some control genes. Using these p53-spotted oligonucleotide arrays, an examination was carried out of the temporal patterns of gene transcription following p53 induction in EB-1 cells. Sample RNAs were isolated from EB-1 and the parental EB cells treated with 100 μm zinc chloride for 0 (control), 2, 4, 8, 12, and 24 hr. RNAs transcribed from the EB-1 cDNA were labeled with Cy5 and mixed with EB-derived RNAs labeled with Cy3. The expression level of each gene was then represented by a relative ratio of its intensity in EB-1 cells to that in the control EB cells.
The temporal profiles of gene expression were quite diverse among the 400 genes. To classify these expression profiles, we performed cluster analysis using the GENE CLUSTER program (Eisen et al. 1998). Genes were selected for this analysis if their expression levels in EB-1 cells following p53 induction were different from that in EB control by at least a factor of 2.5 in at least two time points. With these criteria, 69 genes were included in the cluster analysis shown in Figure 3. These genes fall into eight clusters with distinct expression kinetics following p53 induction (Fig. 4). The genes found in each cluster are summarized in Table 1.
Figure 3.
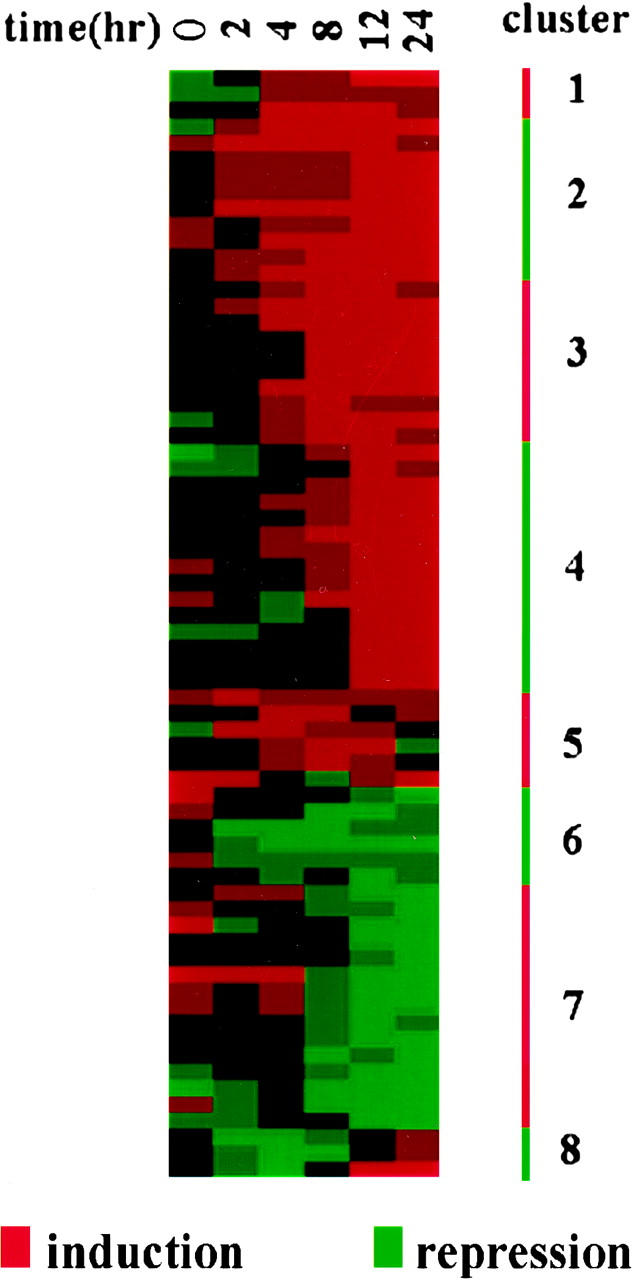
Cluster analysis of gene expression profiles following p53 induction. A total of 69 genes selected for this analysis were clustered into several groups on the basis of the similarity of their expression profiles by a reported procedure (Eisen et al. 1998). The expression level of each gene is represented by the ratio of gene intensity in EB-1 over that in EB and is shown in a horizontal strip. The degree of redness represents level of induction, whereas that of greenness represents level of repression.
Figure 4.

Average expression profiles for each gene cluster. The expression profile of each gene in a cluster was normalized onto a 0–10 scale and then averaged. Eight clusters were derived from the cluster analysis, showing different induction kinetics.
Table 1.
List of genes in different clusters
| Accession no.
|
Definition
|
Accession no.
|
Definition
|
Accession no.
|
Definition
|
|---|---|---|---|---|---|
| cluster 1 | cluster 4 | ||||
| M65199 | endothelin 2 | M31159 | IGFBP-3 | J02947 | superoxide dismutase 3; extracellular |
| AF090950 | negative growth regulator MyD118 | X57348 | 14-3-3ς Protein | M68520 | cyclin-dependent kinase 2 |
| AF016266 | TRAIL receptor 2 (KILLER/DR5) | X70340 | TGF-α | U35143 | retinoblastoma-binding protein |
| cluster 2 | AF026246 | endogenous retrovirus H and E sequence | cluster 7 | ||
| AB000584 | TGF-β superfamily protein | X62571 | keratin 17 | D28480 | DNA replication licensing factor cdc47 homolog |
| U03106 | p21/waf1 | R42736 | ESTs; similar to KIAA0835 protein | M14630 | prothymosin α |
| Z18951 | caveolin | Y00503 | keratin 19 | AA644331 | ESTs; similar to cyclin B2 |
| X06268 | collagen; type II α1 | D85815 | human DNA for rhoHP1, | L13687 | ADP-ribosylation factor-like protein 2 (ARL2) |
| AF059611 | nuclear matrix protein NRP/B (NRPB) | M26152 | serum amyloid a protein precursor | M12623 | human non-histone chromosomal protein HMG-17 |
| X54156 | tumor protein p53 | M13755 | interferon-induced 17-kD protein | U31278 | mitotic feedback control protein Madp2 homolog |
| M60974 | GADD45 | M26062 | interleukin-2 receptor β chain (IL-2Rb) | D14657 | human KIAA0101 gene; |
| L13463 | regulator of G-protein signaling 2 G0S8 | Z11502 | ANNEXIN XIII | X62534 | high-mobility group 2 (HMG2) |
| L36069 | potassium channel α subunit | U28369 | human semaphorin V mRNA; | D83987 | human mRNA for KIAA0030 |
| cluster 3 | M32011 | neutrophil NADPH oxidase 2 | X75252 | prostatic binding protein | |
| M59371 | tyrosine-protein kinase receptor eck | M36263 | Estradiol 17 β-dehydrogenase 1 | U16799 | ATPase; Na+/K+ transporting; beta 1 polypeptide |
| AF010309 | Homo sapiens Pig3 | cluster 5 | L25931 | lamin B receptor | |
| J05192 | actin; α2 | D30751 | BMP4 | X74330 | DNA primase polypeptide 1 |
| M67454 | apoptosis (APO-1/FAS) | M99564 | P protein (melanocyte- specific transporter) | J04088 | topoisomerase (DNA) II alpha |
| L14577 | cystathionine-β-synthase | X14787 | thrombospondin 1 | M15024 | Myb proto-oncogene protein |
| M74178 | macrophage stimulating protein (msp) | L13738 | human activated p21cdc42Hs kinase (ack) | L32866 | human effector cell protease receptor-1 (EPR-1) |
| K02403 | complement component 4A | AB002058 | HUMAN P2XM | cluster 8 | |
| AF065388 | H. sapiens tetraspan NET-1 mRNA | AF010316 | H. sapiens Pig12 (PIG12) | Y11525 | CCAAT/enhancer binding protein (C/EBPα) |
| M21389 | keratin 5 | cluster 6 | L07597 | ribosomal protein S6 kinase; 90 kD; polypeptide 2 | |
| M16006 | plasminogen activator inhibitor (PAI-1) | J04173 | phosphoglycerate mutase 1 (brain) | X77567 | H. sapiens mRNA for InsP3 5-phosphatase |
| U14417 | Ral GDP dissociation stimulator | L25665 | possible GTP-binding protein hsr1 | ||
| X15880 | collagen; type VI; α1 |
p53 is a transcriptional regulator for many genes. Because of differences in binding affinities for different promoters and mode of regulation, p53-responsive genes are likely to show different kinetics in their expression profiles after p53 induction, as was observed in this cluster analysis. The transcriptional activation of some genes was obvious as early as 2 hr after stimulation with 100 μm zinc chloride. These genes, represented in cluster 2, include known p53-activated targets such as p21/Waf1/Cip1 and GADD45. Genes in clusters 1 and 3 showed intermediate response and were activated between 4 and 8 hr of incubation with zinc. The late genes, shown in cluster 4, were not activated until after 8 hr of zinc induction. Cluster 5 contains genes that were transiently induced by p53, however the expression profiles were quite heterogeneous among them. Some genes like BMP4 and cdc42Hs kinase (ack) were activated as early as 2 hr, whereas P2XM was not activated until 8 hr after addition of zinc.
The p53-repressed genes also exhibited heterogeneous kinetics in their response to p53 induction, with early responsive genes in cluster 6, late responsive genes in cluster 7, and some transiently repressed genes in cluster 8. Genes in cluster 8 also displayed very heterogeneous profiles.
To determine the reliability of these results, this experiment was repeated, but the mRNA levels were measured using RT–PCR. As shown in Figure 5, the expression profiles measured by these two independent methods were very similar, even though the absolute value for each mRNA level might be different between the two measurements.
Figure 5.

Independent measurements of selected gene expression levels by RT–PCR (left) and p53-spotted oligonucleotide arrays (right). Expression levels measured by RT–PCR were normalized to GAPDH and plotted relative to the level in control cells. Both methods gave very similar results in the gene expression profiles.
Some genes, such as Bak and IGγ, were activated by p53 to a lesser extent, and therefore were not included in this analysis. Some of the well-known p53-regulated genes were also absent in this study, either because the oligonucleotides on the chip do not allow efficient hybridization (such as human mdm2 gene) or simply because these genes are not represented on the chips (such as Bax).
Different functional groups of p53-regulated genes
p53 regulates various cellular responses upon activation. To examine its effect in detail, we classified the p53-responsive genes into different functional groups, as shown in Table 2.
Table 2.
Different functional groups of p53-regulated genes
| Function
|
Gene definition
|
p53 regulation
|
Response
|
Response to 50 μm zinc
|
UVa
|
γa
|
|---|---|---|---|---|---|---|
| Apoptosis | Fas/APO-1 | activation | intermediate | − | 0 | 3 |
| KILLER/DR5 | activation | intermediate | + | 0 | 1 | |
| Oxidative stress | Homo sapiens Pig3 | activation | intermediate | − | 0 | 3 |
| cystathionine-β-synthase | activation | intermediate | − | 0 | 1 | |
| serum amyloid a protein precursor | activation | late | − | 0 | 1 | |
| neutrophil cytosolic factor 2 | activation | late | − | 0 | 0 | |
| H. sapiens Pig12 (PIG12) | activation | transient | − | 0 | 0 | |
| superoxide dismutase 3; extracellular | repression | early | + | 2 | 2 | |
| Cell cycle | p21/Waf1 | activation | early | + | 1 | 1 |
| GADD45 | activation | early | + | 2 | 2 | |
| regulator of G-protein signaling 2 G0S8 | activation | early | + | 1 | 0 | |
| negative growth regulator MyD118 | activation | intermediate | + | 0 | 1 | |
| 14-3-3ς protein | activation | late | + | 1 | 2 | |
| cyclin-dependent kinase 2 | repression | early | + | 0 | 0 | |
| retinoblastoma-binding protein (RbAp46) | repression | early | + | 0 | 1 | |
| prothymosin α | repression | late | + | 0 | 0 | |
| ESTs; similar to CYCLIN B2 | repression | late | − | 0 | 1 | |
| MAD2 mitotic feedback protein Madp2 | repression | late | + | 0 | 0 | |
| lamin B receptor | repression | late | + | 2 | 3 | |
| DNA replication | DNA primase polypeptide 1 | repression | late | + | 0 | 1 |
| topoisomerase (DNA) II α | repression | late | − | 0 | 1 | |
| DNA replication licensing factor | repression | late | − | 0 | 0 | |
| chromosomal protein HMG-17 | repression | late | + | 0 | 1 | |
| high-mobility group 2 (HMG2) | repression | late | − | 0 | 0 | |
| ECM | collagen; type II α1 | activation | early | − | 0 | 2 |
| collagen; type VI α1 | activation | intermediate | + | 0 | 2 | |
| Cell migration | macrophage stimulating protein (msp) | activation | intermediate | − | 0 | 0 |
| plasminogen activator inhibitor (PAI-1) | activation | intermediate | − | 0 | 1 | |
| Nuclear matrix | nuclear matrix protein NRP/B (NRPB) | activation | early | + | 0 | 0 |
| Cytoskeleton | actin; α2 | activation | intermediate | − | 0 | 0 |
| keratin 5 | activation | intermediate | − | 0 | 0 | |
| keratin 17 | activation | late | − | 0 | 1 | |
| keratin 19 | activation | late | − | 1 | 1 | |
| Membrane protein | caveolin | activation | early | − | 0 | 0 |
| ANNEXIN XIII | activation | late | + | 1 | 2 | |
| Signal transduction | tyrosine-protein kinase receptor eck | activation | intermediate | − | 0 | 2 |
| Ral GDP dissociation stimulator | activation | intermediate | + | 0 | 0 | |
| interleukin-2 receptor β chain (IL-2Rb) | activation | late | + | 0 | 3 | |
| human DNA for rhoHP1, | activation | late | + | 0 | 1 | |
| HUMAN P2XM | activation | transient | + | 0 | 1 | |
| p21cdc42Hs kinase(ack) | activation | transient | + | 0 | 0 | |
| possible GTP-binding protein hsr1 | repression | early | + | 0 | 1 | |
| effector cell protease receptor-1 (EPR-1) | repression | late | − | 2 | 0 | |
| ADP-ribosylation factor-I (ARL2) | repression | late | + | 0 | 2 | |
| Myb proto-oncogene protein | repression | late | − | 1 | 0 | |
| ribosomal protein S6 kinase 2,90 KD | repression | transient | − | 1 | 0 | |
| InsP3 5-phosphatase | repression | transient | − | 0 | 0 | |
| CCAAT/enhancer binding prot. (C/EBPα) | repression | transient | − | 1 | 2 | |
| Growth factor | TGF-β superfamily protein | activation | early | + | 2 | 3 |
| inhibitor | IGFBP-3 | activation | late | + | 0 | 1 |
| transforming growth factor; α | activation | late | − | 0 | 3 | |
| BMP4 | activation | transient | + | 0 | 1 | |
| Angiogenesis | endothelin 2 | activation | intermediate | − | 0 | 3 |
| thrombospondin 1 | activation | transient | − | 0 | 0 |
Number of treated cell lines showed 2.5-fold or higher changes compared to control.
The first prominent group of genes was involved in cell cycle regulation. On p53 induction in EB-1 cells, many of the early responsive genes were cell cycle regulators, including p21/Waf1/Cip1, GADD45, and G0S8. G0S8 is involved in G0/G1 switch (Siderovski et al. 1994). A negative growth regulator MyD118, which is a GADD45 homolog (Liebermann and Hoffman 1998), and 14-3-3 protein ς, which aids in a G2 arrest after γ irradiation (Hermeking et al. 1997), were activated at a delayed time. Meanwhile, CDK2 and RbAp46, the latter of which is a Rb-binding protein (Guan et al. 1998), were down-regulated as early as 2 hr after zinc induction. Among the delayed repressed gene products (cluster 7), most were involved in cell cycle progression, such as a Cyclin B2 homolog and MAD2, or in DNA replication, such as topoisomerase II α and DNA primase.
The second group of genes activated appeared to play important roles in apoptosis and oxidative stress response, and these genes were activated at intermediate-to-late stages. For example, both Fas/APO-1 and KILLER/DR5 were induced around 4 hr following zinc stimulation. Activation of these death receptors by their cognate ligands triggers apoptosis in various cell types (Wu et al. 1997; Muller et al. 1998). In addition, p53 also activates genes regulating cellular responses to oxidative stress, such as Pig3 (Polyak et al. 1997) and neutrophil NADPH oxidase (Lomax et al. 1989). Meanwhile, superoxide dismutase 3, an important enzyme to eliminate reactive oxygen species from the cell, was immediately repressed following p53 induction.
Among the p53-regulated genes, many encode components of the extracellular matrix and their modifying enzymes, cytoskeleton, and nuclear matrix proteins. The responses vary in this group, for example, two collagen genes and plasminogen activator inhibitor (PAI-1), an inhibitor for the proteases that degrade extracellular matrix, were activated at early-to-intermediate time points, whereas cytoskeleton genes, such as actin and cytokeratins (which might terminally differentiate cells) were activated at a later stage. Interestingly, NRP/B, a nuclear matrix protein that interacts with the retinoblastoma protein (p110Rb) (Kim et al. 1998), was activated only 2 hr after p53 induction. Both caveolin and Annexin XIII are membrane proteins that are involved in vesicle trafficking within a cell (Fiedler et al. 1995; Parton 1996); yet these two genes were activated at very different stages (Table 2).
The fourth group includes genes that participate in the signal transduction pathways within a cell. They include cell surface receptors (Eck, P2XM, and EPR-1), intracellular effectors (Ack, Hsr1, Arl2), and DNA-binding proteins (C/EBPα). The kinetics of this group of genes exhibits a great deal of heterogeneity, indicating that several different signaling pathways might be activated in the same cell at different times. In particular, a group of small GTPases and their associated factors, such as human rhoHP1, Cdc42HS kinase Ack, and Ral, a GDP dissociation stimulator, were regulated by p53 (Table 2). These genes belong to the Ras and Rho family, members of which regulate diverse functions, including actin cytoskeletal organization, mitogen-activated protein kinase cascades, cell cycle progression, and even cellular transformation.
Two of the p53-regulated genes were involved in angiogenesis, endothelin 2, and thrombospondin 1, the latter of which is a potent inhibitor of angiogenensis. Several growth factors and growth factor inhibitors, including IGFBP-3, TGF-α, and two TGF-β family members, were also activated by p53, suggesting p53 might regulate the cells adjacent to a p53-activated cell in the body.
Dose-dependent gene activation by p53
The level of p53 induction in EB-1 cells depends on the concentration of zinc chloride in the culture medium (Fig. 1B). This permitted an investigation of the effect of p53 levels on gene transcription. Zinc (50 μm) could induce p53 protein to a level that was comparable with that of endogenous p53 induced by γ irradiation in another colon carcinoma cell line HCT116 (Fig. 1B,C; data not shown), whereas 100 μm zinc induced p53 protein to a much higher concentration (Fig. 1B). The expression profiles of the 69 genes were compared in cells stimulated with either 50 or 100 μm zinc chloride. A total of 24 genes were activated and 15 genes were repressed in both 50 and 100 μm zinc-induced cells. Some sample expression profiles are shown in Figure 6A, and the complete list is available at our web site. Most of the temporal patterns of these genes were similar between the two measurements, although a few genes, such as GADD45, SOD3, and CDK2, showed slight delays in their response to 50 μm zinc. Among the genes that were not responsive to 50 μm zinc induction, but were induced at 100 μm zinc chloride, some of them have been shown to contain p53-binding sites, such as Pig3 (Fig. 6B). The different responses among p53-regulated genes to low-level p53 could reflect the difference in p53 protein's affinity for these promoters. It is also possible that additional cellular events occurred in response to different stimuli and modified p53's activity or altered proteins that cooperate with p53 in its transcriptional regulation.
Figure 6.

Gene regulation by different levels of p53. (A) Genes in this group were responsive to p53 at both high and low levels. (B) Genes in this group only responded to high level of p53. Selected gene expression profiles from each group are shown for both 100 and 50 μm zinc-induced cells.
Gene expression following radiation in different cancer cell lines
Irradiation of a cell causes DNA damage and induces growth arrest in certain cells or apoptosis in others. The cellular response to radiation is quite complicated and involves coordinated activities from many genes, some of which are p53-mediated. The gene expression patterns in cells after UV and γ irradiation were examined using the p53-spotted oligonucleotide arrays. Cell lines selected for this experiment included a breast cancer cell line MCF-7, a teratocarcinoma cell line Tera-1, and a neuroblastoma cell line IMR32. All of these lines contain wild-type p53 and the level of steady-state p53 increased on UV or γ irradiation (Fig. 1C; data not shown). We also included two cell lines with null p53, a human colon tumor cell line EB and a human lung cancer cell line H1299. Sample RNAs were isolated from control cells and UV- or γ-irradiated cells, which were then labeled and hybridized to the p53 spotted oligonucleotide arrays. The expression level of each gene was represented by the ratio of its intensity in irradiated cells over that in the control.
To decipher the different gene expression patterns following UV and γ irradiation in different cell lines, a two-way cluster analysis of selected genes was performed. A total of 112 genes were selected for this analysis in four UV-treated cell lines and four γ-irradiated cell lines together with EB-1 cells at four different time points after zinc-mediated p53 induction. On the first dimension of this analysis we found that four clusters were formed (Fig. 7): γ-irradiated cells with wild-type p53, UV-treated cells with wild-type p53, EB-1 cells following p53 induction, and cells with null p53. The cellular transcriptional patterns following γ treatment were very similar to each other in cells with wild-type p53, and this was also the case for the UV-radiated cells. The gene expression patterns in the p53 null cell lines were distinct from those of the cell lines with wild-type p53. Although UV and γ radiation induced changes in certain gene expressions in p53 null cell lines, these changes were different from those of cells with wild-type p53. Thus the overall transcription pattern of a cell depends on the nature of radiation and p53 status in the cell.
Figure 7.
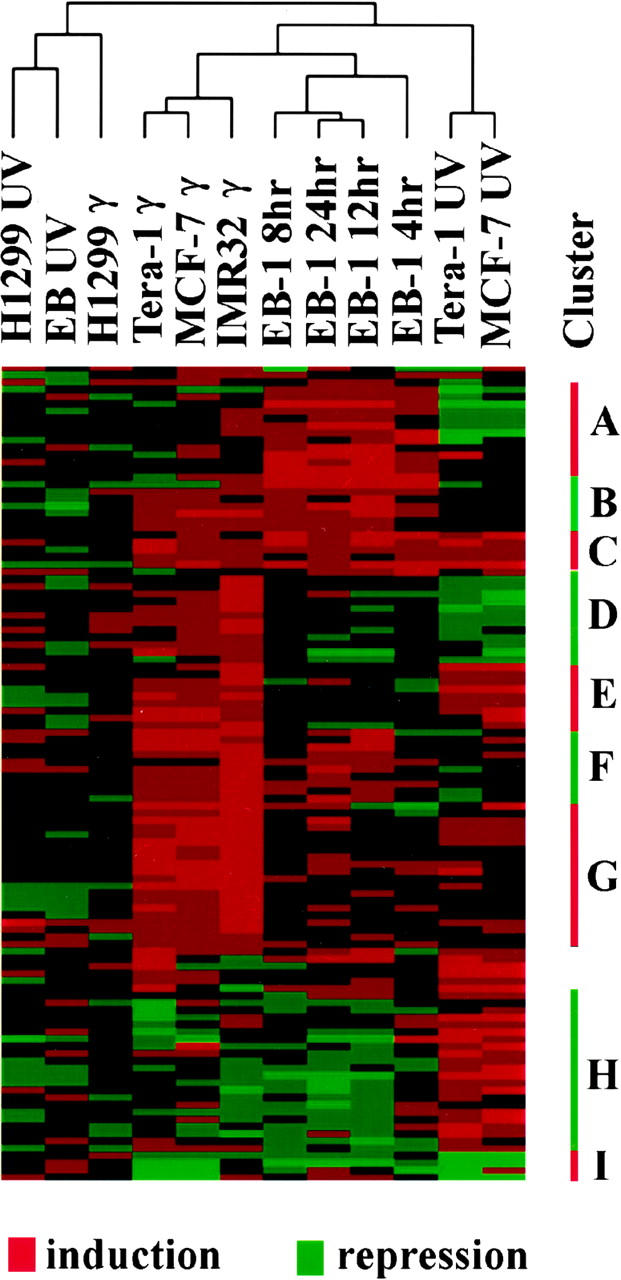
Cluster analysis of gene expression patterns after UV or γ irradiation. A total of 112 genes and 5 different cell lines following indicated radiation were included in this analysis. This analysis was performed in two dimensions, with one for cell lines and radiation, the other one for 112 genes. Cells with wild-type p53 fall into distinct clusters following different irradiation. The 112 genes were clustered into 9 groups with different expression patterns in cells with wild-type p53 following different types of radiation.
On the second dimension, the 112 genes included in this analysis fall into 9 clusters in cells with wild-type p53 (Fig. 7). A partial summary is shown in Table 2, and the complete list of genes in each cluster is available on our web site. Only a small number of genes were induced in several cell types following UV radiation, γ radiation, and zinc-induced p53 activation (cluster C), including p21/Waf1/Cip1, GADD45, Bak, and a TGF-β family member. Similar results were seen for repressed genes following all three treatments (cluster I), which included Lamin B receptor, superoxide dismutase 3, and C/EBP α. Many genes activated by p53 in EB-1 cells were not detected in either UV- or γ-irradiated cells. These genes were grouped in cluster A, including G0S8, Cytokeratin 5, and Ral GDP dissociation stimulator. On the other hand, quite a few genes that were not detected in EB-1 cells were activated in either UV-irradiated cells (cluster H), or γ-treated cells (clusters D and G), or both (cluster E). This result suggested that activation of these genes by a specific type of radiation likely requires other cellular factors in addition to p53. Furthermore, it was known that p53 protein itself was differentially modified following different types of DNA damage, and such modifications may lead to activation of different downstream genes (Banin et al. 1998; Canman et al. 1998; Khanna et al. 1998; Lu et al. 1998). Most of these changes in gene expressions were not observed in p53 null cell lines. This indicates that most of the gene regulations observed in this study were very likely to be p53 dependent.
Although the cellular transcription profiles of γ- or UV-treated cells were similar to each other, for each individual gene, the response was quite heterogeneous. As shown in Table 2, many genes were only regulated in some γ- or UV-irradiated cells, but not in others. This heterogeneity suggested cell line- or cell type-specific p53 responses.
Discussion
It is now clear that the p53 protein integrates a large number of signals in a cell from various classes of DNA damage, levels of ribonucleoside triphosphate pools, the presence of activated oncogenes, etc. (Levine 1997; Prives and Hall 1999). The p53 protein is kept at low levels via its short half-life, but in response to cellular stress the half-life of p53 increases, p53 protein levels rise, and the protein is activated for transcription probably via phosphorylation, acetylation, or other modifications. Different types of DNA damage activate distinct protein kinases (e.g., γ radiation activates ATM/ATR), which phosphorylate distinct serine or threonine residues on the p53 protein (Banin et al. 1998; Canman et al. 1998; Khanna et al. 1998). Thus, it is possible that the p53 transcriptional response will depend on the nature of the DNA damage. The experiments presented here provide three experimental situations: (1) increased p53 levels and no DNA damage (zinc inducible); (2) UV damage; and (3) γ irradiation. Interestingly, the genes that are transcribed in a p53-dependent manner differ markedly between these three situations, having a small subset in common. Thus, the results presented here support the model where distinct stressful situations in a cell are reported to p53 in a distinct fashion via protein modifications and the responses are different. This is clearly the case for p53 sensing an activated oncogene. Myc or E2F-1 activation increases the levels of p19ARF, which in turn inhibits MDM-2 nuclear cytoplasmic shuttling and increases the half-life of p53 (de Stanchina et al. 1998; Zindy et al. 1998; Tao and Levine 1999; Zhang and Xiong 1999). γ-Radiation (mediated by ATM/ATR protein kinase) and apurinic acid sites (mediated by Ref-1) represent distinct pathways to p53 activation (Jayaraman et al. 1997; Canman et al. 1998).
Another explanation for the heterogeneous responses of gene transcription to p53 activation by diverse agents could be that p53 protein levels in a cell are different when diverse stress signals are used. Indeed, experiments presented here demonstrate different p53 levels in different cells in response to diverse signals (Fig. 1), and different levels of p53 in a cell can regulate some genes but not others (Fig. 6). The p53 DNA responsive element is quite degenerate in its sequence requirements and the binding constant of the p53 to different elements could play a role here. If things were as simple as this, one might expect that genes responsive to 50 μm zinc chloride (i.e., with high binding constants) would form the same set of genes that produce p53-dependent mRNAs very rapidly after p53 induction (Fig. 4, cluster 2). That is not the case (Table 2), and so it seems likely that the great diversity in the kinetics of p53-responsive genes (Fig. 4; Table 1) is likely regulated by additional factors (i.e., mRNA half-life, transport of mRNA, etc.). In addition, it is not clear whether these genes are direct transcriptional targets of p53, even though the regulations were observed following p53 activation. It is likely that some of the late regulated genes may be indirect targets of p53.
This is the first paper to demonstrate a clear set of p53-repressed genes under physiological conditions (i.e., without DNA transfection). There were a number of cytoskeletal functions in the category of repressed genes. The Map-4 gene has previously been identified as a p53-repressed gene, and it may play a role in the phenotype of p53 induction (Murphy et al. 1996). A number of experiments have indicated that p53-mediated repression of some genes could lead to or contribute to apoptosis.
This manuscript confirms a number of p53-responsive genes and extends the list. About 1%–2% of the genes tested were p53 inducible and 1% was p53 repressible. Many of these genes have been shown to be involved in apoptosis (Fas/APO-1, Killer/DR5), or cell cycle arrest (p21/Waf1/Cip1, GADD45, G0S8, 14-3-3ς, RbAp46, MAD2, etc.). These are expected p53 responses. Equally interesting, however, is the growing list of proteins made by the cell in response to p53 induction, which are secreted growth factors and inhibitors, extracellular matrix proteins, or proteins regulating angiogenesis (see Table 2). These proteins suggest a role for p53 in influencing adjacent cells in a tumor or in altering metastatic potential of tumor cells. Several of the proteins regulated by p53 deal with oxidative stress as first shown by Vogelstein and his colleagues (Polyak et al. 1997), whereas a wide variety of signal transduction functions are also represented in the p53-mediated response (Table 2).
What is clear and impressive about the collection of p53-responsive gene expression patterns reported here is their heterogeneity, depending on p53 protein levels in a cell, the type of stress employed to induce p53, the cell type or cell line employed, and the kinetics with which p53-responsive genes are observed. It will clearly take additional time and systematic studies to elucidate a complete catalog of these genes expressed in each experimental situation. This heterogeneity might well explain the differences between responses to chemotherapy or radiation by tumor cell or tissue types. The role of p53 in both the origins of tumors and the response to chemotherapy indicates that these types of studies will surely be useful in developing a more rational approach to cancer treatments.
Materials and methods
Cell culture
The human colon carcinoma cell line EB-1, containing a stably transfected p53 transgene under the transcriptional control of the metallothionein promoter, and its parental cell line EB were described previously (Shaw et al. 1992). These cells were maintained in RPMI media with 10% FBS. For EB-1 cells this media was supplemented with 0.5 gram/liter geneticin (GIBCO BRL). For p53 induction, EB-1 and control EB cells were cultured in the presence of 50, 100, or 200 μm zinc chloride for the time points indicated, and p53 induction was monitored by Western analysis using the monoclonal antibody 1801. The human breast carcinoma cell line MCF-7 was maintained in the RPMI media with 10% FBS. The lung cancer cell line H1299, colon carcinoma HCT116, teratocarcinoma Tera1, and neuroblastoma IMR32 were all maintained in DMEM with 10% FBS. Cells were treated with 20 J of UV, and RNA samples were isolated 6 hr after the irradiation. For γ irradiation, all cells were treated with 6 Gy of γ-ray, and RNAs were isolated 4 hr later, except for MCF-7, which was treated with 10 Gy of γ-ray and RNAs were isolated 3 hr later. High levels of p53 were induced at these time points in each cell line.
DNA microarray
Human GeneChip 6500
Oligonucleotides of defined sequence were synthesized onto glass using a light-directed, solid-phase combinatorial chemistry approach that has been described previously (Human GeneChip 6500, Affymetrix) (Lockhart et al. 1996; McGall et al. 1996; Wodicka et al. 1997). In addition to the cDNAs and ESTs of interest, each array contains oligomers designed to hybridize with control mRNA samples. These control features are used to assure uniform response across the four microarrays used in this experiment. For each experimental mRNA sample, the average intensity of all genes is adjusted to the same value to compensate for array-related differences in hybridization efficiency and signal intensity. In these experiments, the average intensity of all genes was set to 46 intensity units.
P53-array
The spotted oligonucleotide arrays contain oligonucleotides for the interrogation of 400 genes. These 400 genes include 161 from our initial screening, 159 reported p53 targets, together with 80 housekeeping genes as control. Three oligomers of 50 nucleotides were used to represent each gene based on the sequence reliability and uniqueness. For hybridization, total RNA was isolated from cells and reverse transcribed using a primer containing oligo(dT) and a T7 promoter. The resulting cDNAs were then in vitro transcribed using Megascript kit (Ambion), followed by reverse transcription using random hexamers in the presence of Cy3– or Cy5–dUTP (Amersham). The labeled targets were hybridized to the p53-spotted oligonucleotide arrays, and the fluorescent intensity at each element was read by a laser scanner (ScanArray 3000, General Scanning). Some sense oligonucleotides were printed on the arrays to control for background, and the average intensity from these sense oligonucleotides on each array was subtracted from the integrated intensity of each element on the array. The resulting intensity for each element was then normalized across different arrays by adjusting the average intensity of all genes to 100 units.
Data analysis
For data from Human GeneChip 6500, variation was appreciable at intensities of ≤10. Therefore, these low values were judged to contain a significant component of noise and were transformed to 10. The ratio of gene intensity in EB-1 cells to that in EB was used to represent the p53-mediated induction, and the reciprocal ratio was employed as p53-mediated repression. Only those genes with threefold induction or repression in the presence of zinc and an induced intensity of ≥100 were selected in developing the list of p53-responsive genes, because large experimental variations were observed for genes with intensity <100 or expression ratio <3.
For data from p53 arrays, the ratio of intensity in EB-1 cells to that in EB cells was used to represent the expression level of each gene. For the cluster analysis of temporal patterns, only those with expression level in EB-1 cells deviated from that in EB cells by at least a factor of 2.5 in at least two time points were selected for the analysis. For the cluster analysis of gene expression following radiation, 112 genes were selected based on the criteria that levels of expression changed by at least 2.5-fold in at least four samples. The relative intensity for each gene selected was log2 transformed before input into the cluster program.
RT–PCR
Total RNA (2 μg) was reverse transcribed from oligo-dT primers, and the resulting cDNA was amplified by PCR using gene-specific primers. Signal intensities were monitored by densitometric analysis using NIH Image software. Each time course was normalized against GAPDH and transformed into a ratio of the level in EB-1 cells to that in controls. The data was plotted after log2 transformation.
Statistical Analysis
Statistical significance was examined with the χ2 test with the null hypothesis rejected at P < 0.02, employing Statistica software (Statsoft, Tulsa).
Acknowledgments
The colon carcinoma cell lines EB-1 and EB were a kind gift from Dr. N. Kley. The GENE CLUSTER program was kindly provided by Dr. M.B. Eisen. We thank Ghassan Ghandour and Uri Alon for data analysis help. We acknowledge with appreciation a grant from the National Cancer Institute (POl-CA41086).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL alevine@rockvax.rockefeller.edu; FAX (212) 327-8900.
References
- Alon U, Barkai N, Notterman DA, Gish K, Ybarra S, Mack D, Levine AJ. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc Natl Acad Sci. 1999;96:6745–6750. doi: 10.1073/pnas.96.12.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SJ, Markowitz S, Fearon ER, Willson JK, Vogelstein B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science. 1990;249:912–915. doi: 10.1126/science.2144057. [DOI] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Bargonetti J, Manfredi JJ, Chen X, Marshak DR, Prives C. A proteolytic fragment from the central region of p53 has marked sequence-specific DNA-binding activity when generated from wild-type but not from oncogenic mutant p53 protein. Genes & Dev. 1993;7:2565–2574. doi: 10.1101/gad.7.12b.2565. [DOI] [PubMed] [Google Scholar]
- Bennett WP, Hollstein MC, Hsu IC, Sidransky D, Lane DP, Vogelstein B, Harris CC. Mutational spectra and immunohistochemical analyses of p53 in human cancers. Chest. 1992;101:19S–20S. doi: 10.1378/chest.101.3_supplement.19s. [DOI] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- de Stanchina E, McCurrach ME, Zindy F, Shieh SY, Ferbeyre G, Samuelson AV, Prives C, Roussel MF, Sherr CJ, Lowe SW. E1A signaling to p53 involves the p19(ARF) tumor suppressor. Genes & Dev. 1998;12:2434–2442. doi: 10.1101/gad.12.15.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Eliyahu D, Michalovitz D, Eliyahu S, Pinhasi-Kimhi O, Oren M. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc Natl Acad Sci. 1989;86:8763–8767. doi: 10.1073/pnas.86.22.8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer G, Bargonetti J, Zhu H, Friedman P, Prywes R, Prives C. Wild-type p53 activates transcription in vitro. Nature. 1992;358:83–86. doi: 10.1038/358083a0. [DOI] [PubMed] [Google Scholar]
- Fiedler K, Lafont F, Parton RG, Simons K. Annexin XIIIb: A novel epithelial specific annexin is implicated in vesicular traffic to the apical plasma membrane. J Cell Biol. 1995;128:1043–1053. doi: 10.1083/jcb.128.6.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S, Jang SK. Presence of a potent transcription activating sequence in the p53 protein. Science. 1990;249:1046–1049. doi: 10.1126/science.2144363. [DOI] [PubMed] [Google Scholar]
- Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57:1083–1093. doi: 10.1016/0092-8674(89)90045-7. [DOI] [PubMed] [Google Scholar]
- Guan LS, Rauchman M, Wang ZY. Induction of Rb-associated protein (RbAp46) by Wilms' tumor suppressor WT1 mediates growth inhibition. J Biol Chem. 1998;273:27047–27050. doi: 10.1074/jbc.273.42.27047. [DOI] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell. 1997;1:3–11. doi: 10.1016/s1097-2765(00)80002-7. [DOI] [PubMed] [Google Scholar]
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- Iwabuchi K, Li B, Bartel P, Fields S. Use of the two-hybrid system to identify the domain of p53 involved in oligomerization. Oncogene. 1993;8:1693–1696. [PubMed] [Google Scholar]
- Iyer VR, Eisen MB, Ross DT, Schuler G, Moore T, Lee JCF, Trent JM, Staudt LM, Hudson J, Jr, Boguski MS, Lashkari D, Shalon D, Botstein D, Brown PO. The transcriptional program in the response of human fibroblasts to serum. Science. 1999;283:83–87. doi: 10.1126/science.283.5398.83. [DOI] [PubMed] [Google Scholar]
- Jayaraman L, Murthy KG, Zhu C, Curran T, Xanthoudakis S, Prives C. Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes & Dev. 1997;11:558–570. doi: 10.1101/gad.11.5.558. [DOI] [PubMed] [Google Scholar]
- Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, Vogelstein B. Identification of p53 as a sequence-specific DNA-binding protein. Science. 1991;252:1708–1711. doi: 10.1126/science.2047879. [DOI] [PubMed] [Google Scholar]
- Khanna KK, Keating KE, Kozlov S, Scott S, Gatei M, Hobson K, Taya Y, Gabrielli B, Chan D, Lees-Miller SP, Lavin MF. ATM associates with and phosphorylates p53: Mapping the region of interaction. Nat Genet. 1998;20:398–400. doi: 10.1038/3882. [DOI] [PubMed] [Google Scholar]
- Kim TA, Lim J, Ota S, Raja S, Rogers R, Rivnay B, Avraham H, Avraham S. NRP/B, a novel nuclear matrix protein, associates with p110(RB) and is involved in neuronal differentiation. J Cell Biol. 1998;141:553–566. doi: 10.1083/jcb.141.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Liebermann DA, Hoffman B. MyD genes in negative growth control. Oncogene. 1998;17:3319–3329. doi: 10.1038/sj.onc.1202574. [DOI] [PubMed] [Google Scholar]
- Lin J, Chen J, Elenbaas B, Levine AJ. Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes & Dev. 1994;8:1235–1246. doi: 10.1101/gad.8.10.1235. [DOI] [PubMed] [Google Scholar]
- Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- Lomax KJ, Leto TL, Nunoi H, Gallin JI, Malech HL. Recombinant 47-kilodalton cytosol factor restores NADPH oxidase in chronic granulomatous disease. Science. 1989;245:409–412. doi: 10.1126/science.2547247. [DOI] [PubMed] [Google Scholar]
- Lozano G, Liu G. Mouse models dissect the role of p53 in cancer and development. Semin Cancer Biol. 1998;8:337–344. doi: 10.1006/scbi.1998.0096. [DOI] [PubMed] [Google Scholar]
- Lu H, Taya Y, Ikeda M, Levine AJ. Ultraviolet radiation, but not gamma radiation or etoposide-induced DNA damage, results in the phosphorylation of the murine p53 protein at serine-389. Proc Natl Acad Sci. 1998;95:6399–6402. doi: 10.1073/pnas.95.11.6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack DH, Vartikar J, Pipas JM, Laimins LA. Specific repression of TATA-mediated but not initiator-mediated transcription by wild-type p53. Nature. 1993;363:281–283. doi: 10.1038/363281a0. [DOI] [PubMed] [Google Scholar]
- Malkin D, Li FP, Strong LC, Fraumeni JF, Jr, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- McGall G, Labadie J, Brock P, Wallraff G, Nguyen T, Hinsberg W. Light-directed synthesis of high-density oligonucleotide arrays using semiconductor photoresists. Proc Natl Acad Sci. 1996;93:13555–13560. doi: 10.1073/pnas.93.24.13555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalovitz D, Halevy O, Oren M. Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant of p53. Cell. 1990;62:671–680. doi: 10.1016/0092-8674(90)90113-s. [DOI] [PubMed] [Google Scholar]
- Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M, Krammer PH. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med. 1998;188:2033–2045. doi: 10.1084/jem.188.11.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M, Hinman A, Levine AJ. Wild-type p53 negatively regulates the expression of a microtubule-associated protein. Genes & Dev. 1996;10:2971–2980. doi: 10.1101/gad.10.23.2971. [DOI] [PubMed] [Google Scholar]
- Parton RG. Caveolae and caveolins. Curr Opin Cell Biol. 1996;8:542–548. doi: 10.1016/s0955-0674(96)80033-0. [DOI] [PubMed] [Google Scholar]
- Pavletich NP, Chambers KA, Pabo CO. The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes & Dev. 1993;7:2556–2564. doi: 10.1101/gad.7.12b.2556. [DOI] [PubMed] [Google Scholar]
- Pietenpol JA, Tokino T, Thiagalingam S, el-Deiry WS, Kinzler KW, Vogelstein B. Sequence-specific transcriptional activation is essential for growth suppression by p53. Proc Natl Acad Sci. 1994;91:1998–2002. doi: 10.1073/pnas.91.6.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- Prives C, Hall PA. The p53 pathway. J Pathol. 1999;187:112–126. doi: 10.1002/(SICI)1096-9896(199901)187:1<112::AID-PATH250>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Raycroft L, Wu HY, Lozano G. Transcriptional activation by wild-type but not transforming mutants of the p53 anti-oncogene. Science. 1990;249:1049–1051. doi: 10.1126/science.2144364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Seto E, Usheva A, Zambetti GP, Momand J, Horikoshi N, Weinmann R, Levine AJ, Shenk T. Wild-type p53 binds to the TATA-binding protein and represses transcription. Proc Natl Acad Sci. 1992;89:12028–12032. doi: 10.1073/pnas.89.24.12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw P, Bovey R, Tardy S, Sahli R, Sordat B, Costa J. Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proc Natl Acad Sci. 1992;89:4495–4499. doi: 10.1073/pnas.89.10.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siderovski DP, Heximer SP, Forsdyke DR. A human gene encoding a putative basic helix-loop-helix phosphoprotein whose mRNA increases rapidly in cycloheximide-treated blood mononuclear cells. DNA Cell Biol. 1994;13:125–147. doi: 10.1089/dna.1994.13.125. [DOI] [PubMed] [Google Scholar]
- Tao W, Levine AJ. P19(ARF) stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc Natl Acad Sci. 1999;96:6937–6941. doi: 10.1073/pnas.96.12.6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger T, Nau MM, Segal S, Minna JD. p53: A transdominant regulator of transcription whose function is ablated by mutations occurring in human cancer. EMBO J. 1992;11:1383–1390. doi: 10.1002/j.1460-2075.1992.tb05183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Reed M, Wang Y, Mayr G, Stenger JE, Anderson ME, Schwedes JF, Tegtmeyer P. p53 domains: Structure, oligomerization, and transformation. Mol Cell Biol. 1994;14:5182–5191. doi: 10.1128/mcb.14.8.5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Reed M, Wang P, Stenger JE, Mayr G, Anderson ME, Schwedes JF, Tegtmeyer P. p53 domains: Identification and characterization of two autonomous DNA-binding regions. Genes & Dev. 1993;7:2575–2586. doi: 10.1101/gad.7.12b.2575. [DOI] [PubMed] [Google Scholar]
- Wodicka L, Dong H, Mittmann M, Ho MH, Lockhart DJ. Genome-wide expression monitoring in Saccharomyces cerevisiae. Nat Biotechnol. 1997;15:1359–1367. doi: 10.1038/nbt1297-1359. [DOI] [PubMed] [Google Scholar]
- Wu GS, Burns TF, McDonald ER, III, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Markowitz S, Wu G, el-Deiry WS. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet. 1997;17:141–143. doi: 10.1038/ng1097-141. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y. Mutations in human ARF exon 2 disrupt its nucleolar localization and impair its ability to block nuclear export of MDM2 and p53. Mol Cell. 1999;3:579–591. doi: 10.1016/s1097-2765(00)80351-2. [DOI] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes & Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]