

Abstract
Medulloblastomas are among the most common malignancies in childhood, and they are associated with substantial mortality and morbidity. The molecular pathogenesis as well as the ontogeny of these neoplasms is still poorly understood. We have generated a mouse model for medulloblastoma by Cre–LoxP-mediated inactivation of Rb and p53 tumor suppressor genes in the cerebellar external granular layer (EGL) cells. GFAP–Cre-mediated recombination was found both in astrocytes and in immature precursor cells of the EGL in the developing cerebellum. GFAP–Cre;RbLoxP/LoxP;p53−/− or LoxP/LoxP mice developed highly aggressive embryonal tumors of the cerebellum with typical features of medulloblastoma. These tumors were identified as early as 7 weeks of age on the outer surface of the molecular layer, corresponding to the location of the EGL cells during development. Our results demonstrate that loss of function of RB is essential for medulloblastoma development in the mouse and strongly support the hypothesis that medulloblastomas arise from multipotent precursor cells located in the EGL.
Keywords: Cre–LoxP system, medulloblastoma, Rb, GFAP, p53, astrocyte
Astrocytes are the most abundant non-neuronal cells in the central nervous system (CNS) and participate in a wide variety of physiologic and pathologic processes. In humans this cell type most frequently undergoes neoplastic transformation in the adult brain. Although some genetic events in the neoplastic transformation of astrocytes have been identified, relatively little is known about the pathways involved in the development and progression of glial tumors.
The glial fibrillary acidic protein (GFAP) gene encodes an intermediate filament cytoskeletal protein that is restricted primarily to astroglia in the adult CNS (Bignami et al. 1972). Mouse GFAP promoter sequences have been extensively characterized and used to express heterologous genes in transgenic mice (Toggas et al. 1994; Johnson et al. 1995). We reasoned that expressing the Cre recombinase of the Cre–LoxP system (for review, see Rajewsky et al. 1996) under control of this promoter would enable us to study the role of the tumor suppressor genes involved in the pathogenesis of gliomas.
There is evidence that loss of function of the tumor suppressor gene P53 is one of the initiator events in the neoplastic transformation of astrocytes. Families with germ line mutations of P53 have an incidence of CNS tumors of 13%, most of which (73%) are astrocytomas (Kleihues et al. 1997). The pattern of mutations found in sporadic and inherited brain tumors is similar (Fulci et al. 1998). p53 knockout mice have been generated and extensively characterized (Donehower et al. 1992; Jacks et al. 1994), but no increased incidence of glial tumors have been reported in the short life span of these highly tumor prone mice. Astrocytes derived from these p53 knockout mice, however, undergo spontaneous immortalization (Bogler et al. 1995; Yahanda et al. 1995).
The retinoblastoma gene was the first tumor suppressor gene to be cloned (Dryja et al. 1986; Friend et al. 1986; Lee et al. 1987a,b) and has been found mutated in a variety of human sporadic and hereditary tumors (for review, see Weinberg 1995). The retinoblastoma gene product, RB, is not only required for regulating entry into the cell cycle but also for terminal differentiation of different cell types, including CNS precursors (Lee et al. 1994). There is evidence from in vitro studies for the involvement of RB in differentiation and maturation of neurons and glia. Retinoic acid treated p19 embryonal carcinoma cells failed to differentiate into neurons and glia upon functional inactivation of RB by E1A (Slack et al. 1995). Moreover, loss of RB has been implicated in glial malignancies. A proportion of children with the inherited form of retinoblastoma will develop brain tumors later in life, and alterations of chromosome 13 have been reported in glioma cell lines as well as in about one-third of malignant gliomas (Hamel et al. 1993).
Although inactivation of the Rb gene in the mouse germ line leads to embryonic lethality at day 12.5 of gestation (Clarke et al. 1992; Jacks et al. 1992; Lee et al. 1992) as a result of major defects in the hematopoietic and nervous system, mice heterozygous for Rb or mice chimeric for wild-type and Rb-deficient cells develop pituitary tumors early in life (Maandag et al. 1994; Williams et al. 1994). Therefore, the consequences of lack of RB on glial differentiation and neoplastic transformation cannot be studied in these mice.
We have chosen to investigate the role of RB in the neoplastic transformation of astrocytes using the Cre–LoxP system. To assess a possible cooperation in tumorigenesis between RB and p53, we examined the effects of GFAP–Cre-mediated Rb inactivation in a p53-null background. Moreover, to overcome the limitations linked to the short life span of the p53 knockout mice, we used p53 conditional mutant mice, and we studied the selective inactivation of this tumor suppressor gene in astrocytes either alone or in combination with Rb.
We report here that RB is not required for the normal maturation of astrocytes and that the independent inactivation of p53 or RB is not sufficient to cause the neoplastic transformation of these cells in vivo. However, we found that expression of the GFAP–Cre transgene is not restricted to mature astrocytes. Precursor cells located in the external granular layer (EGL) of the cerebellum also express Cre recombinase driven from the GFAP promoter. Lack of Rb in these cells in combination with either a somatic or a germ-line p53 inactivation leads to medulloblastomas, highly aggressive embryonal tumors of the cerebellum. No glial tumors have been found in these mice.
Results
Generation and characterization of GFAP–Cre transgenic mice
Transgenic mice were generated in which Cre expression was under the control of the GFAP promoter (Johnson et al. 1995). Progeny resulting from the pronuclear injection of the GFAP–Cre construct (Fig. 1A) were screened for integration of the transgene. Transgenic lines were derived from all 12 founder mice (FVB inbred). Two lines, Tg2 and Tg6, expressing Cre mRNA in the brain (Fig. 1B) were selected for further analysis. To investigate the tissue specific expression and the functionality of the transgene, we crossed F1 GFAP–Cre Tg2 and Tg6 mice with transgenic LacZ indicator mice (ACZL and/or R26R) in which LacZ expression is only present in Cre-expressing cells and their descendants (Fig. 1C) (Akagi et al. 1997; Soriano 1999). PCR analysis on double transgenic animals showed brain specific recombination of the indicator transgene in both lines (Fig. 1D). Testis, intestine, and peripheral nerves also showed recombination, in agreement with previous reports on GFAP expression outside the CNS (McLendon and Bigner 1994). In situ enzymatic staining for β-galactosidase in combination with immunostaining for GFAP on brains of adult double transgenic mice revealed specific staining of distinct subpopulations of astrocytes. Numerous fibrillary astrocytes located in the white matter (Fig. 2A), protoplasmic astrocytes located in different areas of the cerebral (Fig. 2B) and cerebellar cortex, the Bergmann glia of the cerebellum, subpial astrocytes, and ependymal cells (Fig. 2C) were positive for both LacZ and GFAP. Surprisingly, not only astrocytes in the regions described above but also certain subpopulations of neurons [CA3 and CA4 portion of the neuronal ribbon of the hippocampus (Fig. 2D), the dentate gyrus, some neuronal subpopulations of the cortical molecular layer, and scattered cerebellar granular cells] were β-galactosidase positive albeit GFAP negative. Both Tg2 and Tg6 lines showed this staining pattern. Endogenous β-galactosidase activity was ruled out by the absence of staining in single transgenic littermates.
Figure 1.

Structure of GFAP–Cre transgene and characterization of the transgenic mice. (A) Partial structure of the GFAP–Cre transgene. (B) Northern blot analysis of Cre expression on poly(A)+ RNA from three independent transgenic lines. A 1.6-kb mRNA corresponding to the predicted transcript from the GFAP–Cre transgene was detected in Tg2 and Tg6 mice. Control hybridization with hGAPDH probe shows an equal loading of RNA in each lane. (C) Genomic structure of the transgene in the ACZL mice. Cre-mediated recombination leads to juxtaposition of the lacZ gene to the chicken β-actin promoter. (D) PCR analysis of recombination on GFAP–Cre;ACZL double transgenic mice. A 580-bp product for the recombined allele is present in brain, muscle (likely originating from peripheral myelinated nerve fibers), and intestine (enteric glia). The upper band represents the nonrecombined allele. The location of the primers AG2 and Z3 is illustrated in C.
Figure 2.
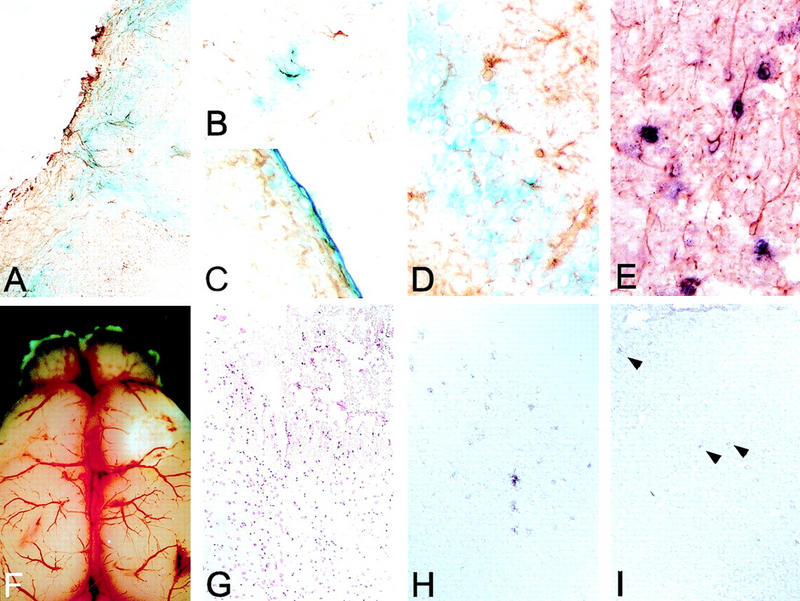
In situ analysis of recombination in GFAP–Cre;ACZL double transgenic mice. (Upper row) Astrocytes double positive for LacZ staining (blue reaction product) and GFAP (brown stain) located in the brain stem (A) and in the cerebral cortex (B); double positive ependymal cells lining the ventricular wall (C) and neurons positive for LacZ staining and negative for GFAP in the CA4 portion of the neuronal ribbon of the hippocampus (D); double labeling of Cre mRNA (black) and GFAP protein (brown) in fibrillary white matter astrocytes. (E). (Lower row) Brain specimen 24 hr after cryolesioning. Macroscopical appearance of the lesioned brain (F) and histological analysis of the lesioned area on frontal sections stained with H&E (G) are shown. (H) Up-regulation of Cre expression in reactive astrocytes located in the necrotic area and surrounding the lesion as demonstrated by Cre mRNA in situ hybridization. In contrast, there is only little basal Cre expression in the cerebral cortex of an unlesioned animal (I).
In situ hybridization analysis revealed a specific expression of the Cre recombinase in astrocytes (Fig. 2E), subependymal cells, and scattered cells located in the CA4 layer of the hipppocampus. This apparently ectopic expression might be explained by a transient Cre expression in multipotent precursor cells located in these regions (Palmer et al. 1997; Eriksson et al. 1998). Differentiation and maturation of these precursor cells would result in loss of Cre expression and sustained LacZ expression. To further assess the specificity of the Cre expression, we performed cryolesions to up-regulate GFAP-directed transgene expression (Johnson et al. 1995). In the mammalian CNS, GFAP expression is up-regulated in astrocytes reacting to pathologic processes. Cold lesions of the brain surface effectively induce astrocyte reaction and hence GFAP expression (Phillips et al. 1995). Cre in situ hybridization was performed on brain sections from GFAP–Cre mice, sacrificed 24 hr after cryolesioning (Fig. 2F,G). As shown in Figure 2H, reactive astrocytes in the vicinity of the lesion contained abundant Cre mRNA. Although the nontreated cortical gray matter contains only few Cre expressing astrocytes (Fig. 2I), their number was significantly increased after cryolesion. These findings suggest that Cre expression follows the specific expression behavior typical of the GFAP promoter.
Cell type-specific inactivation of Rb in the CNS
Mice carrying conditional Rb alleles were generated using an insertion type targeting vector in which two LoxP sequences were inserted into the introns surrounding exon 19 of Rb (Fig. 3A). Excision of the region flanked by the LoxP sites results in truncation of the RB protein that is functionally equivalent to a null allele (Vooijs et al. 1998; Vooijs and Berns 1999).
Figure 3.





Analysis of Rb and p53 recombination in different areas of the adult mouse brain. (A) Structure of the Rb floxed allele: LoxP sites were inserted into the introns surrounding exon 19. (B) A 260-bp PCR product representing the recombined Rb allele (primers Rb212 and Rb18) is present in a subset of cells in all analyzed areas of the brain. The 283-bp product represents the floxed allele (primers Rb19E and Rb18). The location of the primers used for the analysis (Rb212, Rb19E, and Rb18) is illustrated in A. Structure of the p53 floxed allele (C) and PCR analysis of recombination in different areas of the brain (D,E) are shown. The 584-bp band represents the floxed p53 allele (primers 10F and 10R), and the 612-bp band represents the recombined p53 allele (primers 1F and 10R). (M) Molecular marker; (T) tail; (OB) olfactory bulb; (Cx) cortex; (Hc) hyppocampus; (Ce) cerebellum; (Bs) brain stem.
We crossed GFAP–Cre Tg2 mice with homozygous RbLoxP/LoxP mice to obtain GFAP–Cre;RbLoxP/+ mice. These mice were subsequently bred to RbLoxP/LoxP mice to obtain GFAP–Cre;RbLoxP/LoxP mice. These mice were healthy and fertile and showed no sign of CNS disease.
A cohort of 22 GFAP–Cre;RbLoxP/LoxP mice was kept under observation for up to 1 year of age (Table 1), and the brains were examined histologically at different time points (two mice at 1, 3, 5, and 7 months, respectively, and 14 mice at 1 year of age). PCR analysis for Rb recombination (Fig. 3B) and in situ-hybridization for Cre mRNA (data not shown) were performed on selected brain specimens at various time points and confirmed the presence of astrocytes devoid of RB. Histological analysis did not reveal any evidence for dysplastic astrocytes or small neoplastic lesions.
Table 1.
Occurrence of medulloblastoma in compound mutant mice
| Genotype
|
Total no. of mice
|
Medulloblastomas in mice sacrificed at 8 months, 1 year, or when symptoms occurred
|
Medulloblastomas in nonsymptomatic mice sacrificed at earlier time point
|
|---|---|---|---|
| GFAP–Cre;RbLoxP/LoxP | 22 | 0/14a | 0/8 |
| GFAP–Cre;RbLoxP/LoxP;p53+/− | 13 | 0/13b | N.D. |
| GFAP–Cre;RbLoxP/+;p53−/− | 4 | 1/4c (106d) | N.D. |
| GFAP–Cre;RbLoxP/LoxP;p53−/− | 7 | 5/6c (76d–108d) | 1/1 |
| GFAP–Cre;RbLoxP/+;p53+/− | 15 | 0/15b | N.D. |
| GFAP–Cre;p53LoxP/LoxP | 11 | 0/10b | 0/1 |
| GFAP–Cre;RbLoxP/LoxP;p53LoxP/+ | 5 | 0/5b | N.D. |
| GFAP–Cre;RbLoxP/+;p53LoxP/Lox P | 12 | 6/8bc (87d–196d) | 0/4 |
| GFAP–Cre;RbLoxP/LoxP;p53LoxP/LoxP | 12 | 9/9c (86d–118d) | 1/3 |
| GFAP–Cre;RbLoxP/+;p53LoxP/+ | 15 | 0/15b | N.D. |
The total number of mice analyzed for each genotype and the occurrence of medulloblastomas at different time points is presented.
Mice sacrificed at 1 year of age.
Mice sacrificed at 8 months of age.
Mice sacrificed when symptoms occurred; time range in days is shown in parenthesis.
(N.D) Not determined.
Medulloblastomas in mice with a somatic Rb inactivation in a p53-null background
In humans, RB mutations have been found in high-grade astrocytic tumors (Hamel et al. 1993), and the loss of its function is considered to be involved in the progression from the benign form to the highly malignant astrocytoma (Kleihues et al. 1993). Therefore, the lack of an initiation step might be responsible for the lack of neoplastic transformation of Rb-null mutant astrocytes. As there is evidence that loss of p53 function can act as an initiator event in low-grade astrocytoma pathogenesis (von Deimling et al. 1992; Ohgaki et al. 1993), we asked whether there is cooperation between these two tumor suppressor genes in inducing neoplastic transformation of astrocytes. We therefore crossed GFAP–Cre;RbLoxP/LoxP mice with p53-deficient mice (Donehower et al. 1992). Thirteen mice with one intact copy of the p53 allele (GFAP–Cre;RbLoxP/LoxP;p53+/−) were kept under observation for up to 8 months (Table 1), when they had to be sacrificed because of the development of lymphomas and different types of sarcomas (Harvey et al. 1995). Histological analysis of the brains of these mice showed no evidence for cellular atypia or neoplastic lesions of glial origin.
Seven GFAP–Cre;RbLoxP/LoxP mice in a p53−/− background had to be sacrificed around 3–4 months of age (Table 1) because they developed similar tumors as mice with one inactivated p53 allele. Three GFAP–Cre;RbLoxP/LoxP;p53−/− mice, however, presented with symptoms of CNS disease. They circled, held their heads to one side, and showed tremor, gait disturbance with ataxia, and loss of balance. These three mice and the additional four mice without neurological symptoms (all of them between 3 and 4 months of age) were sacrificed, and their brains were analyzed histologically. In all but one case, we detected tumors in the cerebellum (Fig. 4A,B). These tumors consisted of densely packed, polygonal cells with scant cytoplasm and hyperchromatic nuclei (Fig. 4C). The tumor cells were arranged in sheets and rows and occasionally formed pseudorosette structures. There was high mitotic activity throughout the tumor. These hypercellular tumors were located in the vermis or close to the cerebellar peduncles and showed an invasive growth pattern with ill-defined margins. Multifocal occurrence of these tumors was occasionally noted, although in these cases a secondary spread of the tumor could not be ruled out. Localization and morphological appearance of these tumors showed in all instances striking similarity to medulloblastomas, malignant tumors arising in the cerebellum of children.
Figure 4.
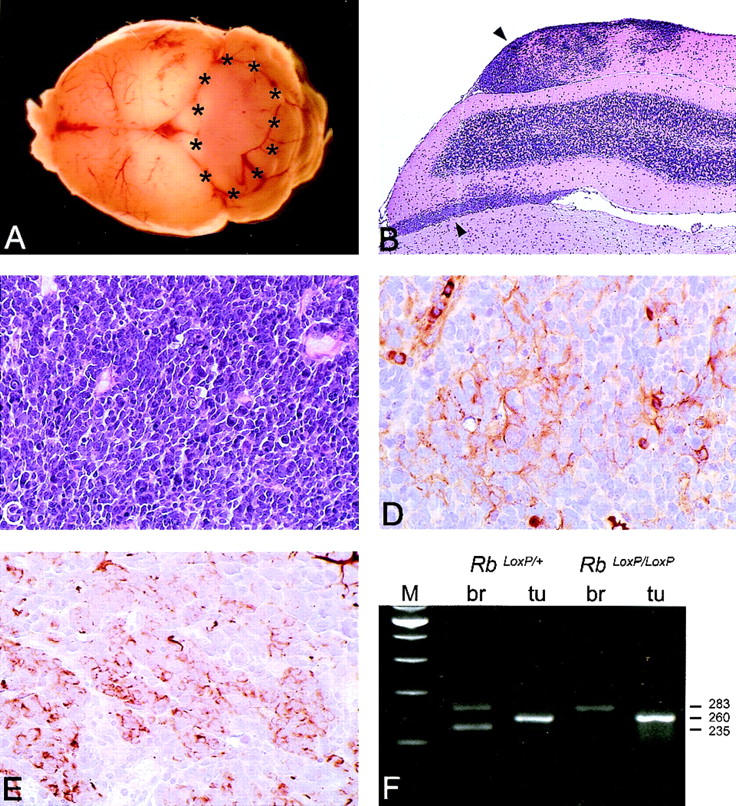
Histopathological analysis of medulloblastomas in GFAP–Cre;RbLoxP/LoxP;p53−/− mice. (A) Macroscopical appearance of a medulloblastoma arising in the cerebellar vermis of a 16-week-old mouse. (B) Low-power magnification of a tumor diffusely infiltrating in a cerebellar folia and in the subarachnoidal space (upper and lower arrows, respectively; H&E). (C) High-power magnification of a hypercellular tumor consisting of densely packed, polygonal cells with scant cytoplasm and hyperchromatic nuclei (H&E). Areas of early neuronal (MAP-2; D) and glial (GFAP; E) differentiation are detectable. (F) PCR analysis of Rb recombination in tumors arising in GFAP–Cre;RbLoxP/+;p53−/− and GFAP–Cre;RbLoxP/LoxP;p53−/− mice. The 283-, 260-, and 235-bp products correspond to the floxed (nonrecombined), the recombined, and the wild-type allele, respectively. The primers used for the analysis (Rb212, Rb19E, and Rb18) are the same as described in Fig. 3.
To find further support for their similarity to human medulloblastomas, we looked for the expression of glial and neuronal markers. Microtubule-associated protein (MAP-2), an early expressed neuronal antigen, was detected in the tumor cells (Fig. 4D), whereas other neuronal markers of advanced differentiation (neuron-specific enolase and synaptophysin) were not expressed (data not shown). GFAP expression was found in clusters of neoplastic cells, characterized by a thin rim of immunoreactivity around the nucleus (Fig. 4E). These cells were clearly discernible from reactive astrocytes with thin, elongated processes and a brighter, scattered nucleus. These findings indicate early neuronal and glial differentiation and suggest an origin from multipotential, uncommitted precursor cells.
To test whether the floxed Rb alleles were recombined in the tumors, we performed a PCR analysis on DNA extracted from tumor tissue and from adjacent normal CNS tissue. This analysis revealed that both floxed Rb alleles were recombined in the tumor tissue (Fig. 4F). In addition to mice with both floxed Rb alleles in the p53-null background, there was one mouse with only one floxed Rb allele in the p53-null background (GFAP–Cre;RbLoxP/+;p53−/−) that also showed signs of CNS disease. Autopsy and histological examination of the brain showed a medulloblastoma morphologically indistinguishable from those described above. PCR analysis performed on neoplastic and normal tissue of this mouse showed that the floxed Rb allele was recombined and the wild-type allele was lost (Fig. 4F).
Histological examination of other brain areas of these mice revealed hydrocephalus in some cases (as a result of obstruction of the fourth ventricle) but no sign of other malignancies.
No medulloblastomas and no glial malignancies in GFAP–Cre;p53LoxP/LoxP mice
Because the short lifespan of the p53 conventional knockout mice might have masked the development of medulloblastomas and precluded the development of other CNS tumors such as gliomas, we crossed GFAP–Cre mice with mice carrying conditional p53 alleles (Fig. 3C). These mice were generated by inserting LoxP sites in intron 1 and intron 10 of the p53 gene using a subsequent targeting strategy. Mice homozygous for the floxed allele were monitored for up to 1 year of age and did not show increased incidence of diseases. On the other hand, p53Δ2–10 mice, obtained after crossing with mice expressing Cre in the germ-line (Deleter–Cre; Schwenk et al. 1995) were showing the same tumor spectrum of p53 conventional knockout mice (J. Jonkers and A. Berns, in prep.).
GFAP–Cre;p53LoxP/LoxP mice were healthy and fertile and were kept under observation for up to 8 months of age. The presence of astrocytes lacking p53 was assessed on selected brain specimens with PCR-based analysis of recombination (Fig. 3D,E) and in situ hybridization for Cre mRNA (data not shown). The histological analysis on the brains of these animals reveals neither medulloblastomas nor malignancies of glial origin.
Conditional inactivation of both p53 and Rb leads to medulloblastomas
To ask whether GFAP–Cre-mediated somatic inactivation of both p53 and Rb would lead to medulloblastomas, we crossed p53LoxP/LoxP mice with GFAP–Cre;RbLoxP/LoxP to obtain GFAP–Cre;RbLoxP/+;p53LoxP/+ mice. These mice were subsequently crossbred to obtain GFAP–Cre;RbLoxP/LoxP;p53LoxP/LoxP mice.
Between 3 and 4 months of age all compound mutants (9/9) developed medulloblastomas with the same histopathologic features described previously.
Six out of 8 mice with one floxed Rb allele and two floxed p53 alleles (GFAP–Cre;RbLoxP/+;p53LoxP/LoxP), kept under observation for up to 8 months, developed medulloblastomas as well, although in some cases, after longer latency (Table 1). PCR analysis was performed on four out of six tumors in which a good separation between neoplastic and adjacent normal tissue was present. In three out of four tumors, the floxed Rb allele was recombined and the wild type allele was lost. In one case, the wild-type and the floxed alleles were still detected in addition to a strong recombination band, most likely this was due to contaminating non-neoplastic cells.
EGL precursor cells are the cells of origin of medulloblastomas
One GFAP–Cre;RbLoxP/LoxP;p53−/− mouse and three GFAP–Cre;RbLoxP/LoxP;p53LoxP/LoxP mice were sacrificed at 2 months of age without showing neurological disease, and the entire cerebella was analyzed by serial sections. In the GFAP–Cre;RbLoxP/LoxP;p53−/− cerebellum, we identified three early neoplastic lesions located on the outer surface of the molecular layer (Fig. 5a), where the EGL cells are located during late embryonal and early postnatal development. RNA in situ hybridization showed strong Cre expression in the neoplastic cells (Fig. 5b). Similar lesions were found in one out of three GFAP–Cre;RbLoxP/LoxP;p53LoxP/LoxP cerebella analyzed.
Figure 5.
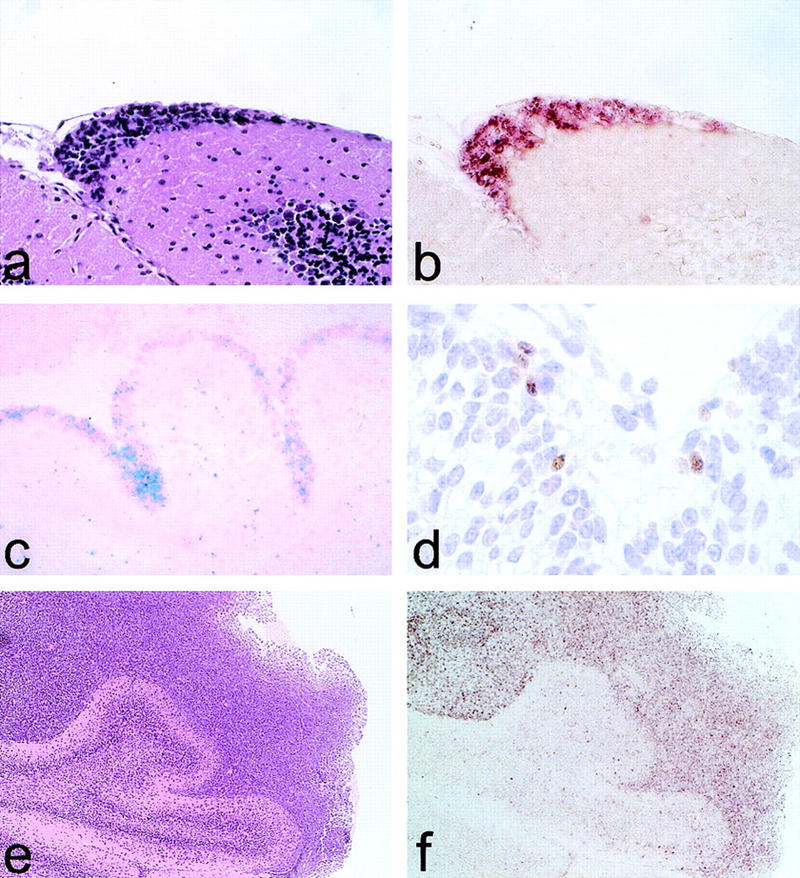
Analysis of the origin of medulloblastomas in GFAP–Cre;RbLoxP/LoxP;p53−/−. Early neoplastic lesions on the outer cerebellar surface of a 7-week-old mutant mouse (H&E; A) and in situ hybridization for Cre mRNA (B) are shown. LacZ-positive EGL cells in the cerebellum of a newborn GFAP–Cre mouse crossed to a LacZ indicator mouse show LacZ staining/Nuclear Red (C), and high-power magnification shows scattered Cre-expressing EGL precursor cells (D). Low-power magnification of a medulloblastoma surrounding cerebellar lamellae (H&E; E) and Math-1 expression in the tumor cells RNA in situ hybridization (F) are shown.
Newborn mice double transgenic for GFAP–Cre and a LacZ indicator construct (R26R) showed LacZ staining not only in the developing forebrain and in the spinal ganglia but also in the EGL cells of the cerebellum (Fig. 5c). Moreover, scattered precursor cells located in the EGL of the same animals are expressing Cre, as shown in Figure 5d. This finding suggests that precursor cells of the EGL activate the GFAP promoter and thus induce Cre expression and Cre-mediated recombination. An ectopic expression of the GFAP–Cre construct in the EGL precursor cells, however, cannot be excluded.
Math1 (Akazawa et al. 1995; Ben-Arie et al. 1996), the mouse homolog of the Drosophila gene atonal, encodes a basic helix–loop–helix transcription factor that is specifically expressed in the precursor cells of the EGL and is required in vivo for the genesis of granule cells (Ben-Arie et al. 1997).
We performed in situ hybridization for Math1 on selected cases of medulloblastomas arising in GFAP–Cre;RbLoxP/LoxP;p53−/− or LoxP/LoxP mice, and we observed strong Math1 expression (Fig. 5e,f) in all tumors analyzed. The mature granule cells located in the IGL as well as the subependymal matrix cells did not express Math1, in agreement with previous reports (Ben-Arie et al. 1996, 1997).
These results strongly support our conclusions that these tumors arise from the granule cell precursors retained on the cerebellar surface.
Discussion
GFAP, an intermediate filament protein, is expressed in differentiated and mature astrocytes in the mammalian brain (Eng 1985; Bignami et al. 1992). A detailed expression analysis of neuronal and glial markers during the histogenesis of the human cerebellar cortex, however, described GFAP expression in scattered multipotent precursor cells located in the ventricular zone of the cerebellar anlage at 8 weeks of gestation (Yachnis et al. 1993). Furthermore, a recent study identified ependymal cells, known to express GFAP, as multipotent neural stem cells of the adult mammalian CNS (Johansson et al. 1999).
We report here that GFAP-driven expression of Cre is detectable in newborn mice not only in the developing forebrain and in spinal ganglia but also in precursor cells located in the EGL of the developing cerebellum. Although expression of the Cre transgene was mainly restricted to astrocytes in the brain of adult mice, LacZ expression was also detectable in other cell types. This could simply be due to a stochastic expression of the GFAP–Cre construct independently of the integration site, because we find the same expression pattern in two independently generated transgenic lines. On the other hand, a transient GFAP expression in precursor cells resulting in Cre-mediated recombination of the floxed transgene is an intriguing alternative hypothesis. In ACZL mice this would lead to LacZ expression in the lineage of the recombined precursor cell. After commitment of these precursor cells to a neuronal lineage, there would be sustained LacZ expression albeit undetectable Cre expression.
GFAP expression has never been shown in the EGL of the developing cerebellum, although a low level of expression cannot be excluded. Delaney et al. (1996) generated transgenic mice expressing the thymidine kinase (tk) gene under the control of the GFAP promoter. Treatment of transgenic newborn mice with gancyclovir leads to a conversion of the drug into a toxic metabolite in cells expressing tk. These mice developed ataxia, and histological examination revealed a reduced size of the cerebellum and severe depletion of granule cells. This finding was explained by a deleterious effect on the radial glia (Bergmann glia) that normally guides external granule cells toward the internal granular layer. In the light of our experimental findings, however, this effect might be explained as a result of tk-mediated death of GFAP-expressing precursor cells of the EGL.
The developmental expression of GFAP-driven Cre recombinase in specific subpopulations of precursor cells renders GFAP–Cre mice particularly useful for analyzing the effects of tumor suppressor gene inactivation on the ontogeny of CNS tumors. In addition, GFAP–Cre mice may be instrumental in studying genes involved in the development and maturation of precursor cells or in the analysis of astrocyte-specific genes upon stimulation of mature astrocytes by pathologic stimuli.
Medulloblastoma, a malignant embryonal tumor of the cerebellum, accounts for ∼20% of primary brain tumors in children. Little is known about the molecular genetic events that contribute to the development of this tumor. Mutations of the P53 gene (Malkin et al. 1990; Cogen and McDonald 1996), amplification of c-MYC (Badiali et al. 1991) and N-MYC (Tomlinson et al. 1994), up-regulation of different PAX genes (Kozmik et al. 1995), and, more recently, lack of PATCHED (PTC) expression (Goodrich et al. 1997) have been implicated in the pathogenesis of these neoplasms. Mouse models for medulloblastomas are limited, a subset of mice heterozygous for ptc develop these tumors (Goodrich et al. 1997), and medulloblastomas have been induced in rats by transplantation of in vitro-transformed CNS precursor cells (Wiestler et al. 1992).
We describe here a new mouse model for medulloblastoma obtained by somatic inactivation of Rb in the EGL precursor cells of the developing cerebellum of p53-null mutant mice. The tumors observed in the cerebellum of GFAP–Cre;RbLoxP/LoxP;p53−/− or LoxP/LoxP mice share a number of characteristics with the human tumors. These include onset in young animals, location in corresponding areas of the cerebellum, highly aggressive behavior with typical local infiltration, and a mixed neuronal–glial differentiation pattern.
The ontogeny of medulloblastomas has been a controversial issue since the original description by Bailey and Cushing in 1925. Two hypotheses have been discussed most extensively: The first postulates that these tumors arise from the subependymal matrix cells of the fourth ventricle, which are precursor cells giving rise to neurons and glia in the adult CNS (Yachnis et al. 1994). An alternative hypothesis assumes an uncontrolled proliferation of precursor cells of the EGL (Vandenberg et al. 1987).
Because expression of Cre and recombination occur in scattered EGL cells before neoplastic transformation, we conclude that in our mouse model medulloblastomas arise from multipotent granule cell precursors retained on the cerebellar surface. The expression of Math1, a specific EGL cell marker, in the tumors provides an independent support to our conclusions.
While we observed medulloblastomas in GFAP–Cre;RbLoxP/LoxP;p53−/− or LoxP/LoxP mice as young as 7 weeks, we never found these tumors in GFAP–Cre;RbLoxP/LoxP or GFAP–Cre;p53LoxP/LoxP or in p53−/− mice. In line with the latter observation, only one medulloblastoma has been described in a series of 93 p53−/− mice (Donehower et al. 1992), and no such tumors have been described in another series of 49 animals (Jacks et al. 1994).
Interestingly, not only recombination of the floxed Rb allele but also loss of the wild-type allele was observed in medulloblastomas arising in GFAP–Cre;RbLoxP/+;p53−/− or LoxP/LoxP mice. Hence, loss of function of RB is essential for medulloblastoma development in the mouse. The fact that these tumors are not found in GFAP–Cre;RbLoxP/LoxP;p53+/− mice suggests that p53 deficiency may act as an initiator event in the ontogeny of these tumors. Alternatively, RB deficiency may stimulate excessive cell proliferation that, in normal conditions, might be counteracted by p53-induced apoptosis. Consequently, in a p53-null background, EGL precursor cells that have lost RB will fail to undergo apoptosis and ultimately give rise to neoplasms.
Although medulloblastomas represent 16% of brain tumors found in patients with germ line P53 mutations (Li-Fraumeni syndrome), loss of P53 is not commonly found in sporadic medulloblastomas. Additionally, RB has almost never been found mutated or lost in human medulloblastomas. Simultaneous mutations of both RB and P53 thus appear to be rare events in these tumors. The lack of RB mutations in human medulloblastomas could be explained by mutations involving other components of the RB pathway, although a study on a small series of 16 medulloblastomas failed to detect loss of p16 and/or Cyclin D and CDK4 amplification (Sato et al. 1996).
Alternatively, p53 and RB can be functionally inactivated and therefore contribute to the etiopathogenesis of medulloblastomas even if no mutations are found at the DNA level. SV40 Tag has been recently shown to be expressed (Huang et al. 1999) and to form specific complexes with p53 and RB in 33% of medulloblastoma cases (Zhen et al. 1999).
We never detected gliomas in GFAP–Cre;RbLoxP/LoxP mice kept under observation for up to 1 year of age. Our findings in GFAP–Cre;ACZL double transgenic mice clearly show recombination in astrocytes; moreover, we reproducibly detected astrocytes expressing Cre in different brain areas of GFAP–Cre;RbLoxP/LoxP adult mice.
GFAP–Cre;p53LoxP/LoxP mice, kept under observation for up to 8 months of age, never developed gliomas either. On the other hand, malignant gliomas arise between 1 and 4 months of age in 20% of GFAP–v-src transgenic mice (Weissenberger et al. 1997), and well differentiated neoplasms between 4 and 5 months of age in 9% of p19ARF−/− mice (Kamijo et al. 1999). Speculations on the possible causes of these differences between GFAP–Cre;p53LoxP/LoxP and p19ARF−/− mice are premature because the size of our cohort of mice is smaller and the genetic background different. A time course observation of p19ARF−/− mice in the same genetic background as that of the GFAP–Cre;p53LoxP/LoxP is currently under way.
We never observed glial malignancies in our p53/Rb combined mutants. These results might imply that mouse astrocytes are remarkably resistant to neoplastic transformation brought about by loss of p53 and RB. On the other hand, the shortened life span of the mice, which had to be sacrificed because of the development of medulloblastomas, might have precluded the study of late effects of p53 and RB combined deficiency in mature astrocytes.
Materials and methods
Construction of plasmids and generation of transgenic mice
The GFAP–Cre transgene was constructed from the GFAP–LacZ plasmid (Mucke et al. 1991). This construct contains not only a 2-kb fragment encompassing the GFAP promoter region but also positive and negative regulatory sequences located in the following introns (Miura et al. 1990).
The Cre open reading frame with an amino-terminal nuclear localization signal (Gu et al. 1993) was obtained as a NotI fragment after subcloning in a modified pBR vector. This fragment was cloned into the unique NotI cloning site of the GFAP construct. The final construct contained 2 kb of GFAP 5′-flanking sequences, 89 residues of GFAP exon 1, 0.2 kb of SV40 DNA with splice sites, a 1-kb Cre insert, and 0.2 kb of SV40 DNA with a poly(A) signal, followed by the rest of the GFAP gene. The construct was then released from the backbone, purified by agarose gel electrophoresis and electroelution, and injected into fertilized FVB/N mouse oocytes.
Screening of transgenic mice
Transgenic GFAP–Cre founder mice were identified by Southern blot analysis. DNA was extracted from tails according to standard protocols, digested with EcoRV and probed with a 1-kb Cre-containing fragment. After founders were established, genotyping was performed by PCR. Tail tip DNA was amplified with primers Cre1 (5′-ACCAGCCAGCTATCAACTC-3′) and Cre2 (5′-TATACGCGTGCTAGCGAAGATCTCCATCTTCCAGCAG-3′) yielding a 269-bp product. Thermocycling conditions consisted of 30 cycles of 30 sec at 94°C, 30 sec at 56°C, and 50 sec at 72°C. Reactions contained 200 ng of template DNA, 0.5 μm primers, 100 μm dNTPs, 9% glycerol, 2.5 units of Taq polymerase, 1.8 mm MgCl2, 1× PCR buffer (GIBCO BRL) in a 20-μl volume.
ACZL and R26R mice were screened by PCR using the same conditions described above and primers LZ1 (5′-CGTCACACTACGTCTGAACG-3′) and LZ2 (5′-CGACCAGATGATCACACTCG-3′). Tail DNA from RbLoxP mice and p53 mutant mice was amplified for 30 cycles (30 sec at 94°C, 30 sec at 58°C, and 50 sec at 72°C) using primers Rb18 (5′-GGCGTGTGCCATCAATG-3′) and Rb19 (5′-AACTCAAGGGAGACCTG-3′), p53a (5′-GCCTTCTATCGCCTTCTTGACGAGT-3′), p53b (5′-CGACCTCCGTTCTCTCTCCTCTCTT-3′), p53c (5′-AGACGCACAAACCAAAACAAAATTACA-3′) and p53–int10–fwd (5′-AAGGGGTATGAGGGACAAGG-3′), p53–int10–rev (5′-GAAGACAGAAAAGGGGAGGG-3′), respectively. The composition of the PCR mix was identical for all reactions.
Analysis of transgene transcription
Expression of the transgene was analyzed by Northern blotting. Total RNA was prepared from snap-frozen tissue using Trizol Reagent Total RNA isolation reagent (GIBCO BRL). The PolyATract System (Promega) was used to enrich for poly(A)+ RNA. Northern blot was performed according to standard protocols using 5 μg of poly(A)+ RNA. The probes used were a 1-kb CRE fragment and a 1.3-kb hGAPDH fragment.
PCR analysis of recombination
PCR analysis of Cre-mediated recombination in the ACZL mice was performed on genomic DNA extracted from different organs according to published protocols and followed by phenol–chloroform extraction. Thermocycling conditions consisted of 35 cycles of 30 sec at 94°C, 50 sec at 57°C, and 50 sec at 72°C yielding a 580-bp product for the recombined allele. The primers used were AG2 (5′-CTGCTAACCATGTTCATGCC-3′) and LZ3 (5′-GGCCTCTTCGCTATTACG-3′); the composition of the PCR mix was as described above.
Microdissection of tumors and of adjacent normal brain tissue was performed using a laser capture microdissection system (Arcturus). Ten-micrometer-thick sections from formalin-fixed paraffin-embedded material were used after dewaxing in xylene and drying. The dissected material was transferred to 50 μl of proteinase K buffer and incubated at 37°C overnight, after which the proteinase K was inactivated for 10 min at 96°C. Five microliters were used for subsequent PCR amplification using primers Rb212 (5′-GAAAGGAAAGTCAGGGACATTGGG-3′), Rb18 (5′-GGCGTGTGCCATCAATG-3′), and Rb19E (5′-CTCAAGAGCTCAGACTCATGG-3′) yielding a 283-bp product for the unrecombined RbLoxP allele, a 260-bp product for the recombined RbΔ19 allele, and a 235-bp fragment for the wild-type Rb allele (Vooijs et al. 1998). The PCR cycling profile consisted of 35 cycles of 30 sec at 94°C, 30 sec at 58°C, and 50 sec at 72°C. The PCR mix consisted of 0.5 μm primers, 100 μm dNTPs, 9% glycerol, 2.5 units of Taq polymerase, and 1× PCR buffer (GIBCO BRL) in a 20-μl volume. A hot start was performed using Invitrogen Hot-wax MgCl2 beads (1.5 mm). PCR products were analyzed on a 3% NuSieve agarose gel.
PCR analysis of Cre-mediated recombination in the p53LoxP/LoxP mice was performed on genomic DNA extracted from different areas of the brain and followed by phenol–chloroform extraction. Thermocycling conditions consisted of 30 cycles of 30 sec at 94°C, 30 sec at 58°C, and 50 sec at 72°C yielding a 612-bp product for the recombined allele. The primers used were p53–int1–fwd (5′-CACAAAAACAGGTTAAACCCA-3′) and p53–int10–rev (5′-GAAGACAGAAAAGGGGAGGG-3′); the composition of the PCR mix was as described above.
Cryolesion
Mice were anesthetized, their scalps excised, and the parietal region of the head was exposed. A metal rod, cooled in liquid nitrogen, was applied to the skull for 45 sec. The skin wound was closed by interrupted sutures. Animals were sacrificed 24 hr or 3 days after cryolesioning; brains were dissected out, sliced coronally, and fixed in 4% PFA–PBS or frozen in OCT.
RNA in situ hybridization
In situ hybridization for Cre RNA was performed on selected brain sections. A 0.8-kb NruI–XbaI fragment of the cre coding region was directionally cloned in an EcoRV–XbaI cut pSP72 vector. Digoxygenin-labeled antisense probe was synthesized from the BglII cleaved construct using the Sp6 polymerase. Math1 antisense and sense probes were synthesized from the XhoI and ERI respectively cleaved ICTd construct (a gift of H. Zoghbi, HHMI, Baylor College of Medicine, Houston, TX) using T7 and T3 polymerase. The RNA in situ hybridization on frozen and on formalin-fixed paraffin-embedded material was essentially performed as described (Marino et al. 1995).
For subsequent GFAP immunolabeling, sections were processed as described below. The sections were mounted in glycerol gelatin.
Detection of β-galactosidase activity
Embryos were dissected into cold PBS and kept on ice. When all the embryos were collected, they were changed to cold 4% PFA–PBS and rocked at 4°C for 1–4 hr depending on the age of the embryo. Adult brains were dissected out, cooled at 4°C for 5 min, and sliced coronally into 2-mm slices. The slices were fixed in 4% PFA–PBS at 4°C for 2 hr. The β-galactosidase staining was performed according to standard procedure.
Selected brains were frozen and embedded in OCT compound, in a mixture of dry ice and 2-methylbutane. They were then cut with a cryostat in 6- and 12-μm sections and stained according to standard procedures. For subsequent GFAP immunolabeling, sections were processed as described below. The sections were mounted in glycerol gelatin.
Histological analysis
Whole mouse brains were fixed for at least 24 hr in 4% paraformaldehyde in PBS. Coronal slices of ∼2-mm thickness were dehydrated through graded alcohols and embedded in paraffin. Sections of 4-μm nominal thickness were mounted on coated slides and routinely stained with hematoxilin and eosin (H&E).
Immunohistochemistry for GFAP (polyclonal, 1:500; DAKO), Synaptophysin (polyclonal, 1:50; DAKO), MAP-2 (monoclonal, 5 mg/ml; Boehringer Mannheim), NSE (monoclonal, 1:200; DAKO), and CRE (polyclonal, 1:14,000; Novagen) was performed on selected sections. Biotinylated secondary antibodies (goat anti-rabbit and rabbit anti-mouse; DAKO) were used at a dilution of 1:800. Visualization was achieved using biotin/avidin–peroxidase (DAKO) and diaminobenzidine as a chromogen.
Acknowledgments
We thank Karin van Veen and Frank Matthesius for assisting in the generation and genotyping of the mice, Nell Bosnie and Fina van der Ahe for the daily care of the mice, and Dennis Hoogervorst for the preparation of histological slides. L. Mucke and L. Donehower kindly provided the GFAP promoter construct and the p53 knockout mice, respectively. S.M. is a recipient of a Marie Curie Research Fellowship of the European Community (ERBFMBICT972620).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL tberns@nki.nl; FAX 31 20 5122011.
References
- Akagi K, Sandig V, Vooijs M, van der Valk M, Giovannini M, Strauss M, Berns A. Cre-mediated somatic site-specific recombination in mice. Nucleic Acids Res. 1997;25:1766–1773. doi: 10.1093/nar/25.9.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akazawa C, Ishibashi M, Shimizu C, Nakanishi S, Kageyama R. A mammalian helix-loop-helix factor structurally related to the product of Drosophila proneural gene atonal is a positive transcriptional regulator expressed in the developing nervous system. J Biol Chem. 1995;270:8730–8738. doi: 10.1074/jbc.270.15.8730. [DOI] [PubMed] [Google Scholar]
- Badiali M, Pession A, Basso G, Andreini L, Rigobello L, Galassi E, Giangaspero F. N-myc and c-myc oncogenes amplification in medulloblastomas. Evidence of particularly aggressive behavior of a tumor with c-myc amplification. Tumori. 1991;77:118–121. doi: 10.1177/030089169107700205. [DOI] [PubMed] [Google Scholar]
- Bailey P, Cushing H. Medulloblastoma cerebelli: Common type midcerebellar glioma of childhood. Arch Neurol Psych. 1925;14:192–223. [Google Scholar]
- Ben-Arie N, Bellen HJ, Armstrong DL, McCall AE, Gordadze PR, Guo Q, Matzuk MM, Zoghbi HY. Math1 is essential for genesis of cerebellar granule neurons. Nature. 1997;390:169–172. doi: 10.1038/36579. [DOI] [PubMed] [Google Scholar]
- Ben-Arie N, McCall AE, Berkman S, Eichele G, Bellen HJ, Zoghbi HY. Evolutionary conservation of sequence and expression of the bHLH protein Atonal suggests a conserved role in neurogenesis. Hum Mol Genet. 1996;5:1207–1216. doi: 10.1093/hmg/5.9.1207. [DOI] [PubMed] [Google Scholar]
- Bignami A, Eng LF, Dahl D, Uyeda CT. Localization of the glial fibrillary acidic protein in astrocytes by immunofluorescence. Brain Res. 1972;43:429–435. doi: 10.1016/0006-8993(72)90398-8. [DOI] [PubMed] [Google Scholar]
- Bignami A, Perides G, Asher R, Dahl D. The astrocyte–extracellular matrix complex in CNS myelinated tracts: A comparative study on the distribution of hyaluronate in rat, goldfish and lamprey. J Neurocytol. 1992;21:604–613. doi: 10.1007/BF01187120. [DOI] [PubMed] [Google Scholar]
- Bogler O, Huang HJ, Cavenee WK. Loss of wild-type p53 bestows a growth advantage on primary cortical astrocytes and facilitates their in vitro transformation. Cancer Res. 1995;55:2746–2751. [PubMed] [Google Scholar]
- Clarke AR, Maandag ER, van Roon M, van der Lugt NM, van der Valk M, Hooper ML, Berns A, te Riele H. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–330. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- Cogen PH, McDonald JD. Tumor suppressor genes and medulloblastoma. J Neurooncol. 1996;29:103–112. doi: 10.1007/BF00165523. [DOI] [PubMed] [Google Scholar]
- Delaney CL, Brenner M, Messing A. Conditional ablation of cerebellar astrocytes in postnatal transgenic mice. J Neurosci. 1996;16:6908–6918. doi: 10.1523/JNEUROSCI.16-21-06908.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CJ, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Dryja TP, Rapaport JM, Joyce JM, Petersen RA. Molecular detection of deletions involving band q14 of chromosome 13 in retinoblastomas. Proc Natl Acad Sci. 1986;83:7391–7394. doi: 10.1073/pnas.83.19.7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng LF. Glial fibrillary acidic protein (GFAP): The major protein of glial intermediate filaments in differentiated astrocytes. J Neuroimmunol. 1985;8:203–214. doi: 10.1016/s0165-5728(85)80063-1. [DOI] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork ET, Alborn AM, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus [see comments] Nat Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, Dryja TP. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643–646. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- Fulci G, Ishii N, Van ME. p53 and brain tumors: From gene mutations to gene therapy. Brain Pathol. 1998;8:599–613. doi: 10.1111/j.1750-3639.1998.tb00187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Gu H, Zou YR, Rajewsky K. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 1993;73:1155–1164. doi: 10.1016/0092-8674(93)90644-6. [DOI] [PubMed] [Google Scholar]
- Hamel W, Westphal M, Shepard HM. Loss in expression of the retinoblastoma gene product in human gliomas is associated with advanced disease. J Neurooncol. 1993;16:159–165. doi: 10.1007/BF01324703. [DOI] [PubMed] [Google Scholar]
- Harvey M, Vogel H, Lee EY, Bradley A, Donehower LA. Mice deficient in both p53 and Rb develop tumors primarily of endocrine origin. Cancer Res. 1995;55:1146–1151. [PubMed] [Google Scholar]
- Huang H, Reis R, Yonekawa Y, Lopes JM, Kleihues P, Ohgaki H. Identification in human brain tumors of DNA sequences specific for SV40 large T antigen. Brain Pathol. 1999;9:33–42. doi: 10.1111/j.1750-3639.1999.tb00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse [see comments] Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Johansson CB, Momma S, Clarke DL, Risling M, Lendahl U, Frisen J. Identification of a neural stem cell in the adult mammalian central nervous system. Cell. 1999;96:25–34. doi: 10.1016/s0092-8674(00)80956-3. [DOI] [PubMed] [Google Scholar]
- Johnson WB, Ruppe MD, Rockenstein EM, Price J, Sarthy VP, Verderber LC, Mucke L. Indicator expression directed by regulatory sequences of the glial fibrillary acidic protein (GFAP) gene: In vivo comparison of distinct GFAP-lacZ transgenes. Glia. 1995;13:174–184. doi: 10.1002/glia.440130304. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999;59:2217–2222. [PubMed] [Google Scholar]
- Kleihues P, Burger PC, Scheithauer BW. The new WHO classification of brain tumours. Brain Pathol. 1993;3:255–268. doi: 10.1111/j.1750-3639.1993.tb00752.x. [DOI] [PubMed] [Google Scholar]
- Kleihues P, Schauble B, zur Hausen A, Esteve J, Ohgaki H. Tumors associated with p53 germline mutations: A synopsis of 91 families. Am J Pathol. 1997;150:1–13. [PMC free article] [PubMed] [Google Scholar]
- Kozmik Z, Sure U, Ruedi D, Busslinger M, Aguzzi A. Deregulated expression of PAX5 in medulloblastoma. Proc Natl Acad Sci. 1995;92:5709–5713. doi: 10.1073/pnas.92.12.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis [see comments] Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- Lee EY, Hu N, Yuan SS, Cox LA, Bradley A, Lee WH, Herrup K. Dual roles of the retinoblastoma protein in cell cycle regulation and neuron differentiation. Genes & Dev. 1994;8:2008–2021. doi: 10.1101/gad.8.17.2008. [DOI] [PubMed] [Google Scholar]
- Lee WH, Bookstein R, Hong F, Young LJ, Shew JY, Lee EY. Human retinoblastoma susceptibility gene: Cloning, identification, and sequence. Science. 1987a;235:1394–1399. doi: 10.1126/science.3823889. [DOI] [PubMed] [Google Scholar]
- Lee WH, Shew JY, Hong FD, Sery TW, Donoso LA, Young LJ, Bookstein R, Lee EY. The retinoblastoma susceptibility gene encodes a nuclear phosphoprotein associated with DNA binding activity. Nature. 1987b;329:642–645. doi: 10.1038/329642a0. [DOI] [PubMed] [Google Scholar]
- Maandag EC, van der Valk M, Vlaar M, Feltkamp C, O'Brien J, van der Lugt N, Berns A, te Riele H. Developmental rescue of an embryonic-lethal mutation in the retinoblastoma gene in chimeric mice. EMBO J. 1994;13:4260–4268. doi: 10.1002/j.1460-2075.1994.tb06746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkin D, Li FP, Strong LC, Fraumeni JJ, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms [see comments] Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- Marino S, Kretschmer C, Brandner S, Cavard C, Zider A, Briand P, Isenmann S, Wagner EF, Aguzzi A. Activation of HIV transcription by human foamy virus in transgenic mice. Lab Invest. 1995;73:103–110. [PubMed] [Google Scholar]
- McLendon RE, Bigner DD. Immunohistochemistry of the glial fibrillary acidic protein: Basic and applied considerations. Brain Pathol. 1994;4:221–228. doi: 10.1111/j.1750-3639.1994.tb00837.x. [DOI] [PubMed] [Google Scholar]
- Miura M, Tamura T, Mikoshiba K. Cell-specific expression of the mouse glial fibrillary acidic protein gene: Identification of the cis- and trans-acting promoter elements for astrocyte-specific expression. J Neurochem. 1990;55:1180–1188. doi: 10.1111/j.1471-4159.1990.tb03123.x. [DOI] [PubMed] [Google Scholar]
- Mucke L, Oldstone MB, Morris JC, Nerenberg MI. Rapid activation of astrocyte-specific expression of GFAP-lacZ transgene by focal injury. New Biol. 1991;3:465–474. [PubMed] [Google Scholar]
- Ohgaki H, Eibl RH, Schwab M, Reichel MB, Mariani L, Gehring M, Petersen I, Holl T, Wiestler OD, Kleihues P. Mutations of the p53 tumor suppressor gene in neoplasms of the human nervous system. Mol Carcinog. 1993;8:74–80. doi: 10.1002/mc.2940080203. [DOI] [PubMed] [Google Scholar]
- Palmer TD, Takahashi J, Gage FH. The adult rat hippocampus contains primordial neural stem cells. Mol Cell Neurosci. 1997;8:389–404. doi: 10.1006/mcne.1996.0595. [DOI] [PubMed] [Google Scholar]
- Phillips MJ, Weller RO, Kida S, Iannotti F. Focal brain damage enhances experimental allergic encephalomyelitis in brain and spinal cord. Neuropathol Appl Neurobiol. 1995;21:189–200. doi: 10.1111/j.1365-2990.1995.tb01050.x. [DOI] [PubMed] [Google Scholar]
- Rajewsky K, Gu H, Kuhn R, Betz UA, Muller W, Roes J, Schwenk F. Conditional gene targeting. J Clin Invest. 1996;98:600–603. doi: 10.1172/JCI118828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Schauble B, Kleihues P, Ohgaki H. Infrequent alterations of the p15, p16, CDK4 and cyclin D1 genes in non-astrocytic human brain tumors. Int J Cancer. 1996;66:305–308. doi: 10.1002/(SICI)1097-0215(19960503)66:3<305::AID-IJC6>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack RS, Skerjanc IS, Lach B, Craig J, Jardine K, McBurney MW. Cells differentiating into neuroectoderm undergo apoptosis in the absence of functional retinoblastoma family proteins. J Cell Biol. 1995;129:779–788. doi: 10.1083/jcb.129.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice [see comments] Nature. 1994;367:188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- Tomlinson FH, Jenkins RB, Scheithauer BW, Keelan PA, Ritland S, Parisi JE, Cunningham J, Olsen KD. Aggressive medulloblastoma with high-level N-myc amplification. Mayo Clin Proc. 1994;69:359–365. doi: 10.1016/s0025-6196(12)62221-6. [DOI] [PubMed] [Google Scholar]
- Vandenberg SR, Herman MM, Rubinstein LJ. Embryonal central neuroepithelial tumors: Current concepts and future challenges. Cancer Metastasis Rev. 1987;5:343–365. doi: 10.1007/BF00055377. [DOI] [PubMed] [Google Scholar]
- von Deimling A, Eibl RH, Ohgaki H, Louis DN, von Ammon K, Petersen I, Kleihues P, Chung RY, Wiestler OD, Seizinger BR. p53 mutations are associated with 17p allelic loss in grade II and grade III astrocytoma. Cancer Res. 1992;52:2987–2990. [PubMed] [Google Scholar]
- Vooijs M, Berns A. Developmental defects and tumor predisposition in Rb mutant mice. Oncogene. 1999;18:5293–5303. doi: 10.1038/sj.onc.1202999. [DOI] [PubMed] [Google Scholar]
- Vooijs M, van der Valk M, te Riele H, Berns A. Flp-mediated tissue-specific inactivation of the retinoblastoma tumor suppressor gene in the mouse. Oncogene. 1998;17:1–12. doi: 10.1038/sj.onc.1202169. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Weissenberger J, Steinbach JP, Malin G, Spada S, Rulicke T, Aguzzi A. Development and malignant progression of astrocytomas in GFAP-v-src transgenic mice. Oncogene. 1997;14:2005–2013. doi: 10.1038/sj.onc.1201168. [DOI] [PubMed] [Google Scholar]
- Wiestler OD, Brustle O, Eibl RH, Radner H, Aguzzi A, Kleihues P. Retrovirus-mediated oncogene transfer into neural transplants. Brain Pathol. 1992;2:47–59. [PubMed] [Google Scholar]
- Williams BO, Schmitt EM, Remington L, Bronson RT, Albert DM, Weinberg RA, Jacks T. Extensive contribution of Rb-deficient cells to adult chimeric mice with limited histopathological consequences. EMBO J. 1994;13:4251–4259. doi: 10.1002/j.1460-2075.1994.tb06745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachnis AT, Rorke LB, Lee VM, Trojanowski JQ. Expression of neuronal and glial polypeptides during histogenesis of the human cerebellar cortex including observations on the dentate nucleus. J Comp Neurol. 1993;334:356–369. doi: 10.1002/cne.903340303. [DOI] [PubMed] [Google Scholar]
- Yachnis AT, Rorke LB, Trojanowski JQ. Cerebellar dysplasias in humans: Development and possible relationship to glial and primitive neuroectodermal tumors of the cerebellar vermis. J Neuropathol Exp Neurol. 1994;53:61–71. doi: 10.1097/00005072-199401000-00008. [DOI] [PubMed] [Google Scholar]
- Yahanda AM, Bruner JM, Donehower LA, Morrison RS. Astrocytes derived from p53-deficient mice provide a multistep in vitro model for development of malignant gliomas. Mol Cell Biol. 1995;15:4249–4259. doi: 10.1128/mcb.15.8.4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen HN, Zhang X, Bu XY, Zhang ZW, Huang WJ, Zhang P, Liang JW, Wang XL. Expression of the simian virus 40 large tumor antigen (Tag) and formation of tag-p53 and tag-pRb complexes in human brain tumors. Cancer. 1999;86:2124–2132. [PubMed] [Google Scholar]